

Abstract
Of the nearly 3 million Americans diagnosed with epilepsy, approximately 30% are unresponsive to current medications. Recent data has shown that early postnatal transplantation of interneuronal precursor cells increases GABAergic inhibition in the host brain and dramatically suppresses seizure activity in epileptic mice. In this review, we will highlight findings from seizure-prone mice and humans that demonstrate the link between dysfunctional GABAergic inhibition and hyperexcitability. In particular, we will focus on rodent models of temporal lobe epilepsy (TLE), the most common and difficult to treat form of the disease, and interneuronopathies, an emerging classification. A wealth of literature showing a causal link between reduced GABA-mediated inhibition and seizures has directed our efforts to recover the loss of inhibition via transplantation of interneuronal precursors. Numerous related studies have explored the anticonvulsant potential of cell grafts derived from a variety of brain regions, yet the mechanism underlying the effect of such heterogeneous cell transplants is unknown. In discussing our recent findings and placing them in context with what is known about epilepsy, and how related transplant approaches have progressed, we hope to initiate a frank discussion of the best path toward the translation of this approach to patients with intractable forms of epilepsy.
Keywords: epilepsy, seizures, transplantation, GABA, interneuron progenitor
Introduction
Surges of abnormal electrical discharge in the brain lead to the spontaneously occurring and recurrent seizures that define epilepsy. These seizures vary from staring spells, or absence seizures, lasting only a few seconds to full body convulsions, or tonic clonic seizures that can last for several minutes to hours. Given the toll of human suffering due to epilepsy, the need for more effective treatments is undeniable. At present, the most common treatment for these patients is the use of anti-seizure medications many of which act by globally increasing GABA-mediated inhibition (Porter, 1997). As with all medications, antiepileptic drugs can have unpleasant side effects that can severely affect a patient’s quality of life. Such side effects vary depending on the medication, but include nausea, weight loss or gain, impaired vision, drowsiness and dizziness. It is also a concern that exposure of the immature child’s brain to powerful anti-seizure medications, which work by inhibiting ubiquitously expressed GABA receptors or membrane-bound ion channels, will have irreversible consequences on cognitive development (Lagae, 2006; Mandelbaum et al., 2009). Each year hundreds of patients who are unresponsive to traditional noninvasive treatments opt to undergo surgery to remove a restricted region of the brain from which seizures are thought to originate. One of the most common procedures – a resection of the temporal lobe or “temporal lobectomy” which contains hippocampal structures - most commonly lead to depression and deficits in both verbal and non-verbal memory. In rare cases, patients who undergo unilateral or bilateral resections of hippocampal structures suffer amnesia (Polkey, 2009). With regard to postoperative seizure outcome, a large fraction of patients reported that their condition had improved as defined by a cessation or a worthwhile reduction in seizures and improved quality of life. However, more than 10% of surgical patients experienced no improvement in their condition or a worsening of seizures (Engel Jr., 2009). Although pharmaceutical and surgical interventions provide seizure relief in the majority of epileptic patients, thousands of patients continue to experience uncontrollable seizures or only partial seizure control. Children with more severe forms of pediatric epilepsy are particularly vulnerable to the long-term consequences of these intractable seizures. Such patients experience frequent seizures, in some cases several hundred per day, that can impair brain development and cognition (Lee, 1989; Hoie et al., 2006; Carreno et al., 2008).
To develop new treatments for epilepsy, we must understand how a brain becomes hyperexcitable, how this wave of abnormal excitation spreads and, how it can be stopped. Careful studies of both healthy and epileptic animals, and when possible patients, have yielded substantial insight into the many causes of epilepsy. The mechanisms currently thought to underlie epilepsy have been thoroughly and eloquently reviewed in comprehensive texts on the disease (Schwartzkroin, 1995; Engel Jr., 1997; Delgado-Escueta, 1999) and are only briefly summarized here. Epilepsy is caused by an increase in neuronal excitability due to alterations in ion channel function, inhibitory and/or excitatory synaptic transmission, neuronal circuitry, protein levels (e.g. neurosteroids, neuropeptides) and/or the expression of genes encoding for critical proteins (e.g. receptors, trophic factors). These changes generally can occur in response to a brain insult, developmental brain malformation and genetic mutation or follow an unknown origin (i.e., idiopathic).
While acknowledging that the causes of epilepsy are numerous, in this review we will focus on how a loss of inhibition and its subsequent recovery via cell transplantation can lead to and ameliorate seizures, respectively. We will review data from acquired and genetic models of epilepsy suggesting that a reduction in inhibition due to a dysfunctional inhibitory circuit, interneuron loss or compromised interneuron function invariably results in spontaneous seizures. Based on this evidence, we propose that the addition of “new” inhibitory interneurons (with properties mimicking the endogenous interneuron population) to the epileptic brain can have therapeutic effects in a wide variety of epilepsy disorders, and will discuss this and alternative cell-based approaches to suppressing excitability in rodent models of epilepsy.
Acquired models of epilepsy
Temporal lobe epilepsy (TLE) is the most common intractable form of epilepsy. To understand what causes TLE, the development and pathology of the disease have been extensively modeled in rodents (Cavalheiro, 2006; Dudek, 2006; Galanapoulou, 2006; Curia et al., 2008). In the commonly used pilocarpine or kainate models of TLE, a prolonged period of seizure activity, or status epilepticus, is pharmacologically induced in a wild-type rodent. Status epilepticus is followed by a latent period of few or no seizures. Within weeks of the acute seizure episode, surviving rodents begin to exhibit spontaneous recurrent seizures (Shibley & Smith, 2002; Echauz, 2009) that gradually increase in severity over time (Williams et al., 2009). The hippocampi of TLE rodents are characterized by increased adult neurogenesis in a germinal zone of the dentate gyrus (Parent et al., 1997), numerous changes in gene, protein and trophic factor expression (Mudo et al., 1996; Becker et al., 2003; Lund et al., 2008; Brooks-Kayal et al., 2009), mossy fiber sprouting (Buckmaster et al., 2002) and neuronal loss (Turski et al., 1984).
Neuronal loss and mossy fiber sprouting have also been observed in the resected hippocampi of TLE patients. Although patients can lose both excitatory (pyramidal cells in CA1 and CA3) and inhibitory (hilar interneurons) neurons there is a consistent reduction in cells in the GABAergic interneuron-rich hilar region of the dentate gyrus (de Lanerolle et al., 1989; Mathern et al., 1995; Spreafico et al., 1998; Wittner et al., 2001). These anatomical data are further supported by electrophysiological studies which have demonstrated a reduction in GABA-mediated synaptic transmission from granule cells of TLE patients (Williamson et al., 1999). TLE rodents have been used to determine how neuronal loss may affect function within the entorhinal cortex and dentate gyrus, regions which provide and receive, respectively, the majority of excitatory synaptic input to the hippocampus (Segal & Landis, 1974; Steward & Scoville, 1976; Schwartz & Coleman, 1981). In the pilocarpine TLE model, Layer II neurons within the entorhinal cortex exhibit a reduction in inhibitory synaptic transmission and recurrent inhibition, an increase in excitatory synaptic transmission, and increased action potential firing (Kobayashi et al., 2003; Kumar & Buckmaster, 2006; Kumar et al., 2007). Loss of GABA circuitry has also been reported in patients with cortical dysplasia (a refractory form of epilepsy common in children) and certain forms of traumatic brain injury associated with seizures (Spreafico et al., 2000; Golarai et al., 2001; Calcagnotto et al., 2005; Knopp et al., 2008). In addition to loss of synaptic GABAergic inhibition, the dentate gyrus exhibits reduced expression of postsynaptic GABAAR δ and α5 subunits that mediate tonic conductance (Houser & Esclapez, 2003; Peng et al., 2004; Glykys et al., 2008). Consistent with a hypothesized reduction in tonic inhibition, the dentate gyrus of epileptic brains is hyperexcitable and less sensitive to neurosteroids that act on δ subunit containing GABAARs (Peng et al., 2004).
Several recent studies suggest that the epileptic brain attempts to compensate for a lack of inhibition by increasing the inhibitory potential of surviving GABAergic interneuron populations. In a mouse model of TLE, surviving GABAergic interneurons in the dentate hilus grow larger, extend dendrites, sprout more axons and form new synapses (Zhang et al., 2009). Although there are fewer GABAergic interneurons in the epileptic brain, those that remain form more inhibitory synapses than in controls (Thind et al., 2009). This may be a homeostatic mechanism by which interneurons attempt to compensate for interneuron loss, increased excitation and decreased inhibition by forming abundant, albeit dysfunctional, inhibitory connections using the surviving interneurons. That the epileptic brain may have developed an endogenous program to increase inhibition is promising given that interneuronal precursor cell transplantation is based on the same rationale.
Taken together, these data provide valuable insight into how individual neurons contribute to abnormal excitation by receiving more excitatory and less inhibitory inputs, relaying more excitation, and forming new synapses. Studies of the developing and epileptic hippocampus suggest a cellular mechanism by which activity spreads in the hippocampus and further highlight a critical role for GABA-expressing interneurons (Morgan & Soltesz, 2008; Bonifazi et al., 2009). Using a combination of imaging and electrophysiological techniques in the developing hippocampus, Cossart and colleagues have proposed the presence of GABAergic “hub” neurons that connect a large number of cells across long distances. Given the ability of GABAergic inputs to produce giant depolarizing potentials (GDPs) in early development (Ben-Ari et al., 1989), electrophysiological stimulation of a single hub interneuron is able to depolarize a large number of neurons and synchronize network activity (Bonifazi et al., 2009). However, stimulation of other hub neurons slows down oscillations or reduces network activity. The diverse effects of hub neuron activation on network dynamics likely reflect both the excitatory and inhibitory (Khalilov et al., 1999) actions of GABA in early development. Whether GABAergic hub neurons persist in the adult brain and how they might affect network dynamics remains unknown. Computational studies using large-scale realistic models of a seizure-prone adult rat dentate gyrus have started to examine how excitatory hub neurons may generate and propagate hyperexcitability (Morgan & Soltesz, 2008). In the presence of a small number of hub granule cells, minimal granule cell stimulation dramatically increases the number of granule cells that fire, boosts the speed at which activity spreads across the network and doubles the duration for which this activity persists. These studies, described in more detail elsewhere in this volume, will require further experiments to reveal whether hub cells exist in the adult (and epileptic) hippocampus.
Genetic models of interneuronopathy
Unlike rodent models that were designed to mimic specific behavioral and anatomical pathologies exhibited by TLE patients, genetic interneuronopathy models were generated to understand how one or two genes contribute to brain development and function. In many cases, the primary intent of generating these genetic models was not to study epilepsy, and many originate from laboratory groups studying interneuron origins and diversity (as discussed elsewhere in this volume). The relation between each of the genes, inhibition and excitability were explored only after it became evident that the genetic mutation led to a loss of interneurons, synaptic inhibition, and/or interneuron function.
For example, mice lacking the Dlx1 gene were generated in the Rubenstein laboratory to examine interneuron development. Dlx1 encodes a transcription factor expressed in several specific populations of GABAergic interneurons (e.g. SOM+, CR+ and NPY+ interneurons) (Cobos et al., 2005). Previously, Dlx1/2 double mutants were shown to lack 80% of GABA immunoreactive cells and to die within a few hours after birth (Anderson et al., 1997). In contrast, Dlx1 mutants are indistinguishable from wild-type mice at birth and exhibited a selective loss of SOM+, CR+, NPY+, and NOS+ interneurons as they approached reproductive maturity (P30+) (Cobos et al., 2005). The analysis of these mice demonstrated that Dlx1 is required for interneuron survival, not development. Subsequent electrophysiological studies revealed that interneuron loss is accompanied by a reduction in synaptic inhibition and spontaneous electrographic and behavioral seizures. Dlx1 mutant mice can now be used to examine how the loss of selective interneuron subtypes and inhibition changes in the adult brain leads to spontaneous seizures. More recently a Dlx5/6 heterozygote mouse was found to have normal interneuronal densities, at all developmental stages, but a susceptibility to spontaneous epileptic discharge and reduced total EEG power in the 30 to 80 Hz range as young adults (Wang et al., 2010),
A number of other interneuronopathy models have also been generated including, but not limited to, mice with reduced or no expression of the ARX, uPAR, Tlx1, Sox2, cD2, Ppt1 genes. Additional model possibilities, including those based on selective or conditional manipulation of genes required for interneuron genesis and migration, continue to emerge. In each of these mice, a selective loss of interneuron subtypes and, when examined, abnormal electrographic activity have been demonstrated (Monaghan et al., 1997; Gupta et al., 2001; Powell et al., 2001; Roy et al., 2002; Avilion et al., 2003; Powell et al., 2003; Ferri et al., 2004; Jalanko et al., 2005; Kato & Dobyns, 2005; Glickstein et al., 2007; Kielar et al., 2007; Cavallaro et al., 2008; Marsh et al., 2009; Price et al., 2009). These discoveries and the recent identification of an epileptic phenotype and reduced cortical interneuron function in children with ARX mutations prompt a new clinical classification for epilepsy termed ‘interneuronopathy’ (Kato & Dobyns, 2005). As an epilepsy model group, these genetically modified mice can be used to understand how specific interneuron subtypes contribute to overall brain activity and counterbalance hyperexcitability.
Aside from rodent models of interneuronopathy that result in interneuron loss, reducing expression of a voltage-gated Na+ channel interferes with interneuron function and causes spontaneous seizures and death. Mice homozygous for the human mutation in the gene encoding Nav1.1 (Scna−/− mice) begin exhibiting ataxia and seizures on postnatal day 9 (P9) and are dead by P15. Heterozygous mutants (Scna+/− mice) exhibit spontaneous electrographic seizures and sporadic deaths at an older age (P21-27) (Yu et al., 2006) as well as seizures induced by elevated core body temperature (Oakley et al., 2009). Given both the Scna mutation and subsequent phenotypic characteristics of these mice, heterozygous mutants are considered a model of severe myoclonic epilepsy in infancy. Hyperexcitability in mice that are homozygous and heterozygous for the Scna mutation is likely explained by a selective reduction of sodium current density and action potential firing recorded from mutant interneurons (Yu et al., 2006). In contrast, sodium currents recorded from pyramidal cells were no different between wild-type and heterozygous or homozygous mutants. Scna mutants suggest that the selective interference of interneuron excitability is sufficient to induce a severe epileptic phenotype. A common theme that emerges from these studies is the possibility that seizure activity can be suppressed in Scna mutant mice, interneuronopathic mouse models, and acquired TLE models by recovering the loss of GABAergic inhibition through the addition of new interneurons.
Advances in cell transplantation in the treatment of epilepsy
For two decades now, the therapeutic potential of fetal and progenitor cell transplantations in suppressing seizures has been tested with mixed results. It is important to note, that early studies were not able to label grafted cells for subsequent immunohistochemical analysis of cell type as they predated the advent of fluorescently labeled mice (Fine et al., 1990; Holmes et al., 1991) and did not examine spontaneous seizures (Buzsaki et al., 1988; Loscher et al., 1998). Transplantation of fetal locus coeruleus neurons reduced the frequency of ictal spikes induced by subsequent injections of the GABAAR antagonist picrotoxin (Buzsaki et al., 1988). “GABA-rich” substantia nigra tissue grafted into kindled rats transiently increased the stimulation intensity required to induce seizure-like afterdischarge activity (Loscher et al., 1998). Although both of these findings suggest that transplantation increased seizure resistance, a subsequent study to transplant cells genetically engineered to express GAD65 into the piriform cortex of kindled rats did not significantly affect afterdischarge activity (Gernert et al., 2002). Human neural stem cells, a promising source of pluripotent cells, transplanted into the pilocarpine-induced rat model of TLE differentiate into cells that are positive for GABAergic (26%), glutamatergic (2%) or astrocytic (21%) markers. Such grafted cells decrease the percentage of pilocarpine rats that develop behaviorally monitored spontaneous seizures and reduce both seizure frequency and severity (Chu et al., 2004). Like the human stem cell grafts, striatal precursor cell grafts yield a small fraction of GABAergic cells (23%). Nonetheless, striatal precursors transplanted into rats soon after kainate-induced status epilepticus were also associated with a reduction in spontaneous, behaviorally assessed seizures (Hattiangady et al., 2008).
Although these results are encouraging, they leave a number of critical questions unanswered. That is, cells are grafted and the effects on seizure behavior (or seizure-like afterdischarge activity) are monitored but little information is available with regard to what happens to the grafted cells and the host brain post-transplantation. In particular, to interpret the effects of transplants on global brain excitability it is important to know (i) how many of the grafted cells survive in the host brain and how far they migrate from the injection site, (ii) what types of cells (i.e. excitatory, inhibitory, glial, immature) the grafts yield, (iii) whether graft-derived cells functionally integrate into the host brain and (iv) how grafts affect electrographically and behaviorally monitored spontaneous seizure activity. In the absence of such information, the mechanisms by which grafted cells can act as an effective therapeutic option for epilepsy are left unknown. To address these questions, it would be helpful if grafted cells express fluorescent markers that would enable their identification for immunohistochemistry and electrophysiological recordings in the host brain.
As described above, a variety of fetal tissue sources have been grafted including rodent precursor cells of the hippocampus (Shetty & Hattiangady, 2007a), striatum (Loscher et al., 1998), locus coeruleus (Buzsaki et al., 1988; Bengzon et al., 1993), spinal cord (Loscher et al., 1998), medial (Alvarez-Dolado et al., 2006; Baraban et al., 2009) or lateral (Hattiangady et al., 2008) ganglionic eminences, and human neural stem cells (Chu et al., 2004). In addition to fetal tissue, neural progenitor cells derived from embryonic stem cell lines have been used (Li et al., 2007; Carpentino et al., 2008). When cell fate is examined, some of these grafts appear to contain a mixture of excitatory neurons, inhibitory interneurons, glial cells and immature cells (Chu et al., 2004; Carpentino et al., 2008). Such heterogeneity complicates interpretations of how grafts affect host brain activity. The exceptions are transplantations of the medial ganglionic eminence, and fetal CA1 and CA3 regions, which are committed to interneuronal or pyramidal cell fates, respectively (Zaman et al., 2000; Alvarez-Dolado et al., 2006; Baraban et al., 2009).
Fetal CA1 and CA3 transplantations into rodent hippocampus have examined whether host brain variables such as age and health, both clinically relevant factors, affect graft survival. For both “middle-aged” (12–14 month old) and “aged” (22–24 month old) healthy rats, an equal fraction of cells transplanted into the hippocampus survived (18–23% of injected cells) (Zaman & Shetty, 2002). Interestingly, the epileptic hippocampus appears to be a more receptive environment for grafts e.g., a larger percentage of transplanted cells survived in the epileptic versus healthy hippocampi of kainate lesioned rats. When neural progenitors derived from an embryonic cell line were transplanted into the CA3 region, these cells formed tumors in a healthy hippocampus. In contrast, they did not form tumors and migrated longer distances toward the dentate gyrus in rat models of TLE (Carpentino et al., 2008).
In addition to monitoring graft survival and spread, the impact of CA1 and CA3 transplants on the host epileptic brain has been examined. The hippocampi of TLE rodents and patients exhibit CA3 pyramidal cell and hilar interneuron loss as well as a reduction in GABAergic inhibition (see text in section Acquired Models of Epilepsy). Fetal CA3 cell transplants return the number of GAD67+ and calbindin+ interneurons in the TLE hippocampus toward that of control levels (Shetty & Turner, 2000; Shetty & Hattiangady, 2007b). However, it is currently unknown what causes this effect and whether an increase in GABAergic interneuron density translates to increased GABA-mediated inhibition in these animals.
Work from our lab has employed transplantations of the embryonic medial ganglionic eminence (MGE) into the newborn neocortex and hippocampus to understand how grafted cells behave in the host and affect host brain activity as assessed via single cell and cortical electrographic recordings (Alvarez-Dolado et al., 2006; Baraban et al., 2009). The MGE was chosen as the source of donor cells based on previous work from our collaborators and other laboratories featured in this special issue. An array of techniques including fluorescent tracing of MGE cells, in utero fate mapping and genetic models demonstrated that the MGE is the primary source of cortical interneurons (Lavdas et al., 1999; Sussel et al., 1999; Cobos et al., 2001; Wichterle et al., 2001). Further, grafting studies indicate the impressive migratory potential of MGE cells placed on cultured embryonic brain slices and transplanted into the adult striatum and thalamus in vivo (Wichterle et al., 1999). In our studies, MGE cells derived from GFP+ mice were transplanted into the neocortex and hippocampus of P2 wild-type mice (Figure 1). Thirty to sixty days after transplantation (DAT), immunohistochemistry, single cell electrophysiology, electron microscopy and cortical video-electrographic recording were used to assess a number of measures of both grafted-derived cells and the host brain. At 60 DAT, graft-derived cells had dispersed widely away from the injection site (up to 3 mm away in either direction) and had already started to migrate 3 days after injection. GFP+ MGE cells developed the morphological characteristics of a variety of interneuron types (i.e. basket cells, chandelier cells, bipolar cells) and 69% of these cells were GABA+. We expect the number of GABAergic cells is an underestimate given the low signal produced by the GABA primary antibody and that we found no cells with pyramidal cell-like morphology and only a few oligodendrocytes (1.8%). At 30 DAT, graft-derived cells also expressed interneuron markers (e.g. PV, SOM, CR, CB, NPY) in the same ratios reported for native interneuron populations. Not only do graft-derived GFP+ cells exhibit the morphological and neurochemical features of interneurons, they also fire like mature interneurons (e.g. fast-spiking, stuttering, regular spiking non-pyramidal cells). Single cell recordings of host pyramidal cells and interneurons have shown statistically significant increases in phasic and tonic GABAergic currents of grafted mice. Electron microscopy of the grafted brain demonstrated that GFP+ cells form inhibitory synapses onto host pyramidal cells. Finally, in a test of the therapeutic potential of this approach, MGE cells were transplanted into the neocortex of a genetic model of epilepsy lacking a voltage activated K+ channel, Kv1.1−/− mice. These mice were chosen because they exhibit a high frequency of behavioral and electrographic seizures and the age of seizure onset is consistent (Smart et al., 1998; Wenzel et al., 2007). We found that the total number of spontaneous seizures recorded in grafted Kv1.1 mutant animals was reduced by 86%; 2 of 8 MGE graft recipients exhibited no seizures during the recording period. For these assessments, we used dual digital cameras coupled to a cortical electroencephalographic recording system (Pinnacle Technology) and mice were monitored continuously for sessions up to 24 hr. Despite the promising results of these studies, they represent only the first step toward the use of interneuronal precursor transplantation in treating epilepsy.
Figure 1.

A summary of the neonatal MGE cell transplantation protocol. GFP+ MGE cells from E13.5 mice were transplanted into the P2 mouse neocortex. 30–60 days after transplantation (DAT), host mice were used for immunohistochemical, electrophysiological, electron microscopic, electroencephalographic and behavioral analysis. Primary findings are summarized in the text box (right)
In future studies, it is vital to test a more clinically relevant scenario in which cells are transplanted after a rodent exhibits spontaneous seizures. This type of protocol would more closely mimic the clinical condition in which epilepsy patients opt for more invasive procedures only after they have been experiencing recurrent seizures that are not controlled by available medications. To move toward this translational goal, first, MGE cells must be transplanted into the adult mouse brain and replicate the same successes with regard to migration, differentiation, and integration that were observed following neonatal transplants. Wichterle and colleagues have previously shown that MGE cells grafted into the adult brain can migrate long distances (~1.3 mm) away from the transplantation site (Wichterle et al., 1999). Further studies are necessary to determine whether MGE cells transplanted into the adult brain differentiate into GABAergic interneurons that elevate inhibition in the host. Using the adult transplant protocol, MGE cells can then be transplanted into epileptic animals to assess the efficacy of graft-derived interneurons in reducing spontaneous behavioral and electrographic seizure activity in a variety of epilepsy models. Long-term assessment of potential tumorigenicity and evaluation of grafted animals using a battery of behavioral and cognitive tests is also recommended.
Recent work by Alvarez-Dolado and coworkers has utilized an adult transplantation protocol to demonstrate the therapeutic potential of MGE cell grafting in treating a mouse model of seizure susceptibility (Zipancic et al., 2010). MGE cells transplanted into the adult hippocampus migrated, differentiated into GABAergic interneurons, increased synaptic inhibition in the host brain and survived months after grafting (~20% survival rate at 2 months, 4 months and 1 year post transplantation). Grafted MGE cells were able to reduce the frequency of severe, behaviorally monitored seizures that are induced by an injection of a seizure causing agent, pentylenetetrazol.
To examine whether MGE cells reduce the frequency of spontaneous seizures, preferably those that model a human form of epilepsy, we are characterizing MGE cells transplanted into the adult hippocampus. Preliminary recordings of GFP+ graft-derived MGE cells transplanted into the adult hippocampus demonstrate that green cells receive both excitatory and inhibitory synaptic inputs and fire action potentials 47 DAT (Figure 2). It is also worth noting, that we have never observed tumor formations in mice grafted with MGE interneuron progenitor cells (either early postnatal or adult) and no gross changes in behavior have been observed. The therapeutic promise of MGE cell transplantation is not limited to seizure suppression and may extend to a variety of neurological diseases. For example, MGE cells grafted into the adult striatum of Parkinsonian rats differentiate into GABAergic interneurons, receive synaptic inputs and ameliorate motor deficits (Martinez-Cerdeno et al., 2010).
Figure 2.
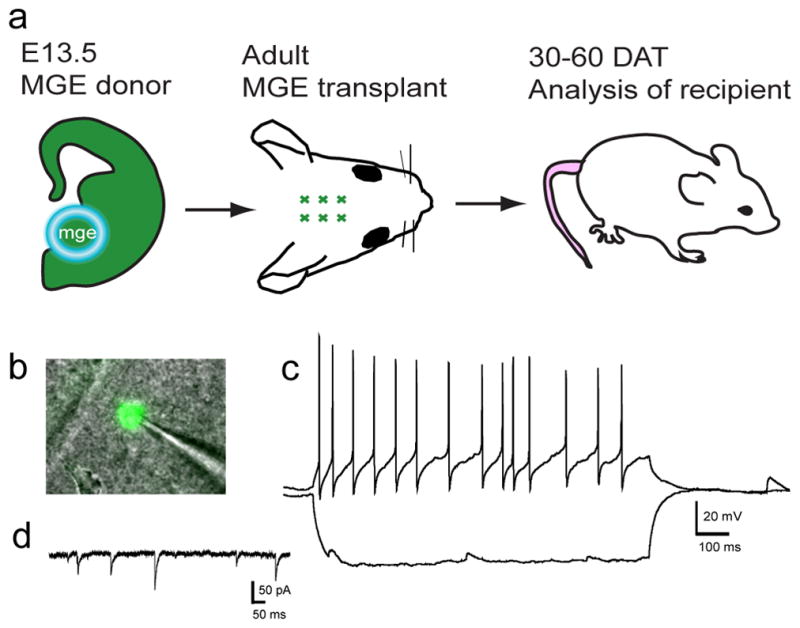
Following adult hippocampal transplantation, GFP+ graft-derived cells become functional neurons. A. An adult CD-1 mouse (P101) received a GFP+ MGE cell graft and was sacrificed 47 DAT for electrophysiological analysis. B. Image of GFP+ cell for which the recordings are shown in C and D. C. Cell fired action potentials with fast afterhyperpolarizations that are characteristic of interneurons. Excitatory postsynaptic potentials (EPSPs) can be seen during and after the hyperpolarizing current step. D. Use of an internal solution containing KGluconate and KCl (Bacci et al., 2003) allowed voltage clamp recordings from the same cell. Spontaneous inhibitory postsynaptic currents (IPSCs) recorded in the presence of glutamate receptor blockers demonstrate that GFP+ cells receive inhibitory synaptic input. Vhold = −60 mV.
Conclusion
Our understanding of epilepsy has made tremendous progress since the disease was first described in 400 B.C. In ancient Grecian and in Biblical times, seizures were thought to be caused by demonic possession and “epileptics” were treated with magic and exorcism (Temkin, 1971). This ideology persisted for several hundreds of years, to the detriment of the treatment of patients and of scientific advancement. Finally in the mid 1800s, the first anticonvulsant medications, bromide salts, were administered followed by phenobarbital in 1912 (Porter, 1997). Our understanding of brain function and the causes of epilepsy continue to evolve and have led to more effective medications that help patients who would otherwise experience no relief from seizures. For example, recent work by Staley, Jensen and colleagues combined our knowledge of how Cl− mediated transmission develops and seizures are generated to administer anti-seizure medications in newborn rats for subsequent use in newborn epileptic patients (Dzhala et al., 2005; Kahle et al., 2009). Given that frequent, severe seizures at a young age can interfere with brain development, treating pediatric epilepsy is of particular importance for cognitive development. Despite the benefits of anti-seizure medications, it is critical that alternative treatments are developed to increase therapeutic options for those patients who are experiencing inadequate relief from seizures. Gene therapy that inhibit surges of electrical brain activity and electrical devices that detect seizure onset are currently being explored (Shoeb et al., 2004; Brooks-Kayal et al., 2009; Foti, 2009; Shoeb et al., 2009).
It is our hope that the transplantation of cells engineered to release GABA or interneuronal precursors with an endogenous ability to release GABA will effectively suppress seizures, first in rodent models then in patients. As we move forward in testing the therapeutic efficacy of cell transplantations, we must be rigorous in our scientific approach. Simply put, it is probably insufficient to transplant cells and assess the effect of grafting on visually monitored seizure-like behaviors. It would be better to examine how grafted cells behave in the host brain and how they affect host brain activity both on a cellular and network level. Network activity can be monitored via long-term video-EEG recordings of grafted animals to accurately assess effects on epilepsy. Such information will enable modifications to both grafted cells and the development of a transplantation protocol that may further enhance seizure suppression and allow us to anticipate or circumvent problems associated with grafting. As cell therapy in Parkinson’s disease has taught us, the road from successful transplantation in neonatal rodents to adult rodents to clinical trials is arduous and requires a clear and careful understanding of how grafted cells effect a recovery in brain function (Bjorklund & Lindvall, 2000; Dunnett et al., 2001).
Acknowledgments
We would like to thank Drs. D.H. Lowenstein and N.M. Barbaro for recommending text on the history and treatment of epilepsy. This work was supported by a NIH Ruth L. Kirschstein National Research Service Award 5F32NS061497-02 (to J.Y.S) and NIH R01 grants NS048528-05 and NS040272-07 (to S.C.B.).
References
- Alvarez-Dolado M, Calcagnotto ME, Karkar KM, Southwell DG, Jones-Davis DM, Estrada RC, Rubenstein JL, Alvarez-Buylla A, Baraban SC. Cortical inhibition modified by embryonic neural precursors grafted into the postnatal brain. J Neurosci. 2006;26:7380–7389. doi: 10.1523/JNEUROSCI.1540-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Avilion AA, Nicolis SK, Pevny LH, Perez L, Vivian N, Lovell-Badge R. Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev. 2003;17:126–140. doi: 10.1101/gad.224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci A, Rudolph U, Huguenard JR, Prince DA. Major differences in inhibitory synaptic transmission onto two neocortical interneuron subclasses. J Neurosci. 2003;23:9664–9674. doi: 10.1523/JNEUROSCI.23-29-09664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraban SC, Southwell DG, Estrada RC, Jones DL, Sebe JY, Alfaro-Cervello C, Garcia-Verdugo JM, Rubenstein JL, Alvarez-Buylla A. Reduction of seizures by transplantation of cortical GABAergic interneuron precursors into Kv1.1 mutant mice. Proc Natl Acad Sci U S A. 2009;106:15472–15477. doi: 10.1073/pnas.0900141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AJ, Chen J, Zien A, Sochivko D, Normann S, Schramm J, Elger CE, Wiestler OD, Blumcke I. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. Eur J Neurosci. 2003;18:2792–2802. doi: 10.1111/j.1460-9568.2003.02993.x. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Lindvall O. Specific functions of grafted locus coeruleus neurons in the kindling model of epilepsy. Exp Neurol. 1993;122:143–154. doi: 10.1006/exnr.1993.1115. [DOI] [PubMed] [Google Scholar]
- Bjorklund A, Lindvall O. Cell replacement therapies for central nervous system disorders. Nat Neurosci. 2000;3:537–544. doi: 10.1038/75705. [DOI] [PubMed] [Google Scholar]
- Bonifazi P, Goldin M, Picardo MA, Jorquera I, Cattani A, Bianconi G, Represa A, Ben-Ari Y, Cossart R. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science. 2009;326:1419–1424. doi: 10.1126/science.1175509. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Raol YH, Russek SJ. Alteration of epileptogenesis genes. Neurotherapeutics. 2009;6:312–318. doi: 10.1016/j.nurt.2009.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Zhang GF, Yamawaki R. Axon sprouting in a model of temporal lobe epilepsy creates a predominantly excitatory feedback circuit. J Neurosci. 2002;22:6650–6658. doi: 10.1523/JNEUROSCI.22-15-06650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G, Ponomareff G, Bayardo F, Shaw T, Gage FH. Suppression and induction of epileptic activity by neuronal grafts. Proc Natl Acad Sci U S A. 1988;85:9327–9330. doi: 10.1073/pnas.85.23.9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagnotto ME, Paredes MF, Tihan T, Barbaro NM, Baraban SC. Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J Neurosci. 2005;25:9649–9657. doi: 10.1523/JNEUROSCI.2687-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentino JE, Hartman NW, Grabel LB, Naegele JR. Region-specific differentiation of embryonic stem cell-derived neural progenitor transplants into the adult mouse hippocampus following seizures. J Neurosci Res. 2008;86:512–524. doi: 10.1002/jnr.21514. [DOI] [PubMed] [Google Scholar]
- Carreno M, Donaire A, Sanchez-Carpintero R. Cognitive disorders associated with epilepsy: diagnosis and treatment. Neurologist. 2008;14:S26–34. doi: 10.1097/01.nrl.0000340789.15295.8f. [DOI] [PubMed] [Google Scholar]
- Cavalheiro EAN-M, MG, Mello LE, Leite JP. The Pilocarpine Model of Seizures. In: Pitkanen AS, PA, Moshe S, editors. Animal Models of Seizures and Epilepsy. Elsevier Academic Press; Burlington, MA: 2006. pp. 433–448. [Google Scholar]
- Cavallaro M, Mariani J, Lancini C, Latorre E, Caccia R, Gullo F, Valotta M, DeBiasi S, Spinardi L, Ronchi A, Wanke E, Brunelli S, Favaro R, Ottolenghi S, Nicolis SK. Impaired generation of mature neurons by neural stem cells from hypomorphic Sox2 mutants. Development. 2008;135:541–557. doi: 10.1242/dev.010801. [DOI] [PubMed] [Google Scholar]
- Chu K, Kim M, Jung KH, Jeon D, Lee ST, Kim J, Jeong SW, Kim SU, Lee SK, Shin HS, Roh JK. Human neural stem cell transplantation reduces spontaneous recurrent seizures following pilocarpine-induced status epilepticus in adult rats. Brain Res. 2004;1023:213–221. doi: 10.1016/j.brainres.2004.07.045. [DOI] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL. Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci. 2005;8:1059–1068. doi: 10.1038/nn1499. [DOI] [PubMed] [Google Scholar]
- Cobos I, Puelles L, Martinez S. The avian telencephalic subpallium originates inhibitory neurons that invade tangentially the pallium (dorsal ventricular ridge and cortical areas) Dev Biol. 2001;239:30–45. doi: 10.1006/dbio.2001.0422. [DOI] [PubMed] [Google Scholar]
- Curia G, Longo D, Biagini G, Jones RS, Avoli M. The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods. 2008;172:143–157. doi: 10.1016/j.jneumeth.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lanerolle NC, Kim JH, Robbins RJ, Spencer DD. Hippocampal interneuron loss and plasticity in human temporal lobe epilepsy. Brain Res. 1989;495:387–395. doi: 10.1016/0006-8993(89)90234-5. [DOI] [PubMed] [Google Scholar]
- Delgado-Escueta AVW, WA, Olsen RW, Porter RJ, editors. Jasper’s Basic Mechanisms of the Epilepsies. Lippincott Williams and Wilkins; Philadelphia, PA: 1999. [Google Scholar]
- Dudek FECS, Williams PA, Grabenstatter HL. Kainate-Induced Status Epilepticus: A Chronic Model of Epilepsy. In: Pitkanen AS, PA, Moshe S, editors. Models of Seizures and Epilepsy. Elsevier Academic Press; Burlington, MA: 2006. pp. 415–432. [Google Scholar]
- Dunnett SB, Bjorklund A, Lindvall O. Cell therapy in Parkinson’s disease - stop or go? Nat Rev Neurosci. 2001;2:365–369. doi: 10.1038/35072572. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Echauz JWS, Litt B. Seizure Analysis and Detection In Vivo. In: Baraban SC, editor. Animal Models of Epilepsy: Methods and Innovations. Humana Press; New York: 2009. pp. 207–209. [Google Scholar]
- Engel J., Jr . Overview of Surgical Treatment of Epilepsy. In: Shorvon SPE, Engel J Jr, editors. The Treatment of Epilepsy. Blackwell Publishing; Hoboken: 2009. [Google Scholar]
- Engel JP, Jr, TA . Epilepsy: A Comprehensive Textbook. Lippincott-Raven Publishers; Philadelphia, PA: 1997. [Google Scholar]
- Ferri AL, Cavallaro M, Braida D, Di Cristofano A, Canta A, Vezzani A, Ottolenghi S, Pandolfi PP, Sala M, DeBiasi S, Nicolis SK. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- Fine A, Meldrum BS, Patel S. Modulation of experimentally induced epilepsy by intracerebral grafts of fetal GABAergic neurons. Neuropsychologia. 1990;28:627–634. doi: 10.1016/0028-3932(90)90038-p. [DOI] [PubMed] [Google Scholar]
- Foti SBR, SJ, Brooks-Kayal AR, McCown TJ. Viral Vector Gene Therapy for Epilpesy. In: Baraban SC, editor. Animal Models of Epilepsy. Humana Press; New York: 2009. [Google Scholar]
- Galanapoulou ASaM, SL . Electrical Kindling in Developing Rats. In: Pitkanen AS, PA, Moshe S, editors. Models of Seizures and Epilepsy. Elsevier Academic Press; Burlington: 2006. pp. 371–377. [Google Scholar]
- Gernert M, Thompson KW, Loscher W, Tobin AJ. Genetically engineered GABA-producing cells demonstrate anticonvulsant effects and long-term transgene expression when transplanted into the central piriform cortex of rats. Exp Neurol. 2002;176:183–192. doi: 10.1006/exnr.2002.7914. [DOI] [PubMed] [Google Scholar]
- Glickstein SB, Moore H, Slowinska B, Racchumi J, Suh M, Chuhma N, Ross ME. Selective cortical interneuron and GABA deficits in cyclin D2-null mice. Development. 2007;134:4083–4093. doi: 10.1242/dev.008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Mann EO, Mody I. Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci. 2008;28:1421–1426. doi: 10.1523/JNEUROSCI.4751-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001;21:8523–8537. doi: 10.1523/JNEUROSCI.21-21-08523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci U S A. 2001;98:13566–13571. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, Shetty AK. Grafting of striatal precursor cells into hippocampus shortly after status epilepticus restrains chronic temporal lobe epilepsy. Exp Neurol. 2008;212:468–481. doi: 10.1016/j.expneurol.2008.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoie B, Mykletun A, Waaler PE, Skeidsvoll H, Sommerfelt K. Executive functions and seizure-related factors in children with epilepsy in Western Norway. Dev Med Child Neurol. 2006;48:519–525. doi: 10.1017/S0012162206001095. [DOI] [PubMed] [Google Scholar]
- Holmes GL, Thompson JL, Huh K, Holmes C, Carl GF. Effect of neural transplants on seizure frequency and kindling in immature rats following kainic acid. Brain Res Dev Brain Res. 1991;64:47–56. doi: 10.1016/0165-3806(91)90208-z. [DOI] [PubMed] [Google Scholar]
- Houser CR, Esclapez M. Downregulation of the alpha5 subunit of the GABA(A) receptor in the pilocarpine model of temporal lobe epilepsy. Hippocampus. 2003;13:633–645. doi: 10.1002/hipo.10108. [DOI] [PubMed] [Google Scholar]
- Jalanko A, Vesa J, Manninen T, von Schantz C, Minye H, Fabritius AL, Salonen T, Rapola J, Gentile M, Kopra O, Peltonen L. Mice with Ppt1Deltaex4 mutation replicate the INCL phenotype and show an inflammation-associated loss of interneurons. Neurobiol Dis. 2005;18:226–241. doi: 10.1016/j.nbd.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Barnett SM, Sassower KC, Staley KJ. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(−) cotransporter NKCC1. J Child Neurol. 2009;24:572–576. doi: 10.1177/0883073809333526. [DOI] [PubMed] [Google Scholar]
- Kato M, Dobyns WB. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: proposal for a new term, “interneuronopathy”. J Child Neurol. 2005;20:392–397. doi: 10.1177/08830738050200042001. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Dzhala V, Ben-Ari Y, Khazipov R. Dual role of GABA in the neonatal rat hippocampus. Dev Neurosci. 1999;21:310–319. doi: 10.1159/000017380. [DOI] [PubMed] [Google Scholar]
- Kielar C, Maddox L, Bible E, Pontikis CC, Macauley SL, Griffey MA, Wong M, Sands MS, Cooper JD. Successive neuron loss in the thalamus and cortex in a mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2007;25:150–162. doi: 10.1016/j.nbd.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopp A, Frahm C, Fidzinski P, Witte OW, Behr J. Loss of GABAergic neurons in the subiculum and its functional implications in temporal lobe epilepsy. Brain. 2008;131:1516–1527. doi: 10.1093/brain/awn095. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wen X, Buckmaster PS. Reduced inhibition and increased output of layer II neurons in the medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2003;23:8471–8479. doi: 10.1523/JNEUROSCI.23-24-08471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Buckmaster PS. Hyperexcitability, interneurons, and loss of GABAergic synapses in entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2006;26:4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar SS, Jin X, Buckmaster PS, Huguenard JR. Recurrent circuits in layer II of medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2007;27:1239–1246. doi: 10.1523/JNEUROSCI.3182-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagae L. Cognitive side effects of anti-epileptic drugs. The relevance in childhood epilepsy. Seizure. 2006;15:235–241. doi: 10.1016/j.seizure.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL. Long-term sequelae of epilepsy. Ann Acad Med Singapore. 1989;18:49–51. [PubMed] [Google Scholar]
- Li T, Steinbeck JA, Lusardi T, Koch P, Lan JQ, Wilz A, Segschneider M, Simon RP, Brustle O, Boison D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain. 2007;130:1276–1288. doi: 10.1093/brain/awm057. [DOI] [PubMed] [Google Scholar]
- Loscher W, Ebert U, Lehmann H, Rosenthal C, Nikkhah G. Seizure suppression in kindling epilepsy by grafts of fetal GABAergic neurons in rat substantia nigra. J Neurosci Res. 1998;51:196–209. doi: 10.1002/(SICI)1097-4547(19980115)51:2<196::AID-JNR8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Lund IV, Hu Y, Raol YH, Benham RS, Faris R, Russek SJ, Brooks-Kayal AR. BDNF selectively regulates GABAA receptor transcription by activation of the JAK/STAT pathway. Sci Signal. 2008;1:ra9. doi: 10.1126/scisignal.1162396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbaum DE, Burack GD, Bhise VV. Impact of antiepileptic drugs on cognition, behavior, and motor skills in children with new-onset, idiopathic epilepsy. Epilepsy Behav. 2009;16:341–344. doi: 10.1016/j.yebeh.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, Brooks-Kayal A, Golden JA. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cerdeno V, Noctor SC, Espinosa A, Ariza J, Parker P, Orasji S, Daadi MM, Bankiewicz K, Alvarez-Buylla A, Kriegstein AR. Embryonic MGE precursor cells grafted into adult rat striatum integrate and ameliorate motor symptoms in 6-OHDA-lesioned rats. Cell Stem Cell. 6:238–250. doi: 10.1016/j.stem.2010.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathern GW, Babb TL, Pretorius JK, Leite JP. Reactive synaptogenesis and neuron densities for neuropeptide Y, somatostatin, and glutamate decarboxylase immunoreactivity in the epileptogenic human fascia dentata. J Neurosci. 1995;15:3990–4004. doi: 10.1523/JNEUROSCI.15-05-03990.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan AP, Bock D, Gass P, Schwager A, Wolfer DP, Lipp HP, Schutz G. Defective limbic system in mice lacking the tailless gene. Nature. 1997;390:515–517. doi: 10.1038/37364. [DOI] [PubMed] [Google Scholar]
- Morgan RJ, Soltesz I. Nonrandom connectivity of the epileptic dentate gyrus predicts a major role for neuronal hubs in seizures. Proc Natl Acad Sci U S A. 2008;105:6179–6184. doi: 10.1073/pnas.0801372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudo G, Jiang XH, Timmusk T, Bindoni M, Belluardo N. Change in neurotrophins and their receptor mRNAs in the rat forebrain after status epilepticus induced by pilocarpine. Epilepsia. 1996;37:198–207. doi: 10.1111/j.1528-1157.1996.tb00012.x. [DOI] [PubMed] [Google Scholar]
- Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA. Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci U S A. 2009;106:3994–3999. doi: 10.1073/pnas.0813330106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkey CE. Complications of Epilepsy Surgery. In: Shorvon SPE, Engel J Jr, editors. The Treatment of Epilepsy. Blackwell Publishing; Hoboken: 2009. [Google Scholar]
- Porter RJM, BS, MacDonald RL, Dichter MA, Dam M, Treiman DM, Chadwick D, Gram L, Pedley TA. Antiepileptic Drugs. In: Engel JP Jr, TA, editors. Epilepsy: A Comprehensive Textbook. Lippincott-Raven Publishers; Philadelphia: 1997. pp. 1381–1382. [Google Scholar]
- Powell EM, Campbell DB, Stanwood GD, Davis C, Noebels JL, Levitt P. Genetic disruption of cortical interneuron development causes region- and GABA cell type-specific deficits, epilepsy, and behavioral dysfunction. J Neurosci. 2003;23:622–631. doi: 10.1523/JNEUROSCI.23-02-00622.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Price MG, Yoo JW, Burgess DL, Deng F, Hrachovy RA, Frost JD, Jr, Noebels JL. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci. 2009;29:8752–8763. doi: 10.1523/JNEUROSCI.0915-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy K, Thiels E, Monaghan AP. Loss of the tailless gene affects forebrain development and emotional behavior. Physiol Behav. 2002;77:595–600. doi: 10.1016/s0031-9384(02)00902-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SP, Coleman PD. Neurons of origin of the perforant path. Exp Neurol. 1981;74:305–312. doi: 10.1016/0014-4886(81)90169-2. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Brain development and epilepsy. Oxford University Press; New York: 1995. [Google Scholar]
- Segal M, Landis S. Afferents to the hippocampus of the rat studied with the method of retrograde transport of horseradish peroxidase. Brain Res. 1974;78:1–15. doi: 10.1016/0006-8993(74)90349-7. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Concise review: prospects of stem cell therapy for temporal lobe epilepsy. Stem Cells. 2007a;25:2396–2407. doi: 10.1634/stemcells.2007-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B. Restoration of calbindin after fetal hippocampal CA3 cell grafting into the injured hippocampus in a rat model of temporal lobe epilepsy. Hippocampus. 2007b;17:943–956. doi: 10.1002/hipo.20311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty AK, Turner DA. Fetal hippocampal grafts containing CA3 cells restore host hippocampal glutamate decarboxylase-positive interneuron numbers in a rat model of temporal lobe epilepsy. J Neurosci. 2000;20:8788–8801. doi: 10.1523/JNEUROSCI.20-23-08788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibley H, Smith BN. Pilocarpine-induced status epilepticus results in mossy fiber sprouting and spontaneous seizures in C57BL/6 and CD-1 mice. Epilepsy Res. 2002;49:109–120. doi: 10.1016/s0920-1211(02)00012-8. [DOI] [PubMed] [Google Scholar]
- Shoeb A, Edwards H, Connolly J, Bourgeois B, Treves T, Guttag J. Patient-specific seizure onset detection. Conf Proc IEEE Eng Med Biol Soc. 2004;1:419–422. doi: 10.1109/IEMBS.2004.1403183. [DOI] [PubMed] [Google Scholar]
- Shoeb A, Pang T, Guttag J, Schachter S. Non-invasive computerized system for automatically initiating vagus nerve stimulation following patient-specific detection of seizures or epileptiform discharges. Int J Neural Syst. 2009;19:157–172. doi: 10.1142/S0129065709001938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the K(V)1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Battaglia G, Arcelli P, Andermann F, Dubeau F, Palmini A, Olivier A, Villemure JG, Tampieri D, Avanzini G, Avoli M. Cortical dysplasia: an immunocytochemical study of three patients. Neurology. 1998;50:27–36. doi: 10.1212/wnl.50.1.27. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Tassi L, Colombo N, Bramerio M, Galli C, Garbelli R, Ferrario A, Lo Russo G, Munari C. Inhibitory circuits in human dysplastic tissue. Epilepsia. 2000;41(Suppl 6):S168–173. doi: 10.1111/j.1528-1157.2000.tb01576.x. [DOI] [PubMed] [Google Scholar]
- Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Temkin O. The Falling Sickness: A History of Epilepsy from the Greeks to the Beginnings of Modern Neurology. The John Hopkins University Press; Baltimore, MD: 1971. [Google Scholar]
- Thind KK, Yamawaki R, Phanwar I, Zhang G, Wen X, Buckmaster PS. Initial loss but later excess of GABAergic synapses with dentate granule cells in a rat model of temporal lobe epilepsy. J Comp Neurol. 2009;518:647–667. doi: 10.1002/cne.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Bortolotto ZA, Mello LM, Schwarz M, Turski L. Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res. 1984;321:237–253. doi: 10.1016/0006-8993(84)90177-x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Dye CA, Sohal V, Long JE, Estrada RC, Roztocil T, Lufkin T, Deisseroth K, Baraban SC, Rubenstein JL. Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J Neurosci. 2010;30:5334–5345. doi: 10.1523/JNEUROSCI.5963-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Vacher H, Clark E, Trimmer JS, Lee AL, Sapolsky RM, Tempel BL, Schwartzkroin PA. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–2046. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A. Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci. 1999;2:461–466. doi: 10.1038/8131. [DOI] [PubMed] [Google Scholar]
- Wichterle H, Turnbull DH, Nery S, Fishell G, Alvarez-Buylla A. In utero fate mapping reveals distinct migratory pathways and fates of neurons born in the mammalian basal forebrain. Development. 2001;128:3759–3771. doi: 10.1242/dev.128.19.3759. [DOI] [PubMed] [Google Scholar]
- Williams PA, White AM, Clark S, Ferraro DJ, Swiercz W, Staley KJ, Dudek FE. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Patrylo PR, Spencer DD. Decrease in inhibition in dentate granule cells from patients with medial temporal lobe epilepsy. Ann Neurol. 1999;45:92–99. doi: 10.1002/1531-8249(199901)45:1<92::aid-art15>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Wittner L, Magloczky Z, Borhegyi Z, Halasz P, Toth S, Eross L, Szabo Z, Freund TF. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience. 2001;108:587–600. doi: 10.1016/s0306-4522(01)00446-8. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zaman V, Shetty AK. Survival of fetal hippocampal CA3 cell grafts in the middle-aged and aged hippocampus: effect of host age and deafferentation. J Neurosci Res. 2002;70:190–199. doi: 10.1002/jnr.10401. [DOI] [PubMed] [Google Scholar]
- Zaman V, Turner DA, Shetty AK. Survival of grafted fetal neural cells in kainic acid lesioned CA3 region of adult hippocampus depends upon cell specificity. Exp Neurol. 2000;161:535–561. doi: 10.1006/exnr.1999.7304. [DOI] [PubMed] [Google Scholar]
- Zhang W, Yamawaki R, Wen X, Uhl J, Diaz J, Prince DA, Buckmaster PS. Surviving hilar somatostatin interneurons enlarge, sprout axons, and form new synapses with granule cells in a mouse model of temporal lobe epilepsy. J Neurosci. 2009;29:14247–14256. doi: 10.1523/JNEUROSCI.3842-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipancic I, Calcagnotto ME, Piquer-Gil M, Mello LE, Alvarez-Dolado M. Transplant of GABAergic precursors restores hippocampal inhibitory function in a mouse model of seizure susceptibility. Cell Transplant. doi: 10.3727/096368910X491383. [DOI] [PubMed] [Google Scholar]