

Abstract
New anthrax lethal factor inhibitors (LFIs) were designed based upon previously identified potent inhibitors 1a and 2. Combining the new core structures with modifications to the C2-side chain yielded analogs with improved efficacy in the rat lethal toxin model. [BMCL ABSTRACT]
Anthrax disease results from infection with the gram-positive bacteria Bacillus anthracis. Inhalation anthrax is especially dangerous with a survival rate of < 15% if left untreated.2 As the 2001 US postal service attacks demonstrated, mortality from this disease approaches 50% even with antibiotic treatment and intensive care.3 While the pathogenesis of anthrax is not fully understood, it is known that the bacteria secrete three proteins: edema factor (EF) and lethal factor (LF) each of which combine with protective antigen (PA) to form two binary toxins; edema toxin and lethal toxin (ET and LT, respectively). These toxins act as virulence factors and suppress the immune system of the host.4 The protein EF is a Ca2+ /calmodulin-dependant adenylate cyclase which appears to impair immune function, while LF acts as a Zn2+-dependent metalloproteinase and disrupts cell signaling pathways by cleavage of MEK proteins. LT is also considered a primary causative agent leading to death of the host.5 Given the potential for mass casualties by using anthrax as a weapon of bioterrorism, new methods of treating patients infected with B. anthracis leading to improved survival are clearly needed.
One plausible approach is to combine an antibiotic to clear the active infection and stop further release of toxins, with an LF inhibitor (LFI) to block the action of the toxin already present in the body. Indeed, the first in vivo studies to clearly support the use of an LFI in this manner were conducted by scientists at Merck in a rabbit model of anthrax.6 Since the natural incidence of infection by B. anthracis is extremely low and lethality can be very high, phase II and III clinical trials with LFIs are not possible. As a result, development of an antidote for LF intoxication in humans presents a special challenge and must rely on the use of the “animal rule” to demonstrate a potential for efficacy in humans.7 Given this fact, we chose to incorporate an in vivo toxin model early in our screening cascade. The rat LT challenge model is attractive from a drug discovery point of view since it is reproducible, has the resolution needed to rank order compounds being tested in a given study, and death of the animals results specifically from the action of anthrax LF.8 A second benefit of using a pharmacology-based model early in the discovery process is that it provides the potential for early readouts of Structure Activity Relationships (SAR) as they relate to in vivo efficacy, pharmacokinetic (PK) properties, and specific structural features of compound classes being evaluated as potential candidates for pre-clinical studies.
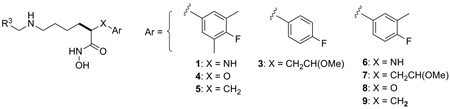
In a previous paper,9 we disclosed the discovery of two compound classes (Figure 1) with high intrinsic potency and good in vivo efficacy in the rat LT model of LF intoxication. Compound 1a, representing the one-atom linking series, was found to be a sub-nanomolar LFI capable of providing 100% protection in the rat LT model when administered IV at 10 mg/kg. Compound 2, a member of the two atom linking series, while not preventing death, did provide an increase the Median Survival Time (MST) of the test animals relative to the vehicle only treated controls. In Part 2 of this series we describe our work toward improving the in vivo efficacy of these LFIs by exploring further structural modifications to these two compound classes, guided in part by in vivo data obtained from the rat LT model.
Figure 1.

Previously identified small molecule LFIs.
Combining 1a and 2 into a generic structure (Figure 2) provided a starting point for targeting structural modifications and determining the impact of these changes on intrinsic potency and in vivo efficacy. These modifications included the synthesis of new core structures by replacing the X-groups in 1a and 2 with either an oxygen atom (4, X=O) or methylene group (5, X=CH2), and exploring alternate substitution patterns on the aryl ring of the core structure (analog series 6, 7, 8, and 9). Also of interest was investigating changes to the C2 side chain in terms of overall length (o and p = 1 to 3), the role of the Y-group (Y=NH vs. O, CH2) and the position of this group relative to the core structure (eg. 3 vs. 2). Summarized below are the results of these studies.
Figure 2.

Structural changes based upon LFIs 1a and 2.
Representative synthetic schemes for the preparation of compounds in series 4 and each of the remaining series depicted in Figure 2 are given in Scheme 1 and the Supplementary Data10 respectively. Analogs in the phenoxyacetic acid series (4: R2=Me; 8: R2=H) were prepared using the asymmetric alkylation route shown in Scheme 1.11 Preparation of the 3-methyl-4-flouro-phenoxyacetic acid from ethyl 2-bromoacetate and conversion to the oxazolidinone derivative 10 followed standard synthetic methods. Asymmetric alkylation of 10 (R2=H) afforded the benzyl protected allylic alcohol 11 (R2=H) in moderate yield.12 When the tri-substituted analog (R2=Me) was used, the yield for this step dropped below 20%. A possible explanation for this result came with the examination of model transition states which suggested the added Me-group on the aryl ring could sterically hinder access of the incoming electrophile to the desired re-face of the enolate. Catalytic hydrogenation to reduce the olefin and deprotect the alcohol was followed by oxidation to provide the aldyehyde 12 in good overall yield. Reductive amination to give the secondary amine followed by treatment of this product with hydroxylamine provide access to LFIs in 4 and 8 directly from the N-acyloxazolidinone.
Scheme 1.

Reagents and conditions: (a) LiHMDS, THF, allyl iodide, −70°C to rt; (b) H2 (1 atm), Pd-C, EtOH, rt; (c) Dess-Martin periodane, CH2Cl2, rt; (d) ArCH2NH2, NaBH(OAc)3, dichloroethane, rt; (e) NH2OH, KCN, MeOH, H2O, THF, rt.
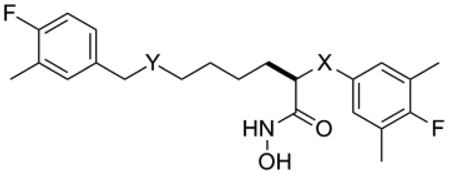
Table 1 provides Ki values for a set of new LFIs that illustrates the effect of the planned modifications (Figure 2) on intrinsic potency against LF. We were pleased to find that when compared to the aniline series (1, X = NH), interchanging of X-groups (cf. 1ad vs. 4a–d, and 5a–d) while holding the C2 side chain constant provided LFIs with similar intrinsic potency (ΔKi ≤ 10x), the one exception being LFI 1a. In the two-atom link series, analogs represented by 3a–d where the C2-side chain of 2 (o=1, p=3) was replaced by that found in series 1 (o=3, p=1; Figure 1) were also prepared. This change resulted in a 10-fold increase in intrinsic potency against LF (cf. 3a vs. 2) and was our first indication that the position of the amino-group of the C2-side chain was important for inhibitor potency. Modifications to the aryl ring of each core structure by removal of a methyl group (6ac, 8a–c, and 9a–c) or the addition of a methyl group (7a–c) had no significant effect on the intrinsic potency. Finally, the extension of the C2 side chain below the N-atom (p = 2, d-series) appeared to slightly decrease potency of these compounds. Taken together, the similar inhibitory activity of these LFIs seen with simple changes in the core structure provides a potential way to modulate the physicochemical (PC) and PK properties of these compounds without sacrificing intrinsic potency against the target.
Table 1.
Anthrax LF inhibitory data for LFIs represented in Figure 2.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| LFI | R3 | LF (FRET) Ki(nM) |
|||||||
| 1 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| a | 3-Me,4-FPh | 0.05 | 1.5 | 2.4 | 0.39 | 0.62 | 1.3 | 3.0 | 1.2 |
| b | 4-FPh | 0.24 | 0.58 | 1.2 | 0.13 | 0.81 | 0.93 | 1.1 | 0.25 |
| c | 4-ClPh | 1.0 | 1.2 | 2.0 | 0.71 | 0.68 | 1.4 | 0.75 | 0.36 |
| d | CH2(4-FPh) | 2.1 | 1.1 | 4.6 | 9.5 | - | - | - | - |
Values are means of three experiments; standard deviation is < 15%; (-) = not prepared
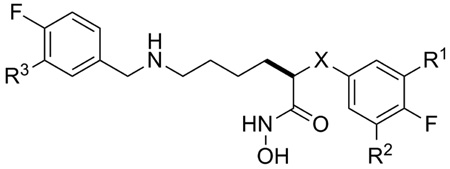
Based on the observation that analogs in the 5-amino series were consistently more potent compared to the 3-amino series (eg. 1a vs.2), we were interested in knowing if the secondary amine was essential for LFI activity (Figure 2, Y=NH vs. O, CH2). The data in Table 2 show a clear preference for the secondary amine at the 5-position of the C2-side chain, followed by oxygen and then a methylene group. Comparing 1a and 5a with their carbon analogs (Y=CH2, 14, 16), reveals a >1000-fold loss in binding affinity (ΔΔG > 4.5 Kcals/mol) and suggests the amine may participate in a directed hydrogen-bond, electrostatic, or specific ionic interaction in the ligand binding site. Given that the LF protease is selective for substrates having Arg or Lys rich sequences on the non-prime side of the catalytic site,13 the preference for a basic amine group in the C2-side chain suggests a similar interaction occurs with this class of LFI during the binding process. While the ether analogs (Y=O) display intrinsic potencies in a range suitable for small molecule drugs,14 the Ki values for the all carbon C2-side chain series (Y=CH2) fall outside this range and were no longer considered targets for analog synthesis.
Table 2.
Effect of the Y-group on intrinsic potency.
 | |||
|---|---|---|---|
| LFI | X | Y | LF (FRET) |
| Ki (nM) | |||
| 1a | NH | NH | 0.05 |
| 13 | NH | O | 30.4 |
| 14 | NH | CH2 | 273 |
| 5a | CH2 | NH | 0.39 |
| 15 | CH2 | O | 25 |
| 16 | CH2 | CH2 | 577 |
Values from three experiments; std deviation < 15%
After exploring a number of modifications to the basic core structure of our LFIs (Figure 2) and discovering that most led to only slight differences in intrinsic potency versus LF, we investigated whether these changes would affect the ability of these LFIs to protect animals in the rat LT challenge model. As discussed above, this model provides a quick read out on the ability of an LFI to block the action of LF in vivo and also relative metabolic stability and distribution properties as it relates to the observed efficacy. All new LFIs were dosed initially at 5.0 mg/kg and those found to provide complete protection at this dose, re-tested at the lower dose of 2.5 mg/kg. Given that inter-experimental variability exists for in vivo models, we normalized the data for each in vivo study (Tables 3 and 4) to the Median Survival Time (MST) of the control animals in each experiment. Parameters used to evaluate LFI efficacy were MST, percent survival, and the presence or absence of morbidity observed in the survivors. In order compare data between tables, we included a standard compound in each study.
Table 3.
Rat LT model survival data for LFIs, experiment 1.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| LFI | Ki (nM) |
X | R1 | R2 | R3 | 5.0 mg/kg | 2.5 mg/kg | ||
| Survivala | rMSTb | Survivala | rMSTb | ||||||
| 1a | 0.05 | NH | Me | Me | Me | 3/3 | - | 0/3 | 4.2 |
| 1b | 0.24 | NH | Me | Me | H | 3/3 | - | 3/3 | - |
| 5a | 0.39 | CH2 | Me | Me | Me | 2/3 | 7.8 | nt | - |
| 6a | 0.62 | NH | Me | H | Me | 0/3 | 5.8 | nt | - |
| 17 | 3.8 | NH | Cl | H | H | 0/3 | 1.4 | nt | - |
| 8a | 3.0 | O | Me | H | Me | 0/3 | 1.3 | nt | - |
| 3a | 1.5 | CH2CH(OMe) | H | H | Me | 1/3 | 5.0 | nt | - |
| 3b | 0.58 | CH2CH(OMe) | H | H | H | 3/3 | - | 0/3 | 5.0 |
Fischer 344 rats were dosed IV with LFIs followed 20 to 30 minutes later by 10µg of LT (10 µg PA + 10 µg LF) dosed IV.
Number of survivors per group. Survival curves for all LFIs versus controls showed statistical significance (P<0.05). When comparing curves for individual LFIs at 5.0 mg/kg, significance (P<0.05) was observed for 1a vs. 6a, 6a vs. 17, and 6a vs. 8a; but was not considered significant for 1a vs. 5a, or 3a vs.3b.
rMST = MST(LFI)/ MST(controls) where vehicle treated controls (MST = 82.3 ± 9.6 minutes, n=9); when less than 50% deaths occurred the average survival time was used: rMST = aveST(LFI)/MST(controls); nt = not tested, (-) = not applicable.
Table 4.
Rat LT model survival data for LFIs, experiment 2.
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| LFI | Ki(nM) | X | R1 | R2 | R3 | o | p | 5.0 mg/kg |
|
| Survivala | rMST b | ||||||||
| 1b | 0.24 | NH | Me | Me | F | 3 | 1 | 2/4 | 17.0 |
| 5b | 0.13 | CH2 | Me | Me | F | 3 | 1 | 1/5 | 13.6 |
| 4b | 1.2 | O | Me | Me | F | 3 | 1 | 0/5 | 2.9 |
| 18 | 2.0 | O | Me | Me | Cl | 3 | 1 | 3/6 | 1.3 |
| 19 | 2.2 | NH | Me | Me | F | 3 | 3 | 1/5 | 2.0 |
| 20 | 4.6 | CH2CH(OMe) | H | H | F | 4 | 1 | 0/4 | 1.4 |
| 21 | 5.0 | CH2CH(OMe) | H | H | F | 4 | 2 | 0/4 | 1.3 |
| 22 | 3.1 | O | Me | Me | F | 4 | 1 | 0/3 | 1.4 |
| 23 | 3.3 | O | Me | Me | F | 4 | 2 | 0/3 | 1.1 |
Fischer 344 rats were dosed IV with LFIs followed 20 to 30 minutes later by 10µg of LT (10 µg PA + 10 µg LF) dosed IV.
Number of survivors per group. Survival curves for all LFIs except 23 versus controls showed statistical significance (P<0.05). When comparing curves for individual LFIs at 5.0 mg/kg, significance (P<0.05) was observed for 1b vs. 4b, 4b vs. 5b, and 4b vs. 22; but was not considered significant for 1b vs. 19, or 4b vs.18.
rMST = MST(LFI)/ MST(controls) where vehicle treated controls (MST = 70.7 ± 5.1 minutes, n=9); when less than 50% deaths occurred the average survival time was used: rMST = aveST(LFI)/MST(controls).
In the first experiment (Table 3), the survival curves10 for all of the LFIs tested were found to be statistically different (P<0.05) relative to the vehicle control group. Since a previous study had shown that LFI 1a was capable of providing 100% protection at 10, 5, and 2.5 mg/kg in the rat LT model,9 LFI 1a served as the standard to which the new analog data were compared. In this experiment, LFI 1a and two other compounds demonstrated complete protection when dosed at 5.0 mg/kg, although 1a was unable to provide full protection the at the 2.5 mg/kg dose. Interestingly, the removal of the methyl group from the meta-position of the terminal phenyl ring (R3 = Me to H) to give 1b led to a significant increase in efficacy providing 100% protection without morbidity at both the 5 and 2.5 mg/kg doses even though the intrinsic potency of 1b is 5-fold lower relative to 1a. This same trend appears to be present in the two atom linking group series where 3b provides qualitatively better protection relative to the methyl homolog 3a (P = 0.12) suggesting that the R3 methyl group poses a metabolic liability with respect to in vivo efficacy. In support of this hypothesis, the C2-side chain present in these compounds fits the classic pharmacophore model supporting CYP2D6 activity; an alkylarylamine with a site of oxidation, in this case the benzylic methyl group, located 5 to 7 Å from the amine nitrogen atom.15 In regard to these experiments, the rat possesses six CYP2D isoforms, four of which are similar to human CYP2D6 in their ability to metabolize xenobiotic molecules.16 This finding alerted us to the possibility that the lower efficacy observed in the a-series relative to the b-series may be due to CYP2D metabolism and should be considered in future inhibitor design. Comparing LFI 5a with 1a suggests that the change in the X-group from NH to CH2 had no measurable effect on activity (5a vs. 1a, P = 0.34). What came as a surprise was the finding that removal of a methyl group (R2) from the core structure aryl ring of 1a to give 6a, while having only a minor effect on intrinsic potency (Table 3), resulted in a dramatic loss of in vivo efficacy (P = 0.026). While this result appears counterintuitive to the metabolic liability expected for decreasing the number of benzylic methyl groups in a molecule, the data for 8a and 17 also support this finding.
In a second experiment using LFI 1b as the standard (Table 4), we were interested in determining the effect of changing the X-group and variations in chain length on in vivo efficacy. With the exception of LFI 23, the survival curves10 for each LFI tested were found to be statistically different (P < 0.05) relative to the control group, though in this case, none of the compounds provided 100% protection. Unlike in the previous experiment where LFI 1b was fully protective at both doses tested, only 50% protection was observed in this study at the 5.0 mg/kg dose. Even with this difference, the data obtained from this experiment still supports certain trends.
For example, when comparing a change in the X-group on efficacy, the data for LFIs 1b, 4b, and 5b suggest that the phenoxyacetic acid derivatives (4, X=O) are less effective in this model than the aniline (1b vs 4b, P=0.011) or carbon series (5b vs 4b, P=0.011). While the limited data available from this experiment and the presence of survivors for 18 make this trend less convincing, comparing the data for 6a versus 8a (Table 3, P=0.025) and subsequent experiments testing this hypothesis (data not shown) provide additional support for this conclusion. The data in Table 4 also suggest that an increase in chain length between the secondary amine nitrogen and the terminal aryl ring does not have a significant impact on in vivo efficacy (cf. 1b, p=1 to 19, p=3; P=0.130). In contrast, adding an additional methylene to the C2 side chain to give 6-amino analogs (o=4) provided LFIs with good intrinsic potency (20 to 23) but no ability to protect against LF intoxication. This effect is best observed by comparing 20 and 21 with compound 3b in Table 3 where complete protection by the latter compound was observed at the same dose of LFI. Further, the 2 to 3-fold increase in survival time when comparing 22 and 23 with 4b, while modest (4b vs. 22, P=0.004) is also supportive of this positional effect. This lack of in vivo efficacy without a commensurate loss of intrinsic potency implies a metabolic liability is associated with the secondary amine at position 6 of the C2 side chain that is absent in the 5-amino series.
In summary, we have used the SAR from in vitro and in vivo experiments to identify structural features in new LFIs with improved efficacy relative to 1a and 2 in the rat LT challenge model. Part 3 of this series will present a closer look into how variation in the X-group and additional changes to the C2-side chain can affect in vivo efficacy in the rat LT challenge model.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health for their support of this work with grants R44 AI052587 and U01 AI078067. Animal studies were supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 2.Friedlander AM. Chapter 22. In: Sidell FR, Takafuji ET, Franz DR, editors. Medical Aspects of Chemical and Biological Warfare. Washington D.C.: TMM Publications; 1997. p. 467. [Google Scholar]
- 3.Jernigan JA, Stephens DS, Ashford DA, Omenaca C, Topiel MS, Galbraith M, et al. Emerg. Infect. Diseases. 2001;7:933. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moayeri M, Leppla SH. Mol. Aspects Med. 2009;30:439. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Klein F, Hodges DR, Mahlandt BG, Jones WI, Haines BW, Lincoln RE. Science. 1962;138:1331. doi: 10.1126/science.138.3547.1331. [DOI] [PubMed] [Google Scholar]; (b) Pezard C, Berche P, Mock M. Infect. Immun. 1991;59:3472. doi: 10.1128/iai.59.10.3472-3477.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shoop WL, Xiong Y, Wiltsie J, Woods A, et al. Proc. Natl. Acad. Sci. USA. 2005;102:7958. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For more information regarding the use of this rule see: US FDA Guidance for Industry. Animal Models — Essential Elements to Address Efficacy Under the Animal Rule. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm078923.pdf.
- 8.Newman ZL, Printz MP, Li S, Crown D, Breen L, Miller-Randolph S, Flodman P, Leppla SH, Moayeri M. PLoS Pathog. 2010;6:e1000906. doi: 10.1371/journal.ppat.1000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao G-S, Kim S, Moayeri M, Cregar-Hernandez L, McKasson L, Margosiak SA, Leppla SH, Johnson AT. Bioorg. Med. Chem. Lett. 2010;20:6850. doi: 10.1016/j.bmcl.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Synthetic routes to compounds 3a, 5a, 13, 14, 15, and 16, along with protocols for the biochemical and in vivo assays, and survival curves for the data given in Tables 3 and 4 are provided as supplementary material.
- 11.Johnson A, Jiao G-S, Kim S. WO 2009/008905 A1
- 12.Evans DA, Ennis MD, Mathre DJ. J. Am. Chem. Soc. 1982;104:1737. [Google Scholar]
- 13.Turk BE, Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC, Cantley LC. Nature Struct. Mol. Biol. 2004;11:60. doi: 10.1038/nsmb708. [DOI] [PubMed] [Google Scholar]
- 14.Overton JP, Al-Lazikani B, Hopkins AL. Nat. Rev. Drug Discov. 2006;5:993. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 15.Smith DA, van de Waterbeemd H, Walker DK. In: Methods and Principles in medicinal Chemistry. Mannhold R, Kubinyi H, Timmerman H, editors. Vol. 13. Weinheim: Wiley-VCH Verlag GmbH; 2001. pp. 78–80. [Google Scholar]
- 16.Venhorst J, ter Laak AM, Commandeur JNM, Funae Y, Hiroi T, Vermeulen NPE. J. Med. Chem. 2003;46:74. doi: 10.1021/jm0209578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.