

Abstract
This article reviews the role of amyloid-β (Aβ) and mitochondria in synaptic damage and cognitive decline found in patients with Alzheimer’s disease (AD). Recent molecular, cellular, animal model, and postmortem brain studies have revealed that Aβ and mitochondrial abnormalities are key factors that cause synaptic damage and cognitive decline in AD. Aβ is reported to accumulate in subcellular compartments and to impair the normal function of neurons in AD patients. Further, recent studies using biochemical methods and electron microscopy have revealed that the accumulation of Aβ at nerve terminals affect synaptic activities, including the release of neurotransmitters and synaptic vesicles. Recent studies of the relationship between mitochondria and Aβ in AD patients suggest that in mitochondria, structural changes caused by Aβ result in increased mitochondrial fragmentation, decreased mitochondrial fusion, mitochondrial dysfunction, and synaptic damage. This paper discusses the latest research on Aβ, mitochondria, age-dependent factors of AD in the brain, and synaptic damage in AD. This paper also briefly discusses potential mitochondrial therapeutics in the treatment of patients with AD.
Keywords: Amyloid beta, Amyloid precursor protein, Synaptic pathology, Mitochondrial therapeutics
INTRODUCTION
Alzheimer’s disease (AD) is a late-onset mental illness that is characterized by the loss of memory and an impairment of multiple cognitive functions [1–3]. The major pathological features in the brains of AD patients are the presence of intra-neurofibrillary tangles and extracellular protein amyloid-β (Aβ) deposits, particularly in the regions related to memory and cognition [4]. Currently, 5 million Americans suffer from AD [5]. It is estimated that by the year 2050, 50% of people worldwide (approximately 370 million) who are 85 years of age or older will be afflicted with AD [4,6]. With such a large, aged population poised to be afflicted, AD has become a major health concern that must be reckoned with. Despite tremendous progress that has been made in AD research, we still do not have early detectable markers, nor agents or drugs that can slow the progression of AD.
Causal factors for Alzheimer’s disease
Genetic mutations in the amyloid-β protein precursor (AβPP), presenilin 1 (PS1), and PS2 genes cause a small proportion (about 2%) of all AD cases (early-onset or familial AD; Table 1); however, causal factors are still unknown for a vast majority of AD patients (late-onset AD). Recent genetic studies have identified several risk factors for late-onset AD, including genetic variants in the sortilin-related receptor 1 gene; clusterin (a protein associated with the clearance of cellular debris); the complement component receptor 1; and apolipoprotein E4 (ApoE ε4) genotype [7–13] (Table 1). In the last decade, tremendous progress has been made in understanding the role of ApoE ε4 involvement in AD progression and pathology [14–21]. In addition, several other factors, including epigenetics, lifestyle, diet, and environmental exposure may contribute to the development of late-onset AD [3]. Recently, oxidative stress and mitochondrial abnormalities have been implicated in the development of late-onset AD [22–26], and aging has been identified as the ‘number 1’ risk factor in AD progression and pathology.
Table 1.
Genes and risk factors involved in Alzheimer’s disease pathogenesis
| Chromosome | Gene | Phenotype/Pathology | Ref. |
|---|---|---|---|
| Genes in Familial AD | |||
| 21 | Amyloid-β protein precursor | Mutations in AβPP are causal factors of FAD and are involved in the increased production of all Aβ | [7] |
| 14 | Presenilin 1 | Mutations in PS1 are causal factors of FAD and are involved in gamma secretase activity and in the increased production of Aβ1-42 | [8] |
| 1 | Presenilin 2 | Mutations in PS2 are causal factors of FAD and are involved in gamma secretase activity and in the increased production of Aβ1-42 | [9] |
| Genes Involved in AD as a Risk Factor | |||
| 19 | Apolipoprotein E allele 4 | E4 polymorphism is a risk factor for late-onset AD and is involved in the increased production of Aβ | [10] |
| 11 | Sortilin related receptor | Involved in increased production of Aβ | [11] |
| 8 | Clusterin | Clusterin is expressed abundantly in the brain and involved in clearing of Aβ from brain to plasma. However, it is also reenter to the brain, and involved in decreased clearance of Aβ | [12,13] |
| 1 | Complement component receptor 1 | Involved in clearance of Aβ. However, variants in complement component receptor 1 interfere with Aβ clearance | [12,13] |
Sites of pathology in Alzheimer’s disease
Anatomical and immunohistochemical analyses of AD postmortem brains and the brains from AD transgenic mice revealed that a neurodegenerative process is initiated in layer 2 of the entorhinal cortex [4]. This process spreads to the hippocampus, temporal cortex, frontoparietal cortex, and, finally, to subcortical nuclei. Interestingly, in AD patients, these regions of the brain are involved in learning, memory, and cognitive functions [4]. Aβ secretion also occurs mainly in these regions, as do Aβ deposits; however, the reasons for Aβ secretion and the formation of Aβ deposits in these areas are not fully understood.
Cellular changes
More than two decades of intense research has revealed that AD is a complex, heterogeneous disease, with multiple cellular changes implicated in its pathogenesis [1,2,4,8,27,28]. Major cellular changes that have been implicated in AD are: 1) Aβ and amyloid cascade events; 2) hyperphosphorylation of tau and intracellular neurofibrillary tangles; 3) synaptic pathology and neuronal loss; 4) mitochondrial structural and functional abnormalities; and 5) inflammatory responses [22,29–46].
Early events in Alzheimer’s disease progression
Although multiple cellular changes have been reported to be involved in AD pathogenesis, synaptic pathology and mitochondrial oxidative damage have been identified as early events in AD progression and pathogenesis [3]. It is generally accepted that an accumulation of Aβ in synapses and in synaptic mitochondria, particularly in neurons affected by AD, cause synaptic degeneration and cognitive decline in AD patients [2,3,47–50].
In this article, we first focus on Aβ, its generation, accumulation, and age-related factors of Aβ; next we focus on mitochondrial structural and functional abnormalities, and synaptic damage in AD progression and pathology.
AMYLOID-β
In AD, Aβ, the 39–43 amino acid residue protein, is a major component of neuritic plaques found in brain regions known to be responsible for learning and memory [51]. Aβ is generated by proteolysis of AβPP by the sequential enzymatic actions of β-site AβPP cleaving enzyme 1 (BACE 1), β-secretase, and γ secretase. In early-onset AD, genetic mutations in AβPP, PS1, and PS2 genes activate β- and γ-secretases, and cleave Aβ [3]. AβPP mutations that flank the Aβ domain increase the production of Aβ42 (Fig 1). In late-onset AD, it has been hypothesized that factors related to oxidative stress may be involved in activating β- and γ-secretases, cleaving AβPP, and releasing Aβ [25] (Fig. 1). Recently, several in vitro and in vivo studies have provided experimental evidence to support to this hypothesis [51–56] (Fig. 1). In both early onset and late-onset AD, levels of Aβ are steady-state and are controlled by the production of Aβ, the clearance of Aβ, and the degradation of Aβ. Decreased clearance of Aβ or the overproduction of Aβ may lead to an accumulation of Aβ in subcellular compartments and may initiate a cascade of events in the brain, a process referred to as “Aβ cascade hypothesis” [57]. Interestingly, Aβ can self-aggregate into multiple forms, ranging from 4 kDa monomers to oligomers and to fibrils. These fibrils eventually form β-pleated sheets, insoluble fibers, and deposits (Fig. 2). Soluble oligomers are the most toxic form of Aβ for neurons. Anatomical analyses of AD postmortem brains and AD transgenic mice revealed that Aβ secretion occurs mainly in the entorhinal cortex, hippocampus, temporal cortex, and frontoparietal cortex of the brain. These areas are important for learning, memory, and cognitive functions. The reasons for Aβ secretion and formation of amyloid deposits in these areas are not fully understood. However, it is possible that the capability of Aβ clearance is low in these areas or that oxidative stress is high, in which case oxidatively damaged neurons may produce more Aβ. This loop—increased production of Aβ and decreased clearance—may lead to an excessive accumulation of Aβ in brains.
Figure 1.

Amyloid-β secretion in AD neurons. In early-onset AD, mutations AβPP, PS1, and PS2 genes activate β- and γ-secretases, cleave Aβ. In late-onset AD, oxidative stress related factors (O2•, H2O2, OH), risk factors including ApoE4, sortilin related receptor 1, clusterin, and complement component either activate secretases or decrease Aβ clearance in AD neurons. The cleaved Aβ accumulate in subcellular compartments, including mitochondria, endoplasmic reticulum, Golgi-network, lysosomes, and multivesicular bodies, and disrupt the functions of these subcellular organelles and damage neuronal function.
Figure 2.
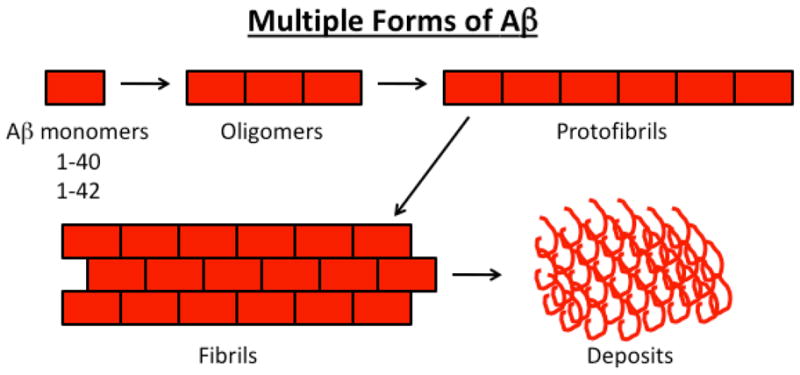
Formation of amyloid-β species and deposits in AD brain.
Recent studies have revealed that Aβ is secreted wherever AβPP and the β- and γ-secretases are present. AβPP, and the β- and γ-secretases, are localized to several cellular compartments, including the endoplasmic reticulum, plasma membrane, trans-Golgi network, and multivesicular bodies [2,3]. Because of the presence of AβPP and the β- and γ-secretases in these subcellular compartments, Aβ is known to be present in these regions. In addition, several studies reported that Aβ is localized to mitochondrial membranes [37,38,46].
Age-dependent increase in the production of Aβ
Multiple lines of evidence suggest that aging is a key factor for the increased production of Aβ and the decrease in Aβ-degrading enzymes in the AD brain. A time-course analysis of Aβ in AD transgenic mouse lines revealed that Aβ levels and deposits increase in AD-affected regions of the brain in an age-dependent manner [2,38]. In addition, studies of postmortem brains from aged humans with mild cognitive impairment (MCI) and patients with AD found an age-dependent increase of Aβ levels in the aged persons with MCI and in AD patients [58]. These studies suggest that aging plays a key role in the production and accumulation of Aβ in the brains of AD patients and in AD transgenic mice.
In the last decade, a large body of research has been devoted to understanding Aβ toxicity, particularly intracellular Aβ. It is now generally accepted that extracellular Aβ deposits are the by-products of AD pathology. Recent studies of AD patients and AD transgenic mice found intracellular Aβ present in AD-affected brain regions [38,58,59–67]. Aβ1-42 participates mainly in fibrillogenesis and the formation of Aβ deposits. It is generally accepted that intracellular Aβ has been found to precede extracellular Aβ deposits in AD brains [68]. In addition, several studies of AD transgenic mice reported that intracellular Aβ accumulates early in AD progression [2,3].
Aging and deceased levels of Aβ-degrading enzymes
Several molecular and cellular studies have revealed that Aβ-degrading enzymes, including nephrilysin (NEP) and the insulin-degrading enzyme (IDE), decrease as disease progresses in AD patients [69–72].
Hellström-Lindahl and colleagues [69] investigated whether decreased NEP levels contribute to the accumulation of Aβ in AD patients and in aged persons without AD. Protein levels of NEP were reduced in the temporal and frontal cortex of brains from AD patients and aged patients without AD. They found an inverse correlation between NEP and insoluble Aβ levels in both groups, suggesting that NEP is involved in the clearance of Aβ. The observed, age-dependent decline in NEP may be related to the increased levels of Aβ found in aged patients without AD, during normal aging. \
Mohajeri et al. [70] measured NEP levels in AD mice. Neuronal upregulation of NEP in young AD transgenic mice expressing the mutant AβPP led to the reduction of Aβ levels and the delayed formation of Aβ deposits. In contrast, a comparable increase of NEP levels in the brains from aged AβPP mice (swe) with pre-existing Aβ deposits did not result in a significant reduction of plaque pathology. They suggested that the use of NEP for AD therapy might be most effective early in the course of AD pathophysiology since NEP is age-dependent.
Apelt and collaborators [71] measured the levels of mRNA and proteins of NEP in AβPP transgenic mice during postnatal maturation and aging. NEP levels were decreased in the cerebral cortex of mice 2–22 months old, independent of their transgene status. Immunocytochemistry revealed few NEP-positive dystrophic neurites around Aβ plaques and an up-regulation of NEP in plaque-surrounding reactive astrocytes, which suggests a role for Aβ deposit-mediated astrogliosis in Aβ degradation.
Iwata et al. [72] sought to determine whether spatial changes in NEP correlate with Aβ in AD-affected regions of brains from AD transgenic mice. When NEP levels in various brain regions of 10-, 80-, and 132-week-old AD transgenic mice were evaluated by an NEP-dependent, endopeptidase-activity assay and Western blot quantitative analysis, a clear change in NEP levels was observed in the hippocampal formation, levels reduced by 20% at 132 weeks, compared to the 10-week group. In addition, quantitative immunohistochemical analysis confirmed the reduction of NEP levels in the outer molecular layer and in the polymorphic layer of the dentate gyrus, and in the stratum lucidum of the hippocampus, by 56%, 82%, and 83% respectively in the mice at 132 weeks, compared to the 10-week group. NEP levels were decreased at the terminal zones in axons of the lateral perforant path and in mossy fibers. These are also the brain sites that exhibit disease pathology in mutant AβPP transgenic mice and synaptic loss in AD patients.
Similar to NEP, IDE is also an important enzyme that is involved in Aβ clearance and that has been found to be decreased in the brains of aged persons without AD [73,74]. The concentration of IDE and its activity were significantly decreased in the hippocampus in the brains from aged humans without MCI, compared to humans who were mildly cognitively impaired and aged persons who were considered at high risk to develop AD [73]. Membrane-bound IDE concentrations and IDE activity in the hippocampus continued to decrease as the patients progressed from MCI to mild-severe AD. Most interestingly, IDE activity in thehippocampal membrane negatively correlated with Aβ42 in the brains from aged persons with MCI and with AD. Findings from the Zhao et al. [73] study suggest that interventions aimed at promoting membrane-bound IDE activities in the brain of aged persons with MCI may help to prevent the onset and possibly the progression of AD through mechanisms involving the clearance of monomeric Aβ from the brain.
Farris and colleagues [74] recently studied the connection between IDE gene and Aβ using a rat model for IDE. In a well-characterized rat model of type 2 diabetes mellitus, they found naturally occurring IDE missense mutations, which decreased catalytic efficiency, and they found a significant deficit (about 15 to 30%) in the degradation of both insulin and Aβ. Endogenously secreted Aβ40 and Aβ42 were significantly elevated in primary neuronal cultures from animals with the IDE mutations. These researchers concluded that naturally occurring, partial loss-of-function mutations in IDE were sufficient to cause diabetes mellitus 2 and impaired neuronal regulation of Aβ levels. However, they noted that the brain apparently compensates for the partial deficit during the life span of the rat [74].
MITOCHONDRIA AND ALZHEIMER’S DISEASE
Mitochondrial dysfunction in AD pathogenesis was described two decades ago, but its underlying mechanisms were not clear until recently. Mitochondrial dysfunction has been found and described in postmortem brains from patients with AD [39,75–77], in their platelets [78], in AD transgenic mice [34,37,38,79–81], and in cell lines that express mutant AβPP and/or cells treated with Aβ [82–84].
Increasing evidence suggests that mitochondrial abnormalities play a large role in AD pathogenesis. Decreased mitochondrial enzymes, including cytochrome oxidase activity, pyruvate dehydrogenase, and α-ketodehydrogenase were found in fibroblasts, lymphoblasts, and postmortem brains from AD patients and age-matched control subjects [reviewed in 3]. Further, a recent study described abnormal mitochondrial dynamics in fibroblasts from AD patients, indicating that impaired mitochondrial dynamics are involved in AD pathogenesis [85]. Several other studies found increased free radical production, lipid peroxidation, oxidative DNA damage, oxidative protein damage, decreased ATP production, and decreased cell viability in brains from AD patients compared to those from age-matched control subjects [39,75–77,86].
In the 1990s, Swerdlow and colleagues [88] studied mitochondrial function using a cytoplasmic hybrid (cybrid) approach to determine the role of mitochondrial DNA (mtDNA) in AD pathogenesis [87,88]. They isolated platelets from AD and age-matched control subjects, and fused those platelets with both human neuroblastoma (SH-SY5Y) cells and human teratocarcinoma (NT2) cells depleted of their endogenous mtDNA [87,89]. The cells lacking mtDNA did not exhibit mitochondrial functional activities; that is, only those cells with mtDNA exhibited intact mitochondrial functional activities. However, cells containing cybrids of AD and control subjects exhibited mitochondrial respiratory activities, with a difference between the cybrid cell lines containing AD subject mitochondria and cybrid cell lines with control subject mitochondria. AD cybrid cell lines exhibited increased Aβ42 production and mitochondrial dysfunction: the cytochrome oxidase activity was lower, free radical production and oxidative stress markers were elevated, calcium homeostasis was altered, the mitochondrial membrane potential was reduced, and apoptosis pathways were altered [88]. Findings from these cybrids studies further support that mitochondria are involved in AD pathogenesis.
In other studies, increased mitochondrial DNA changes were found in postmortem brain tissue from AD patients and aged-matched control subjects, compared to DNA changes in brain tissue from young control subjects without AD [90,91]. These findings suggest that the accumulation of mitochondrial DNA in AD pathogenesis is age-related.
The Reddy laboratory [34,92] and others [93–95] found that mitochondrial encoded genes were abnormally expressed in AD postmortem brains and in those from AD transgenic mice. Recently, we [34] investigated gene expression profiles in brain slices from AβPP transgenic mice at 3 stages of AD progression: long before (2 months of age), immediately before (5 months), and after (18 months) the appearance of Aβ plaques in the cerebral cortex [34]. We compared those profiles to those of age-matched wild-type mice. Our analysis revealed that the genes related to mitochondrial energy metabolism and apoptosis were upregulated in the 2-month-old AβPP transgenic mice and that the same genes were upregulated in these mice at 5 and 18 months of age. In another study, we found decreased cytochrome oxidase, increased free radicals, and increased carbonyl proteins in the 2-month-old AβPP transgenic mice compared to the age-matched wild-type mice [38]. Taken together, these results suggest that mitochondrial energy metabolism is impaired by mutant AβPP and/or Aβ, and that the upregulation of mitochondrial genes may be a compensatory response to this impairment. Further, we found abnormal mitochondrial gene expression in the 2-month-old AβPP transgenic mice, suggesting that mitochondrial dysfunction is an early event in AD progression.
Using quantitative RT-PCR techniques, the Reddy laboratory also analyzed mRNA expression in 11 mitochondrial-encoded genes from the frontal cortex of 3 subject groups: patients with early AD, patients with definite AD, and age-matched control subjects [92]. This analysis revealed a down-regulation of mitochondrial genes in complex I of electron transport chain genes in both early and definite AD brain specimens, but not in the control subjects. In the brain specimens from both the early and definite AD patients, complex I showed a down-regulation of mitochondrial genes, but complexes III and IV showed increased mRNA expressions, suggesting a great demand for energy production in the brains from AD patients. These results suggest that mitochondrial dysfunction is an early event in AD progression and continues into later-stage AD progression, and that abnormal mitochondrial gene expression may be a compensatory response to mutant AβPP- and Aβ-initiated mitochondrial toxicity.
Further, the Reddy laboratory [38] and others [37,39,41,46,96] found that AβPP and Aβ are localized to mitochondrial membranes and is responsible for generating free radicals and initiating mitochondrial dysfunction. Other groups found presequence peptidase, a peptidase that is known to degrade Aβ species, in the mitochondria of AD neurons [97], further supporting the association of Aβ with mitochondria and mitochondrial dysfunction in AD. In addition, recent studies of mitochondrial structure and of neuronal cells expressing mutant AβPP in brain tissues from AD patients found that Aβ fragments mitochondria and causes structural changes in neurons [55,98,99].
Overall, findings from these studies suggest that mitochondrial abnormalities occur early in AD progression.
AβPP, Aβ, AND ABNORMAL MITOCHONDRIAL DYNAMICS IN ALZHEIMER’S DISEASE
Increasing evidence suggests that mutant AβPP and/or Aβ overexpression cause mitochondrial fragmentation in neurons affected AD [98–100]. In a recent gene expression study of AD transgenic mice, the Reddy laboratory [34] found increased expression of mitochondrial-encoded genes in AD affected regions of the brain that may be due to the excessive production of mitochondria. This overproduction of mitochondria may be due to mutant AβPP and Aβ toxicity in neurons affected by AD [34,38,99].
The relationship between the overexpression of mutant AβPP and Aβ, and the increased production of mitochondria is supported by several studies.
AβPP, and monomeric and oligomeric forms of Aβ have been found in mitochondrial membranes [37–39,46,79,96,101]. In the Lustbader et al. study [79], they found Aβ normally interacting with the mitochondrial matrix protein ABAD, leading to mitochondrial dysfunction. Caspersen et al. [37] found an accumulation of Aβ in the mitochondria from postmortem brain specimens of AD patients and AβPP transgenic mice. Recently, the Reddy laboratory found Aβ monomers and oligomers in mitochondria isolated from the cerebral cortex of AβPP transgenic mice [38] and from N2a cells expressing AβPP. Our digitonin fractionation analysis of isolated mitochondria from AβPP transgenic mice revealed Aβ in the outer and inner membranes and matrix of mitochondria. Our study also found that mitochondrial Aβ decreases cytochrome oxidase activity and increases free radicals and carbonyl proteins. Yao et al. [46] found Aβ mitochondrial membranes in cortical tissues from triple transgenic mice.
Using confocal and electron microscopy, and human neuroblastoma (M17) cells transfected with wild-type or mutant AβPP, Wang and coworkers [98] investigated the effects of AβPP and Aβ on mitochondrial structural changes. Confocal and electron microscopic analysis revealed that about 40% of M17 cells overexpressing wild-type AβPP and more than 80% of M17 cells overexpressing mutant AβPP displayed alterations in mitochondrial morphology, particularly fragmented mitochondria. They also found that increased levels of Fis1 are critical for mitochondrial fission in AβPPwt and AβPPswe M17 cells. The overexpression of AβPP and/or Aβ-derived diffusible ligand treatment also led to mitochondrial fragmentation and morphological changes.
Using electron and confocal microscopy, gene expression analysis, and biochemical methods, the Reddy laboratory studied mitochondrial structure and function, and neurite outgrowth in neurons treated with Aβ [99]. In neurons treated with only Aβ, we found increased expressions of mitochondrial fission genes (Drp1 and Fis1) and decreased expressions of fusion genes (Mfn1, Mfn2, and Opa1), indicating abnormal mitochondrial dynamics in AD neurons. mRNA expression of antioxidant enzyme-encoded genes (peroxiredoxins 1–6) was significantly decreased in neurons treated with Aβ relative to untreated neurons. Our electron microscopy of neurons treated with Aβ revealed a significant increase in mitochondrial fragmentation, further supporting abnormal mitochondrial dynamics. We also found significantly decreased neurite outgrowth and decreased mitochondrial function in cells treated with Aβ [99]. These findings suggest that Aβ fragments mitochondria and causes abnormal mitochondrial dynamics, leading to mitochondrial dysfunction.
Zhao and colleagues [102] studied the effects of wild-type and an arctic form of Aβ42 using neurons from adult flies. They performed extensive time-course analyses to determine the function and structure of both axon and presynaptic terminals of individual neurons. They found Aβ accumulated intracellularly, and they found a wide range of age-dependent changes, including the depletion of presynaptic mitochondria, a slow-down of bi-directional transports of axonal mitochondria, decreased synaptic vesicles, increased large vacuoles, and elevated synaptic fatigue. These structural and functional synaptic changes correlated with age-dependent deficits in the motor behavior of the flies. Such changes were accelerated in flies expressing the arctic form of Aβ. The depletion of presynaptic mitochondria was the earliest phenotype that they were able to detect in the fly. Zhao et al. [102] determined this depletion was not caused by the change in axonal transport of mitochondria. They also found a dramatic reduction in the number of axonal mitochondria and also a significant increase in their size, in aged Aβ-expressing flies, suggesting a global depletion of mitochondria in the neuron and an impairment of mitochondria fission. These results suggest that Aβ accumulation depletes presynaptic and axonal mitochondria, leading to other presynaptic deficits.
Taken together, these findings suggest Aβ enters mitochondria and causes abnormal mitochondrial dynamics in neurons that are known to be affected in AD, and that such abnormal mitochondrial dynamics cause mitochondrial dysfunction and abnormal mitochondrial trafficking in AD neurons.
Abnormal mitochondrial trafficking in AD
In a process called mitochondrial trafficking, mitochondria travel along the axons and dendrites to supply energy to nerve terminals for normal neural communication; then they travel back to the cell body [103]. Mitochondria are transported from the cell body back to nerve terminals via an anterograde mechanism and from nerve terminals to the cell body via a retrograde mechanism. In healthy neurons, anterograde and retrograde transport of mitochondria are equal and active. In AD neurons, both anterograde and retrograde transport of mitochondria are slow because of the presence of large number of defective and functionally inactive mitochondria [104–105]. As discussed earlier, AD mitochondria with an accumulation of Aβ disrupt mitochondrial function and inhibit ATP production. These Aβ-laden mitochondria are not able to supply sufficient levels of energy to the nerve terminals, which may impair neurotransmission and may ultimately result in synaptic damage, neurodegeneration, and cognitive decline in AD patients [103].
Aβ, SYNAPTIC ALTERATIONS, AND MITOCHONDRIAL DAMAGE IN ALZHEIMER’S DISEASE
Synapses are the sites of high-energy demand [3]. Synaptic damage is considered the earliest cellular event in AD pathogenesis, and synaptic loss is the best correlate of cognitive impairment in AD [29–31,106]. Damaged synapses due to insufficient mitochondrial ATP lead to synaptic degeneration [3]. Synapses and neurites are mostly damaged in the vicinity of Aβ plaques [107,108].
In healthy subjects, synaptic terminals transmit signals between cells in order to process information. During aging, the number of synapses and their transmissions of signals dramatically decrease [109, 110]. The decrease in synapses has been documented in different brain regions of aged persons, supporting the hypothesis that synaptic changes are ubiquitous features of normal brain aging [3].
Synaptic loss and Alzheimer’s disease
Several studies using electron microscopy and AD postmortem brains revealed a loss of synapses in the hippocampus of AD patients compared the number of synapses in control subjects [39,111,112]. This loss correlates with cognitive decline in AD patients. Bretoni-Freddari et al. [111] studied the number of synapses per neurons in cerebellar and hippocampal brain tissues from adult and aged control subjects and from AD-affected and unaffected brain tissues in patients with AD. The synapse-to-neuron ratio varied according to the brain regions from which the samples were taken and the individual’s health. No significant differences in the synapse-to-neuron ratio were found in samples taken from the cerebellum of adult and aged persons without AD and of aged AD patients. However, in the hippocampal samples, the synapse-to-neuron ratio decreased more than 50% in the adult and aged persons without AD, compared to the ratio in the aged AD patients.
In several studies investigating the extent that synaptic loss correlates with cognitive decline in AD patients [31,112] researchers found a 25–30% decrease in synapses in the cortex and a 15–35% decrease in synapses per cortical neuron, suggesting that synaptic loss in AD patients may correlate more with cognitive decline than with the number of Aβ plaques, neurofibrillary tangles, neuronal loss, or the extent of cortical gliosis.
Loss of synaptic proteins and Alzheimer’s disease
Using immunoblotting and immunohistochemical analyses to determine synaptic proteins, several studies revealed decreased levels of presynaptic (synaptophysin) and postsynaptic proteins (synaptopodin and PSD95) in AD patients compared to age-matched control subjects [113–118], suggesting that presynaptic and postsynaptic proteins are critically involved in AD progression. Further, immunoblotting analysis of postmortem brain tissues from the cerebral cortex of AD transgenic mice also revealed decreased levels synaptic in AD transgenic mice [119], suggesting that the loss of synaptic proteins are confined to brain regions known to be affected in AD.
Oligomeric Aβ, synaptic damage, and impairments in long-term potentiation
Several recent in vitro and in vivo studies revealed that oligomeric Aβ is responsible for a decrease in the long-term potentiation (LTP) and disruption of synaptic plasticity [63,120–125], in addition to Aβ pathology. Intracellular Aβ was detected in 5XFAD mice 1.5 months of age, and cognitive impairments and LTP abnormalities were found in 5XFAD mice 5–6 months of age [63]. In a triple transgenic mouse model of AD, intracellular Aβ was found in mice 3 months of age and LTP impairments in mice 6 months of age, indicating that intracellular Aβ may be critical for synaptic damage and cognitive impairments. In a well-characterized AD transgenic mouse model (Tg2576), significantly decreased synapse density was observed in the outer molecular layer of the dentate gyrus in mice 6–9 months of age and in layers II and III of the cortex in mice 15–18 months of age [106]. These results suggest that synaptic changes caused by soluble Aβ may contribute to the loss of synapses and of synaptic proteins and may be responsible for cognitive decline in AD patients.
In addition to synaptic pathology [120–125], mitochondrial alterations were also observed in several lines of transgenic mice [37,38,46], indicating that both synaptic damage and mitochondrial abnormalities, particularly the accumulation of Aβ in mitochondria, may have a role in triggering cognitive deficits in AD transgenic mice. Recent mitochondrial and synaptic studies revealed that mitochondria are distributed abnormally, suggesting that abnormal mitochondrial distribution may contribute to synaptic damage in AD [102,126]
Further, using electron microscopy and a rapid Golgi method, Baloyannis and collaborators [127] investigated synaptic alterations, including synapses and dendritic spines in subjects with early AD and in control subjects. They found substantial loss of synapases and synaptic alterations in the medial geniculate bodies in neurons from the AD subjects. In particular, mitochondrial alterations and fragmentation of Golgi apparatus were seen in the neurons of the medial geniculate bodies and of the inferior colliculi. These findings suggest that mitochondrial and synaptic alterations in the medial geniculate bodies and inferior colliculi are involved in the impairment of neuronal communication and symbolic sound perception in the early stages of AD progression.
Overall, findings from studies of postmortem brains from AD patients and AD transgenic mice revealed synaptic damage in neurons affected by AD, and suggest that an accumulation of oligomeric Aβ at synapses and synaptic mitochondria cause synaptic damage and cognitive impairments. Therefore, therapeutics targeting Aβ and mitochondria may be helpful in reducing the progression of AD in AD patients.
MITOCHONDRIAL THERAPEUTICS
Currently, several laboratories are involved in developing therapeutic strategies to delay or prevent progression and development of AD. Several groups are focused on developing and testing antioxidants that target mitochondria [128–132] and several others are involved in developing and testing drugs that target Aβ, such as drugs that involve immunotherapy, and β-and γ-secretase inhibitors. Both approaches have shown promise at preclinical levels, meaning they have been successful in AD transgenic mice but essentially unsuccessful in clinical trials. Since synaptic damage and mitochondrial dysfunction have been reported as early pathogenic events associated with aging and AD, it may be possible to treat these pathogenic events by: 1) developing molecules that treat mitochondria (by targeting ROS); these molecules would decrease free radical production and oxidative damage, and boost overall mitochondrial function, which would ultimately increase synaptic branching of neurons; such increased branching would increase neural communication; and 2) therapeutically boosting ATP levels in mitochondria, which would ultimately increase synaptic outgrowth and neuronal connectivity.
Given the significant involvement of mitochondrial dysfunction in aging and AD, it seems reasonable to delay their progression in patients with neurodegenerative diseases such as AD, through antioxidant treatment or a diet supplemented diet with antioxidants. However, recent studies of AD patients’ intake of natural antioxidants gave mixed results. Findings from some epidemiologic studies point to the increased intake of antioxidant vitamins, including vitamin E, vitamin C, and beta carotene, to possibly reduce the risk of developing AD [3]. However, findings from other studies do not. They found that their antioxidant approaches did not decrease the risk of developing AD in elderly people, which suggests that antioxidant approaches will not be effective in treating neurodegenerative diseases because naturally occurring antioxidants, such as vitamins E and C, do not cross the blood-brain barrier and so cannot reach the relevant sites of free radical generation [3]. To overcome these problems and to better assess whether antioxidant approaches may be valuable therapeutic treatments, improved delivery of antioxidants to the brains of AD patients is needed.
In the last decade, tremendous progress has been made in developing mitochondrially-targeted antioxidants that are capable of crossing the blood-brain. To increase the delivery of antioxidants into mitochondria, several antioxidants have been developed: triphenylphosphonium-based antioxidants (MitoQ, MitoVitE, and MitoPBN);the cell-permeable, small peptide-based antioxidant SS31; and mitochondrial-permeability transition pore inhibitors such as Dimebon [128–133]. Several laboratories across the world are investigating neuroprotective molecules including those that can target antioxidants to mitochondria, such as SS31, MitoQ, and Dimebon, but the research is at its infant stages and is currently focused on animal models of AD.
CONCLUSIONS AND FUTURE DIRECTIONS
Increasing evidence suggests that Aβ, mitochondrial dysfunction, and synaptic damage are critically involved in AD progression and development. The latest research into AD revealed that Aβ and mitochondrial abnormalities are key factors that cause synaptic damage in AD neurons. Aβ is reported to accumulate in subcellular compartments and to impair the normal function of neurons. Further, recent in vitro and in vivo studies of Aβ using biochemical methods and electron microscopy revealed that the accumulation of Aβ at nerve terminals damages synaptic activities, including the release of neurotransmitters and synaptic vesicles. Further, recent discoveries of mitochondria in AD suggest that structural changes in mitochondria, including increased mitochondrial fragmentation and decreased mitochondrial fusion, are critical factors associated with mitochondrial dysfunction and synaptic damage in AD. Despite tremendous progress that has been made in AD research, we still do not have drugs or other agents to prevent or slow down disease progression. Further, we still do not know the precise toxic effects that are caused by Aβ and mitochondrial abnormalities at synapses, particularly in neurons, that are involved in cognitive decline. Further research is needed to develop drugs capable of crossing the blood-brain barrier and targeting mitochondria, and to develop the agents to boost mitochondrial function and decrease Aβ toxicity and improve synaptic branching and cognitive functions in elderly people and patients with AD.
Acknowledgments
The research presented in this paper was supported by National Institutes of Health (AG028072, AG026051, and RR00163), the Alzheimer’s Association (IIRG-09-92429), and Medivation, Inc.
References
- 1.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 3.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy PH, McWeeney S. Mapping cellular transcriptosomes in autopsied Alzheimer’s disease subjects and relevant animal models. Neurobiol Aging. 2006;27:1060–1077. doi: 10.1016/j.neurobiolaging.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Alzheimer Association Report. Alzheimer’s Disease Facts and Figures. 2009. [Google Scholar]
- 6.Salloway S. Current and future treatments for Alzheimer’s disease. CNS Spectr. 2009;14:4–7. doi: 10.1017/s1092852900024895. discussion 16–8. [DOI] [PubMed] [Google Scholar]
- 7.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 8.Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, White JA, Bonnycastle L, Weber JL, Alonso ME, et al. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science. 1992;258:668–671. doi: 10.1126/science.1411576. [DOI] [PubMed] [Google Scholar]
- 9.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 10.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, Chen F, Shibata N, Lunetta KL, Pardossi-Piquard R, Bohm C, Wakutani Y, Cupples LA, Cuenco KT, Green RC, Pinessi L, Rainero I, Sorbi S, Bruni A, Duara R, Friedland RP, Inzelberg R, Hampe W, Bujo H, Song YQ, Andersen OM, Willnow TE, Graff-Radford N, Petersen RC, Dickson D, Der SD, Fraser PE, Schmitt-Ulms G, Younkin S, Mayeux R, Farrer LA, St George-Hyslop P. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossù P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanché H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alpérovitch A, Lathrop M, Amouyel P European Alzheimer’s Disease Initiative Investigators. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 13.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O’Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004;25:641–650. doi: 10.1016/j.neurobiolaging.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhong N, Weisgraber KH. Understanding the basis for the association of apoE4 with Alzheimer’s disease: opening the door for therapeutic approaches. Curr Alzheimer Res. 2009;6:415–418. doi: 10.2174/156720509789207921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbalip.2010.02.007. in press. [DOI] [PubMed] [Google Scholar]
- 19.Urosevic N, Martins RN. Infection and Alzheimer’s disease: the APOE epsilon4 connection and lipid metabolism. J Alzheimers Dis. 2008;13:421–435. doi: 10.3233/jad-2008-13407. [DOI] [PubMed] [Google Scholar]
- 20.Struble RG, Cady C, Nathan BP, McAsey M. Apolipoprotein E may be a critical factor in hormone therapy neuroprotection. Front Biosci. 2008;13:5387–5405. doi: 10.2741/3088. [DOI] [PubMed] [Google Scholar]
- 21.Bookheimer S, Burggren A. APOE-4 genotype and neurophysiological vulnerability to Alzheimer’s and cognitive aging. Annu Rev Clin Psychol. 2009;5:343–362. doi: 10.1146/annurev.clinpsy.032408.153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 23.Swerdlow RH, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: an update. Exp Neurol. 2009;218:308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010 doi: 10.3233/JAD-2010-100339. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reddy PH. Amyloid precursor protein-mediated free radicals and oxidative damage: implications for the development and progression of Alzheimer’s disease. J Neurochem. 2006;96:1–13. doi: 10.1111/j.1471-4159.2005.03530.x. [DOI] [PubMed] [Google Scholar]
- 26.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 27.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 28.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 29.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 30.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 31.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 32.Smith MA, Perry G. Free radical damage, iron, and Alzheimer’s disease. J Neurol Sci. 1995;134 (Suppl):92–94. doi: 10.1016/0022-510x(95)00213-l. [DOI] [PubMed] [Google Scholar]
- 33.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 35.Perry G, Raina AK, Nunomura A, Wataya T, Sayre LM, Smith MA. How important is oxidative damage? Lessons from Alzheimer’s disease. Free Radic Biol Med. 2000;28:831–834. doi: 10.1016/s0891-5849(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 36.Zhu X, Su B, Wang X, Smith MA, Perry G. Causes of oxidative stress in Alzheimer disease. Cell Mol Life Sci. 2007;64:2202–2210. doi: 10.1007/s00018-007-7218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 38.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 39.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 41.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, Savaskan E, Czech C, Götz J, Eckert A. Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice. Proc Natl Acad Sci U S A. 2009;106:20057–20062. doi: 10.1073/pnas.0905529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 45.Manczak M, Mao P, Nakamura K, Bebbington C, Park B, Reddy PH. Neutralization of granulocyte macrophage colony-stimulating factor decreases amyloid beta 1–42 and suppresses microglial activity in a transgenic mouse model of Alzheimer’s disease. Hum Mol Genet. 2009;18:3876–3893. doi: 10.1093/hmg/ddp331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Venkitaramani DV, Chin J, Netzer WJ, Gouras GK, Lesne S, Malinow R, Lombroso PJ. Beta-amyloid modulation of synaptic transmission and plasticity. J Neurosci. 2007;27:11832–11837. doi: 10.1523/JNEUROSCI.3478-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM, Lin MT, Gouras GK. Synaptic activity reduces intraneuronal Abeta, promotes APP transport to synapses, and protects against Abeta-related synaptic alterations. J Neurosci. 2009;29:9704–9713. doi: 10.1523/JNEUROSCI.2292-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bayer TA, Wirths O. Intracellular accumulation of amyloid beta – a predictor for synaptic dysfunction and neuronal loss in Alzheimer’s disease. Front Ag Neurosci. 2010;2:8. doi: 10.3389/fnagi.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 52.Tamagno E, Guglielmotto M, Aragno M, Borghi R, Autelli R, Giliberto L, Muraca G, Danni O, Zhu X, Smith MA, Perry G, Jo DG, Mattson MP, Tabaton M. Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem. 104:683–695. doi: 10.1111/j.1471-4159.2007.05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quiroz-Baez R, Rojas E, Arias C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem Int. 2009;55:662–670. doi: 10.1016/j.neuint.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 54.Jo DG, Arumugam TV, Woo HN, Park JS, Tang SC, Mughal M, Hyun DH, Park JH, Choi YH, Gwon AR, Camandola S, Cheng A, Cai H, Song W, Markesbery WR, Mattson MP. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2008.07.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen C, Chen Y, Liu H, Zhang K, Zhang T, Lin A, Jing N. Hydrogen peroxide promotes Abeta production through JNK-dependent activation of gamma-secretase. J Biol Chem. 2008;283:17721–17730. doi: 10.1074/jbc.M800013200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oda A, Tamaoka A, Araki W. Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J Neurosci Res. 2010;88:1137–1145. doi: 10.1002/jnr.22271. [DOI] [PubMed] [Google Scholar]
- 57.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 58.Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging. 2005;26:1235–1244. doi: 10.1016/j.neurobiolaging.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 59.Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, Suzuki H, Araki W, noue H, Shirotani K, Takahashi K, Gallyas F, Tabira T. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- 60.Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;306:116–120. doi: 10.1016/s0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- 61.Wirths O, Multhaup G, Czech C, Feldmann N, Blanchard V, Tremp G, Beyreuther K, Pradier L, Bayer TA. Intraneuronal APP/A beta trafficking and plaque formation in beta-amyloid precursor protein and presenilin-1 transgenic mice. Brain Pathol. 2002;12:275–286. doi: 10.1111/j.1750-3639.2002.tb00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 65.Van Broeck B, Vanhoutte G, Pirici D, Van Dam D, Wils H, Cuijt I, Vennekens K, Zabielski M, Michalik A, Theuns J, De Deyn PP, Van der Linden A, Van Broeckhoven C, Kumar-Singh S. Intraneuronal amyloid beta and reduced brain volume in a novel APP T714I mouse model for Alzheimer’s disease. Neurobiol Aging. 2008;29:241–252. doi: 10.1016/j.neurobiolaging.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 66.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 67.Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 68.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 69.Hellström-Lindahl E, Ravid R, Nordberg A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: inverse correlation with A beta levels. Neurobiol Aging. 2008;29:210–221. doi: 10.1016/j.neurobiolaging.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 70.Mohajeri MH, Kuehnle K, Li H, Poirier R, Tracy J, Nitsch RM. Anti-amyloid activity of neprilysin in plaque-bearing mouse models of Alzheimer’s disease. FEBS Lett. 2004;562:16–21. doi: 10.1016/S0014-5793(04)00169-3. [DOI] [PubMed] [Google Scholar]
- 71.Apelt J, Ach K, Schliebs R. Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci Lett. 2003;339:183–186. doi: 10.1016/s0304-3940(03)00030-2. [DOI] [PubMed] [Google Scholar]
- 72.Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of Abeta-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70:493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- 73.Zhao Z, Xiang Z, Haroutunian V, Buxbaum JD, Stetka B, Pasinetti GM. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol Aging. 2007;28:824–830. doi: 10.1016/j.neurobiolaging.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 74.Farris W, Mansourian S, Leissring MA, Eckman EA, Bertram L, Eckman CB, Tanzi RE, Selkoe DJ. Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am J Pathol. 2004;164:1425–1434. doi: 10.1016/s0002-9440(10)63229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 76.Maurer I, Zierz S, Möller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 77.Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 78.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 79.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;16:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 80.Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- 81.Eckert A, Hauptmann S, Scherping I, Rhein V, Muller-Spahn F, Götz J, Muller WE. Soluble beta-amyloid leads to mitochondrial defects in amyloid precursor protein and tau transgenic mice. Neurodegener Dis. 2008;5:157–159. doi: 10.1159/000113689. [DOI] [PubMed] [Google Scholar]
- 82.Kaminsky YG, Kosenko EA. Effects of amyloid-beta peptides on hydrogen peroxide-metabolizing enzymes in rat brain in vivo. Free Radic Res. 2008;42:564–573. doi: 10.1080/10715760802159057. [DOI] [PubMed] [Google Scholar]
- 83.Schmidt C, Lepsverdize E, Chi SL, Das AM, Pizzo SV, Dityatev A, Schachner M. Amyloid precursor protein and amyloid beta-peptide bind to ATP synthase and regulate its activity at the surface of neural cells. Mol Psychiatry. 2008;13:953–969. doi: 10.1038/sj.mp.4002077. [DOI] [PubMed] [Google Scholar]
- 84.Matsumoto K, Akao Y, Yi H, Shamoto-Nagai M, Maruyama W, Naoi M. Overexpression of amyloid precursor protein induces susceptibility to oxidative stress in human neuroblastoma SH-SY5Y cells. J Neural Transm. 2006;113:125–135. doi: 10.1007/s00702-005-0318-0. [DOI] [PubMed] [Google Scholar]
- 85.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol. 2008;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett JP, Jr, Davis RE, Parker WD., Jr Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 88.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–3428. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- 89.Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11:133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 91.Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 93.Chandrasekaran K, Giordano T, Brady DR, Stoll J, Martin LJ, Rapoport SI. Impairment in mitochondrial cytochrome oxidase gene expression in Alzheimer disease. Brain Res Mol Brain Res. 1994;24:336–340. doi: 10.1016/0169-328x(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 94.Chandrasekaran K, Hatanpää K, Brady DR, Rapoport SI. Evidence for physiological down-regulation of brain oxidative phosphorylation in Alzheimer’s disease. Exp Neurol. 1996;142:80–88. doi: 10.1006/exnr.1996.0180. [DOI] [PubMed] [Google Scholar]
- 95.Simonian NA, Hyman BT. Functional alterations in Alzheimer’s disease: selective loss of mitochondrial-encoded cytochrome oxidase mRNA in the hippocampal formation. J Neuropathol Exp Neurol. 1994;53:508–512. doi: 10.1097/00005072-199409000-00010. [DOI] [PubMed] [Google Scholar]
- 96.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, Eneqvist T, Tjernberg L, Ankarcrona M, Glaser E. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281:29096–290104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 98.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Manczak M, Mao P, Calkins M, Cornea A, Reddy PA, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against Abeta toxicirt in Alzheimer’s disease neurons. J Alzheimer Dis. 2010 doi: 10.3233/JAD-2010-100564. (being reviewed/in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ, Wang YH, YangCheng HY, Zhu HL, Sun XJ, Huang FD. Expression of beta-amyloid Induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. 2010;30:1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Reddy PH. Amyloid beta, mitochondrial structural and functional dynamics in Alzheimer’s disease. Exp Neurol. 2009;218:286–292. doi: 10.1016/j.expneurol.2009.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wang X, Perry G, Smith MA, Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis. 2010;7:56–59. doi: 10.1159/000283484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hamos JE, DeGennaro LJ, Drachman DA. Synaptic loss in Alzheimer’s disease and other dementias. Neurology. 1989;39:355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- 107.Dong H, Martin MV, Chambers S, Csernansky JG. Spatial relationship between synapse loss and beta-amyloid deposition in Tg2576 mice. J Comp Neurol. 2007;500:311–321. doi: 10.1002/cne.21176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Meyer-Luehmann M, Mielke M, Spires-Jones TL, Stoothoff W, Jones P, Bacskai BJ, Hyman BT. A reporter of local dendritic translocation shows plaque-related loss of neural system function in APP-transgenic mice. J Neurosci. 2009;29:12636–12640. doi: 10.1523/JNEUROSCI.1948-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scheff SW, Anderson KJ, DeKosky ST. Strain comparison of synaptic density in hippocampal CA1 of aged rats. Neurobiol Aging. 1985;6:29–34. doi: 10.1016/0197-4580(85)90068-5. [DOI] [PubMed] [Google Scholar]
- 110.Scheff SW, Scott SA, DeKosky ST. Quantitation of synaptic density in the septal nuclei of young and aged Fischer 344 rats. Neurobiol Aging. 1991;12:3–12. doi: 10.1016/0197-4580(91)90032-f. [DOI] [PubMed] [Google Scholar]
- 111.Bertoni-Freddari C, Fattoretti P, Casoli T, Meier-Ruge W, Ulrich J. Morphological adaptive response of the synaptic junctional zones in the human dentate gyrus during aging and Alzheimer’s disease. Brain Res. 1990;517:69–75. doi: 10.1016/0006-8993(90)91009-6. [DOI] [PubMed] [Google Scholar]
- 112.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 113.Sze CI, Troncoso JC, Kawas C, Mouton P, Price DL, Martin LJ. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–944. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 114.Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W, Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. discussion 173–180. [DOI] [PubMed] [Google Scholar]
- 115.Yao PJ, Morsch R, Callahan LM, Coleman PD. Changes in synaptic expression of clathrin assembly protein AP180 in Alzheimer’s disease analysed by immunohistochemistry. Neuroscience. 1999;94:389–394. doi: 10.1016/s0306-4522(99)00360-7. [DOI] [PubMed] [Google Scholar]
- 116.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mukaetova-Ladinska EB, Xuereb JH, Garcia-Sierra F, Hurt J, Gertz HJ, Hills R, Brayne C, Huppert FA, Paykel ES, McGee MA, Jakes R, Honer WG, Harrington CR, Wischik CM. Lewy body variant of Alzheimer’s disease: selective neocortical loss of t-SNARE proteins and loss of MAP2 and alpha-synuclein in medial temporal lobe. Scientific World Journal. 2009;9:1463–1475. doi: 10.1100/tsw.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Head E, Corrada MM, Kahle-Wrobleski K, Kim RC, Sarsoza F, Goodus M, Kawas CH. Synaptic proteins, neuropathology and cognitive status in the oldest-old. Neurobiol Aging. 2009;30:1125–1134. doi: 10.1016/j.neurobiolaging.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 120.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 122.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 123.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 124.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;109(Suppl 1):153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Breyhan H, Wirths O, Duan K, Marcello A, Rettig J, Bayer TA. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol. 2009;117:677–685. doi: 10.1007/s00401-009-0539-7. [DOI] [PubMed] [Google Scholar]
- 127.Baloyannis SJ, Costa V, Mauroudis I, Psaroulis D, Manolides SL, Manolides LS. Dendritic and spinal pathology in the acoustic cortex in Alzheimer’s disease: morphological and morphometric estimation by Golgi technique and electron microscopy. Acta Otolaryngol. 2007;127:351–354. doi: 10.1080/00016480601126986. [DOI] [PubMed] [Google Scholar]
- 128.Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- 129.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 130.Reddy PH. Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J Biomed Biotechnol. 2006;2006:31372. doi: 10.1155/JBB/2006/31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Reddy PH. Mitochondrial dysfunction in aging and Alzheimer’s disease: strategies to protect neurons. Antioxid Redox Signal. 2007;9:1647–1658. doi: 10.1089/ars.2007.1754. [DOI] [PubMed] [Google Scholar]
- 132.Reddy PH. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromolecular Med. 2008;10:291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D Dimebon investigators. Effect of Dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: a randomised, double-blind, placebo-controlled study. Lancet. 2008;372:207–215. doi: 10.1016/S0140-6736(08)61074-0. [DOI] [PubMed] [Google Scholar]