

Abstract
The mechanisms that regulate peripheral nervous system (PNS) gliogenesis are incompletely understood. For example, gut neural crest stem cells (NCSCs) do not respond to known gliogenic factors, suggesting that yet-unidentified factors regulate gut gliogenesis. To identify new mechanisms, we performed gene expression profiling to identify factors secreted by gut NCSCs during the gliogenic phase of development. These cells highly expressed leucine-rich glioma inactivated 4 (Lgi4) despite the fact that Lgi4 has never been implicated in stem cell function or enteric nervous system development. Lgi4 is known to regulate peripheral nerve myelination (having been identified as the mutated gene in spontaneously arising claw paw mutant mice), but Lgi4 is not known to play any role in PNS development outside of peripheral nerves. To systematically analyze Lgi4 function, we generated gene-targeted mice. Lgi4-deficient mice exhibited a more severe phenotype than claw paw mice and had gliogenic defects in sensory, sympathetic, and enteric ganglia. We found that Lgi4 is required for the proliferation and differentiation of glial-restricted progenitors throughout the PNS. Analysis of compound-mutant mice revealed that the mechanism by which Lgi4 promotes enteric gliogenesis involves binding the ADAM22 receptor. Our results identify a new mechanism regulating enteric gliogenesis as well as novel functions for Lgi4 regulating the proliferation and maturation of glial lineage cells throughout the PNS.
Introduction
The neural crest is a heterogeneous collection of progenitors, including multipotent neural crest stem cells (NCSCs) and restricted progenitors, that give rise to the neurons and glia of the PNS (Le Douarin, 1986; Fraser and Bronner-Fraser, 1991; Stemple and Anderson, 1992; Henion and Weston, 1997). These neurons and glia constitute sensory, sympathetic, parasympathetic, and enteric ganglia as well as peripheral nerves. Although the regulation of neurogenesis has been elucidated to a considerable extent (Anderson et al., 1997), comparatively less is known about the regulation of gliogenesis.
Some of the cell-extrinsic factors that regulate gliogenesis have been identified. Notch ligands instruct NCSCs to undergo gliogenesis (Morrison et al., 2000) and Notch signaling is necessary for normal gliogenesis in the PNS (Wakamatsu et al., 2000; Taylor et al., 2007). Neuregulin (Nrg) instructs NCSCs to undergo glial lineage determination (Shah et al., 1994; Morrison et al., 1999), and then promotes the proliferation, survival, and maturation of glial lineage cells (Dong et al., 1995; Topilko et al., 1997). Nrg is necessary for gliogenesis in vivo (Meyer and Birchmeier, 1995; Riethmacher et al., 1997). These gliogenic factors interact with each other and with other lineage determination factors to combinatorially regulate NCSC differentiation (Shah and Anderson, 1997; Paratore et al., 2001; Joseph et al., 2004).
Known gliogenic factors cannot fully explain PNS gliogenesis. Neither Notch ligands nor Nrg cause embryonic day 14.5 (E14.5) gut NCSCs to undergo gliogenesis in culture despite the fact that these cells undergo gliogenesis in vivo at this stage of development (Bixby et al., 2002) and are capable of forming glia in diverse PNS locations after transplantation into chick embryos (Mosher et al., 2007). This suggests there are yet-unidentified factors that promote PNS gliogenesis. Moreover, clusters of neural crest cells exhibit a much greater gliogenic response to Nrg compared with single, isolated neural crest cells (Paratore et al., 2001). This suggests that unknown autocrine or paracrine factors secreted by neural crest cells can augment the gliogenic response to Nrg.
Lgi4 is secreted by Schwann cells and regulates peripheral nerve myelination (Bermingham et al., 2006) by binding to the A disintegrin and metalloproteinase 22 (ADAM22) receptor expressed by neurons (Fukata et al., 2006; Sagane et al., 2008; Ozkaynak et al., 2010). Adam22-deficient mice also exhibit defects in peripheral nerve myelination (Sagane et al., 2005). Lgi4 is mutated in spontaneously arising claw paw (clp) mutant mice, which exhibit a characteristic arthrogryposis-like forelimb posture phenotype caused by delayed peripheral nerve myelination (Koszowski et al., 1998; Darbas et al., 2004; Bermingham et al., 2006). Claw paw mutant mice have a small insertion in the Lgi4 gene, which disrupts Lgi4 splicing, leading to a mutant form of the Lgi4 protein that lacks exon 4 (Bermingham et al., 2006). Many claw paw mice die soon after birth but some survive to adulthood as nerve myelination gradually recovers (Darbas et al., 2004). Despite their importance in nerve myelination, Lgi4 and ADAM22 are not known to regulate PNS development outside of peripheral nerves.
We discovered that Lgi4 was highly expressed by gut NCSCs during the gliogenic phase of gut development. We generated Lgi4-deficient mice (Lgi4LacZ/LacZ) and found that they exhibited a defect in peripheral nerve myelination attributable to a defect in Schwann cell differentiation, similar to claw paw mice (Lgi4clp/clp); however, Lgi4LacZ/LacZ mice had a more severe phenotype and all died within 3 weeks of birth. We discovered that Lgi4LacZ/LacZ mice had defects in glial-restricted progenitor proliferation and glial differentiation in enteric, sympathetic, and sensory ganglia. Lgi4 deficiency reduced the numbers of enteric and satellite glia in these ganglia and impeded their acquisition of a mature morphology. Adam22-deficient mice and Lgi4LacZ/LacZAdam22−/− compound-mutant mice had similar gliogenic defects as Lgi4LacZ/LacZ mice in the enteric nervous system, suggesting that Lgi4 promotes gliogenesis by binding ADAM22 in multiple regions of the developing PNS. Our results identify a new mechanism that regulates enteric gliogenesis and new functions for Lgi4 and ADAM22 regulating gliogenesis throughout the PNS.
Materials and Methods
Mice.
To generate Lgi4LacZ/LacZ (Lgi4tm1Sjm) mice, BAC (bacterial artificial chromosome) clones containing the Lgi4 genomic locus were purchased (Invitrogen), and a targeting vector was constructed using bacterial recombineering (Copeland et al., 2001; Liu et al., 2003). Bruce 4.G9 embryonic stem (ES) cells [a subline of Bruce4 selected for improved genetic stability (Köntgen et al., 1993; Hughes et al., 2007)] were electroporated with the targeting construct, positively selected with G418 (Invitrogen), and negatively selected with gancyclovir (cytovene from Syntex) (for the targeting strategy, see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Correctly targeted ES cell clones were identified by Southern blot, and their chromosome numbers were confirmed to be euploid. Three independent ES cell clones were injected into blastocysts obtained from B6(Cg)-Tyrc-2J/J mice (The Jackson Laboratory). The resulting male ES cell/mouse chimeras were crossed with B6(Cg)-Tyrc-2J/J mice to obtain germline transmission. After germline transmission, the neo cassette was removed by crossing with B6.Cg-Tg(ACTFLPe)9205Dym/J mice (Rodríguez et al., 2000). Lgi4LacZ/LacZ mice and Adam22−/− mice (Sagane et al., 2005) were housed at the University of Michigan Unit for Laboratory Animal Medicine, an Association for Assessment and Accreditation of Laboratory Animal Care-accredited facility that follows the National Research Council's Guide for the Care and Use of Laboratory Animals.
Cell culture and self-renewal assay.
Neural crest stem and progenitor cells were isolated and cultured as described in previous studies (Molofsky et al., 2005; Joseph et al., 2008; Nishino et al., 2008). For adherent cultures, PNS cells were enzymatically dissociated and plated at clonal density (0.33 cells/μl = 500 cells per 35 mm well), in six-well plates (Corning) that had been sequentially coated with 150 μg/ml poly-d-lysine (Biomedical Technologies) and 20 μg/ml laminin (Sigma-Aldrich). For the nonadherent culture of neurospheres, PNS cells were plated at 0.67–1.33 cells/μl (1000–2000 cells per 35 mm well) in ultra-low-binding six-well plates (Corning). Cells were initially cultured for 8–10 d in “self-renewal medium” to promote the formation of undifferentiated colonies. This medium contained a 5:3 mixture of DMEM-low/Neurobasal medium, supplemented with 20 ng/ml recombinant human basic fibroblast growth factor (bFGF) (R&D Systems), 20 ng/ml IGF1 (R&D Systems), 15% chick embryo extracts (CEEs), 1% N2 supplement (Invitrogen), 2% B27 supplement (Invitrogen), 50 mm 2-mercaptoethanol, 35 μg/ml (110 nm) retinoic acid (Sigma-Aldrich), and penicillin/streptomycin (Lonza Walkersville). Cultures were subsequently refed with “differentiation medium” and allowed to grow for an additional 4–7 d. Differentiation medium contained 10 ng/ml (instead of 20 ng/ml) bFGF, 5% fetal bovine serum (Invitrogen), no IGF, and no CEE. After being grown in self-renewal medium, neurospheres were routinely transferred to adherent cultures containing differentiation medium before being stained to assess multilineage differentiation (see below). All cultures were maintained at 37°C in 6% CO2 incubators.
To measure self-renewal, individual PNS neurospheres were replated (one per well) for 72 h onto adherent plates to allow the spheres to spread out over the culture dish, facilitating dissociation. Individual adherent colonies were then dissociated with trypsin and collagenase (four parts 0.05% trypsin-EDTA plus one part 10 mg/ml collagenase IV) for 3 min at 37°C followed by trituration. Two thousand dissociated cells were replated per well of a six-well plate. Secondary neurospheres were cultured and differentiated, and the number of multilineage secondary colonies generated by each primary colony was counted.
Isolation of neural tissues.
E13.5 embryonic PNS tissues [dorsal root ganglion (DRG), sympathetic chain, and gut] were dissected into ice-cold PBS and dissociated by incubating for 4 min at 37°C in trypsin/EDTA (Lonza Walkersville; product 17-161E; diluted 1:10 in Ca-, Mg-free HBSS) plus 0.25 mg/ml type 4 collagenase (Worthington). Postnatal day 0 (P0) gut plexus/outer muscle layer cells were minced and dissociated for 15 min at 37°C in 0.025% trypsin/EDTA (Invitrogen; 25300-054) plus 1 mg/ml type 4 collagenase (Worthington) in Ca-, Mg-free HBSS. After centrifuging, the cells were gently triturated, filtered through nylon screen (45 mm; Sefar America), counted by hemocytometer, and resuspended in staining medium before being sorted or plated. Staining medium was L15 medium containing 1 mg/ml BSA (Sigma-Aldrich; A-3912), 10 mm HEPES, pH 7.4, and penicillin/streptomycin (Lonza Walkersville).
Antibodies and immunohistochemistry.
PNS tissues were fixed in 4% paraformaldehyde overnight, and then cryoprotected in 30% sucrose, embedded in OCT compound, and frozen. Ten micrometer sections were cut, and then preblocked for 1 h at room temperature in blocking solution (PBS containing 5% goat serum, 0.2% bovine serum albumin, and 0.5% Triton X-100), incubated with primary antibody at 4°C overnight, followed by washing, and incubation in secondary antibody for 1 h at room temperature. Primary antibodies included those against Krox20 (Covance; PRB-236P; 1:1000), Periaxin (gift from Dr. P. Brophy, University of Edinburgh, Edinburgh, UK; 1:400) (Gillespie et al., 1994), Peripherin (Millipore; 1:1000), β-galactosidase (gift from Dr. T. Glaser, University of Michigan, Ann Arbor, MI), Tuj1 (Covance; MMS-435P; 1:1000), brain-specific fatty acid binding protein (BFABP) (gift from Dr. T. Muller, Max Delbruck Center, Berlin, Germany; 1:2000) (Kurtz et al., 1994), p75 (Millipore; AB1554; 1:1000), Sox10 (R&D Systems; 20B7; 1:400), GFAP (BD Biosciences Pharmingen; 1:400), phosphohistone H3 (pH3) (Cell Signaling Technology; 1:200), HuC/D (1:200; Invitrogen), and NeuN (Millipore; MAB377; 1:1000). For secondary antibodies, Alexa Fluor 488- or 555-conjugated antibodies were used (Invitrogen; 1:1000). Slides were counterstained in 2.5 μg/ml 4′,6-diamino-2-phenylindole dihydrochloride (DAPI) for 10 min at room temperature, and then mounted using ProLong antifade solution (Invitrogen).
For in situ hybridization to detect Adam9, Adam11, Adam22, and Adam23 transcripts, E14.5 embryos or P0 pups were fixed in 4% paraformaldehyde overnight, cryoprotected in 30% sucrose, embedded in OCT compound, and frozen. Ten micrometer sections were cut, pretreated with 2 μg/ml proteinase K at 37°C for 20 min, with 0.2N HCl for 10 min at room temperature, with 0.1 m triethanolamine-HCl for 10 min at room temperature, and hybridized with digoxigenin-labeled Adam antisense probe at 55°C overnight. The next day, sections were washed with 2× SSC for 30 min at 55°C, with 0.2× SSC for 40 min, blocked with 20% goat serum for 1 h, and incubated with anti-digoxigenin-AP (alkaline phosphatase) Fab fragment (1:2000; Roche) for 60 min at room temperature. Sections were washed with Tris-buffered saline, pH 9.5, with 0.1% Tween 20 for 30 min at room temperature, and incubated with 0.5 μl/ml NBT (nitroblue tetrazolium chloride) plus 3.5 μl/ml BCIP (5-bromo-4-chloro-3′-indolylphosphatase p-toluidine salt) (Roche).
For 5-bromo-4-chloro-3-indolyl-β-d-balactoside (X-gal) staining, E10.5 and E13.5 mouse embryo or P0 pups were fixed with 1% paraformaldehyde/0.2% glutaraldehyde for 10 min at 4°C. Then whole embryos (E10.5) or cryosections (E13.5 and P0) were incubated in staining solution at 37°C for 4–16 h.
Genotyping.
Genotyping was performed by PCR using GoTag Flexi DNA polymerase (Promega), following the manufacturer's instructions. The primers for Lgi4 genotyping were as follows: 5′-GCATCCCACGGAGATGTAGT-3′ (common sense primer), 5′-CAACCTGCACCTTTCCAAAT-3′ (antisense primer for the detection of the wild-type allele), and 5′-GTTGTGGCGGATCTTGAAGT-3′ (antisense primer for the detection of the Lgi4LacZ allele). Primers for Adam22 genotyping were as follows: 5′-TGAGTTGGGCAGAACTGAGTCACTG-3′ (common sense primer), 5′-AGGAATTGCAAAGAAGAGCCTGTGAC-3′ (antisense primer for the detection of the wild-type allele), and 5′-CATGCTCCAGACTGCCTTGGGAAAAG-3′ (antisense primer for the detection of Adam22− allele). The PCR cycle was 94°C for 2 min, followed by 35 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 1 min, and followed by 72°C for 5 min.
Immunocytochemistry.
PNS neurospheres were replated into adherent cultures in which the plates had been coated with poly-d-lysine and laminin. The neurospheres were allowed to differentiate for 7–9 d, and then fixed in acid ethanol for 20 min at −20°C, washed, blocked, and triply labeled with antibodies against peripherin (Millipore Bioscience Research Reagents; AB1530; 1:1000), GFAP (Sigma-Aldrich; G-3893; 1:200), and smooth muscle actin (SMA) (Sigma-Aldrich; A-2547; 1:200) as described previously (Kruger et al., 2002). Cells were stained for 10 min at room temperature with 10 μg/ml DAPI (Sigma-Aldrich; D-8417) to visualize nuclei.
To study the proliferation of glial progenitors, cells were fixed in 70% ethanol for 30 min at −20°C and stained with antibody against phosphohistone H3 (Cell Signaling Technology; 9701; 1:100). Cells were counterstained for 10 min at room temperature with 10 μg/ml DAPI (Sigma-Aldrich; D-8417).
For X-gal staining of neurospheres, E13.5 PNS neurospheres were fixed with 1% paraformaldehyde/0.2% glutaraldehyde for 5 min at 4°C and incubated for 1 h at 37°C in staining solution: PBS containing 2 mm X-gal (Invitrogen), 2 mm MgCl2, 5 mm potassium ferrocyanide, 5 mm potassium ferricyanide, and 0.02% NP-40. In some cases, neurospheres were transferred to plates coated with poly-d-lysine and laminin, and cultured for 3 d in self-renewal medium. Cells were fixed with 1% paraformaldehyde for 5 min at 4°C and incubated in staining solution at 37°C for 1 h to assess LacZ expression. Cells were then blocked and stained with anti-Nestin antibody (Millipore; MAB353; 1:400) for 1 h at room temperature, followed by Alexa Fluor 488-conjugated anti-mouse IgG1 antibody (Invitrogen; 1:500).
Electron microscopy.
Nerves were dissected and fixed in 2.5% glutaraldehyde overnight at 4°C. Nerves were rinsed with PBS and refixed in 1% OsO4 (Electron Microscopy Sciences) for 1 h at room temperature. Nerves were then dehydrated in an ethanol series (30, 50, 70, 95, and 100% ethanol) before infiltrating and embedding in Spurr resin (Bozzola and Russell, 1992). Semithin sections (1 μm) were cut with a glass knife. Thin sections (70 nm) were cut with a diamond knife. Some sections were stained with uranyl acetate to reveal the basal lamina. Sections were examined with a transmission electron microscope (Phillips CM-100).
Binding assay.
For immunoprecipitation, 293T cells were cotransfected with Lgi-4-FLAG and HA-tagged ADAMs (ADAM9-HA, ADAM11-HA, ADAM22-HA, and ADAM23-HA; gift from Dr. M. Fukata, National Institute for Science, Okazaki, Japan). Twenty-four hours after transfection, cells were harvested, suspended in lysis buffer [20 mm Tris, 137 mm NaCl, 10% glycerol, 1% NP-40, 2 mm EDTA, and protease inhibitor mixture (Complete mini-tablet; Roche)] for 1 h at 4°C, and incubated with protein G-agarose at 4°C for 60 min to absorb nonspecific binding. After eliminating protein G-agarose by centrifugation, small fractions of supernatants were saved as input, and the rest was incubated overnight at 4°C with either anti-FLAG antibody (M2; Sigma-Aldrich; 1:500) or anti-HA antibody (Sigma-Aldrich; H6908; 1:500). The next day, immunoprecipitated fractions were harvested by incubating with protein G-agarose for 60 min at 4°C, washed with washing buffer, and boiled for 5 min with SDS sample loading buffer. SDS-PAGE was done in 4–20% Tris-glycine gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked in Tris-buffered saline with 0.05% Tween 20 and 5% milk, incubated with primary and secondary antibodies, and washed following standard procedures. Horseradish peroxidase-conjugated secondary antibodies were detected by chemiluminescence (ECL Plus; GE Healthcare).
For the cell surface binding assay, COS7 cells were seeded onto poly-d-lysine-coated coverslips and cotransfected with Lgi-4-FLAG and HA-tagged ADAMs. Twenty-four hours after transfection, cells were fixed with 2% paraformaldehyde for 10 min at 4°C and blocked with PBS containing 2 mg/ml BSA for 10 min at 4°C. Cells were immediately stained with anti-FLAG antibody (1:1000), followed by Alexa Fluor 488-conjugated anti-mouse IgG1 antibody (Invitrogen; 1:500). Then cells were permeabilized with 0.1% Triton X-100 for 10 min, blocked, and stained with anti-HA antibody (1:1000), followed by Alexa Fluor 555-conjugated anti-mouse IgG1 antibody (Invitrogen; 1:500).
Results
Lgi4 is expressed by neural crest and glial lineage cells throughout the PNS
To identify new mechanisms by which secreted factors regulate PNS development, we compared the gene expression profiles of highly purified p75+α4+ NCSCs isolated from the E14.5 rat gut to whole fetal RNA (Iwashita et al., 2003). The most highly upregulated secreted gene product in NCSCs compared with whole fetal RNA was Lgi4 (Table 1). By microarray analysis and quantitative reverse transcription-PCR, this gene was 35 ± 18- and 44 ± 17-fold, respectively, upregulated in gut NCSCs compared with whole fetal RNA using three independent samples of each cell population for each type of analysis. We therefore wondered whether Lgi4 regulates PNS development beyond its known role in peripheral nerve myelination.
Table 1.
Genes that encode secreted proteins and that are significantly more highly expressed in gut NCSCs compared with whole fetal RNA by gene expression profiling (fold change, >3.0) (Iwashita et al., 2003)
| Gene symbol | Unigene ID | NCSC | Fetus | NCSC/fetus |
|---|---|---|---|---|
| Leucine-rich glioma inactivated 4 (Lgi4) | Rn0.143830 | 5583 | 177 | 31.5 |
| Chemokine (C-X-C motif) ligand 14 (Cxcl14) | Rn0.3130 | 477 | 56 | 4.8 |
| Angiopoietin 2 (Angpt2) | Rn0.138360 | 452 | 44 | 4.5 |
| Interleukin 33 (Il33) | Rn0.106849 | 384 | 61 | 3.8 |
| Taxilin-α (Txlna) | Rn0.217158 | 887 | 244 | 3.6 |
| Cortistatin (Cort) | Rn0.6204 | 362 | 77 | 3.6 |
| Placental growth factor (Pgf) | Rn0.6960 | 576 | 160 | 3.6 |
| Bone morphogenetic protein 7 (Bmp7) | Rn0.18030 | 1388 | 405 | 3.4 |
| Secreted protein, acidic, cystein-rich (Sparc) | Rn0.98989 | 702 | 228 | 3.1 |
| Midkine (Mdk) | Rn0.17447 | 1780 | 588 | 3.0 |
To systematically examine Lgi4 expression, we generated a gene-targeted mouse in which LacZ was knocked into the Lgi4 genomic locus by homologous recombination (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). LacZ was inserted in-frame with the Lgi4 start codon so that LacZ expression could be used to infer the Lgi4 expression pattern. Lgi4LacZ/+ mice were born with expected Mendelian ratios (see Fig. 3C), survived into adulthood, and were developmentally grossly normal. X-gal staining of Lgi4LacZ/+ embryos indicated that Lgi4 was expressed by migrating neural crest cells (Fig. 1A) and then by neural crest-derived cells throughout the developing PNS but rarely by cells outside of the PNS (Fig. 1B). At E10.5, Lgi4 was expressed in the trigeminal, facial, dorsal root, and sympathetic ganglia, as well as in peripheral nerves, and gut (Fig. 1B). At later stages of PNS development (E13.5 to P0), Lgi4 expression was maintained in the myenteric plexus of the gut, in DRGs, in sympathetic ganglia, in peripheral nerves, and in parasympathetic ganglia (Fig. 1C).
Figure 3.
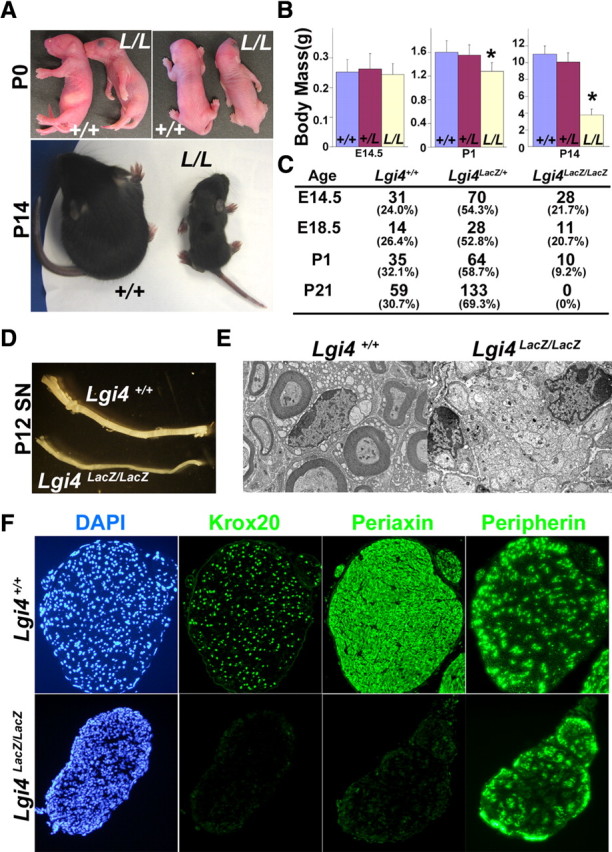
Lgi4LacZ/LacZ mice exhibit abnormal forelimb posture, peripheral nerve hypomyelination, growth retardation, and neonatal death. A, Lgi4LacZ/LacZ mice (L/L) exhibited an arthrogryposis-like forelimb posture abnormality at P0 and growth retardation relative to littermate controls (+/+) by P14. B, The body mass of Lgi4LacZ/LacZ mice was similar to littermate controls at E14.5 but significantly reduced at P1 and P14 (*p < 0.01 by Student's t test; n = 12–18 mice/genotype at E14.5, 18–27 at P0, and 9–11 at P14). Error bars indicate SD. C, Genotyping the progeny of Lgi4LacZ/+ intercrosses revealed expected numbers of Lgi4LacZ/LacZ mice at E14.5 and E18.5, but many Lgi4LacZ/LacZ mice died immediately after birth and no Lgi4LacZ/LacZ mice survived to P21. D, E, Sciatic nerves from P12 Lgi4LacZ/LacZ mice were thinner (D) and hypomyelinated (E) relative to sciatic nerves from littermate controls. F, Immunostaining of transverse sections through P12 Lgi4LacZ/LacZ and control sciatic nerves revealed little Krox20 or Periaxin staining in the absence of Lgi4, but normal peripherin staining.
Figure 1.
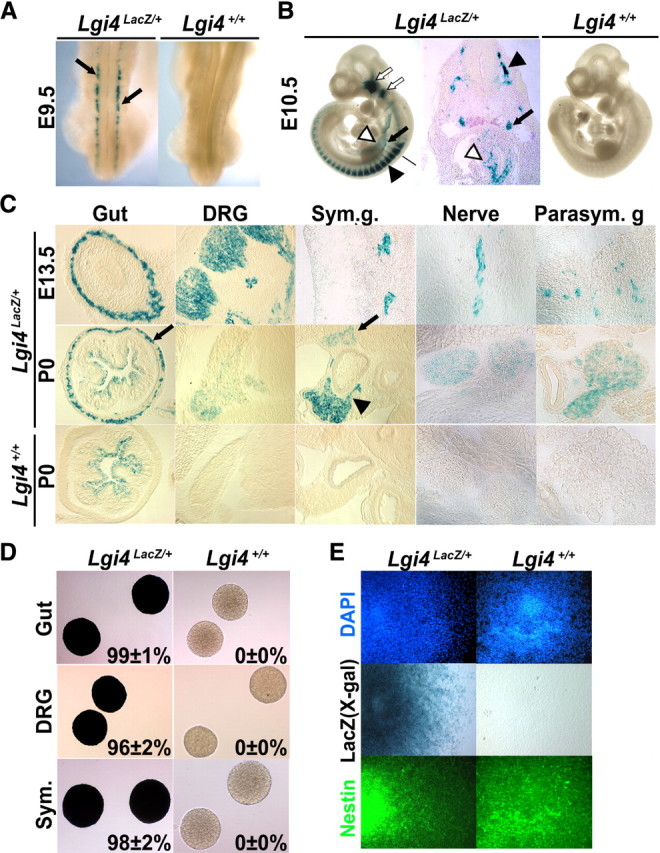
Lgi4 is expressed by neural crest stem cells and other undifferentiated neural crest cells throughout the developing PNS. A, Dorsal view of X-gal-stained E9.5 Lgi4LacZ/+ and Lgi4+/+ embryos revealed Lgi4 expression (blue; arrows) by migrating trunk neural crest cells. B, X-gal staining of E10.5 Lgi4LacZ/+ and Lgi4+/+ embryos revealed Lgi4 expression by cranial ganglia (open arrows), DRGs (black arrowhead), sympathetic ganglia (black arrow), and migrating neural crest cells within the gut (open arrowhead). The cross section of the Lgi4LacZ/+ embryo was taken at the level of the black bar indicated in the photo at left. C, X-gal staining of sections from E13.5 and P0 Lgi4LacZ/+ and Lgi4+/+ mice revealed Lgi4 expression in the myenteric plexus (gut; arrow), DRGs, sympathetic ganglia [at P0, the arrowhead indicates sympathetic ganglion (Sym. g.) and the arrow indicates celiac ganglion], peripheral nerve, and parasympathetic ganglion (Parasym. g.). Note the bacterial LacZ activity in the lumen of the P0 gut. D, Virtually all neurospheres cultured from the gut, DRGs, and sympathetic ganglia of E13.5 Lgi4LacZ/+ mice stained with X-gal (mean ± SD X-gal stained is indicated in each panel; n = 3 independent experiments). E, Lgi4LacZ/+ neurospheres transferred to adherent cultures stained positively for X-gal and Nestin.
To confirm Lgi4 expression in undifferentiated neural crest cells, we cultured cells from E13.5 gut, DRG, and sympathetic ganglia in nonadherent cultures at clonal density. Cells from each region of the developing PNS formed neurospheres in culture, consistent with our previous demonstration that each of these locations contain NCSCs that can be propagated in culture as neurospheres (Bixby et al., 2002; Taylor et al., 2007; Joseph et al., 2008). Almost all spheres from each region of the developing PNS stained with X-gal, indicating Lgi4 expression, in contrast to spheres from control littermates (Fig. 1D). We confirmed that X-gal-stained spheres contained neural stem/progenitor cells by staining with Nestin (Fig. 1E).
At birth (P0), when most neural crest cells had differentiated, Lgi4 expression became restricted to a subpopulation of cells in each ganglion (see the reduced X-gal staining in ganglia at P0 compared with E13.5 in Fig. 1C). To assess which cells expressed Lgi4 at P0, we stained sections through the gut, DRGs, and superior cervical (sympathetic) ganglia with antibodies against LacZ and the neuronal markers HuC/D or NeuN, or the glial marker BFABP. In each region of the newborn PNS, we detected LacZ staining in glial cells but not in neurons (Fig. 2A–C). Our data indicate that Lgi4 is initially expressed by undifferentiated neural crest cells throughout the developing PNS but that, by birth, expression becomes restricted to glial cells.
Figure 2.
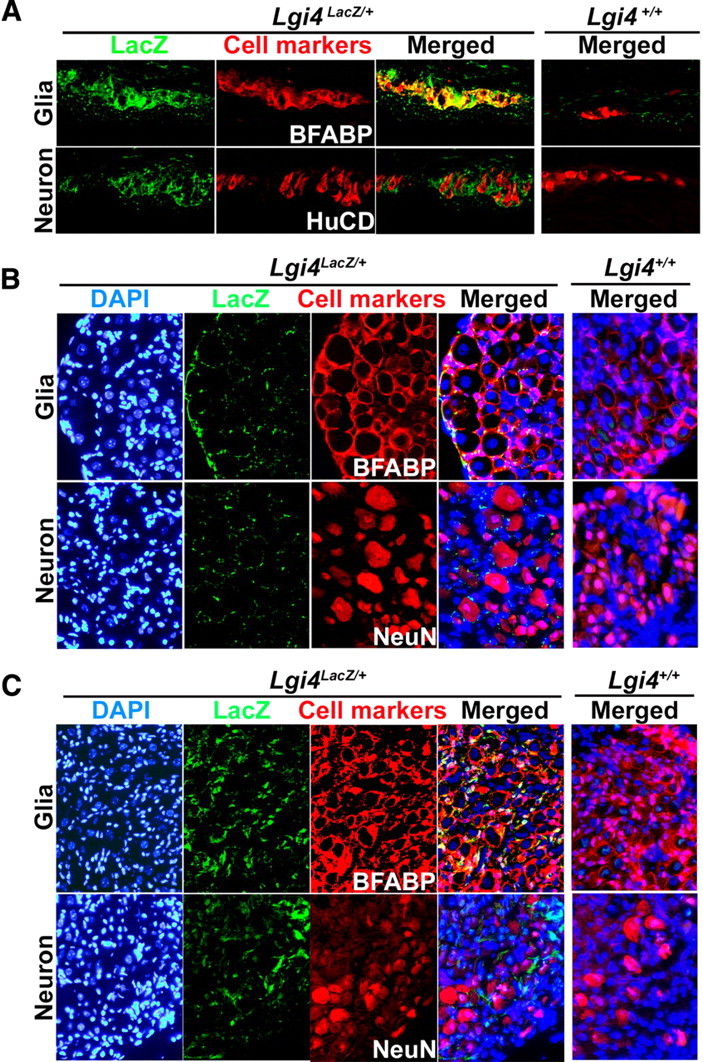
Lgi4 is expressed by glia in the myenteric plexus, DRGs, and sympathetic ganglia of adult Lgi4LacZ/+ mice. Immunofluorescence analysis of the LacZ expression pattern in the myenteric plexus (A), DRGs (B), and sympathetic ganglia (C) of young adult Lgi4LacZ/+ mice showed overlapping staining with the glial marker BFABP but not with the neuronal markers HuC/D or NeuN. Nuclei were visualized using DAPI staining (blue).
Lgi4LacZ/LacZ mice exhibit a more severe phenotype than Lgi4clp/clp mice
To examine the function of Lgi4 in PNS development, we analyzed Lgi4LacZ/LacZ mice. Lgi4LacZ/LacZ mice exhibited a forelimb posture (arthrogryposis-like) phenotype similar to the defining phenotype in claw paw mutant (Lgi4clp/clp) mice (Bermingham et al., 2006) (Fig. 3A). Lgi4LacZ/LacZ mice also exhibited severe growth retardation after birth. Lgi4LacZ/LacZ fetuses had normal body mass at E14.5, but were significantly smaller than littermate controls at P1, and approximately one-third the mass of littermate controls by P14 (Fig. 3A,B). Most of the Lgi4LacZ/LacZ mice died immediately after birth and no Lgi4LacZ/LacZ mice survived to P21 (Fig. 3C). This complete lethality within 3 weeks of birth contrasts with the phenotype of Lgi4clp/clp mice, which sometimes survive into adulthood (Koszowski et al., 1998; Darbas et al., 2004; Bermingham et al., 2006). This suggests that the Lgi4LacZ targeted allele gives a complete loss of Lgi4 function, whereas the splicing defect in the Lgi4clp allele gives a partial loss of function.
Sciatic nerves of Lgi4LacZ/LacZ mice and littermate controls were examined at P12 to assess whether Lgi4LacZ/LacZ nerves exhibit a hypomyelination phenotype similar to Lgi4clp/clp nerves (Koszowski et al., 1998; Darbas et al., 2004; Bermingham et al., 2006). Lgi4LacZ/LacZ nerves were thinner than control nerves (Fig. 3D), and examination by electron microscopy revealed a severe defect in myelination (Fig. 3E). The myelination defect in Lgi4LacZ/LacZ nerves was further confirmed by staining with antibodies against Krox20 and Periaxin, which exhibit delayed expression within Lgi4clp/clp nerves. In Lgi4LacZ/LacZ nerves, Krox20 and Periaxin expression in Schwann cells were dramatically reduced at P12 compared with control nerves (Fig. 3F). The reduction in Krox-20 expression was further confirmed by quantitative PCR (0.08 ± 0.02-fold in P12 Lgi4LacZ/LacZ nerves compared with control nerves; three mice per genotype). The expression of Oct-6, a marker of immature Schwann cells, did not change in the Lgi4-deficient sciatic nerves (1.15 ± 0.12-fold; three mice per genotype), consistent with a previous report (Darbas et al., 2004). The neuronal marker peripherin also did not differ between Lgi4LacZ/LacZ and control nerves (Fig. 3F). Lgi4LacZ/LacZ and control nerves had similar numbers of endoneurial cells in transverse nerve sections (312 ± 54 cells per section in control, and 296 ± 38 cells in Lgi4LacZ/LacZ nerves). Since Schwann cells represent the vast majority of endoneurial cells (Joseph et al., 2004), these data suggest that normal numbers of Schwann cells are generated in the absence of Lgi4 but that these cells fail to fully differentiate to a myelinating phenotype.
Lgi4 deficiency impairs glial differentiation in vitro
To assess whether Lgi4 regulates NCSCs function, we cultured NCSCs from the gut, DRG, and sympathetic chain of E13.5 Lgi4LacZ/LacZ mice and littermate controls (Fig. 4A). To minimize fusion between neurospheres, we cultured cells at very low density in nonadherent cultures (one cell per microliter of medium), and then transferred individual neurospheres to adherent secondary cultures to determine the fraction of neurospheres capable of multilineage differentiation into peripherin+ neurons, GFAP+ glia, and SMA+ myofibroblasts. The percentage of dissociated cells that formed neurospheres that underwent multilineage differentiation was slightly lower in Lgi4LacZ/LacZ mice compared with wild-type mice, although the difference was not statistically significant (Fig. 4B). Lgi4LacZ/LacZ neurospheres also did not significantly differ from control neurospheres in terms of diameter or self-renewal potential (Fig. 4B). Self-renewal potential was assessed by dissociating individual neurospheres, and then subcloning to secondary cultures to determine the number of multilineage secondary colonies that could be subcloned from each primary stem cell colony. These data suggest that Lgi4 deficiency did not significantly affect NCSC frequency or self-renewal potential.
Figure 4.
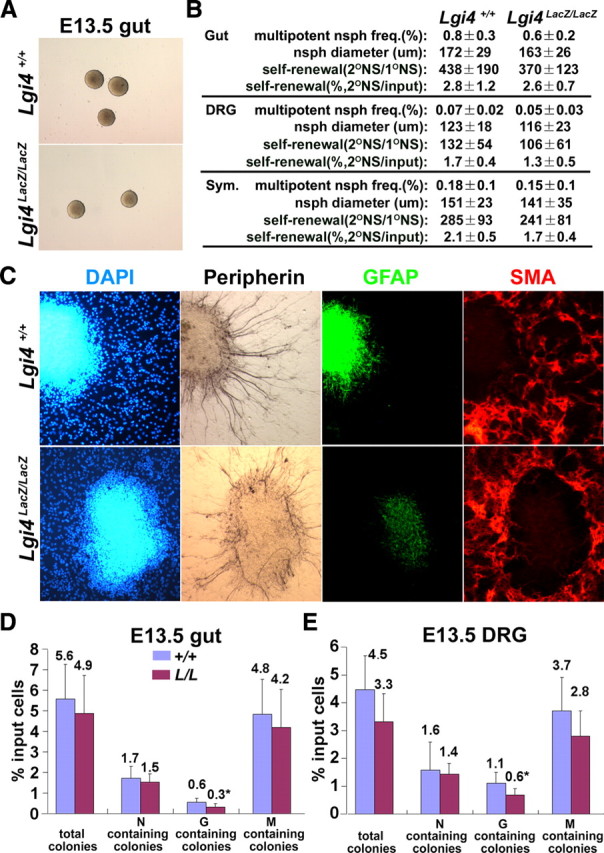
Lgi4 is not required for neural crest stem cell formation or self-renewal but is required for normal glial differentiation in culture. A, Gut cells from E13.5 Lgi4LacZ/LacZ mice formed neurospheres with normal morphology after 10 d in nonadherent, low-density (∼1 cell/μl) cultures. B, Lgi4 deficiency did not affect the percentage of cells from E13.5 gut, DRGs, or sympathetic ganglia that formed multilineage neurospheres, neurosphere diameter, or self-renewal potential (number or percentage of cells from individual primary neurospheres that formed multipotent secondary neurospheres on subcloning; n = 5 independent experiments). C, Multilineage Lgi4LacZ/LacZ NCSC colonies (cultured from E13.5 gut) did not differ from control colonies in terms of neurons (peripherin+) or myofibroblasts (SMA+) but had fewer GFAP+ glia and less pronounced GFAP staining. D, E, Adherently cultured gut (D) and DRG (E) cells from E13.5 Lgi4LacZ/LacZ embryos formed normal numbers of total colonies, neuron (N)-containing colonies, and myofibroblast (M)-containing colonies, but significantly fewer glia (G)-containing colonies compared with littermate control cells. Cells were plated at clonal density (500 cells per 35 mm dish) such that individual cells could form spatially distinct colonies (*p < 0.05; 4 independent experiments). Error bars indicate SD.
We noticed that GFAP staining was consistently reduced in Lgi4LacZ/LacZ NCSC colonies compared with control NCSC colonies. In contrast, we did not detect any effect of Lgi4 deficiency on peripherin or SMA staining (Fig. 4C). The reduction in GFAP staining appeared to reflect a reduced number of GFAP+ cells within Lgi4LacZ/LacZ colonies as well as reduced staining intensity in GFAP+ cells. We observed similar reductions in the number and staining intensity of GFAP+ cells within NCSC colonies cultured from fetal DRG and sympathetic chain (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), as well as from P0 gut (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). These results suggested that Lgi4LacZ/LacZ mice had defects in gliogenesis throughout the PNS, not just in developing peripheral nerves.
To quantify the difference in GFAP staining, we cultured cells from the guts and DRGs of E13.5 Lgi4LacZ/LacZ embryos and littermate controls in clonal adherent cultures. We also cultured cells from the guts of P0 Lgi4LacZ/LacZ mice and littermate controls. Under these conditions, NCSCs form multilineage colonies and restricted progenitors form glia-only or neuron-only colonies (Joseph et al., 2008; Nishino et al., 2008). The total number of colonies formed by Lgi4LacZ/LacZ cells did not significantly differ from control cells (Fig. 4D; supplemental Fig. 3B, available at www.jneurosci.org as supplemental material); therefore, neural crest stem/progenitor cells were not grossly depleted or impaired in their ability to survive in culture in the absence of Lgi4. However, in all cases, multilineage colonies and restricted progenitor colonies contained fewer GFAP+ cells in the absence of Lgi4. Significantly fewer colonies contained GFAP+ glia in the Lgi4LacZ/LacZ cultures, but the total number of colonies that contained neurons or myofibroblasts was not significantly affected by Lgi4 deficiency (Fig. 4D,E; supplemental Fig. 3B, available at www.jneurosci.org as supplemental material). These data suggest that Lgi4 is required for the generation of normal numbers of glia in multiple regions of the PNS.
Fewer glial cells are generated throughout the PNS in vivo in the absence of Lgi4
To test whether Lgi4 deficiency affects enteric gliogenesis in vivo, we examined sections from the guts of Lgi4LacZ/LacZ mice and littermate controls. Lgi4LacZ/LacZ embryos had normal numbers of p75+ neural crest stem/progenitor cells migrating through the foregut and midgut at E10.5 (Fig. 5A,B) consistent with our data indicating that Lgi4 is not required for the generation or self-renewal of gut NCSCs (Fig. 4B). The numbers of Tuj1+ neurons in the foregut, midgut, and hindgut were also normal in Lgi4LacZ/LacZ embryos at E10.5 and at E18.5 (Fig. 5C,D), suggesting that enteric neural crest migration and neurogenesis were not grossly affected by the absence of Lgi4. In contrast, we observed significantly fewer BFABP+ glial cells per section through the foregut, midgut, and hindgut of Lgi4LacZ/LacZ embryos at E18.5 (no gliogenesis was detected at E10.5) (Fig. 5C,D). This demonstrates that fewer enteric glia are generated in vivo in the absence of Lgi4, consistent with what we had observed from gut NCSCs in culture.
Figure 5.
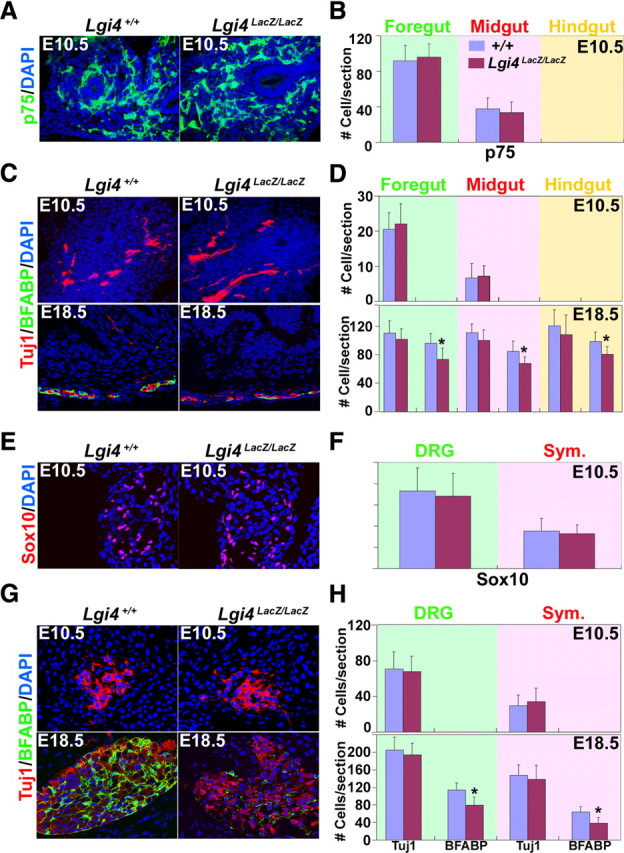
Lgi4LacZ/LacZ mice have normal numbers of undifferentiated progenitors and neurons but fewer glia throughout the PNS. A, Transverse sections of guts from E10.5 control and Lgi4LacZ/LacZ embryos were stained for p75 (a marker of neural crest stem/progenitor cells). B, The number of p75+ cells that colonized the gut at E10.5 did not differ between control and Lgi4LacZ/LacZ embryos (mean ± SD of number of p75+ cells per section; 4 sections per gut region/mouse and 3 mice/genotype). C, Transverse sections of the gut from E10.5 or E18.5 control and Lgi4LacZ/LacZ embryos were stained with antibodies against neuronal (Tuj1) and glial (BFABP) markers. D, The number of neurons (Tuj1+) per gut section was similar in control and Lgi4LacZ/LacZ embryos at E10.5 and E18.5. No glia (BFABP+) were observed in E10.5 gut sections, but the number of glia per section was significantly lower in Lgi4LacZ/LacZ embryos at E18.5 (*p < 0.05; 4–6 sections/gut region per mouse and 4–5 mice/genotype). E, Transverse sections of sympathetic ganglia from E10.5 control and Lgi4LacZ/LacZ mouse embryos were stained with antibody against Sox10 (a marker of neural crest stem/progenitor cells at this stage of development). F, The number of Sox10+ cells per section did not differ between E10.5 control and Lgi4LacZ/LacZ embryos in DRG or sympathetic ganglia (4 sections/mouse and 3 mice/genotype). G, Transverse sections of sympathetic ganglia from E10.5 and E18.5 control and Lgi4LacZ/LacZ embryos were stained with antibodies against neuronal (Tuj1) and glial (BFABP) markers. H, The number of neurons (Tuj1+) per section through DRGs and sympathetic ganglia was similar in control and Lgi4LacZ/LacZ embryos at E10.5 and E18.5. No glia (BFABP+) were observed in E10.5 sections, but the number of glia per section was significantly lower in Lgi4LacZ/LacZ embryos at E18.5 (*p < 0.05; 4–6 sections/mouse and 4–6 mice/genotype). Error bars indicate SD.
We also examined sections through DRGs and sympathetic ganglia at E10.5 and E18.5. At E10.5, there was no difference between Lgi4LacZ/LacZ embryos and littermate controls in the number of Sox10+ neural crest cells per section from DRGs or sympathetic ganglia (Fig. 5E,F). This suggests that undifferentiated neural crest cells were able to migrate normally into DRGs and sympathetic ganglia in the absence of Lgi4. We also observed no difference in the number of Tuj1+ neurons per section from the DRGs and sympathetic ganglia of E10.5 or E18.5 Lgi4LacZ/LacZ mice compared with littermate controls (Fig. 5G,H). This suggested that neurogenesis was grossly normal in the absence of Lgi4. In contrast, the number of BFABP+ glia per sections was significantly reduced in both DRGs and sympathetic ganglia from E18.5 Lgi4LacZ/LacZ embryos compared with littermate controls (no gliogenesis was detected at E10.5) (Fig. 5G,H). These data suggest that Lgi4 is not required for the generation or maintenance of NCSCs in DRGs or sympathetic ganglia, but is required to generate normal numbers of glia in these ganglia.
Glial cells fail to adopt a mature phenotype in the absence of Lgi4
To assess whether the differentiation of glial cells to a mature phenotype is affected by Lgi4 deficiency outside of peripheral nerves, we examined the myenteric plexus of P0 Lgi4LacZ/LacZ mice and littermate controls. Lgi4LacZ/+ mice exhibited a dense, mesh-like myenteric plexus when whole mounts of the outer plexus/muscle layers were stained with X-gal (Fig. 6A). In contrast, the myenteric plexus from Lgi4LacZ/LacZ mice appeared sparse with fewer connections among ganglia (Fig. 6A). Since the number of neurons within the myenteric plexus was normal in the Lgi4LacZ/LacZ gut (Fig. 5D) and neuronal differentiation appeared normal in culture (Fig. 4C), we hypothesized that the sparseness of the plexus was primarily attributable to glial defects. To assess this, we stained newborn gut plexus/muscle layers with antibodies against GFAP, BFABP, or TuJ1. Both GFAP and BFABP staining were greatly reduced within the myenteric plexus of Lgi4LacZ/LacZ mice compared with controls (Fig. 6B). Higher magnification images indicated that enteric glia within the myenteric plexus were smaller and had fewer processes in the absence of Lgi4 (Fig. 6B). In contrast, TuJ1 staining appeared relatively normal, although there again appeared to be fewer connections among ganglia in the Lgi4LacZ/LacZ gut (Fig. 6B). The enteric glia in Lgi4LacZ/LacZ mice therefore failed to adopt a normal, mature morphology.
Figure 6.
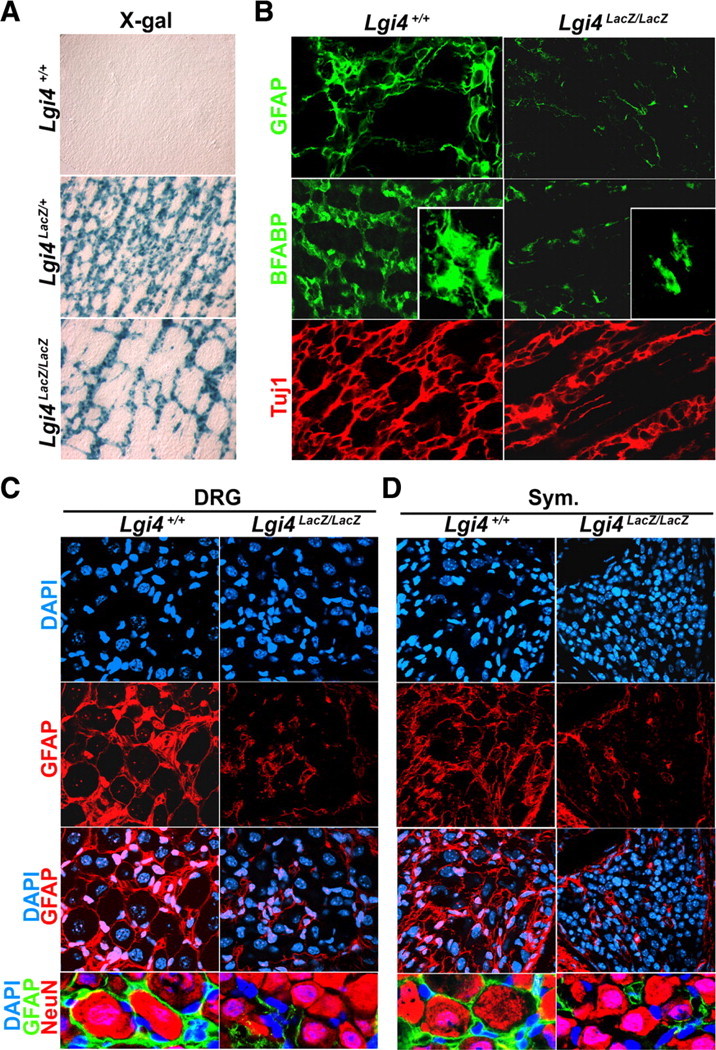
Lgi4 is required for glial cells to acquire a normal, mature morphology throughout the PNS in vivo. A, Whole-mount X-gal staining of outer plexus/muscle layers from the P0 gut of Lgi4LacZ/LacZ mice and littermate controls. The Lgi4LacZ/LacZ gut had a sparse myenteric plexus with fewer connections among ganglia relative to the Lgi4LacZ/+ gut. B, Whole-mount immunostaining of outer plexus/muscle layers from the P0 gut of Lgi4LacZ/LacZ mice and littermate controls with antibodies against the glial markers GFAP and BFABP, and the neuronal marker TuJ1. GFAP+ and BFABP+ staining was sparse in the Lgi4LacZ/LacZ gut. Higher magnification images of glial cells (inset; BFABP) suggested that Lgi4LacZ/LacZ glia were smaller with fewer and shorter processes. C, D, GFAP immunostaining in DRGs (C) and sympathetic ganglia (D) in sections from P4 wild-type mice showed a honeycomb-like pattern that enwrapped neurons, but in sections from P4 Lgi4LacZ/LacZ mice there were fewer glia with shorter processes that did not enwrap neurons.
Similar defects were observed in satellite cells in the DRGs and sympathetic ganglia of P4 Lgi4LacZ/LacZ mice and littermate controls. In wild-type DRGs, GFAP+ satellite glia adopted a honeycomb-like pattern that completely wrapped around sensory neurons. In contrast, within Lgi4LacZ/LacZ DRGs, the GFAP+ satellite cells appeared sparse, had short processes compared with wild-type satellite cells, and usually did not wrap around the neurons (Fig. 6C) (for lower power images showing the entire ganglion, see supplemental Fig. 4, available at www.jneurosci.org as supplemental material). By quantitative PCR, we detected a significant reduction of Erm expression in Lgi4LacZ/LacZ DRGs (0.58 ± 0.11-fold in Lgi4LacZ/LacZ DRGs compared with control ganglia; p < 0.05; three mice per genotype). Since Erm is expressed by satellite glia but not by Schwann cells (Hagedorn et al., 2000), this is consistent with our immunofluorescence analysis in indicating a defect in satellite glia in Lgi4LacZ/LacZ DRGs. The same unusual glial morphology was observed in P4 Lgi4LacZ/LacZ sympathetic ganglia: GFAP+ cells again appeared to be sparse and had short processes that failed to wrap around neurons in contrast to what was observed in control sympathetic ganglia (Fig. 6D). These data indicate that satellite glia fail to adopt a normal, mature morphology throughout the developing PNS in the absence of Lgi4.
Lgi4 is required for the proliferation of glial-restricted progenitors
To investigate the mechanisms by which Lgi4 regulates gliogenesis, we first tested whether Lgi4 acts instructively on NCSCs to promote glial fate determination. To test this, we generated 293T cells that secreted recombinant Lgi4 protein that could be concentrated from conditioned medium (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). We sorted p75+α4+ NCSCs from the E14.5 rat sciatic nerve and added either Lgi4 conditioned medium, control conditioned medium, or Neuregulin1-β1 (Nrg) to the cultures. We used rat sciatic nerve NCSCs in these experiments because they are more sensitive to the instructive effects of gliogenic factors than mouse NCSCs and can be isolated with a higher degree of purity than mouse NCSCs (Morrison et al., 1999). Nrg robustly promoted the generation of glia-only colonies at the expense of multilineage colonies (Fig. 7A), consistent with our previous demonstration that Nrg instructs sciatic nerve NCSCs to form glial-restricted progenitors (Morrison et al., 1999). In contrast, Lgi4 conditioned medium did not significantly affect colony composition relative to control conditioned medium (Fig. 7A). Lgi4 conditioned medium also did not affect colony composition when added to cultures of mouse gut NCSCs (data not shown). We therefore observed no evidence that Lgi4 could promote glial lineage determination.
Figure 7.
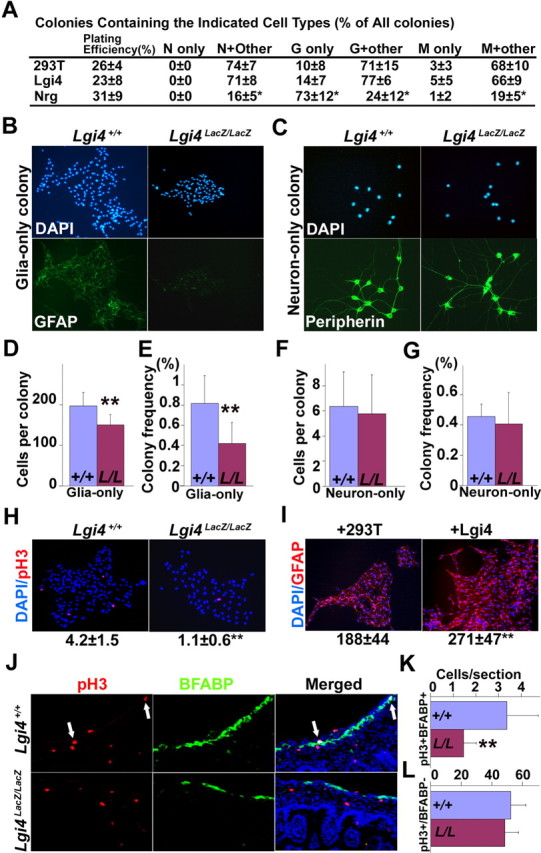
Lgi4 is required for the proliferation of glial-restricted progenitors but not neuronal-restricted progenitors. A, p75+α4+ rat sciatic nerve NCSCs were cultured at clonal density for 10 d in standard medium supplemented with control conditioned medium (293T), Lgi4 conditioned medium, or recombinant Neuregulin-1. Lgi4 did not affect colony composition, in contrast to Neuregulin-1, which instructed glial differentiation by NCSCs at the expense of multilineage differentiation. B–H, Dissociated muscle/plexus layer cells from P0 wild-type and Lgi4LacZ/LacZ gut were cultured at clonal density for 10 d in adherent cultures. Lgi4LacZ/LacZ cells formed smaller glia-only colonies with less GFAP staining than control cells (B) but normal neuron-only colonies (C). The number of cells per glia-only colony (D) and the frequency of glia-only colonies (E) were significantly reduced in cultures of Lgi4LacZ/LacZ cells (**p < 0.05). In contrast, the number of cells per neuron-only colony (F) and frequency of neuron-only colonies (G) was not affected by Lgi4 deficiency. H, The frequency of pH3+ cells per glia-only colony was significantly decreased in Lgi4LacZ/LacZ colonies (**p < 0.05; see mean ± SD below images). I, Muscle/plexus layer cells from P0 wild-type gut were cultured for 10 d in standard medium supplemented with control conditioned medium or Lgi4 conditioned medium. Lgi4 significantly increased the number of cells per glia-only colony (**p < 0.05; see mean ± SD below images). J–L, E16.5 gut sections from wild-type and Lgi4LacZ/LacZ mice revealed a significantly lower frequency of pH3+BFABP+ cells (J, white arrows) in the Lgi4LacZ/LacZ gut (K) (**p < 0.05) but a normal frequency of pH3+BFABP− cells (L). Error bars indicate SD.
We then tested whether Lgi4 promotes gliogenesis by acting selectively to promote the proliferation or survival of glial-restricted progenitors. To test the effect of Lgi4 on glial-restricted progenitors, we cultured dissociated cells from E13.5 or P0 gut, or from E13.5 DRG, sympathetic ganglia, or sciatic nerve from Lgi4LacZ/LacZ mice and littermate controls in adherent cultures at clonal density (such that individual cells formed spatially distinct colonies). Lgi4LacZ/LacZ gut cells formed significantly fewer and significantly smaller glia-only colonies compared with control cells (Fig. 7B,D,E; supplemental Fig. 6A, available at www.jneurosci.org as supplemental material). Lgi4LacZ/LacZ sympathetic ganglion cells, but not sciatic nerve cells, also formed significantly fewer and significantly smaller glia-only colonies compared with control cells (supplemental Fig. 6A,C, available at www.jneurosci.org as supplemental material). Lgi4LacZ/LacZ DRG cells formed fewer and smaller glia-only colonies, but the difference compared with control cells was not statistically significant (supplemental Fig. 6A,C, available at www.jneurosci.org as supplemental material). The frequency, size, and peripherin staining of neuron-only colonies from all regions of the developing PNS were not affected by Lgi4 deficiency (Fig. 7C,F,G). These results suggest that Lgi4 is required for the expansion of glial-restricted progenitors in gut and sympathetic ganglia, and perhaps in DRG, but not in sciatic nerve.
To study the mechanism by which Lgi4 promotes the expansion of glial-restricted progenitors, we tested whether Lgi4 deficiency affected survival or proliferation within glial-restricted progenitor colonies. To assess the frequency of cell death, we stained cultures with an antibody against activated-caspase-3 but found only rare cells undergoing cell death within glia-only colonies, regardless of whether the colonies were formed by Lgi4LacZ/LacZ or control cells (data not shown). We also stained sections through the gut and DRGs of E13.5 Lgi4LacZ/LacZ mice and littermate controls but did not detect any effect of Lgi4 deficiency on the frequency of activated caspase-3+ cells in vivo (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). We were therefore unable to attribute the differences in gliogenesis in vitro or in vivo to cell death in the absence of Lgi4.
In contrast, we did observe effects of Lgi4 on the proliferation of glial-restricted progenitors. We observed a significantly lower frequency of dividing cells within Lgi4LacZ/LacZ compared with control glia-only colonies based on the frequency of pH3+ cells (Fig. 7H). We also observed a significantly lower frequency of dividing cells within Lgi4LacZ/LacZ compared with control glia-only colonies from E13.5 DRGs and sympathetic ganglia, but not from sciatic nerve (supplemental Fig. 6B, available at www.jneurosci.org as supplemental material). Glia-only colonies from E13.5 gut also exhibited a reduction, but this was not statistically significant (supplemental Fig. 6B, available at www.jneurosci.org as supplemental material). Addition of Lgi4 conditioned medium to cultures of wild-type gut cells significantly increased the number of cells within glia-only colonies (Fig. 7I), without affecting the size or composition of neuron-only colonies (data not shown). We also stained E16.5 gut sections from Lgi4LacZ/LacZ mice and littermate controls with antibodies against pH3 and BFABP. Significantly fewer pH3+BFABP+ cells were observed in Lgi4LacZ/LacZ gut sections compared with control gut sections (Fig. 7J,K). In contrast, we observed no difference between Lgi4LacZ/LacZ and control gut sections in the frequency of pH3+ cells that did not express BFABP (Fig. 7L). These results suggest that Lgi4 is required for the proliferation of glial-restricted progenitors from gut, sympathetic ganglia, and DRGs, but that it is not required for the proliferation of neuronal progenitors from these ganglia or for the proliferation of glial progenitors from peripheral nerve.
ADAM22 is a receptor for Lgi4 that regulates enteric gliogenesis
A very recent study demonstrated that Lgi4 promotes peripheral nerve myelination by binding ADAM22 (Ozkaynak et al., 2010), but ADAM22 is not known to regulate other aspects of PNS development. We confirmed that Lgi4 bound to ADAM11, 22, and 23 in immunoprecipitation and cell surface binding assays but not to an unrelated family member, ADAM9 (Fig. 8A,B). Adam22 and Adam23 were expressed throughout the PNS at E14.5 and P0, including within DRGs, sympathetic ganglia, and myenteric plexus (Fig. 8C; supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Adam9 expression was not detected in the PNS and Adam11 expression was detected in developing DRGs and sympathetic ganglia, but not in the myenteric plexus (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). These results suggested that Lgi4 could potentially have multiple receptors but that ADAM22 and 23 were the best candidates to mediate the effects on Lgi4 throughout the developing PNS.
Figure 8.
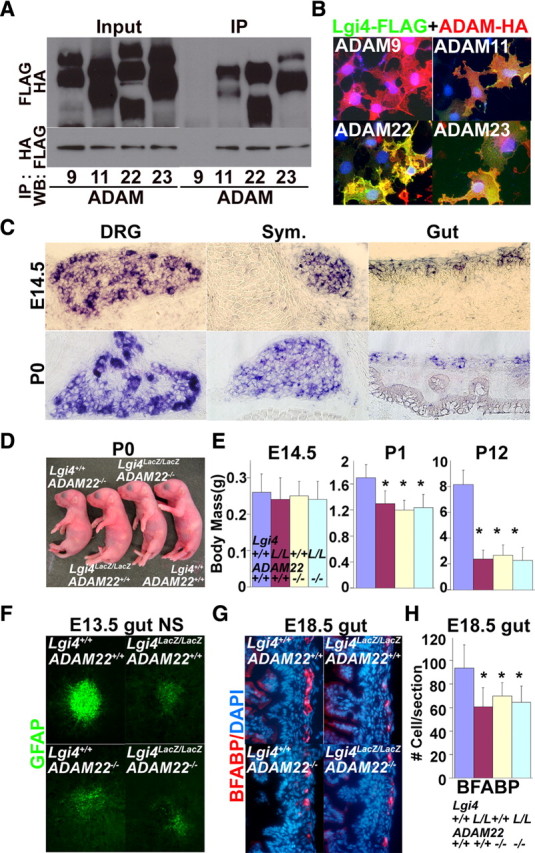
ADAM22 is a physiological receptor for Lgi4 that mediates the effect of Lgi4 on PNS gliogenesis. A, B, Lgi4 binds to ADAM11, ADAM22, and ADAM23, but not ADAM9. A, FLAG-tagged Lgi4 was cotransfected with HA-tagged ADAMs into 293T cells. Immunoprecipitation with anti-FLAG pulled down ADAM11, ADAM22, and ADAM23, but not ADAM9. Immunoprecipitation with anti-HA antibody pulled down Lgi4 when ADAM11, ADAM22, or ADAM23 were expressed but not when ADAM9 was expressed. Input, Before immunoprecipitation; IP, after immunoprecipitation. B, Immunofluorescence analysis indicated that FLAG-tagged Lgi4 overexpressed in COS7 cells was retained on the cell surface when HA-tagged-ADAM11, ADAM22, or ADAM23 were coexpressed but not when ADAM9 was expressed. C, In situ hybridization indicated that Adam22 is expressed in DRGs, sympathetic ganglia, and myenteric plexus of E14.5 and P0 mice. Adam11 and Adam23 were also expressed in certain locations within the PNS (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). D, At birth, Adam22−/− and Lgi4LacZ/LacZAdam22−/− mice exhibited an arthrogryposis-like forelimb posture abnormality similar to Lgi4LacZ/LacZ mice. E, Adam22−/− and Lgi4LacZ/LacZAdam22−/− mice exhibited postnatal growth retardation similar to Lgi4LacZ/LacZ mice. Lgi4LacZ/LacZ and Adam22−/− mutations did not have additive effects on growth. *p < 0.01. F, Compared with wild-type NCSC colonies, E13.5 gut NCSC colonies from Adam22−/− and Lgi4LacZ/LacZAdam22−/− mice generated smaller numbers of glial cells with less pronounced GFAP staining, similar to Lgi4LacZ/LacZ NCSCs. G, H, Compared with wild type, E18.5 gut sections from Adam22−/− and Lgi4LacZ/LacZAdam22−/− mice had smaller numbers of glial cells, similar to Lgi4LacZ/LacZ NCSCs (*p < 0.05; 4–5 mice/genotype with 4–6 sections/mouse). Lgi4LacZ/LacZ and Adam22−/− mutations did not have additive effects on gliogenesis, suggesting that Lgi4 and ADAM22 act in the same pathway. Error bars indicate SD.
To formally test whether ADAM22 mediates Lgi4 function, we performed side-by-side comparisons of the phenotypes of Lgi4LacZ/LacZ mice, Adam22−/− mice, and Adam22−/−Lgi4LacZ/LacZ compound-mutant mice. Most Adam22−/− mice and Adam22−/−Lgi4LacZ/LacZ mice died soon after birth and all of these mice died within 3 weeks after birth, just like Lgi4LacZ/LacZ mice (supplemental Fig. 9, available at www.jneurosci.org as supplemental material). Newborn Adam22−/− and Adam22−/−Lgi4LacZ/LacZ compound-mutant mice exhibited arthrogryposis-like (claw paw) phenotypes similar to Lgi4LacZ/LacZ mice (Fig. 8D). We observed similar degrees of peripheral nerve hypomyelination in newborn Adam22−/− mice, Lgi4LacZ/LacZ mice, and Adam22−/− Lgi4LacZ/LacZ mice: in each case, Krox20 expression, Periaxin expression, and axonal myelination were dramatically reduced (supplemental Fig. 10, available at www.jneurosci.org as supplemental material). We also observed similar degrees of postnatal growth retardation in Adam22−/− mice and Adam22−/−Lgi4LacZ/LacZ double-mutant mice compared with Lgi4LacZ/LacZ mice (Fig. 8E). Adam22−/− and Adam22−/−Lgi4LacZ/LacZ mice were significantly smaller than wild-type controls, but were not significantly different from Lgi4LacZ/LacZ mutants at P1 or P12 (Fig. 8E). Thus, Adam22−/− mice have a gross phenotype similar to Lgi4LacZ/LacZ mice in terms of hypomyelination, growth retardation, and premature death. Moreover, these phenotypes in Adam22−/−Lgi4LacZ/LacZ compound-mutant mice are similar to those in Adam22−/− and Lgi4LacZ/LacZ single-mutant mice.
To test whether ADAM22 mediates the effect of Lgi4 on gliogenesis in the enteric nervous system, we cultured NCSCs from the E13.5 gut of Adam22−/− Lgi4LacZ/LacZ compound-mutant mice as well as Adam22−/− and Lgi4LacZ/LacZ single-mutant mice. In each case, Adam22−/−Lgi4LacZ/LacZ NCSC colonies, Adam22−/− NCSC colonies, and Lgi4LacZ/LacZ NCSC colonies contained fewer GFAP+ glia with less pronounced GFAP staining compared with wild-type colonies (Fig. 8F). Yet peripherin+ neurons and SMA+ myofibroblasts appeared normal in colonies of all genotypes. These results indicated that Adam22-deficient gut NCSCs exhibit a gliogenic defect in culture, similar to Lgi4LacZ/LacZ NCSCs.
To test whether ADAM22 mediates the effect of Lgi4 on enteric gliogenesis in vivo, we cut sections through the guts of E18.5 Adam22−/−Lgi4LacZ/LacZ compound-mutant mice as well as Adam22−/− and Lgi4LacZ/LacZ single-mutant mice. We observed significant reductions in the number of glia per section from Adam22−/−Lgi4LacZ/LacZ compound-mutant mice, Adam22−/− mice, and Lgi4LacZ/LacZ mice compared with wild-type mice. Yet we observed no additive effect of mutations in Adam22 and Lgi4 on glial cell number. This demonstrates that ADAM22, like Lgi4, regulates enteric gliogenesis, and the similarity of the single-mutant and compound-mutant phenotypes suggests that Lgi4 and ADAM22 act genetically within the same pathway. Our data suggest that ADAM22 is a physiological receptor for Lgi4 in the enteric nervous system.
Discussion
Our results identify a new mechanism that regulates the proliferation and maturation of glial lineage cells throughout the developing PNS. Lgi4LacZ/LacZ NCSCs from gut and DRGs formed fewer GFAP+ glia in culture (Fig. 4C), and unfractionated cells from gut, DRG, and sympathetic chain formed fewer glia-containing colonies overall (Fig. 4D,E; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Consistent with these results in culture, we observed significantly fewer BFABP+ glia in sections through the E18.5 gut, DRG, and sympathetic chain (Fig. 5). Lgi4LacZ/LacZ enteric glia were also smaller, with shorter/fewer processes compared with wild-type glia (Fig. 6B). Similar results were observed in DRGs and sympathetic ganglia in which Lgi4LacZ/LacZ satellite glia failed to wrap around neurons, in contrast to wild-type satellite cells (Fig. 6C,D). Lgi4 is thus required to generate normal numbers of glia as well as mature glial morphologies throughout the PNS.
The failure to generate normal numbers of glia in the absence of Lgi4 is at least partially caused by a defect in the proliferation of glial-restricted progenitors. Although neuronal-restricted progenitors were present in normal frequencies and formed normal sized colonies (Fig. 7C,F,G), glial-restricted progenitors were depleted and formed smaller colonies in the absence of Lgi4 (Fig. 7B,D,E; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Consistent with this, recombinant Lgi4 promoted the proliferation of glial-restricted progenitors in culture (Fig. 7I). Also, Lgi4LacZ/LacZ mice had a lower frequency of BFABP+pH3+ gut cells, but not BFABP−pH3+ cells, compared with littermate controls in vivo (Fig. 7K,L).
Our data suggest Lgi4 promotes the proliferation of glial-restricted progenitors as well as the differentiation of their progeny to a mature phenotype in ganglia throughout the developing PNS. Nrg is also known to promote the proliferation and maturation of glial progenitors (Dong et al., 1995). Indeed, Nrg has distinct effects on cells at multiple stages of the gliogenesis process as it also promotes glial lineage determination by NCSCs in developing nerves (Shah et al., 1994; Morrison et al., 1999; Joseph et al., 2004). One mechanism by which gliogenic factors can have distinct effects on cells at different stages of gliogenesis may be by activating distinct signaling pathways in different cells within the glial lineage. Nrg may promote proliferation by activating MAPK signaling while promoting differentiation by activating PI3-kinase signaling (Taveggia et al., 2005; Bhatheja and Field, 2006). Like Nrg, Lgi4/ADAM22 signaling might activate different intracellular signaling pathways in cells at distinct stages of gliogenesis, promoting the proliferation of immature glial progenitors while promoting the differentiation of their more mature progeny.
Our results also reveal a previously unrecognized role for ADAM22 regulating gliogenesis within the enteric nervous system and likely in other regions of the developing PNS as well. Like Lgi4, ADAM22 is expressed throughout the developing PNS (Fig. 8C) and Lgi4 binds to ADAM22 (Fig. 8A,B). Genetically, Adam22 and Lgi4 appear to function in the same pathway because Adam22−/− and Lgi4LacZ/LacZ mice have similar growth retardation and premature death phenotypes, but these phenotypes are unchanged (not additive) in Adam22−/−Lgi4LacZ/LacZ compound-mutant mice. Adam22−/− mice and Adam22−/−Lgi4LacZ/LacZ compound-mutant mice also exhibit defects in gliogenesis in culture from gut NCSCs and in vivo within the myenteric plexus that are similar to those observed in Lgi4LacZ/LacZ mice (Fig. 8F–H). These data suggest that ADAM22 regulates gliogenesis in the enteric nervous system as a receptor for Lgi4.
ADAM22 is not necessarily the only physiological receptor for Lgi4. Adam11 is expressed in some regions of the developing PNS, and Adam23 is widely expressed in the developing PNS (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Lgi4 could have different receptors in different regions of the PNS, different receptors on different cell types within particular regions of the PNS, or multiple receptors expressed by the same cell types. In principle, different receptors could either have different functions or redundant functions. However, there do not appear to be redundant receptors with respect to nerve myelination or gut gliogenesis as Adam22−/− mice have phenotypes that are very similar to Lgi4LacZ/LacZ mice in these regions of the PNS.
Another issue concerns the identity of the cells that express ADAM22. In our hands, available antibodies against ADAM22 do not give clean staining that distinguishes between different ADAM family members. This makes it difficult to distinguish precisely which cells express ADAM22. Our analysis of in situ hybridization patterns indicates that Adam22 is expressed by neurons in multiple regions of the PNS, consistent with a recent study that observed Adam22 expression by sensory neurons in DRGs (Ozkaynak et al., 2010). It is more difficult to confidently assess whether Adam22 is expressed by glia because glia are much smaller, often cluster around neurons, and in situ hybridization does not necessarily give single-cell resolution. This raises two possibilities for ADAM22 function. One possibility is that ADAM22 is expressed by neurons, which secrete factors that promote gliogenesis throughout the PNS on stimulation by Lgi4 secreted by glial lineage cells. The other possibility is that Lgi4 can directly act on glial-restricted progenitors in some regions of the PNS. Consistent with this possibility, our in vitro results suggest that Lgi4 can directly promote the proliferation of glial-restricted progenitors in colonies that do not contain neurons (Fig. 7B,D,E). Resolution of these issues will require the development of better tools to monitor ADAM22 expression.
Our results show that Lgi4/ADAM22 signaling has an unanticipated role regulating gliogenesis throughout the PNS, promoting the generation and differentiation of enteric glia and satellite cells in DRGs and sympathetic ganglia. Although Lgi4 and ADAM22 were previously known to regulate peripheral nerve myelination (Sagane et al., 2005; Bermingham et al., 2006; Ozkaynak et al., 2010), they were not known to regulate PNS development outside of nerves and were not known to regulate the proliferation of glial lineage cells. By promoting the proliferation and maturation of glial lineage progenitors, Lgi4 collaborates with glial lineage determination factors such as Nrg and Notch ligands, to promote the generation of glia throughout the PNS.
Footnotes
This work was supported by the Howard Hughes Medical Institute and National Institute of Neurological Disorders and Stroke Grant NS-040750-10. Flow cytometry was partially supported by the University of Michigan (UM) Comprehensive Cancer (UMCC) National Institutes of Health (NIH) Grant CA46592. Antibody production was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant NIH5P60-DK20572 [Michigan Diabetes Research and Training Center (MDRTC)]. Gene-targeted mouse production was partially supported by the MDRTC, UMCC, and UM–Gastrointestinal Hormone Research Core Center NIH Grant 5P30DK034933. J.N. was supported by a postdoctoral fellowship from the Japan Society for the Promotion of Science. Thanks to David Adams and Martin White for flow cytometry, to Elizabeth Smith (UM Hybridoma Core) for antibody production, and to Elizabeth Hughes and Keith Childs for the generation of Lgi4 knock-out mice. Thanks to P. Brophy (University of Edinburgh), T. Glaser (University of Michigan), and T. Muller (Max Delbruck Center) for sharing antibodies. Thanks to Dr. Masaki Fukada and Yoko Fukada for sharing ADAM-HA expression vectors.
References
- Anderson DJ, Groves A, Lo L, Ma Q, Rao M, Shah NM, Sommer L. Cell lineage determination and the control of neuronal identity in the neural crest. Cold Spring Harb Symp Quant Biol. 1997;62:493–504. [PubMed] [Google Scholar]
- Bermingham JR, Jr, Shearin H, Pennington J, O'Moore J, Jaegle M, Driegen S, van Zon A, Darbas A, Ozkaynak E, Ryu EJ, Milbrandt J, Meijer D. The claw paw mutation reveals a role for Lgi4 in peripheral nerve development. Nat Neurosci. 2006;9:76–84. doi: 10.1038/nn1598. [DOI] [PubMed] [Google Scholar]
- Bhatheja K, Field J. Schwann cells: origins and role in axonal maintenance and regeneration. Int J Biochem Cell Biol. 2006;38:1995–1999. doi: 10.1016/j.biocel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Bixby S, Kruger GM, Mosher JT, Joseph NM, Morrison SJ. Cell-intrinsic differences between stem cells from different regions of the peripheral nervous system regulate the generation of neural diversity. Neuron. 2002;35:643–656. doi: 10.1016/s0896-6273(02)00825-5. [DOI] [PubMed] [Google Scholar]
- Bozzola JJ, Russell LD. Electron microscopy: principles and techniques for biologists. Boston: Jones and Bartlett Publishers; 1992. [Google Scholar]
- Copeland NG, Jenkins NA, Court DL. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet. 2001;2:769–779. doi: 10.1038/35093556. [DOI] [PubMed] [Google Scholar]
- Darbas A, Jaegle M, Walbeehm E, van den Burg H, Driegen S, Broos L, Uyl M, Visser P, Grosveld F, Meijer D. Cell autonomy of the mouse claw paw mutation. Dev Biol. 2004;272:470–482. doi: 10.1016/j.ydbio.2004.05.017. [DOI] [PubMed] [Google Scholar]
- Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR. Neu differentiation factor is a neuron-glia signal and regulates survival, proliferation and maturation of rat Schwann cell precursors. Neuron. 1995;15:585–596. doi: 10.1016/0896-6273(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Fraser SE, Bronner-Fraser M. Migrating neural crest cells in the trunk of the avian embryo are multipotent. Development. 1991;112:913–920. doi: 10.1242/dev.112.4.913. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- Gillespie CS, Sherman DL, Blair GE, Brophy PJ. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994;12:497–508. doi: 10.1016/0896-6273(94)90208-9. [DOI] [PubMed] [Google Scholar]
- Hagedorn L, Paratore C, Brugnoli G, Baert JL, Mercader N, Suter U, Sommer L. The Ets domain transcription factor Erm distinguishes rat satellite glia from Schwann cells and is regulated in satellite cells by neuregulin signaling. Dev Biol. 2000;219:44–58. doi: 10.1006/dbio.1999.9595. [DOI] [PubMed] [Google Scholar]
- Henion PD, Weston JA. Timing and pattern of cell fate restrictions in the neural crest lineage. Development. 1997;124:4351–4359. doi: 10.1242/dev.124.21.4351. [DOI] [PubMed] [Google Scholar]
- Hughes ED, Qu YY, Genik SJ, Lyons RH, Pacheco CD, Lieberman AP, Samuelson LC, Nasonkin IO, Camper SA, Van Keuren ML, Saunders TL. Genetic variation in C57BL/6 ES cell lines and genetic instability in the Bruce4 C57BL/6 ES cell line. Mamm Genome. 2007;18:549–558. doi: 10.1007/s00335-007-9054-0. [DOI] [PubMed] [Google Scholar]
- Iwashita T, Kruger GM, Pardal R, Kiel MJ, Morrison SJ. Hirschsprung disease is linked to defects in neural crest stem cell function. Science. 2003;301:972–976. doi: 10.1126/science.1085649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer D, Anderson DJ, Morrison SJ. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 2004;131:5599–5612. doi: 10.1242/dev.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, Conway SJ, Parada LF, Zhu Y, Morrison SJ. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008;13:129–140. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köntgen F, Süss G, Stewart C, Steinmetz M, Bluethmann H. Targeted disruption of the MHC class-II AA gene in C57BL/6 mice. Int Immunol. 1993;5:957–964. doi: 10.1093/intimm/5.8.957. [DOI] [PubMed] [Google Scholar]
- Koszowski AG, Owens GC, Levinson SR. The effect of the mouse mutation claw paw on myelination and nodal frequency in sciatic nerves. J Neurosci. 1998;18:5859–5868. doi: 10.1523/JNEUROSCI.18-15-05859.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger GM, Mosher JT, Bixby S, Joseph N, Iwashita T, Morrison SJ. Neural crest stem cells persist in the adult gut but undergo changes in self-renewal, neuronal subtype potential, and factor responsiveness. Neuron. 2002;35:657–669. doi: 10.1016/s0896-6273(02)00827-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Zimmer A, Schnütgen F, Brüning G, Spener F, Müller T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development. 1994;120:2637–2649. doi: 10.1242/dev.120.9.2637. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM. Cell line segregation during peripheral nervous system ontogeny. Science. 1986;231:1515–1522. doi: 10.1126/science.3952494. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C. Multiple essential functions of neuregulin in development. Nature. 1995;378:386–390. doi: 10.1038/378386a0. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–749. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, Anderson DJ. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101:499–510. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- Mosher JT, Yeager KJ, Kruger GM, Joseph NM, Hutchin ME, Dlugosz AA, Morrison SJ. Intrinsic differences among spatially distinct neural crest stem cells in terms of migratory properties, fate determination, and ability to colonize the enteric nervous system. Dev Biol. 2007;303:1–15. doi: 10.1016/j.ydbio.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkaynak E, Abello G, Jaegle M, van Berge L, Hamer D, Kegel L, Driegen S, Sagane K, Bermingham JR, Jr, Meijer D. Adam22 is a major neuronal receptor for Lgi4-mediated Schwann cell signaling. J Neurosci. 2010;30:3857–3864. doi: 10.1523/JNEUROSCI.6287-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratore C, Goerich DE, Suter U, Wegner M, Sommer L. Survival and glial fate acquisition of neural crest cells are regulated by an interplay between the transcription factor Sox10 and extrinsic combinatorial signaling. Development. 2001;128:3949–3961. doi: 10.1242/dev.128.20.3949. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the erbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- Rodríguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- Sagane K, Hayakawa K, Kai J, Hirohashi T, Takahashi E, Miyamoto N, Ino M, Oki T, Yamazaki K, Nagasu T. Ataxia and peripheral nerve hypomyelination in ADAM22-deficient mice. BMC Neurosci. 2005;6:33. doi: 10.1186/1471-2202-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4:387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NM, Anderson DJ. Integration of multiple instructive cues by neural crest stem cells reveals cell-intrinsic biases in relative growth factor responsiveness. Proc Natl Acad Sci U S A. 1997;94:11369–11374. doi: 10.1073/pnas.94.21.11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell. 1994;77:349–360. doi: 10.1016/0092-8674(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973–985. doi: 10.1016/0092-8674(92)90393-q. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P, Chao MV, Falls DL, Role L, Salzer JL. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47:681–694. doi: 10.1016/j.neuron.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MK, Yeager K, Morrison SJ. Physiological Notch signaling promotes gliogenesis in the developing peripheral and central nervous systems. Development. 2007;134:2435–2447. doi: 10.1242/dev.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topilko P, Murphy P, Charnay P. Embryonic development of Schwann cells—multiple roles for neuregulins along the pathway. Mol Cell Neurosci. 1997;8:71–75. doi: 10.1006/mcne.1996.0045. [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y, Maynard TM, Weston JA. Fate determination of neural crest cells by Notch-mediated lateral inhibition and asymmetrical cell division during gangliogenesis. Development. 2000;127:2811–2821. doi: 10.1242/dev.127.13.2811. [DOI] [PubMed] [Google Scholar]