

Abstract
Background
Alcohol use disorders (AUDs) are associated with an increased susceptibility to a variety of common and devastating pulmonary diseases including community- and hospital-acquired pneumonias, as well as the acute respiratory distress syndrome (ARDS). Alveolar macrophages play an important role in preventing the development of these disorders through maintaining lung sterility and resolving lung inflammation. Although alcohol exposure has been associated with aberrant alveolar macrophage function in animal models, the clinical relevance of these observations in humans is not established. Therefore, we sought to determine the effects of AUDs on human alveolar macrophage gene expression.
Methods
Whole genome microarray analysis was performed on alveolar macrophages obtained by bronchoalveolar lavage from a test cohort of subjects with AUDs (n=7), and controls (n=7) who were pair-matched on age, gender, and smoking. Probe set expression differences in this cohort were validated by real time reverse transcription-polymerase chain reaction (RT RT-PCR). Functional analysis with web-based bioinformatics tools was utilized with microarray data to assess differentially expressed candidate genes (p<0.01) based on alcohol consumption. Alveolar macrophage mRNA samples from a second cohort of subjects with AUDs (n=7) and controls (n=7) were used to confirm gene expression differences related to AUDs. Results: In both the test and confirmatory cohorts, AUDs were associated with upregulation of alveolar macrophage gene expression related to apoptosis, including perforin-1, granzyme A, and CXCR4 (fusin). Pathways governing the regulation of progression through cell cycle and immune response were also affected, as was upregulation of gene expression for mitochondrial superoxide dismutase. Overall, 12 genes’ expression was affected by AUDs independent of smoking.
Conclusions
AUDs are associated with unique changes in human alveolar macrophage gene expression. Novel therapies targeting alveolar macrophage gene expression in the setting of AUDs may prove to be clinically useful in limiting susceptibility for pulmonary disorders in these individuals.
Keywords: pneumonia, acute lung injury, apoptosis, perforin, human
Introduction and Background
Alveolar macrophages are a major component of pulmonary innate immunity with a spectrum of roles in the pathophysiology of pneumonia and acute lung injury (ALI). They are characterized by the ability to avidly phagocytose pathogens in the terminal airways, and have the ability to recruit granulocytes from the systemic circulation via generating chemotactic mediators when an infectious inoculum is overwhelming (Nelson and Summer, 1998; Toews et al., 1979). Additionally, these cells also function in the ingestion of apoptotic granulocytes in the setting of ALI to promote resolution of lung inflammation (Haslett, 1999).
The association between alcohol use disorders (AUDs), including alcohol abuse and alcohol dependence, and an increased susceptibility for lung diseases including severe bacterial pneumonias and ALI have long been appreciated in the intensive care unit setting (Moss et al., 1996; Moss et al., 1999; van der Poll and Opal, 2009). When bacterial pneumonia occurs in patients with AUDs, a variety of deleterious outcomes may be expected, including greater symptom severity, increased hospital length of stay, increased need for intensive care unit admission, and worse survival (de Roux et al., 2006; Fernandez-Sola et al., 1995). In patients with ALI and AUDs, increased mortality (Moss et al., 1996) and the development of non-pulmonary organ dysfunction (Moss et al., 1999) are frequently observed. The effects of alcohol consumption on lung that contribute to the predisposition for these pulmonary disorders are incompletely understood. Better delineation of alcohol-specific abnormalities in the lung could lead to novel therapies to decrease the incidence and severity of pneumonia and ALI in at-risk individuals who abuse alcohol.
The effect of alcohol on alveolar macrophages may explain the observed association between AUDs and lung diseases including bacterial pneumonia and ALI. Studies conducted in animal models have determined that alcohol consumption limits the ability of alveolar macrophages to produce cytokines important in host defense (Standiford and Danforth, 1997), decreases their ability to express receptors for granulocyte-monocyte colony stimulating factor (GMCSF) (Joshi et al., 2005; Joshi et al., 2006), decreases their ability to phagocytose bacteria (Brown et al., 2004; Ping et al., 2007) and impairs their terminal differentiation into mature cells (Brown et al., 2009). Moreover, increased apoptosis of alveolar macrophages has also been reported in this setting that is associated with the excessively oxidized pulmonary milieu elicited by alcohol (Ping et al., 2007). Importantly, studies in human subjects have confirmed intrapulmonary oxidative stress in the setting of AUDs (Moss et al., 2000; Yeh et al., 2007). However, the effect of AUDs on human alveolar macrophage functions, including apoptosis, has been limited. While prior animal model observations demonstrating the effect of alcohol on alveolar macrophages have potentially important implications in our understanding of the pathophysiology of pulmonary diseases in AUDs, such as bacterial pneumonia and ALI, without translational human studies our understanding of their relevance to human disease is limited.
We postulated that examining the transcriptome of alveolar macrophages would help identify gene expression affected by AUDs that could contribute to the predisposition for bacterial pneumonia and ALI in this population. Based on prior observations, we hypothesized that there would be differences in alveolar macrophage gene expression relevant to oxidative stress pathways (Brown et al., 2004; Moss et al., 2000) and to programmed cell death or apoptosis (Ping et al., 2007) among subjects with AUDs compared to pair-matched controls. We validated differences in gene expression observed in microarray with real-time reverse transcription polymerase chain reaction (real time RT-PCR) experiments. Further, we sought to confirm expression differences identified in the first cohort of AUD subjects and controls with a second set of experiments in samples from a separate cohort of AUD subjects and pair-matched controls.
Materials and methods
Subject management
Subject screening, recruitment, and enrollment
Subjects with alcohol use disorders (AUDs) were recruited from the Denver Comprehensive Addictions Rehabilitation and Evaluation Services (Denver CARES) center, an inpatient facility affiliated with Denver Health and Hospital System in Denver, CO. Control subjects without AUDs were recruited from the Denver VA Medical Center’s smoking cessation clinic, and via approved flyers posted on the University of Colorado Denver’s medical campus. Institutional review boards at all participating sites approved this study and all subjects provided written informed consent prior to participating in this protocol.
Subjects with alcohol abuse were eligible to participate if they met all of the following criteria at study entry: 1) an Alcohol Use Disorders Identification Test (AUDIT) score of ≥8 for men, or ≥5 for women 2) alcohol use within the seven days prior to enrollment, and 3) age of ≥21. The AUDIT questionnaire is a standardized survey to detect current and previous alcohol abuse that has been validated in a variety of clinical settings (Reinert and Allen, 2002).
In an effort to minimize the effects of co-morbidities, AUD subjects and controls were ineligible to participate in the study if they met any of the following criteria: 1) prior medical history of liver disease (documented history of cirrhosis, total bilirubin ≥2.0 mg/dL, or albumin < 3.0), 2) prior medical history of gastrointestinal bleeding (due to the concern of varices), 3) prior medical history of heart disease (documentation of ejection fraction < 50%, myocardial infarction, or severe valvular dysfunction), 4) prior medical history of renal disease (end-stage renal disease requiring dialysis, or a serum creatinine ≥2 mg/dL), 5) prior medical history of lung disease defined as an abnormal chest radiograph or spirometry (percent predicted forced vital capacity [FVC] or forced expiratory volume in 1 second [FEV1] <75%), 6) concurrent illicit drug use defined as a positive toxicology screen, 7) prior history of diabetes mellitus, 8) prior history of HIV infection, 9) failure of the patient to provide informed consent, 10) pregnancy, 11) abnormal nutritional risk index (Detsky et al., 1984). Potential subjects >55 years of age were also excluded to minimize the presence of concomitant but asymptomatic co-morbidities.
Control subjects were pair-matched to AUD subjects on the basis of age, gender, and current tobacco use. This strategy was chosen to ultimately allow the use of a paired statistical analysis to examine variability within each AUD subject/control subject pair, while eliminating between-subject variability. Using this type of an analysis enhanced our ability to detect differences in alveolar macrophage gene expression most likely related to alcohol use disorders. Further, to meet eligibility as a control, control subjects must have had AUDIT scores <3, and not have consumed alcohol within the prior month.
Bronchoscopy with bronchoalveolar lavage (BAL)
Bronchoscopy procedures were performed in the inpatient Clinical and Translational Research Center (CTRC) at the University of Colorado Hospital. All procedures were performed utilizing telemetry monitoring and standard conscious sedation protocols as previously described (Hunninghake et al., 1979). The bronchoscope was wedged into a subsegment of either the right middle lobe or the lingula. Three 50 mL aliquots of sterile, room temperature 0.9% NaCl were sequentially instilled (150 mL total) and recovered with gentle aspiration. BAL fluid was then transported to the laboratory immediately in sterile 50 mL conical tubes for processing.
Laboratory Protocols
Cell count, differential, and viability from BAL samples
Using a small aliquot of BAL fluid, cell count per mL was determined by microscopy. Diff-quick (Andwin Scientific, Addison, IL) staining was performed. A minimum of 200 cells were evaluated to determine the differential cell types in all samples. Viability of the cells was determined by Trypan blue exclusion.
RNA Extraction
Cells in BAL fluid were pelleted (900 × g for 5 minutes), and the supernatant was removed. Cell pellets were stored at −80°C. After all samples were collected, RNA was extracted using QIAshredder columns and the RNeasy Mini Kit (Qiagen, Valencia, CA). The protocol utilized the on-column DNase digestion with the RNase-free DNase Set (Qiagen, Valencia, CA). The on-column DNase digestion was also performed per the manufacturer’s instructions. To elute the RNA, 30μl of the RNase-free water provided was pipetted onto the column, which was incubated for 10 minutes at room temperature before centrifugation. The second elution step was performed using the first eluate and also incubated for 10 minutes at room temperature before centrifugation. After elution, the RNA quantity and quality was determined using the NanoDrop Spectrophotometer (Thermo Scientific, Wilmington, DE).
Microarray experiments
Microarray analysis was performed on samples from consecutive subjects with AUDs and pair-matched controls in half of the total subjects (referred to as the Test Cohort). RNA quality was determined by capillary gel electrophoresis using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The MessageAmp Premier RNA Amplification Kit (Ambion, Austin, TX) was used to synthesize and fragment the antisense RNA (aRNA). The aRNA was hybridized to Affymetrix Human Genome U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA) and the arrays were washed and scanned using the manufacturer’s protocols. This array assesses expression of 54,675 genes in the human genome.
Real-time RT-PCR
For all available samples, 1μg of total RNA was reverse transcribed utilizing the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Foster City, CA). The manufacturer’s protocol was followed, and the cDNA was quantified using the NanoDrop Spectrophotometer. For the quantitative real time-PCR, 100ng of cDNA was brought up to 9μl with nuclease-free water and combined with 1μL of TaqMan Gene Expression Assay and 10μl of TaqMan Gene Expression Master Mix (Applied Biosystems, Foster City, CA). Assays (including a no template control) were performed in triplicate under standard real time-PCR conditions (50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles of 95°C for 15 seconds followed by 60° for 1 minute) using a 7300 Real Time PCR System (Applied Biosystems, Foster City, CA) with sequence detection software. Inventoried TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) were used to measure mRNA expression levels for TATA box binding protein (TBP, Assay ID Hs00920497_m1), CXCR4 (fusin, Assay ID Hs00237052_m1), perforin 1 (PERF1, Assay ID Hs00169473_m1), L-selectin (SELL, Assay ID Hs01046459_m1), baculoviral IAP repeat-containing 3 (BIRC3, Assay ID Hs00154109_m1), granzyme A (GZMA, Assay ID Hs00196206_m1), mediator complex subunit 28 (MED28, Assay ID Hs00740863_m1), pro-opiomelanocortin (POMC, Assay ID Hs00174947_m1), and retinol dehydrogenase 10 (RDH10, Assay ID Hs00174947_m1). TBP was used as the endogenous control.
Statistical analyses
Microarray analyses
All analyses utilizing data from microarray experiments were performed using the R Statistical Computing Software, version R2.9 (R Foundation for Statistical Computing, Vienna, Austria, 2009). Data from the microarray experiments was subjected to RMA normalization (Irizarry et al., 2003) and gene filtering procedure to remove genes with very little variation between the AUDs and control groups. After filtering, 7819 genes remained to undergo statistical analysis. Differentially expressed genes were identified using a 2 way ANOVA model (alcohol consumption × smoking) setting the false discovery rate (FDR) to 0.32 or less. We chose genes with a minimum of 1.4 times-fold change in expression difference between AUD and control groups to be verified using real time RT-PCR. Raw expression values for 10 standard housekeeping genes were also examined to select the endogenous control to use in real time RT-PCR calibration. The endogenous control ultimately chosen (TBP) had similar gene expression across all subjects and controls.
Functional classification
We performed functional analysis using DAVID (Huang et al., 2009a), a web-based Database for Annotation, Visualization and Integrated Discovery, using differentially expressed candidate genes (p<0.01) based on alcohol consumption. Use of this bioinformatics source provides a comprehensive set of functional annotation tools for investigators to understand biological meaning behind large lists of genes, including the identification of enriched biological themes, particularly gene ontology (GO) terms, as well as to discover enriched functional-related gene groups.
Real-time RT-PCR
Relative quantitation of mRNA expression between AUD subjects and controls was performed utilizing the comparative CT method. First, triplicate values of each target gene and reference gene were averaged to obtain mean values. Next, the mean amount of target gene expressed by AUD subjects was normalized to TBP expression (the endogenous reference (housekeeping) gene) for each individual subject to obtain the ΔCTAUD. Target gene expression by control subjects was similarly normalized to the endogenous reference gene expression (ΔCTCONTROL). Utilizing the ΔCTAUD and ΔCTCONTROL values, a paired t test analysis was performed to evaluate differences in normalized gene expression between AUD subjects and pair-matched controls within the test cohort and the confirmatory cohort separately. Then, to determine fold expression of mRNA by AUD subjects compared to controls, the normalized control subjects’ values were utilized as calibrators. Each AUD subject’s normalized target gene expression was calibrated to its specific matched control subject’s normalized target gene expression via the equation: , also referred to as ΔΔCT. The mean and standard error for all subject and control pairs’ ΔΔCT was calculated, and the amount of target gene expression in subjects with AUDs relative to control reported using the equation, 2−ΔΔCT (Pfaffl, 2001). Finally, the mean fold-expression differences in target genes between the test and confirmatory cohorts were compared utilizing a t test analysis. A p value of <0.05 was considered significant.
Results
Subjects were identified and enrolled for both cohorts between January 2007 and May 2009. Subjects with AUDs were identified and enrolled exclusively at the Denver CARES facility. Smoking control subjects were enrolled at the Denver VA Medical Center, while non-smoking control subjects were enrolled on the University of Colorado-Anschutz Medical Center campus.
Initial Test Cohort
The test cohort consisted of 7 subjects with AUDs and 7 controls. Subjects in the test cohort were exclusively men. AUD subjects and their controls were otherwise pair-matched in terms of age, tobacco use, and gender (table 1). Spirometry values were also similar between groups. The AUDIT values of all of the individuals with AUDs indicated a current diagnosis of alcohol abuse or dependence, while the AUDIT scores for the controls were under the limit of values indicating AUDs. AUD subjects drank almost daily, and had been consuming alcohol 30 years on average.
Table 1.
Demographics and clinical characteristics of subjects with alcohol use disorders (AUDs) and control subjects for test and confirmation cohorts.
| Test Cohort | Confirmation Cohort | |||
|---|---|---|---|---|
| AUD Subjects, n=7 | Control Subjects, n=7 | AUD Subjects, n=7 | Control Subjects, n=7 | |
| Age, mean | 44 ± 6 | 37 ± 11 | 42 ± 9 | 38 ± 12 |
| Gender, no. women/men | 0/7 | 0/7 | 5/2 | 5/2 |
| Tobacco use, % | 5/7 (71%) | 5/7 (71%) | 4/7 (57%) | 4/7 (57%) |
| AUDIT score* | 27 ± 9 | <1 | 26 ± 7 | <1 |
| Days/week consuming alcohol | 5 ± 2 | -- | 6 ± 1 | -- |
| Years consuming alcohol | 30 ± 6 | -- | 25 ± 6 | |
| FEV1, % predicted | 94 ± 9 | 85 ± 7 | 93 ± 11 | 90 ± 12 |
| FVC, % predicted | 97 ± 9 | 93 ± 7 | 96 ± 15 | 93 ± 16 |
| BAL cell count per mL (in millions) | 21 ± 12 | 25 ± 21 | 19 ± 10 | 24 ± 24 |
| Monos/macrophages, % | 91 ± 3 | 93 ± 4 | 91±2 | 96 ± 2 |
| Lymphocytes, % | 6 ± 3 | 6 ± 4 | 7±2 | 3 ± 1 |
| Neutrophils, % | 3 ± 2 | 2 ± 1 | 2±1 | 2 ± 1 |
The Alcohol Use Disorders Identification Test (AUDIT) score is calculated from a validated 10 question survey to identify current alcohol use that has been utilized in a variety of clinical settings. AUD was defined as an AUDIT score of ≥8 in men, and ≥5 in women. FEV1=forced expiratory volume in the first second of expiration; FVC=forced vital capacity; BAL=bronchoalveolar lavage.
Cell counts and differentials on the subjects’ and controls’ BAL samples did not differ significantly. Notably, the percentage of alveolar macrophages in samples from subjects and controls exceeded 90%. Smokers tended to have higher cell counts present in BAL fluid compared to controls, but this did not significantly differ between the groups (p=0.08). Viability of cells in BAL exceeded 98%.
Effect of alcohol use disorders on gene expression in alveolar macrophages by microarray
After filtering the data, two-way analysis of variance (ANOVA) modeling was performed on both alcohol and smoking effects. The F statistics revealed 470 differentially expressed probe sets at a false discovery rate (FDR) of 0.32. Further analysis showed that 373 of the 470 probe sets were uniquely significant in smoking effects (79.4%), while 12 of the 470 probe sets were distinctly significant in alcohol effects (2.6%). Among these 470, 85 (18.0%) probe sets were common on both effects (figure 1).
Figure 1.
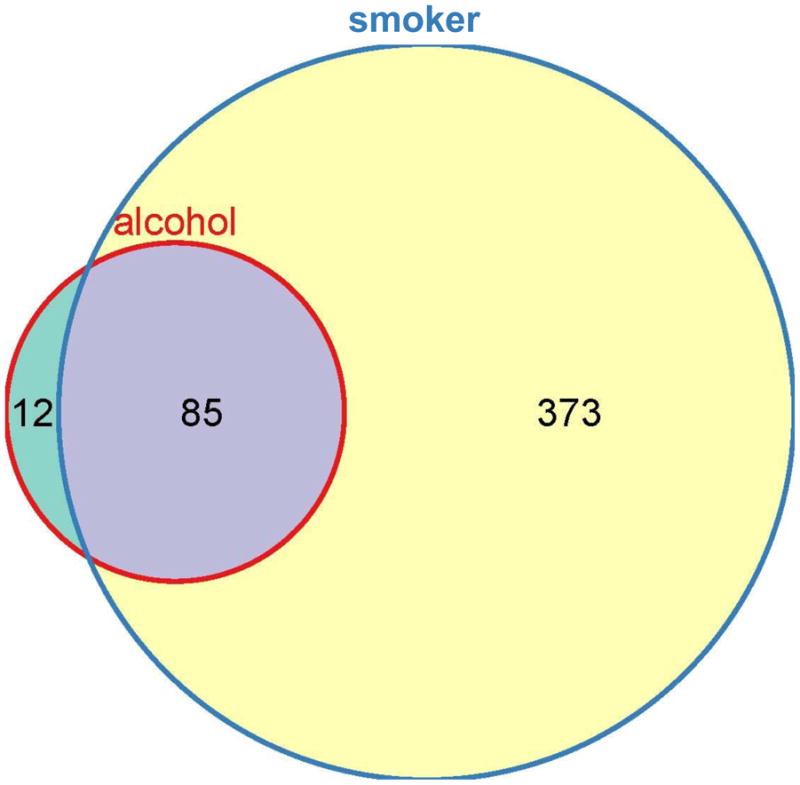
Venn diagram illustrating the relative contribution of alcohol use disorders (red circle) and smoking (blue circle) on differences in microarray gene expression in the test cohort. The test cohort consisted of 7 subjects with alcohol use disorders, and 7 pair-matched controls. Two-way ANOVA modeling was performed on both alcohol and smoking effects. The F statistics revealed 470 total differentially expressed probesets at FDR of 0.32. Further analysis showed that 373 of the 470 probesets were uniquely significant in smoking effects (79.4%, illustrated in yellow), while 12 of the 470 probesets were distinctly significant in alcohol effects (2.6%, illustrated in green). Among these 470, 85 (18.0%, illustrated in gray) probesets were common on both effects.
To extend the biological search space for the effect of AUDs on probe sets, given that subjects were a priori pair matched on the basis of smoking, we relaxed the threshold criterion by setting the p value for the group of 470 differentially expressed probe sets to ≤0.01 to include additional probe sets whose expression might be influenced by alcohol and smoking effects. This new specification resulted in the addition of 29 probe sets (initially included in the group of 85 common on both alcohol and smoking), bringing the total number of probe sets to 41. These probe sets remained constrained to an FDR of 0.32. Twenty-seven of these probe sets were upregulated in the setting of AUDs (table 2) while 14 were downregulated (table 3).
Table 2.
Microarray gene expression results for probe sets upregulated in the setting of alcohol use disorders.
| Symbol | Description | GenBank ID | P value | Fold expression difference |
|---|---|---|---|---|
| MED28* | mediator complex subunit 28 | BF064162 | 0.000478 | 1.40 |
| TNFSF10 | tumor necrosis factor (ligand) superfamily, member 10 | AW474434 | 0.000515 | 1.72 |
| SELL* | selectin L | NM_000655 | 0.000569 | 2.77 |
| GBP1 | guanylate binding protein 1, interferon-inducible, 67kDa | NM_002053 | 0.001092 | 1.57 |
| TRAM2* | translocation associated membrane protein 2 | AI986461 | 0.002274 | 1.30 |
| ASCL2 | achaete-scute complex homolog 2 (Drosophila) | AI393930 | 0.002483 | 1.50 |
| PRF1* | perforin 1 (pore forming protein) | AI445650 | 0.002721 | 3.32 |
| TRIB3 | tribbles homolog 3 (Drosophila) | NM_021158 | 0.003693 | 2.04 |
| CXCR4* | chemokine (C-X-C motif) receptor 4 | AJ224869 | 0.004592 | 1.84 |
| GCH1 | GTP cyclohydrolase 1 | NM_000161 | 0.004869 | 1.86 |
| IRF1 | interferon regulatory factor 1 | AW392551 | 0.004875 | 1.50 |
| VEGFA | vascular endothelial growth factor A | AF022375 | 0.004979 | 2.37 |
| SOD2 | superoxide dismutase 2, mitochondrial | X15132 | 0.006047 | 1.72 |
| PRF1* | perforin 1 (pore forming protein) | NM_005041 | 0.006355 | 1.99 |
| MTHFD2 | methylenetetrahydrofolate dehydrogenase (NADP+dependent) 2, methenyltetrahydrofolate cyclohydrolase | NM_006636 | 0.007039 | 1.36 |
| BIRC3 | baculoviral IAP repeat-containing 3 | U37546 | 0.007734 | 2.15 |
| IFITM2 | interferon induced transmembrane protein 2 (1–8D) | NM_006435 | 0.007963 | 1.59 |
| PDGFB* | platelet-derived growth factor beta polypeptide (simian sarcoma viral (v-sis) oncogene homolog) | AU150748 | 0.008461 | 1.43 |
| NDRG1 | N-myc downstream regulated 1 | NM_006096 | 0.00885 | 1.61 |
| GZMA | granzyme A (granzyme 1, cytotoxic T-lymphocyte-associated serine esterase 3) | NM_006144 | 0.009277 | 5.41 |
| IFITM1 | interferon induced transmembrane protein 1 (9–27) | AA749101 | 0.009562 | 4.12 |
| CD8B | CD8b molecule | NM_004931 | 0.01044 | 1.84 |
| GADD45B | growth arrest and DNA-damage-inducible, beta | AF087853 | 0.011215 | 1.54 |
| AF052160 | 0.011826 | 2.04 | ||
| CCT5 | chaperonin containing TCP1, subunit 5 (epsilon) | BF197357 | 0.011852 | 1.76 |
| IFITM1 | interferon induced transmembrane protein 1 (9–27) | NM_003641 | 0.01214 | 3.15 |
| RGS14 | regulator of G-protein signaling 14 | AF037195 | 0.012859 | 1.45 |
Probe sets that are distinctly different in the setting of AUDs, independent of smoking.
Table 3.
Microarray gene expression results for probe sets downregulated in the setting of alcohol use disorders.
| Symbol | Description | GenBank ID | P value | Fold expression difference |
|---|---|---|---|---|
| C19orf12 | chromosome 19 open reading frame 12 | AL568826 | 0.000199 | −1.30 |
| CROCCL1* | ciliary rootlet coiled-coil, rootletin-like 1 | BC006312 | 0.000286 | −1.60 |
| POMC* | proopiomelanocortin | NM_000939 | 0.000406 | −2.20 |
| AK1 | adenylate kinase 1 | BC001116 | 0.00047 | −1.72 |
| C4orf33* | chromosome 4 open reading frame 33 | NM_173487 | 0.000911 | −1.50 |
| AW044646 | 0.002531 | −5.86 | ||
| SART3 | squamous cell carcinoma antigen recognized by T cells 3 | AW173076 | 0.003985 | −1.27 |
| RDH10 | retinol dehydrogenase 10 (all-trans) | NM_172037 | 0.004578 | −1.57 |
| ALAS1* | aminolevulinate, delta-, synthase 1 | NM_000688 | 0.004966 | −1.38 |
| ARSK | arylsulfatase family, member K | AI243677 | 0.005426 | −1.30 |
| CYSLTR1 | cysteinyl leukotriene receptor 1 | NM_006639 | 0.005739 | −1.44 |
| HMG20B* | high-mobility group 20B | AF288679 | 0.007491 | −1.35 |
| CCNE2 | cyclin E2 | AF112857 | 0.01043 | −1.44 |
| NBN | nibrin | AI796269 | 0.010587 | −1.40 |
Probe sets that are distinctly different in the setting of AUDs, independent of smoking
We performed pathway analysis on these 41 probe sets using the DAVID bioinformatics webtool to more closely examine the biological relevance of AUDs in the context of alveolar macrophages. Processes found to be influenced by AUDs in these cells (linked to gene ontology terms) included programmed cell death/apoptosis. Both caspase-mediated apoptotic pathways and granzyme A-mediated apoptotic pathways were affected. Additional processes influenced by AUDs included the regulation of progression through cell cycle, immune response, and cellular localization (table 4). One probe in this group of 41 was associated with oxidative stress: mitochondrial superoxide dismutase.
Table 4.
Pathways in alveolar macrophages identified to be altered in the setting of alcohol use disorders.
| GO Term(s) | Genes in pathway | Pathway Fold-enrichment | FDR | P Value |
|---|---|---|---|---|
| Programmed cell death/Apoptosis | VEGF, BIRC-3, PRF-1, GADD45B, TRIB3, SOD2, TNFSF10, CXCR4, GZMA, GCH1 | 5.6 | 0.1 | .00004 |
| Regulation of progression through cell cycle | IRF1, VEGF, PDGF, GADD45B, CCNE2, IFITM1, NBN | 5.8 | 1.8 | .00093 |
| Immune response | IRF1, CD8B, GBP1, IFITM2, TNFSF10, CXCR4, GZMA, IFITM1 | 3.7 | 7.2 | .0039 |
| Cellular localization | PDGF, SELL, CXCR4 | 12.2 | 30.5 | .019 |
Focusing on the twelve probe sets differentially expressed in association with AUDs independent of smoking history, an unsupervised hierarchical clustering analysis was performed. It was determined that the clustering of these probe sets resulted in separation of AUD subjects and matched controls into two distinct groups (figure 2). Five of the 12 AUD-associated probe sets were characterized with a disulfide bond as a unique sequence feature (UP_SEQ_FEATURE: disulfide bond), including pro-opiomelanocortin, platelet derived growth factor (PDGF)-β, perforin-1, selectin-L, and CXCR4. None of the 12 AUD-associated probe sets specifically referenced to oxidative stress.
Figure 2.

Unsupervised hierarchical clustering image utilizing 12 probe sets determined by two-way ANOVA modeling with microarray data to be differentially regulated based on alcohol use disorders. Utilizing these probe sets in clustering analysis resulted in the subjects separating out into two groups that differed based on history of AUDs, regardless of smoking history. Gene symbols corresponding to each of these probes are listed on the right.
Real-time RT-PCR was utilized to confirm mRNA expression differences in this group of 12 probe sets whose expression ratios differed by >1.4-fold between AUD subjects and controls. Real time RT-PCR was also performed on granzyme A (GZMA) and baculoviral IAP repeat-containing 3 (BIRC-3) based their association in pathways identified by DAVID analysis, and for retinol dehydrogenase 10 (RDH10) given its role in the biosynthesis of retinoic acid (Vitamin A), an important regulator of gene expression whose synthesis is known to be influenced by alcohol (Leo MA et al. 1982). For a majority of genes whose expression was observed by microarray to be upregulated in the setting of AUDs, differences in gene expression via real time RT-PCR confirmed significant fold-expression differences between AUD subjects and controls for these selected genes (Test Cohort results, table 5). The most striking increases in expression were observed for the genes perforin-1 (p<0.05), granzyme A (p<0.01), selectin-L (p<0.01), and baculoviral IAP repeat-containing 3 (p<0.03) (figure 3); however, increased expression of CXCR4 in the setting of AUDs was noted as well (p<0.0001). Of these genes with upregulated expression, granzyme A’s was the most variable across the test cohort (i.e. SEM approximated the mean). Since this variability appeared to be driven by a single AUD/control subject pair, we performed additional analyses of gene expression differences for granzyme A excluding data from this pair. In this latter analysis, we observed that granzyme A expression remained significantly higher in subjects with AUDs (p<0.02, table 5; granzyme A data utilized in figure 3). Finally, although MED28 was significantly increased in expression in those with AUDs by microarray, its increased expression was not confirmed by real time RTPCR (p=0.88).
Table 5.
Fold gene expression differences (mean ± SEM) in AUD subjects versus controls by RT-RTPCR in test and confirmatory cohorts.
| Gene Symbol | Test Cohort Fold Expression Difference, AUDs Subjects vs Controls | Test Cohort p Value | Confirmatory Cohort Fold Expression Difference, AUD Subjects vs Controls | Confirmatory Cohort p Value |
|---|---|---|---|---|
| CXCR4 | 1.77±0.14 | 0.0001 | 1.58±0.14 | 0.003 |
| PRF-1 | 5.98±2.39 | 0.05 | 6.87±2.39 | 0.13 |
| SELL | 3.25±0.62 | 0.01 | 2.00±0.62 | 0.04 |
| MED28 | 1.02±0.10 | 0.88 | 0.94±0.10 | 0.44 |
| GZMA* | 6.38±5.12 (4.50±1.43) | 0.01 (0.02) | 13.16±5.12 (6.38±1.43) | 0.31 (0.08) |
| BIRC3 | 2.74±0.48 | 0.03 | 2.06±0.48 | 0.01 |
| RDH10* | 0.92±1.81 (0.69±0.12) | 0.17 (0.07) | 2.82±1.81 (0.27±0.12) | 0.23 (0.20) |
Values included in parentheses exclude one outlier pair for the specified cohort.
Figure 3.

Fold-differences in mRNA expression by real time RT-PCR expression for selected genes in AUD subjects compared to pair-matched control subjects. Probe sets to validate by real time RT-PCR were chosen after microarray analysis revealed up- or down- regulation in the setting of alcohol use disorders, with fold-expression differences of greater than 1.4. Real time RT-PCR was initially performed on samples from the test cohort (14 total subjects; n=7 with AUD, n=7 pair-matched controls, illustrated to the left). Samples from 14 subjects in the confirmation cohort (n=7 with AUD, n=7 pair-matched controls, illustrated to the right) were also subjected to real time RT-PCR utilizing identical probes to confirm test cohort observations in an independent sample. Mean fold expression differences for chemokine (C-X-C motif) receptor 4 [CXCR4], perforin-1 [PRF1], selectin L [SELL], granzyme A [GZMA], and baculoviral IAP repeat-containing 3 [BIRC-3] were determined to be upregulated in the setting of alcohol use disorders in both cohorts. Expression values did not significantly differ between the test and confirmatory cohorts for each of these upregulated genes (p>0.05 for comparison between test and confirmatory cohorts). Mean fold expression differences for retinol dehydrogenase 10 [RDH10] tended to be downregulated among those with AUDs, and was significantly higher in the test cohort (p<0.03). No significant difference in mediator complex subunit 28 [MED28] expression was observed in association with AUDs (mean fold expression approximately 1 for both test and confirmatory cohorts). Colored bars represent mean fold-expression differences from AUD subjects versus controls for the different genes, with black bars representing the SEM. Data illustrated for GZMA and RDH10 excludes one outlier pair (sample size n=6 with AUD, n=6 pair-matched controls).
In terms of genes whose expression was determined by microarray to be downregulated in the setting of AUDs, we were able to detect expression of pro-opiomelanocortin via real time RT-PCR for only 2 of 7 pairs in the test cohort despite repeated experiments with two different probes; therefore, microarray gene expression differences could not be confirmed. Expression of retinol dehydrogenase 10 by real time RT-PCR did not differ significantly between AUD subjects and controls in the test cohort (p=0.17), but as with granzyme A, much variation in retinol dehydrogenase 10 expression was noted across this cohort (SEM two-fold greater than actual mean, table 5). By again excluding one outlier AUD/control pair with differences in gene expression the furthest away from the mean, and re-analyzing the data, a trend for decreased gene expression was observed among AUD subjects (p=0.07) that paralleled the microarray result (table 5; retinol dehydrogenase 10 data utilized in figure 3).
Confirmation Cohort
The confirmation cohort consisted of 7 subjects with AUDs and 7 control subjects chosen to match individual AUD subjects in terms of age, gender, and tobacco use. Since these experiments were conducted to confirm the observations made previously in the test cohort, we included women and increased the percentage of non-smokers to enhance the generalizability of our results. The two groups were balanced in terms of age, tobacco use, and gender (table 1). As in the test cohort, spirometry values were similar between groups. Additionally, AUDIT scores in the AUD subjects indicated alcohol dependence, with subjects drinking alcohol nearly daily for twenty-five years on average. Cell counts and differentials on the subjects’ and controls’ BAL samples did not differ significantly in the confirmation cohort, and a predominance of monocytes/macrophages in BAL (>90%) was observed.
Real-time RT-PCR was performed on mRNA samples from this cohort using probe sets for genes determined to differ based on alcohol consumption that were identical to those used in experiments on test cohort samples. Despite the fact that this confirmation cohort contained a larger number of women, and fewer smokers, fold expression differences observed were similar in direction and magnitude to the test cohort (p value comparing mean expression values not significantly different between cohorts, figure 3), with the exception of retinol dehydrogenase 10, whose fold expression differences were significantly higher in the confirmation cohort (p<0.03, figure 3). Examining gene expression differences between AUD subject and control pairs within the confirmation cohort, significantly upregulated gene expression in the setting of AUDs was observed for selectin-L (p<0.04), baculoviral IAP repeat-containing 3 (p<0.01), and CXCR4 (0.003), while a trend for upregulated expression of perforin-1 (p=0.13) and granzyme A (p=0.08, excluding an outlier pair) was similarly present. Significant differences in RDH10 and MED28 gene expression between AUD subjects and controls were not demonstrable (table 5). These additional observations validate an effect of AUDs on gene expression for selected alveolar macrophage genes.
Discussion
In these investigations, we utilized microarray analysis and real time RT-PCR to examine gene expression in alveolar macrophages from human subjects who were otherwise healthy except for a history of AUDs. Using these data and bioinformatics tools, we determined that AUDs were associated with an increased expression of genes relevant to apoptosis/programmed cell death, and immune response, including perforin-1, granzyme A, and CXCR4. Additionally, AUDs were also associated with modestly decreased expression of retinol dehydrogenase 10, a key enzyme in retinoic acid biosynthesis. One gene was observed to be upregulated in the setting of AUDs that is associated with oxidative stress: mitochondrial superoxide dismutase (SOD-2). Importantly, we confirmed that alterations in gene expression related to AUDs were also present in an independent set of subject samples, further enhancing the validity of our observations. Collectively, these investigations suggest that AUDs can influence alveolar macrophage function, and potentially cellular viability, through their effect on these pathways. Our observations parallel earlier work in animal models and provides a novel mechanism underlying the predisposition of individuals with AUDs to pulmonary disorders including bacterial pneumonia and ALI. The use of similar techniques in alveolar macrophage investigations has contributed to the understanding of the mechanisms of disease caused by air pollutants (Huang et al., 2009b), smoking (Carolan et al., 2009), and asthma (Madore et al., 2010). To our knowledge, such methods have not been utilized to explore differences in alveolar macrophage gene expression in the setting of a systemic insult such as that caused by AUDs.
Several major pathways of programmed cell death and apoptosis have been reported in the literature. These pathways involve the products of genes determined to be differentially regulated by AUDs in our investigations. For example, perforin and granzyme proteins are important mechanistically in a number of apoptotic cascades. Production of perforin is thought to be restricted mainly to natural killer (NK) cells and cytotoxic T lymphocytes (Koizumi et al., 1993), where it is packaged into cytoplasmic granules. When these granules are released, monomers of perforin proteins are able to polymerase and form pores in target cell membranes, providing access for granzyme proteins into the target cell’s cytosol (Liu et al., 1995). Once inside the cell, granzyme B interacts with caspases, a family of cysteine proteases that function in the initiation and execution of many forms of apoptosis. This apoptotic cascade can be blocked in part by the activation of baculoviral IAP-containing proteins that are anti-apoptotic. Alternatively, granzyme A proteins may induce apoptosis via caspase-independent pathways (Fan et al., 2003). While apoptosis can represent a beneficial mechanism to keep airways clear of dead and dying cells (e.g. neutrophils in the resolution of ALI), thereby limiting the release of pro-inflammatory cytokines, it may potentially promote progression of infectious diseases (Lee et al., 2009). For example, apoptosis can eliminate vital host defense cells (such as alveolar macrophages), or contribute to dissemination of infection by delivering pathogens to naïve host phagocytes that are engulfing apoptotic cells (Khelef et al., 1993).
Along with the effect of alcohol on alveolar macrophage apoptosis already mentioned (Ping et al., 2007), a link between chronic alcohol consumption and apoptosis within the cerebral cortex (Ren et al., 2009), and heart (Doser et al., 2009; Fernandez-Sola et al., 2006) have similarly been described. Apoptotic pathways implicated in these tissues appear to be caspase-dependent. Metabolism of acetaldehyde, the reactive and toxic intermediate formed by the breakdown of ethanol (Zhang et al., 2004) is believed to be a contributor in the propagation of apoptosis. Cardiac and central nervous system investigations have provided evidence that overexpression of aldehyde dehydrogenase-2, a mitochondrial enzyme that facilitates acetaldehyde breakdown, reverses apoptosis caused by alcohol exposure (Doser et al., 2009; Fernandez-Sola et al., 2006). Alternatively, oxidative stress elicited by chronic alcohol consumption could also be relevant to this process, as illustrated in in vitro studies using rodent models of AUDs where the addition of the anti-oxidant procysteine was associated with normalization of apoptotic indices by alveolar macrophages (Brown et al., 2004; Ping et al., 2007). Results from our investigations support that AUDs are associated with modest upregulation of alveolar macrophage SOD-2 expression by microarray (1.72-fold difference between AUD subjects and controls), and modest downregulation of retinol dehydrogenase 10, an enzyme integral in the formation of retinoic acid (vitamin A) (Belyaeva et al., 2008) that may contribute to oxidative stress experienced by alveolar macrophages exposed to alcohol, and affect these cells’ viability. For example, investigators have demonstrated that AUDs are associated with enhanced SOD-2 gene expression and enzyme activity in liver in animal models (Koch et al., 1994), but the effect of AUDs on human alveolar macrophages SOD-2 is unknown. However, evidence from human studies supports reduced levels of retinoic acid in serum that may have downstream effects on cell growth and differentiation (Leo and Lieber 1982). For example, retinoic acid has reported anti-proliferative effects mediated through activator protein (AP)-1. In animal models focused on liver disease, AUDs have been associated with inhibition of retinoic acid biosynthesis and enhancement of AP-1 gene expression that may contribute to cell proliferation, inflammation, and ultimately malignant transformation (Wang et al. 1998). Decreased retinoic acid biosynthesis can also influence nuclear factor-kappa B-mediated transcription of pro-inflammatory molecules such as interleukin-6, resulting in a more pronounced response to inducers of inflammatory reactions, including lipopolysaccharide (Austenaa et al. 2009). Exploring gene and protein expression differences in alveolar macrophages that have been subjected to exogenous stimuli (i.e. lipopolysaccharide), including SOD-2, retinol dehydrogenase 10, and others could provide important clues to better understand pathways in the alveolar macrophage inflammatory response that are affected by oxidative stress.
One other intriguing role of perforin is calcium-dependent cell lysis. Structurally, the protein encoded by the perforin 1 gene is structurally and functionally similar to complement component 9 in that it can create transmembrane tubules and non-specifically lyse a variety of cells. Although this protein is thought to be primarily manufactured by NK and cytotoxic T cells as previously mentioned, it has also been reported to be produced by macrophage precursor cells in one investigation (Li et al., 1994) when these cells were stimulated by interleukin-2 in the presence of calcium. Macrophage precursor cells treated in this fashion developed granules containing perforin, and demonstrated tumor cell cytotoxicity along with hemolysis. Another report attests to the effect of alcohol consumption in decreasing perforin expression in human peripheral cytotoxic T cells and NK cells (Perney et al., 2003); however, the effect of alcohol on perforin expression in other cell types has not been reported. It appears possible that up-regulation of perforin expression by alveolar macrophages may lead to non-specific cytolysis of other resident lung cells, thereby altering the milieu in individuals with AUDs, and promoting lung pathology such as bacterial pneumonia.
For these investigations, we focused our microarray validation experiments on those 12 probe sets that were related to alcohol effects exclusive of smoking. We verified that AUDs were associated with upregulation of CXCR4 and selectin-L gene expression by real time RT-PCR. CXCR4 serves as the receptor for stromal cell-derived factor (SDF)-1, and along with granzyme A was linked to the immune response pathway by DAVID analysis. CXCR4 and selectin-L are also linked to cellular localization, referring to any process by which a cell is transported to, and/or maintained in, a specific location. Alterations in lymphocyte selectin-L expression in the setting of AUDs have been reported that could potentially affect the cell trafficking or other adhesion-dependent functions (Cook et al., 1994). The relationship between AUDs and CXCR4 expression on immune cells has not been specifically explored. CXCR4 is known to be a co-receptor for certain HIV strains (Fauci, 1996). Its expression on macrophages may be increased in the setting of tuberculosis (Hoshino et al. 2004). This association is intriguing given the epidemiologic association between AUDs, HIV and tuberculosis (Lonnroth et al., 2008). Verification of gene expression interferon-induced transmembrane proteins (IFITM)-1 and -2, both of which were significantly upregulated in the setting of AUDs by microarray, could provide additional insight into the effects of alcohol on alveolar macrophage immune response.
We were somewhat surprised that oxidative stress genes with altered expression in the setting of AUDs were limited to SOD-2. Enrolling a larger number of subjects and controls could have increased our power to detect gene expression differences of smaller magnitude. However, it would also have contributed to increased between-subject heterogeneity, and potentially diluted our ability to detect differences solely related to AUDs. We purposely chose to enroll a group of AUD subjects and controls who were pair-matched on the basis of smoking, gender, and age, and differed only on alcohol use history. Therefore, in our statistical analyses (both for microarray and real time-RTPCR) we could focus on within AUD subject versus control differences, and minimize the effects of other confounders present between subjects (e.g. smoking). Also, we additionally examined a confirmatory cohort of pair-matched subjects with AUDs and controls that had a different composition of smokers and gender than the test cohort to enhance the generalizability of our test cohort observations. Since gene expression differences in this second cohort paralleled what was observed in the test cohort despite their demographic differences, we believe that our reported results truly reflect gene expression differences in alveolar macrophages that are elicited by AUDs. Another potential factor that might have been influential in the gene expression differences observed between AUD subjects and controls is our collecting alveolar macrophage RNA from freshly obtained cells not subjected to additional stimulus with exogenous agents prior to gene expression experiments. Recent investigations have reported modest macrophage gene expression differences in smoking versus non-smoking subjects in the absence of other exogenous stimulation. However, by treating cells with cigarette smoke extract or lipopolysaccharide, gene expression alterations can be far more striking (Doyle et al. 2010; Kent et al. 2010). Similar experiments examining the effect of exogenous stimuli on alveolar macrophage gene expression in the setting of AUDs would be relevant to define alterations that occur in situations of stress (e.g. pulmonary infections).
These observations, while novel, are not without certain limitations. We sought to confirm gene expression differences observed in our microarray experiments by real time RT-PCR. We strengthened the applicability and generalizability of these observations by repeating real time RT-PCR experiments in an independent sample of subjects and controls. Nonetheless, we were not able to confirm all significant differences observed via microarray (e.g. pro-opiomelanocortin and retinol dehydrogenase 10 expression differences). Potential explanations for this include our examining genes with fold-expression differences by microarray that were less robust (near the 1.4-fold difference cutoff), or false positive microarray results for certain genes. Secondly, it is possible that gene expression differences we observed might not be representative of protein levels or activity that actually exists in vivo. Localization of proteins to specific cell types and compartments of the lung as well as their quantitation is warranted. Third, it is possible that our observations were influenced by cell types present within BAL other than alveolar macrophages. Our starting population of cells was determined to be >90% mononuclear phagocytes. These cells were processed within minutes after collection. We opted to not perform additional purification steps as we felt the additional cell purity gained would be offset by alterations in gene expression in an artificial (e.g. cell culture media) milieu while these steps were being performed. Finally, the effect of smoking on gene expression is particularly relevant given the high proportion of subjects with AUDs who also smoke. The effects of smoking and other confounders are inherent in human subjects’ research. Pair matching of our AUD subjects to controls on the basis of smoking, age, and gender were performed to control for these variables in the statistical analysis in order to focus specifically on the effects of AUDs. However, it is important to note that some gene expression differences that we subsequently chose to examine by real time RT-PCR (namely granzyme A, baculoviral IAP repeat-containing 3, and retinol dehydrogenase 10) were common on both AUD and smoking effects. Their expression might have been influenced by smoking as well as AUDs; alternatively, a synergistic effect might have been present. Additionally, while we made every effort to match subjects and controls as precisely as possible, unknown confounders might have altered our results.
In summary, AUDs influence alveolar macrophage gene expression in human subjects. Twelve unique genes were determined to have alterations in their expression independent of smoking, while 85 others were affected by both AUDs and smoking. Upregulation of genes relevant to programmed cell death/apoptosis were most prominently affected, including perforin-1 and granzyme A. Other upregulated genes related to AUDs were relevant to processes including the immune response and trafficking of cells, including selectin-L. Our findings provide novel mechanistic insights to the downstream effects of a systemic, chronic insult, AUDs, on these important immune effector cells. Further investigations to determine the effects of modulating these pathways could have implications for decreasing the incidence of pulmonary infections and ALI in individuals with AUDs.
Acknowledgments
The authors wish to thank clients and staff of the Denver CARES facility for their participation in these investigations.
This work was supported by the National Institutes of Health [K23-AA13918; R01-AA014435, K24-HL089223 and 1UL1RR025780]
References
- Austenaa LM, Carlsen H, Hollung K, Blomhoff HK, Blomhoff R. Retinoic acid dampens LPS-induced NF-kappaB activity: results from human monoblasts and in vivo imaging of NF-kappa B reporter mice. J Nutr Biochem. 2009;20:726–734. doi: 10.1016/j.jnutbio.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Belyaeva OV, Johnson MP, Kedishvili NY. Kinetic analysis of human enzyme RDH10 defines the characteristics of a physiologically relevant retinol dehydrogenase. J Biol Chem. 2008;283:20299–20308. doi: 10.1074/jbc.M800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LA, Harris FL, Ping XD, Gauthier TW. Chronic ethanol ingestion and the risk of acute lung injury: a role for glutathione availability? Alcohol. 2004;33:191–197. doi: 10.1016/j.alcohol.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Brown SD, Gauthier TW, Brown LA. Impaired terminal differentiation of pulmonary macrophages in a Guinea pig model of chronic ethanol ingestion. Alcohol Clin Exp Res. 2009;33:1782–1793. doi: 10.1111/j.1530-0277.2009.01017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carolan BJ, Harvey BG, Hackett NR, O'Connor TP, Cassano PA, Crystal RG. Disparate oxidant gene expression of airway epithelium compared to alveolar macrophages in smokers. Respir Res. 2009;10:111. doi: 10.1186/1465-9921-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook RT, Waldschmidt TJ, Ballas ZK, Cook BL, Booth BM, Stewart BC, Garvey MJ. Fine T-cell subsets in alcoholics as determined by the expression of L-selectin, leukocyte common antigen, and beta-integrin. Alcohol Clin Exp Res. 1994;18:71–80. doi: 10.1111/j.1530-0277.1994.tb00883.x. [DOI] [PubMed] [Google Scholar]
- DeRoux RA, Cavalcanti M, Marcos MA, Garcia E, Ewig S, Mensa J, Torres A. Impact of alcohol abuse in the etiology and severity of community-acquired pneumonia. Chest. 2006;129:1219–1225. doi: 10.1378/chest.129.5.1219. [DOI] [PubMed] [Google Scholar]
- Detsky AS, Baker JP, Mendelson RA, Wolman SL, Wesson DE, Jeejeebhoy KN. Evaluating the accuracy of nutritional assessment techniques applied to hospitalized patients: methodology and comparisons. JPEN J Parenter Enteral Nutr. 1984;8:153–159. doi: 10.1177/0148607184008002153. [DOI] [PubMed] [Google Scholar]
- Doser TA, Turdi S, Thomas DP, Epstein PN, Li SY, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941–1949. doi: 10.1161/CIRCULATIONAHA.108.823799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Doyle I, Ratcliffe M, Walding A, Vanden Bon E, Dymond M, Tomlinson W, Tilley D, Shelton P, Dougall I. Differential gene expression analysis in human monocyte-derived macrophages: impact of cigarette smoke on host defence. Mol Immunol. 2010;47:1058–1065. doi: 10.1016/j.molimm.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell. 2003;112:659–672. doi: 10.1016/s0092-8674(03)00150-8. [DOI] [PubMed] [Google Scholar]
- Fauci AS. Host factors and the pathogenesis of HIV-induced disease. Nature. 1996;384:529–534. doi: 10.1038/384529a0. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sola J, Fatjo F, Sacanella E, Estruch R, Bosch X, Urbano-Marquez A, Nicolas JM. Evidence of apoptosis in alcoholic cardiomyopathy. Hum Pathol. 2006;37:1100–1110. doi: 10.1016/j.humpath.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Fernandez-Sola J, Junque A, Estruch R, Monforte R, Torres A, Urbano-Marquez A. High alcohol intake as a risk and prognostic factor for community-acquired pneumonia. Arch Intern Med. 1995;155:1649–1654. doi: 10.1001/archinte.1995.00430150137014. [DOI] [PubMed] [Google Scholar]
- Haslett C. Granulocyte apoptosis and its role in the resolution and control of lung inflammation. Am J Respir Crit Care Med. 1999;160:S5–11. doi: 10.1164/ajrccm.160.supplement_1.4. [DOI] [PubMed] [Google Scholar]
- Hoshino Y, Tse DB, Rochford G, Prabhakar S, Hoshino S, Chitkara N, Kuwabara K, Ching E, Raju B, Gold JA, Borkowsky W, Rom WN, Pine R, Weiden M. Mycobacterium tuberculosis-induced CXCR4 and chemokine expression leads to preferential X4 HIV-1 replication in human macrophages. J Immunol. 2004;172:6251–6258. doi: 10.4049/jimmunol.172.10.6251. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009a;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang YC, Li Z, Carter JD, Soukup JM, Schwartz DA, Yang IV. Fine ambient particles induce oxidative stress and metal binding genes in human alveolar macrophages. Am J Respir Cell Mol Biol. 2009b;41:544–552. doi: 10.1165/rcmb.2008-0064OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunninghake GW, Gadek JE, Kawanami O, Ferrans VJ, Crystal RG. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am J Pathol. 1979;97:149–206. [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Mitchell PO, Fernainy K, Roman J, Eaton DC, Guidot DM. GM-CSF receptor expression and signaling is decreased in lungs of ethanol-fed rats. Am J Physiol Lung Cell Mol Physiol. 2006;291:L1150–L1158. doi: 10.1152/ajplung.00150.2006. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Ritzenthaler JD, Roman J, Fernandez AL, Eaton DC, Brown LA, Guidot DM. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J Immunol. 2005;175:6837–6845. doi: 10.4049/jimmunol.175.10.6837. [DOI] [PubMed] [Google Scholar]
- Kent LM, Fox SM, Farrow SN, Singh D. The effects of dexamethasone on cigarette smoke induced gene expression changes in COPD macrophages. Int Immunopharmacol. 2010;10:57–64. doi: 10.1016/j.intimp.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Khelef N, Zychlinsky A, Guiso N. Bordetella pertussis induces apoptosis in macrophages: role of adenylate cyclase-hemolysin. Infect Immun. 1993;61:4064–4071. doi: 10.1128/iai.61.10.4064-4071.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch OR, De Leo ME, Borrello S, Palombini G, Galeotti T. Ethanol treatment up-regulates the expression of mitochondrial manganese superoxide dismutase in rat liver. Biochem Biophys Res Commun. 1994;201:1356–1365. doi: 10.1006/bbrc.1994.1853. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Horta MF, Youn BS, Fu KC, Kwon BS, Young JD, Liu CC. Identification of a killer cell-specific regulatory element of the mouse perforin gene: an Ets-binding site-homologous motif that interacts with Ets-related proteins. Mol Cell Biol. 1993;13:6690–6701. doi: 10.1128/mcb.13.11.6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Hartman M, Kornfeld H. Macrophage apoptosis in tuberculosis. Yonsei Med J. 2009;50:1–11. doi: 10.3349/ymj.2009.50.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo MA, Lieber CS. Hepatic vitamin A depletion in alcoholic liver injury. N Engl J Med. 1982;307(10):597–601. doi: 10.1056/NEJM198209023071006. [DOI] [PubMed] [Google Scholar]
- Li H, Pohler U, Strehlow I, Hertig S, Baccarini M, Emmendorffer A, Tschopp J, Lohmann-Matthes ML. Macrophage precursor cells produce perforin and perform Yac-1 lytic activity in response to stimulation with interleukin-2. J Leukoc Biol. 1994;56:117–123. doi: 10.1002/jlb.56.2.117. [DOI] [PubMed] [Google Scholar]
- Liu CC, Persechini PM, Young JD. Perforin and lymphocyte-mediated cytolysis. Immunol Rev. 1995;146:145–175. doi: 10.1111/j.1600-065x.1995.tb00688.x. [DOI] [PubMed] [Google Scholar]
- Lonnroth K, Williams BG, Stadlin S, Jaramillo E, Dye C. Alcohol use as a risk factor for tuberculosis - a systematic review. BMC Public Health. 2008;8:289. doi: 10.1186/1471-2458-8-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madore AM, Perron S, Turmel V, Laviolette M, Bissonnette EY, Laprise C. Alveolar macrophages in allergic asthma: an expression signature characterized by heat shock protein pathways. Hum Immunol. 2010;71:144–150. doi: 10.1016/j.humimm.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M, Bucher B, Moore FA, Moore EE, Parsons PE. The role of chronic alcohol abuse in the development of acute respiratory distress syndrome in adults. JAMA. 1996;275:50–54. [PubMed] [Google Scholar]
- Moss M, Guidot DM, Wong-Lambertina M, Ten HT, Perez RL, Brown LA. The effects of chronic alcohol abuse on pulmonary glutathione homeostasis. Am J Respir Crit Care Med. 2000;161:414–419. doi: 10.1164/ajrccm.161.2.9905002. [DOI] [PubMed] [Google Scholar]
- Moss M, Steinberg KP, Guidot DM, Duhon GF, Treece P, Wolken R, Hudson LD, Parsons PE. The effect of chronic alcohol abuse on the incidence of ARDS and the severity of the multiple organ dysfunction syndrome in adults with septic shock: an interim and multivariate analysis. Chest. 1999;116:97S–98S. [PubMed] [Google Scholar]
- Nelson S, Summer WR. Innate immunity, cytokines, and pulmonary host defense. Infect Dis Clin North Am. 1998;12:555–67. vii. doi: 10.1016/s0891-5520(05)70198-7. [DOI] [PubMed] [Google Scholar]
- Perney P, Portales P, Corbeau P, Roques V, Blanc F, Clot J. Specific alteration of peripheral cytotoxic cell perforin expression in alcoholic patients: a possible role in alcohol-related diseases. Alcohol Clin Exp Res. 2003;27:1825–1830. doi: 10.1097/01.ALC.0000093742.22787.30. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XD, Harris FL, Brown LA, Gauthier TW. In vivo dysfunction of the term alveolar macrophage after in utero ethanol exposure. Alcohol Clin Exp Res. 2007;31:308–316. doi: 10.1111/j.1530-0277.2006.00306.x. [DOI] [PubMed] [Google Scholar]
- Reinert DF, Allen JP. The Alcohol Use Disorders Identification Test (AUDIT): a review of recent research. Alcohol Clin Exp Res. 2002;26:272–279. [PubMed] [Google Scholar]
- Ren J, Babcock SA, Li Q, Huff AF, Li SY, Doser TA. Aldehyde dehydrogenase-2 transgene ameliorates chronic alcohol ingestion-induced apoptosis in cerebral cortex. Toxicol Lett. 2009;187:149–156. doi: 10.1016/j.toxlet.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standiford TJ, Danforth JM. Ethanol feeding inhibits proinflammatory cytokine expression from murine alveolar macrophages ex vivo. Alcohol Clin Exp Res. 1997;21:1212–1217. [PubMed] [Google Scholar]
- Toews GB, Gross GN, Pierce AK. The relationship of inoculum size to lung bacterial clearance and phagocytic cell response in mice. Am Rev Respir Dis. 1979;120:559–566. doi: 10.1164/arrd.1979.120.3.559. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Opal SM. Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet. 2009;374:1543–1556. doi: 10.1016/S0140-6736(09)61114-4. [DOI] [PubMed] [Google Scholar]
- Wang XD, Liu C, Chung J, Stickel F, Seitz HK, Russell RM. Chronic alcohol intake reduces retinoic acid concentration and enhances AP-1 (c-Jun and c-Fos) expression in rat liver. Hepatology. 1998;28:744–750. doi: 10.1002/hep.510280321. [DOI] [PubMed] [Google Scholar]
- Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li SY, Brown RA, Ren J. Ethanol and acetaldehyde in alcoholic cardiomyopathy: from bad to ugly en route to oxidative stress. Alcohol. 2004;32:175–186. doi: 10.1016/j.alcohol.2004.01.005. [DOI] [PubMed] [Google Scholar]