

Abstract
Based on findings that cancer cell clonogens exhibit stem cell features, it has been suggested that cancer stem-like cells are relatively radioresistant owing to different intrinsic and extrinsic factors, including quiescence, activated radiation response mechanisms (e.g., enhanced DNA repair, upregulated cell cycle control mechanisms and increased free-radical scavengers) and a surrounding microenvironment that enhances cell survival mechanisms (e.g., hypoxia and interaction with stromal elements). However, these radiosensitivity features are probably dynamic in nature and come into play at different times during the course of chemo/radiotherapy. Therefore, different molecularly targeted radiosensitization strategies may be needed at different stages of therapy. This article describes potential sensitization approaches based on the dynamics and changing properties of cancer stem-like cells during therapy.
Keywords: cancer cell clonogen, cancer stem-like cell, cancer stem-like cell dynamics, radioresistance determinant, therapeutic strategy
It has long been recognized that tumors are heterogeneous in their radiation response. In some cases, the degree of radiosensitivity was believed to be related to intrinsic properties (e.g., DNA repair capability and proliferation status) and to extrinsic properties (e.g., degree of hypoxia within the tumor) of the tumor cell population, which impacted their ability to withstand radiation insult. Therefore, to better understand features that control radiosensitivity, researchers examined tumor cell populations for these properties and then tried to interfere with these resistant pathways to enhance radioresponse. Alternatively, researchers selected tumors that survived radiation treatment and tried to determine whether the radioresistant phenotype persisted. If radioresistant tumors were transplanted into new hosts and again challenged with radiotherapy, in contrast to the setting of chemotherapy – where chemoresistant clones could be readily selected – transplanted radioresistant tumors generally maintained a similar radiosensitivity profile to that of the original tumor [1–3]. This suggested that, while radiosensitivity is strongly influenced by intrinsic cellular determinants, it is also strongly influenced by a common pathophysiological makeup (e.g., the degree of hypoxia), which impacts radiosensitivity and is present in both the original and transplanted tumors. In other cases, the degree of radiosensitivity was believed to be related to the amount of cellular heterogeneity within the tumor. That is, tumors with a high level of heterogeneity might have increased frequencies of subsets of cells exhibiting radioresistant features and, therefore, might be more radioresistant.
Early studies in radiobiology employed both in vitro and in vivo clonogenic assays to address various determinants of cell and tumor radioresponse (for review, see [4,5]). Early in vitro studies suggested that only a small fraction of freshly harvested tumor cells could initiate clonogenic growth. Similarly, using in vivo transplantation assays in appropriate hosts (mostly transplantation of murine tumors into syngeneic mice), only a small fraction of inoculated cells were found capable of initiating the growth of new tumors. These apparently rare cells were given various names (e.g., tumor clonogens, cancer-initiating cells and cancer stem cells) [6–8]. Different tumors appeared to have different frequencies of tumor-initiating cells. Importantly, as illustrated in our own studies [8], tumor populations that had a higher frequency of clonogens were relatively more radioresistant than tumors with fewer clonogens (Figure 1). Interestingly, the efficiency of these cells to initiate new tumors could be strongly influenced by experimental conditions. For example, by mixing in irradiated cells, compromising the host rejection responses by immunosuppressive strategies, or irradiating the tumor inoculation site (causing a local inflammatory response), the efficiency of tumor initiation was significantly increased [9–11]. Even under these enhanced conditions, different tumors exhibited a broad range of frequencies of tumor-initiating cells. However, in some cases, as few as virtually one cell could initiate a new tumor [11]. These results suggested that different tumors contain different frequencies of clonogenic cells, and the ability of clonogenic cells to initiate new tumors may also depend on the local microenvironment in the host.
Figure 1. Tumor control dose 50% value plotted versus tumor cell dose 50% value for murine tumors.
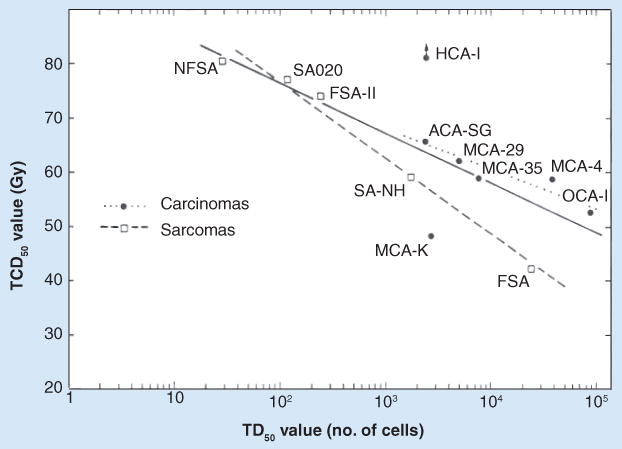
Best-fit lines are shown for each group and for all tumors. TCD50 values decrease as the TD50 values increase.
TCD50: Tumor control dose 50%; TD50: Tumor cell dose 50%.
Reproduced with permission from [8].
The nature of cancer clonogenic cells was not well understood for a number of years owing to a lack of means to identify and characterize these ‘rare’ cells. Early studies using bromo-deoxyuridine (BrdU) pulse-chase labeling technologies suggested that these populations were quiescent in the setting of the developed tumor, yet were drawn into proliferation upon tumor mass reduction [12,13]. Other studies suggested that cancer clonogenic cells exhibited an increased ability to exclude certain compounds (e.g., vital dyes, such as Hoechst 33342) in association with high expression of membrane drug transporters [13]. Using these handles for enrichment and cell-sorting technologies, investigators began to isolate these cells, characterize their unique features and demonstrate an enrichment of clonogenic efficiency in vivo [14,15].
In the hematopoietic setting, it was found that the leukemia clonogenic cells expressed many of the markers found on normal hematopoietic stem cells (e.g., CD34+/CD38-) and that these cells could preferentially regenerate new tumors upon transplantation into suitable hosts [16]. Importantly, the grafts regenerated the heterogeneity found in the original tumor, suggesting that these cells could both initiate new tumors, as well as differentiate into more mature components. Since these properties mirrored that of stem cells in normal organs, the hypothesis evolved that cancer development has many common features with normal organ development, including the ability of the stem cells to self-renew, differentiate and respond in a homeostatic manner when injured.
As reviewed well elsewhere [14,17–19], this notion was extended further into solid tumors, whereby normal stem cell markers (e.g., CD133 for glioblastoma and CD44 for breast cancer) were used to identify and enrich tumor stem cells for further characterization. Interestingly, these studies suggested that these so-called cancer stem-like cells (CSCs) shared many of the same regulatory pathways that are important in normal organ development, raising at least two important possibilities. First, these CSCs were derived from normal organ stem cells that have undergone genetic alterations, leading to the tumor phenotype. Alternatively, these CSCs were tumors cells that had simply reutilized inherent regulatory pathways typical of their tissue of origin. These hypotheses have led to considerable recent controversy over the existence and nature of CSCs [20,21].
Recent studies have suggested that the evident frequency of CSCs is highly dependent on the model system and conditions used in the experiment. For example, in the setting of melanoma, new tumors can be initiated with as few as one cell when model systems are optimized for tumor take (e.g., enhanced immunosuppression) [22]. Moreover, in some settings, cells not expressing supposed CSC markers (e.g., CD133- cells) were also capable of initiating new tumors (albeit with decreased efficiency) and regenerating the same degree of heterogeneity as cells expressing supposed cancer stem cell markers (e.g., CD133+ cells). Part of the problem with interpreting the results of studies using purported CSC-specific markers is that the antibodies being used recognize epitopes on these markers that may reflect post-translational modifications (e.g., glycosylation) [23] or may be accessible in some functional states but not others (e.g., protein folding owing to interactions with other moieties) [24]. As reviewed recently, these findings have led to intermediate concepts [25]. First, not all tumors exhibit typical CSC characteristics. For example, while hematopoietic malignancies may exhibit many self-renewal and differentiation characteristics, similar to that of the normal hematopoietic system, other tumors (e.g., melanoma) may not. Second, tumor cells may exhibit a more plastic phenotype, where they can dynamically pass into and out of the stem cell phenotype but are still capable of initiating new heterogeneous tumors [26,27]. These notions are currently being tested at the genomic, epigenomic and proteomic levels in concert with in vivo transplantation studies.
Molecular characterization & regulation of cancer stem-like cells
The ability to enrich CSCs on the basis of defined markers has led to a characterization of molecular pathways that regulate their self-renewal, differentiation and capability to initiate new tumors. Since studies in some settings suggested that CSCs shared some normal stem cell characteristics, it has been suggested that these CSCs may sit in ‘niches’ or tissue microenvironments, whereby stromal components, including cancer-associated fibroblasts, matrix, other support cells, vasculature and secreted factors, all contribute to the regulation of proliferation, self-renewal and differentiation of the CSCs [28–31]. While different cohorts of regulatory pathways have been described in a variety of tumor settings, certain pathways appear to be frequently involved, including Notch, sonic hedgehog and Wnt [32,33]. Notch activation has been found to be important in maintaining the self-renewal of stem cells in various normal niche settings, possibly associated with the inhibition of differentiation. The Wnt signaling pathway has been thought to maintain self-renewal in various ways, including enhanced proliferation status of stem cells and controlling the ability of stem cells to remain physically associated with the niche [34,35]. Similarly, the hedgehog pathway is also thought to play a role in regulating proliferation and survival of the stem cells [36–38]. In some settings, these pathways are closely related to those pathways observed in the normal organ developmental setting.
The idea that CSCs may exist in microenvironmental niches is also important in understanding the potential dynamic state of CSCs [28,39–41]. For example, just as in normal tissue settings, while CSCs are generally in a quiescent state, they may respond to tumor injury in a homeostatic manner by re-entering the proliferative state with enhanced self-renewal properties. Then, as the tumor bulk expands, feedback signals may be activated that then push for decreased self-renewal (e.g., decreased symmetrical divisions of the CSCs), maturation and a return of CSCs to a more quiescent state. Early evidence supporting this dynamic response to tumor injury came from mouse model systems, where the number and proliferative status of tumor-initiating cells were evaluated by traditional methodologies (i.e., tumor cell dose required to initiate tumors in 50% of animals, tumor control dose 50% [TCD50] and tritiated thymidine suicide). Following radiation and reduction in tumor bulk, CSCs were found to undergo a transient period of accelerated repopulation until the tumor mass was restored [42–45]. Similarly, just as in normal tissue settings, CSCs may exit the niche for various periods of time, lose the expression of purported CSC markers and later return the niche and regain their CSC markers [17,46]. The important concept here is that CSCs probably have a very dynamic, context-associated component; this may be overlooked when trying to characterize inherent properties of CSCs based simply on cell markers.
It is also important to consider the possibility that there exists extensive biological heterogeneity within the CSC population that differentially influences radioresponse. Since most human tumors are believed to have evolved through a progressive, multistep process of genetic and epigenetic events, it is likely that, by the time of tumor presentation, multiple CSC subpopulations exist [25,47]. To examine this possibility, we utilized a recently reported color-coding technology (Brainbow vectors), which permits the visual identification of clonal variants within tumors [48]. When these color-coded populations were grown in vitro, clonal derivatives occupied spatially distinct regions within the growth plane. When these populations were grown as tumor xenografts in nude mice, we observed multicolor tumors, whereby individual clonal variants occupied spatially distinct regions within the tumor with distinguishable growth patterns (e.g., compact vs dispersed clonal patches). This result suggests that each color-coded neighborhood was initiated by a single CSC variant. Interestingly, different CSC variants exhibited different efficiencies of focus initiation, growth rate and differential response to irradiation. This suggests that, even within tumor populations, different subpopulations of CSCs may exhibit heterogeneous biological features that influence ultimate radioresponse.
Factors that influence cancer stem-like cell radiosensitivity
As reviewed extensively elsewhere [5], many factors have been found to determine tumor response to radiotherapy. Intrinsic determinants of radiation sensitivity include DNA repair capability, cell cycle status and regulation of pathways that protect from cellular stress (survival pathways). Within the tumor, physiological context can also influence radiosensitivity. For example, since DNA damage induction is oxygen-dependent, cells sitting in a hypoxic environment are more radioresistant than cells sitting in a well-oxygenated compartment. As described earlier, at the tumor level, the degree of radiosensitivity of the intact tumor correlates with the number of CSCs at the time of treatment. Since radiation is usually given in a fractionated schedule over several weeks, the rate of accelerated repopulation between treatment fractions can also influence tumor radioresponse. Finally, the ability of the remaining surviving cells at the end of radiation therapy to re-establish the CSC niche also influences the final outcome of radiation treatment.
With the identification of markers associated with cells exhibiting the properties of CSCs, and subsequent cell selection based on these markers, a number of investigators have recently examined the aforementioned radiosensitivity determinants in cell populations enriched for CSCs in an attempt to identify targetable pathways to enhance radioresponse. Since these studies have been well reviewed [49–52], we will briefly summarize some of the results. In general, it has been suggested that CSC subpopulations are relatively radioresistant compared with non-CSC subpopulations. For example, CSCs isolated from a variety of tumor settings based on CSC-associated surface markers exhibit lower levels of reactive oxygen species, both before and following radiation treatment, potentially as a result of increased levels of free-radical scavenging substances within the cells (e.g., glutathione and superoxide dismutases) [53]. The result of this would be decreased levels of DNA damage immediately following radiation. Similarly, using indirect measures of DNA damage and its repair (kinetics of phosphorylated H2AX-γ foci and DNA comet assays), enriched CSC cell populations (i.e., CD44+/CD24- breast cancer and CD133+ glioma CSCs) showed enhanced DNA repair capability compared with non-CSC-enriched cell populations [54]. This property was associated with an increased ability to activate DNA damage checkpoint responses following radiation (e.g., activation of Chk1 and Chk2 checkpoint kinases), which serves to slow cell cycle progression and permit repair prior to cell division. Indeed, targeting these pathways using Chk inhibitors enhanced radiation response, both in in vitro and in vivo model systems.
Another property of CSCs that is associated with relative radioresistance is their quiescent status. In general, quiescent cells are more radioresistant than proliferating cells, with G2/M cells being most radiosensitive and late S phase most radioresistant. However, it should be noted that, with fractionated radiation, loss of tumor bulk leads to the re-entry of the CSCs into the cell cycle and a period of accelerated repopulation (Figure 2). More recently, these sets of events were directly visualized based on the purported property of decreased proteasomal activity of CSCs and the use of a proteasome-sensitive living cell dye. In the setting of breast cancer cell mammospheres, the relative frequency of CSCs in the mammosphere populations was found to increase following irradiation during rebound growth, further supporting the idea that CSCs are recruited back into the cell cycle in a homeostatic fashion following radiation [55]. Thus, while these regenerating CSCs may exhibit increased radiosensitivity with subsequent fractions, the ultimate radioresponse at the tumor level is dependent on the balance between enhanced intrinsic radiosensitivity and the rate of CSC self-renewal (e.g., enhanced symmetric divisions of the CSCs) [56]. If the CSCs repopulate quickly between radiation fractions, they can overcome the degree of cell kill with each fraction.
Figure 2. Doses to achieve local control in 50% of cases (tumor control dose 50%) as a function of overall treatment time for squamous cell tumors of the head and neck.
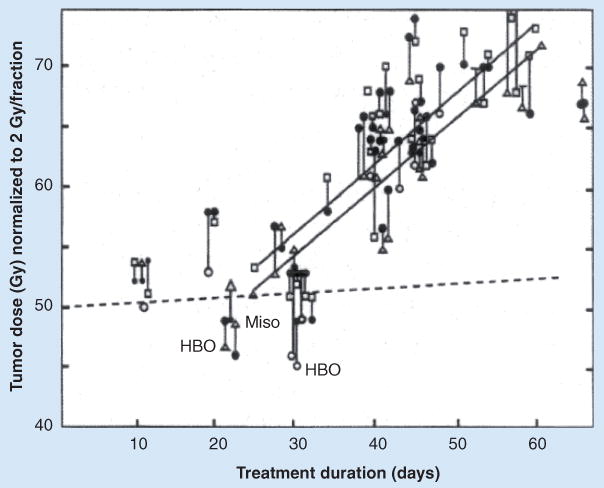
The data points include many published results from the literature, including HBO and the trial of Miso. The dashed line shows the rate of increase in tumor control dose 50% predicted from a 2-month cancer stem-like cell doubling rate.
HBO: High-pressure oxygen trial; Miso: Misonidazole.
Reproduced with permission from [43].
An additional property of CSCs that can affect radiosensitivity is their oxygenation status in the tumor at the time of radiation [57]. It is still unclear where CSCs exist within the tumor. For example, using some CSC cell surface markers (e.g., CD133+) to localize CSCs in brain tumor populations, it has been suggested that the CSCs reside in a perivascular niche region [55,58] and, therefore, would be expected to be well oxygenated unless they have suffered intermittent changes in the level of oxygenation leading to frequent transient periods of hypoxia [59]. On the other hand, our own unpublished pulse-chase studies in one model system to identify the location of label-retaining cells suggest that these cells reside in regions bordering necrosis, implying they have been exposed to chronic hypoxia. Along these lines, prior studies from our group examined tumor subpopulations fractionated on the basis of buoyant density [60,61]. We found that the fractions containing the more oxygenated cells exhibited higher clonogenic capacity but were more radiosensitive. On the other hand, the most radioresistant cell fractions were derived from the more dense (i.e., hypoxic) compartment, but were inherently less clonogenic. Thus, one would have to conclude that CSC subpopulations exist in varied tumor compartments where radioresistance mechanisms may be differentially expressed.
It has also been suggested that CSCs exist in a microenvironment that can impact their survival capability following the stress of radiation treatment [62–64]. For example, direct CSC cellular interactions with surrounding cells have been suggested to upregulate survivin levels and decrease apoptotic response. Similarly, CD133+ glioma CSCs have been found to express increased levels of protective autophagy proteins following irradiation; inhibition of expression of these proteins has been associated with an increased radiation response in in vitro sphere-forming studies [65]. Since there may be heterogeneity between CSC subpopulations within heterogeneous tumors, it is also possible that some CSC subpopulations can impact the radiation response of other CSC subpopulations, either through direct contact or eliciting soluble factors that influence radiation survival pathways (e.g., TGF-α). In addition, niche-associated pathways (e.g., Notch) have been shown to be activated following radiation, resulting in a transition to increased frequencies of symmetric cell divisions that would contribute to a more rapid accelerated repopulation [66]. Notch activation has also been suggested to activate pathways, such as EGF receptor (EGFR), which could impact both DNA repair capability, survival capacity of cells and regeneration kinetics. It has also been demonstrated that the CSC microenvironment may provide local and systemic cytokines, such as EGF, VEGF and FGF, all of which confer radioprotection to tumor cells [63].
Therefore, there is considerable evidence to suggest that, under certain experimental conditions, CSCs exhibit radioresistant features [67]. However, the conclusion of these studies was dependent both on how the CSCs were isolated for study and on how the radioresponse phenotype was defined. As a result, the conclusion that CSCs are inherently radioresistant may be oversimplified for a number of reasons. First, the CSC pool may be more plastic than previously recognized, so cells that do not exhibit CSC features at one point in time may return to this particular CSC state at a subsequent point in time. For example, it has been shown in some experimental settings that isolated CD133+ cells were more radioresistant than isolated CD133- cells. However, a portion of the CD133- population has been shown to still exhibit the capability to initiate new tumors that reiterate the heterogeneity of tumors similar to that initiated by CD133+ cells [25]. Thus, the radioresistant phenotype may not necessarily reflect the intrinsic cancer stem cell potential of the cell but rather the context of that cell at that point in time.
Second, it has to be recognized that the radiosensitivity characteristics of CSCs may be quite dynamic, depending both on their local microenvironment and homeostatic balance within the tumor itself. For example, CSCs may exist within both well-oxygenated and hypoxic regions of the tumor, and this may influence ultimate radioresponse in a complex and dynamic fashion. While hypoxic CSCs in an intact tumor may have an inherent radiosensitive component owing to their long existence in a compromised physiological state, they may also have a radioresistant component owing to their decreased oxygen tension and quiescent status. Thus, while these cells may exhibit an initial radiosensitive component, they may also exhibit a radiation-resistant tail component to the dose–response curve. Moreover, the physiologic status of the hypoxic CSCs dramatically changes once fractionated radiotherapy is initiated, both in terms of oxygenation status and proliferative status. Taken together, the radiation response of CSCs in tumors involves both intrinsic cellular radiosensitivity features, as well as context-associated features, and this complex mixture of factors determines ultimate radiosensitivity, being highly dynamic during fractionated radiotherapy. Thus, therapeutic strategies must take into account this complex and dynamic relationship.
Perspectives on therapeutic implications
As described earlier, many studies have focused on pathways and conditions associated with CSC radioresistance. Therefore, the natural tendency would be to focus on targeting the same radioresistance mechanisms throughout the course of radiotherapy. However, different CSC resistance pathways may play more important roles at different stages of fractionated radiotherapy. For example, relative hypoxia and quiescence of CSCs in the tumor may play a more important radioresistance-determining role at the initiation of radiotherapy compared with later during fractionated radiotherapy, when CSCs undergo increased proliferation and reoxygenation has occurred. Similarly, increased CSC self-renewal mechanisms may play a more prominent role during the accelerated repopulation phase following a substantial reduction of tumor bulk.
There has also been a tendency of investigators to only focus on CSC features that underlie radioresistance and overlook the necessity of treating the tumor bulk. If it is true that, as in normal tissues, homeostatic interactions can physiologically regulate stem cell behavior, then treatment of the tumor bulk could greatly influence the behavior of CSCs and their radioresistant features. For example, kinetic studies suggest that the tumor bulk can have an antiproliferative effect on the CSCs, and that following radiation, re-entry of the CSCs into the proliferative pool only follows a significant reduction in tumor size [45,68]. Thus, only focusing on enhancing the radiotherapeutic effect on the CSCs without addressing the tumor bulk might not take advantage of the radiosensitizing effects associated with the reoxygenation and re-entry of CSCs into the proliferative pool.
It is also important to remember that, in many clinical situations, radiotherapy is given in combination with chemotherapy. For example, in the treatment of most solid tumors, concomitant administration of chemotherapeutic drugs during the course of radiotherapy has been found to enhance clinical outcome (both local tumor control and patient survival), albeit with additional side effects, when compared with radiotherapy alone. Moreover, it is likely that introducing treatments designed to overcome CSC radioresistance mechanisms can also add significant side effects. For example, in the setting of head and neck squamous cell carcinomas, based on positive preclinical data, cetuximab (an anti-EGFR antibody) has been shown to improve the radiotherapeutic outcome of head and neck cancer patients [69]. However, significant cetuximab-associated skin rash was observed in approximately a third of the subjects. Thus, it may be important to identify the greatest window of therapeutic opportunity for addressing each radioresistance mechanism. Interestingly, the greatest benefit of adding cetuximab to fractionated radiotherapy appeared in the group of subjects receiving radiotherapy schedules where radiation was given more frequently than once daily (i.e., hyperfractionated radiotherapy or concomitant boost [where radiotherapy was administered twice daily during the last 12 fractions of a 6-week schedule]) [70], suggesting that increased cetuximab benefit may occur during the period of accelerated repopulation of the CSCs. Thus, as these approaches targeted to overcome CSC resistance mechanisms are added to current standard-of-care chemo/radiotherapy regimens, it will be important to consider the dynamics of CSC radioresistant mechanisms at different stages of tumor treatment (i.e., at the initiation of treatment, during radio/chemotherapy and following the completion of definitive treatment).
At the initiation of therapy, the most likely CSC resistance mechanisms in play include their quiescent status and hypoxia. Several strategies have been initiated recently to overcome these radioresistant mechanisms, including hyperbaric oxygen, vasoactive drugs, erythropoietin, hypoxic cell radiosensitizers and bioreductive drugs [57,71]. However, these approaches so far have resulted in, at most, modest improvements, and sometimes untoward effects. For example, erythropoietin has been administered in combination with radiotherapy to enhance red blood cell (RBC) count and increase the delivery of oxygen to tumors. Unfortunately, while the RBC counts were found to be increased by erythropoietin, this approach may have actually decreased clinical impact, perhaps owing to the presence of erythropoietin receptors on tumor cell populations, whose activation enhanced the ability of these cells to survive radiation insult [72,73].
It has also been demonstrated that, in some settings, the administration of antiangiogenic agents can transiently normalize tumor vascularization by stabilizing pericyte coverage on blood vessels, pruning back abnormal vessels, decreasing extravascular fluid pressure and, thereby, increasing tumor oxygenation [74,75]. If a subfraction of CSCs do exist in a hypoxic component, then this would be predicted to enhance their radiosensitivity [76,77]. Whether this approach has led to improved clinical response has been difficult to determine, since most clinical trials combining radiation and antiangiogenic agents have continued administering the antiangiogenic agents during multiple treatment fractions. More recently, it has been suggested that antiangiogenic treatment can induce feedback pathways in some tumor settings, which would either activate alternative angiogenic pathways or induce tumor cells to undergo an epithelial-to-mesenchymal transition, leading to enhanced metastatic capability [78,79]. For example, chronic antiangiogenic treatment could increase hypoxia, leading to upregulation of the hypoxia-inducible factor (HIF)-lα, and subsequent increased Met expression and activation, leading to enhanced tumor cell migration [80]. Thus, there is increased focus on defining the optimal window of therapeutic opportunity for this strategy and identifying the tumor types most likely to respond in a beneficial manner, as well as characterizing and targeting feedback mechanisms.
There have also been renewed efforts to increase the proliferative component of CSCs prior to the initiation of therapy, especially in the setting of hematopoietic diseases where some of the growth initiators are better known. For example, it was recently reported, in a model system, that the administration of G-CSF induced mobilization and stimulation of human leukemia CSCs into the cell cycle and rendered these cells more responsive to subsequent chemotherapy [81].
While this approach has had mixed clinical impact in prior clinical trials [82,83], and while such an approach has not been reported in solid tumors, there is reason for further exploration. For example, there has been recent interest in the use of EGFs (e.g., KGF) in the prevention of radiation- and chemotherapy-associated mucositis [84]. In these settings, KGF was given post-therapy in order to accelerate the regrowth of the mucosa and decrease the duration of mucositis. While this treatment approach was shown in certain preclinical models to have little impact on the regrowth kinetics of tumors following radiation, it is not clear whether this approach impacted CSC proliferation kinetics. It is also not yet clear which soluble factors control the proliferative status of the CSCs in the intact tumor. For example, since the reduction of tumor bulk results in an increase in the proliferative status of CSCs, it is possible that the tumor bulk elicits factors that directly or indirectly (through the microenvironment) downregulate proliferation of the tumor CSCs. If these factors could be identified, they might serve as an alternative approach to stimulating the proliferative status of quiescent CSC subpopulations prior to the initiation of radiotherapy. At the same time, they might also alter other pathways controlling the radiosensitivity of the CSCs.
As described previously, different CSC radioresistance mechanisms may be more important at different times during the course of radio/chemotherapy. After initial therapy, CSC status is thought to change owing to reoxygenation and feedback homeostatic mechanisms that reinitiate proliferation in CSC subpopulations following a reduction in tumor bulk. While proliferation itself may serve to radiosensitize CSCs, it may also be associated with the activation of signaling pathways that enhance DNA repair and survival following stress, as well as increased capability to grow and outpace the cell killing effects of the therapy.
The phenomenon of accelerated repopulation of CSCs has been appreciated for many years, and a number of strategies have been developed that appear to have clinical benefit. For example, altered radiation schedules (e.g., radiation given more frequently and in a shorter overall time during the period of most active accelerated CSC repopulation or concomitant boost) have been utilized in the treatment of head and neck cancer, with demonstrated benefit both in terms of local control and patient survival [85–88].
Another effective strategy has been the addition of chemotherapeutic agents during the course of radiotherapy (concurrent radio/chemotherapy). More recently, molecularly targeted agents have been added to these approaches to improve outcome. However, whether these approaches directly address CSC radioresistance mechanisms is still not known. For example, in the head and neck cancer trial combining cetuximab and fractionated radiotherapy [69,70], it is possible that the anti-EGFR strategy had additional impact on CSC radioresistance mechanisms during radiotherapy. One possibility is that EGFR antagonism slowed the rate of accelerated repopulation between radiation fractions. A second possibility is that EGFR antagonism inhibited DNA repair and survival pathways of CSCs. While these mechanisms probably also address radioresistant mechanisms of non-CSCs, focusing on these mechanisms at this point in the course of therapy might have a more pronounced therapeutic effect on the CSC population than at the initiation of therapy.
These recent observations suggest that targeting accelerated CSC repopulation following the initiation of radio/chemotherapy is worth further exploration. One approach has been to utilize agents that slow the rate of CSC homeostatic regeneration (e.g., flavopiridol) following a reduction in tumor bulk [89,90]. A second approach has been to combine checkpoint inhibitors to radiosensitize cells by decreasing their capability to repair radiation-induced DNA damage [54]. However, it is not clear whether there is any preferential impact of these strategies on CSCs or non-CSC subpopulations. A third approach has been to alter the self-renewal capabilities of the repopulating CSCs by inhibiting pathways that drive symmetric divisions and prevent differentiation of the CSCs. For example, cyclopamine, an inhibitor of the sonic hedgehog pathway (important in CSC self-renewal), has been explored in preclinical settings and is now in clinical testing [91]. Similar approaches are now underway toward the same goal of slowing self-renewal efficiency during periods of accelerated CSC repopulation (e.g., inhibitors of the Notch and Wnt pathways) [66]. While agents thought to induce differentiation have been utilized in the past with radiation (e.g., retinoids in the treatment of brain cancer) with questionable immediate clinical benefit, there is evidence that such approaches have led to an increment in long-term cure. Thus, there is renewed interest in identifying agents that will push the CSCs toward maturation during the course of radio/chemotherapy and, thus, blunt accelerated repopulation.
A third time frame where different types of CSC-related radioresistance mechanisms may exist is following definitive radio/chemotherapy at the time of minimal residual disease. At this point in time, the few surviving cells may find themselves situated in an environment that may or may not support their long-term survival and ability to regrow and cause tumor recurrence. While the molecular factors that define the ability of residual CSCs to regrow are not well understood, prior studies in model systems provided some hints on the possible mitigating pathways. For example, while irradiation of the tumor bed prior to inoculation of tumor cells has been found to enhance tumor take, the rate of tumor progression appears retarded compared with that observed in the nonirradiated tumor bed [10]. Similarly, the admixture of irradiated tumors with the tumor innoculum enhances the efficiency of tumor take [9,10,92,93]. On the other hand, administration of anti-inflammatory agents (e.g., indomethecin or selective cyclooxygenase [COX-2] inhibitors) has been found to reduce the efficiency of tumor take [94,95]. These observations suggest that inflammatory cytokines and the microenvironmental state can influence the reinitiation efficacy of tumors in the irradiated tumor bed at a time of minimal disease. These activated pathways may be different from those present at the time of minimal residual disease following definitive surgery. Theoretically, this reinitiation phenomenon involves the acquisition of several capabilities by the CSCs, including the ability to survive in a hostile environment, the ability to reinitiate proliferation and the ability to re-establish an adequately vascularized niche. Thus, targeting these pathways following definitive radio/chemotherapy may help to overcome an apparent radioresistance phenotype (i.e., contextual resistance) and improve outcome.
One example of a potential benefit of targeting the residual tumor niche involves the use of the EGFR antibody cetuximab. In a preclinical model system, when cetuximab was combined with radiotherapy and then continued beyond definitive treatment, it provided additional therapeutic benefit, both in terms of delaying tumor regrowth and improving long-term cure (Figure 3) [96]. While this approach had not been included in the prior cetuximab trials in head and neck cancer, it is being examined in current clinical trials. To better understand the mechanisms underlying the long-term benefit of continued cetuximab treatment following definitive therapy, it was recently found that tumor cells growing in an irradiated tumor bed exhibit increased expression of markers typical of an hypoxic microenvironment, including upregulation of HIF-lα, carbonic anhydrase IX, EGFR and VEGF [97]. Treatment with cetuximab in this setting was found to delay xenograft growth in the irradiated tumor bed (Figure 4). Moreover, cells derived from the cetuximab-treated xenografts exhibited reduced proliferation and reduced clonogenicity in vitro. Similar effects have been observed utilizing agents that target the VEGF receptor (VEGFR) [98].
Figure 3. Effect of cetuximab on radiocurability of A431 tumor after fractionated irradiation.

Mice bearing 8-mm tumors in the right hind leg were given cetuximab, local tumor irradiation or both as graded doses of γ-rays delivered twice daily for 7 consecutive days. Cetuximab (1 mg intraperitoneally) was given at 3-day intervals either three times during fractionated radiation or six times, both during and after fractionated radiation. Radiation dose–response curves were generated. Circles, solid line: local tumor control at 130 days after fractionated radiation alone (tumor control dose 50% [TCD50] = 83.1 [95% CI: 73.2–124.8] Gy). Squares, dashed line: cetuximab concurrent with radiation (TCD50 = 46.2 [95% CI: 39.1–57.5] Gy). Triangles, heavily dashed line: cetuximab given during and after radiation (TCD50 = 30.8 [95% CI: 22.2–38.0] Gy). Error bars are 95% CIs on the TCD50.
Reproduced with permission from [96].
Figure 4. Effect of cetuximab on the growth of A431 xenografts in tumor-bed irradiation.

Tumor-bed irradiation of the right hind leg of mice was performed with 20-Gy single-dose γ-irradiation 1 day before subcutaneous injection of A431 tumor cells (TBE). Mice were treated intraperitoneally with cetuximab (C225) in three 1-mg doses in 3-day intervals when tumors reached 5 mm in diameter. The error bars show mean values ± standard error.
Reproduced with permission from [97].
It has also been demonstrated that the hostile (radiation wounded) microenvironment surrounding the residual CSCs leads to the production of factors that attract other cell types to re-establish a healthy supportive niche. For example, it has been recently observed that a variety of cell types are recruited to wounded tissues as part of the healing process. As has been reported for the establishment of tumor metastases, some have reported that HIF-lα is upregulated in the irradiated tumor bed [99], leading to the recruitment of bone marrow-derived cells that help restore the damaged vasculature [100]. Since this process was thought to involve a stromal cell-derived factor 1/CXC chemokine receptor 4 interaction, pharmacologic agents inhibiting these pathways were found to slow or prevent re-establishment of the CSC niche and tumor regrowth [101].
Based on these types of studies, several general treatment approaches have been proposed in the postdefinitive treatment setting. The utilization of antiangiogenic or antivasculogenesis agents may have more impact in this setting than during radio/chemotherapy. While some agents, such as avastin, have not been found to have a dramatic impact on clinical tumor responses, they have been shown to enhance the time to progression when continued past definitive treatment [78]. Another approach that might have a unique benefit in this setting would be the use of agents that alter the self-renewal capability of the residual CSCs. For example, in a human colon cancer model, administration of a neutralizing antibody to δ-like 4 ligand – a membrane-associated ligand for the Notch pathway – was found to decrease the efficiency of tumor engraftment in host recipients [102,103]. On the other hand, it has also been found that chronic δ-like 4 ligand blockage can cause activation of endothelial cells and disruption of homeostasis and tissue atrophy in some normal tissues (e.g., liver, skin, heart and lung) [104]. Thus, the optimal timing and duration of such a therapeutic strategy must be defined.
Except in a few unique settings (e.g., all trans retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia), many differentiating agents have shown minimal impact when used during definitive treatment of tumors; however, they may find promise in the setting of minimal residual disease by decreasing the self-renewal capabilities of CSCs. Similarly, while various immunotherapeutic approaches have had limited impact in the setting of high tumor bulk, the setting of minimal residual disease may provide a better window of therapeutic opportunity to address apparently resistant CSCs. For example, it has been suggested that the presence of a significant tumor bulk can cause tumor anergy (e.g., production of immunosuppressive factors, such as TGF-β or enhanced numbers of regulatory T cells). This anergy may be less pronounced in the setting of minimal numbers of residual CSCs. Interesting, recent tumor vaccine studies have suggested that while these vaccines do not have a significant impact on progression-free survival in patients with reduced tumor burden, they do have an impact on slowing the progression rate and increasing overall survival [105,106]. Whether this approach selectively targets residual-resistant CSCs or progenitor cells is not clear. For example, it was recently reported that cells with many properties of CSCs lacked proteasomal activity [55]. Based on this observation, it was hypothesized that CSCs might be defective in the processing of antigens for MHC class I presentation. If confirmed to be a general phenomenon, it would suggest that CSCs may exist in an immunological sanctuary and might be insensitive to immune-mediated treatment strategies.
Conclusion
While there continues to be considerable controversy regarding the existence and nature of cancer stem cells in different tumor settings, recent studies have suggested that enriched CSCs are relatively radioresistant. These findings have been dependent both on the methods of CSC enrichment and the assays used to estimate radiosensitivity. Taken together, these studies suggested that the radiation response of CSCs in tumors involves both intrinsic cellular radiosensitivity features, as well as context-associated features. However, it needs to be recognized that this complex mixture of radiosensitivity determining factors is probably highly dynamic during fractionated radiotherapy. For example, prior to initiation of therapy, CSCs may exist in a hypoxic environment, but this property may change once therapy is initiated. Similarly, CSCs may be initially quiescent, but will be called into accelerated repopulation following reduction in tumor bulk. In addition, following definitive radio/chemotherapy, the tumor–microenvironment interactions may be quite distinct from what is present prior to the initiation of therapy. Thus, future therapeutic strategies based on targeting potential CSC radioresistance mechanisms must take into account these complex and dynamic processes. whereby different radioresistance pathways may be better targeted at different stages of therapy.
Future perspective
Over the next 5–10 years, we expect that studies on CSC radiosensitivity will focus on several different areas. First, there will be continued efforts to better understand the biology of CSCs and whether the notion of the existence of CSCs is relevant to all tumor types. For example, in some tumors (e.g., leukemia, breast and glioblastoma), there appear to be only a small fraction of cells that exhibit CSC properties; however, in other tumors (e.g., melanoma), the frequency of CSCs may be quite high. An important component of this research direction will focus on the identification of more specific stem cell markers for different tumor types. Similarly, we expect that there will be increased understanding of the intrinsic and extrinsic factors that control the plasticity and maintenance of the CSC state (e.g., expression factors, miRNA expression, post-translational modifications of molecules that control stem cell fate and niche factors that control stem cell renewal). Furthermore, we expect that there will be a search for a better understanding of the extrinsic factors (both from the local microenvironment and elicited by the tumor bulk) that controls the homeostatic balance of the CSCs before, during and after therapy. For example, it has been suggested that the presence of the tumor bulk may elicit cytokines (e.g., TGF-β), which may directly or indirectly influence the proliferative and self-renewal status of CSCs; however, the nature of these factors is not well understood.
A second general area of expected research interest is the identification of specific molecular pathways that influence CSC radiosensitivity, both in terms of intrinsic cellular radiosensitivity (e.g., DNA repair pathways, cell survival pathways and radical scavenger expression), as well as pathways that influence ultimate tumor response after fractionated courses of therapy (e.g., hypoxia, rate of cell regeneration, self renewal capability and ability to reinitiate the tumor niche and regenerate following definitive therapy).
Research progress in the first two areas will lead to the identification of molecular pathways that can influence CSC radiosensitivity at different times during the course of therapy. This would then lead to the use of molecularly targeted approaches to enhance the efficacy of standard radio/chemotherapy regimens. It will be important to recognize that the targetable pathways may play different roles at various times during the course of fractionated therapy. For example, a strategy to enhance radiosensitivity by bringing CSCs out of quiescence (e.g., through the use of growth factors or blocking tumor-bulk cytokines that render CSCs quiescent) may be more beneficial prior to the initiation of therapy than during the period of accelerated repopulation. Similarly, the strategies used to blunt accelerated repopulation may be more beneficial following reduction of tumor bulk than prior to the initiation of therapy. In addition, strategies to target the tumor microenvironment may differ significantly at different times during and following definitive therapy.
It will also be important to consider that the pathways that control both the biological dynamics, as well as radiosensitivity, may differ between tumor types, as well as between different individuals' tumors within a tumor type. Thus, any therapeutic strategy in the long term will need to take into account the biological features that control CSC behavior in each individual tumor (i.e., personalization of therapy). Since molecularly targeted therapies are not without associated side effects, it will be important to identify which patients would likely benefit from particular therapeutic strategies based on their tumor's CSC characteristics. It will also be important to identify targetable pathways that impede CSC function without affecting the normal tissue stem cell fraction, especially when these normal tissue stem cells will undergo homeostatic responses when in the treated field. This may be accomplished if CSCs and normal tissue stem cells are regulated through distinguishable pathways or if their homeostatic response kinetics differ, whereby time windows of therapeutic opportunity may be defined and exploited. All in all, the coalescence of new understandings in these different lines of inquiry will probably lead to a better understanding of the biological behavior of CSCs in different settings, as well as the development of new molecularly targeted therapies to overcome mechanisms of relative CSC radiation resistance.
Footnotes
Financial & competing interests disclosure
This work was supported by National Cancer Institute Grants CA-06294, CA-106451, CA-103830, CA-16672, DE 13157, DAMD 17–02–1–0706 and DAMD 17–1–1–0689. Walter N Hittelman holds the Sophie Caroline Steves Distinguished Professorship in Cancer Research. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
For reprint orders, please contact: reprints@futuremedicine.com
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Redmond KM, Wilson TR, Johnston PG, et al. Resistance mechanisms to cancer chemotherapy. Front Biosci. 2008;13:5138–5154. doi: 10.2741/3070. [DOI] [PubMed] [Google Scholar]
- 2.Suit H. Response to x-irradiation of a tumor recurring after a TCD95 radiation dose. Nature. 1966;211(5052):996–997. doi: 10.1038/211996a0. [DOI] [PubMed] [Google Scholar]
- 3.Ando K, Koike S, Shikita M, et al. Radiosensitivity of late recurrences following radiotherapy of murine fibrosarcomas. Radiat Res. 1988;113(2):334–345. [PubMed] [Google Scholar]
- 4.Hall EJ. Radiobiology for the Radiologist. 5th. Lippincott Williams & Wilkins; NY, USA: 2000. [Google Scholar]
- 5.Milas L, Hittelman WN. Cancer stem cells and tumor response to therapy: current problems and future prospects. Semin Radiat Oncol. 2009;19(2):96–105. doi: 10.1016/j.semradonc.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Shelby P, Buick RN, Tannock I. A critical appraisal of the “human tumor stem-cell assay”. N Engl J Med. 1983;308(3):129–134. doi: 10.1056/NEJM198301203080304. [DOI] [PubMed] [Google Scholar]
- 7.Buick RN. Radiological and clinical implications of the stem cell concept in human malignancy. In: Cory JG, Szentivany A, editors. Cancer Biology and Therapeutics. Plenum Publishers; NY, USA: 1987. pp. 65–76. [Google Scholar]
- 8.Hill RP, Milas L. The proportion of stem cells in murine tumors. Int J Radiat Oncol Biol Phys. 1989;16(2):513–518. doi: 10.1016/0360-3016(89)90353-2. [DOI] [PubMed] [Google Scholar]
- 9.Peters LJ, Hewitt H. The influence of fibrin formation on the transplantability of murine tumor cells: implications for the mechanism of the Revesz effect. Br J Cancer. 1974;29(4):279–291. doi: 10.1038/bjc.1974.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milas L, Ito H, Hunter N, et al. Retardation of tumor growth in mice caused by radiation-induced injury of tumor bed stroma: dependency on tumor type. Cancer Res. 1986;46(2):712–727. [PubMed] [Google Scholar]
- 11.Milas L, Peters LJ. Conditioning of tissues for metastasis formation by radiation and cytotoxic drugs. In: Nicholson GL, Milas L, editors. Cancer Invasion and Metastasis: Biologic and Therapeutic Aspects. Raven Press; NY, USA: 1984. pp. 321–336. [PubMed] [Google Scholar]
- 12.Bickenbach JR, Chism E. Selection and extended growth of murine epidermal stem cells in culture. Exp Cell Res. 1998;244(1):184–195. doi: 10.1006/excr.1998.4163. [DOI] [PubMed] [Google Scholar]
- 13.Goodell MA, Brose K, Paradis G, et al. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med. 1996;183(4):1797–1806. doi: 10.1084/jem.183.4.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordan CT. Cancer stem cell biology: from leukemia to solid tumors. Curr Opin Cell Biol. 2004;16(6):708–712. doi: 10.1016/j.ceb.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 16.Wang JC, Dick JE. Cancer stems cells: lessons from leukemia. Trends Cell Biol. 2005;15(9):494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien CA, Kreso A, Dick JE. Cancer stem cells in solid tumors: an overview. Semin Radiat Oncol. 2009;19(2):71–77. doi: 10.1016/j.semradonc.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 18.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63(18):5821–5828. [PubMed] [Google Scholar]
- 19.Hammati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA. 2003;100(25):5178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea – a paradigm shift. Cancer Res. 2006;66(4):1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 21.Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 2006;66(4):1891–1896. doi: 10.1158/0008-5472.CAN-05-3450. [DOI] [PubMed] [Google Scholar]
- 22.Quintana E, Shackleton M, Sabel MS, et al. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Demonstrates that modifications to xenotransplantation assays can profoundly influence cancer stem-like cell (CSC) frequency estimates such that single tumor cells can be shown to be capable of initiating new tumors. This report challenged the notion that CSCs necessarily represent only a small fraction of cells within tumors.
- 23.Kemper K, Sprick MR, de Bree M, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70(2):719–729. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 24.Cardo-Vila M, Zurita AJ, Giordano RJ, et al. A ligand peptide motif selected from a cancer patient is a receptor-interacting site within human interleukin-11. PLoS ONE. 2008;3(10):E3452. doi: 10.1371/journal.pone.0003452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shackleton M, Quintana E, Fearon ER, et al. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138(5):822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 26.Mani SA, Guo W, Liao MJ, et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010–1012. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 28.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132(4):598–611. doi: 10.1016/j.cell.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voog J, Jones DL. Stem cells and the niche: a dynamic duo. Cell Stem Cell. 2010;6(2):103–115. doi: 10.1016/j.stem.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7(5):349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- 31.Williams DA, Cancelas JA. Leukaemia: niche retreats for stem cells. Nature. 2006;444(7121):827–828. doi: 10.1038/444827a. [DOI] [PubMed] [Google Scholar]
- 32.Molofsky AV, Pardal R, Morrison SJ. Diverse mechanisms regulate stem cell self-renewal. Curr Opin Cell Biol. 2004;16(6):700–707. doi: 10.1016/j.ceb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Henrique D, Hirsinger E, Adam U, et al. Maintenance of neuroepithelial progenitor cells by δ-Notch signaling in the embryonic chick retina. Curr Biol. 1997;7(9):661–670. doi: 10.1016/s0960-9822(06)00293-4. [DOI] [PubMed] [Google Scholar]
- 34.Battle E, Henderson JT, Beghtel H, et al. β-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell. 2002;111(2):251–263. doi: 10.1016/s0092-8674(02)01015-2. [DOI] [PubMed] [Google Scholar]
- 35.Van de Wetering M, Sancho E, Verweij C, et al. The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–250. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 36.Lai K, Kaspar BK, Gage FH, et al. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nat Neurosci. 2003;6(1):21–27. doi: 10.1038/nn983. [DOI] [PubMed] [Google Scholar]
- 37.Kai T, Spradling A. An empty Drosophila stem cell niche reactivates the proliferation of ectopic cells. Proc Natl Acad Sci USA. 2003;100(8):4633–4638. doi: 10.1073/pnas.0830856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evangelista M, Tian H, de Sauvage J. The hedgehog signaling pathway in cancer. Clin Cancer Res. 2006;12(20 Pt 1):5924–5928. doi: 10.1158/1078-0432.CCR-06-1736. [DOI] [PubMed] [Google Scholar]
- 39.Bissell MJ, LaBarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7(1):17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones DL, Wagers AJ. No place like home: anatomy and function of the stem cell niche. Nat Rev Mol Cell Biol. 2008;9(1):11–21. doi: 10.1038/nrm2319. [DOI] [PubMed] [Google Scholar]
- 41.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441(7097):1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 42.Tannock IF. Tumor physiology and drug resistance. Cancer Metastasis Rev. 2001;20(1–2):123–132. doi: 10.1023/a:1013125027697. [DOI] [PubMed] [Google Scholar]
- 43.Withers HR, Taylor JMG, Maciejewski B. The hazard of accelerated tumors clonogen repopulation during radiotherapy. Acta Oncol. 1988;27(2):131–146. doi: 10.3109/02841868809090333. [DOI] [PubMed] [Google Scholar]
- 44.Milas L, Yamada S, Hunter N, et al. Changes in TCD50 as a measure of clonogen doubling time in irradiated and unirradiated tumors. Int J Radiat Oncol Biol Phys. 1991;21(5):1195–1202. doi: 10.1016/0360-3016(91)90276-a. [DOI] [PubMed] [Google Scholar]
- 45.Thames HD, Ruifrock AC, Milas L, et al. Accelerated repopulation during fractionated irradiation of a murine ovarian carcinoma: downregulation of apoptosis as a possible mechanism. Int J Radiat Oncol Biol Phys. 1996;35(5):951–962. doi: 10.1016/0360-3016(96)00256-8. [DOI] [PubMed] [Google Scholar]
- 46.Jaiswal S, Jamieson CHM, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805(1):105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao Y, Valdecanas DP, Liu S, et al. Visualizing lung tumor heterogeneity by color-coding clonal cell subpopulations. Presented at: 101st American Association for Cancer Research Annual Meeting; Washington, DC, USA. 17–21 April 2010. [Google Scholar]
- 49.Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8(7):545–554. doi: 10.1038/nrc2419. [DOI] [PubMed] [Google Scholar]
- 50.Ishii H, Iwatsuki M, Ieta K, et al. Cancer stem cells and chemoradiation resistance. Cancer Sci. 2008;99(10):1871–1877. doi: 10.1111/j.1349-7006.2008.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Woodward WA, Bristow RG. Radiosensitivity of cancer-initiating cells and normal stem cells (or what the Heisenberg uncertainly principle has to do with biology) Semin Radiat Oncol. 2009;19(2):87–95. doi: 10.1016/j.semradonc.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: the 4 R's of radiobiology revisited. Stem Cells. 2010;28(4):639–648. doi: 10.1002/stem.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diehn M, Cho RW, Lobo NA, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ Reports that CSCs express higher levels of free-radical scavenging systems, resulting in lower reactive oxygen species and decreased radiation sensitivity.
- 54.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]; ▪ Demonstrates CD133-expressing glioma stem cells preferentially activate the DNA damage checkpoint, permitting enhanced repair of radiation-induced DNA damage and resulting in increased radioresistance.
- 55.Vlashi E, Kim K, Lagadec C, et al. In vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst. 2009;101(5):350–359. doi: 10.1093/jnci/djn509. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪▪ Reports that cancer-initiating cells exhibit decreased proteasome activity, a property that permits their direct identification, tracking and targeting.
- 56.Pine SR, Ryan BM, Varticovski L, et al. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc Natl Acad Sci USA. 2010;107(5):2195–2200. doi: 10.1073/pnas.0909390107. [DOI] [PMC free article] [PubMed] [Google Scholar]; ▪ Demonstrates that the microenvironment of CD133+ lung CSCs can regulate asymetric cell division and lung cancer cell fate.
- 57.Brown JM. Tumor hypoxia in cancer therapy. Methods Enzymol. 2007;435:297–321. doi: 10.1016/S0076-6879(07)35015-5. [DOI] [PubMed] [Google Scholar]
- 58.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 59.Cardenas-Navia LI, Mace D, Richardson RA, et al. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008;68(14):5812–5819. doi: 10.1158/0008-5472.CAN-07-6387. [DOI] [PubMed] [Google Scholar]
- 60.Grdina DJ, Milas L, Mason KA, Withers HR. Separation of cells from a fibrosarcoma in renografin density gradients. J Natl Cancer Inst. 1974;52(1):253–257. doi: 10.1093/jnci/52.1.253. [DOI] [PubMed] [Google Scholar]
- 61.Grdina DJ, Hittelman WN, White RA, et al. Relevance of density, size and DNA content of tumour cells to the lung colony assay. Br J Cancer. 1977;36(6):659–669. doi: 10.1038/bjc.1977.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Woodward WA, Chen MS, Behbod F, et al. WNT/±catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. 2007;104(2):618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment – tumorigenesis and therapy. Nat Rev Cancer. 2005;5(11):867–875. doi: 10.1038/nrc1735. [DOI] [PubMed] [Google Scholar]
- 64.Ghotra VPS, Puigvert JC, Danen EHJ. The cancer stem cell microenvironment and anti-cancer therapy. Int J Radiat Biol. 2009;85(11):955–962. doi: 10.3109/09553000903242164. [DOI] [PubMed] [Google Scholar]
- 65.Lomonaco SL, Finniss S, Xiang C, et al. The induction of autophagy by γ-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125(3):717–722. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 66.Gupta R, Vyas P, Enver T. Molecular targeting of cancer stem cells. Cell Stem Cell. 2009;5(2):125–126. doi: 10.1016/j.stem.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 67.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. 2008;26(17):2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim JJ, Tannock IF. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat Cancer Rev. 2005;5(7):516–525. doi: 10.1038/nrc1650. [DOI] [PubMed] [Google Scholar]
- 69.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354(6):567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 70.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a Phase III randomized trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11(1):21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]; ▪▪ Reports improved survival in patients with head and neck cancer treated with combined radiation and cetuximab.
- 71.Hardee ME, Arcasoy MO, Blackwell KL, et al. Erythropoietin biology in cancer. Clin Cancer Res. 2006;12(2):332–339. doi: 10.1158/1078-0432.CCR-05-1771. [DOI] [PubMed] [Google Scholar]
- 72.Phillips TM, Kim K, Vlashi E, et al. Effects of recombinant erythropoietin on breast cancer-initiating cells. Neoplasia. 2007;9(12):1122–1129. doi: 10.1593/neo.07694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Overgaard J, Hoff CM, Hansen HS, et al. Randomized study of darbepoietin α as modifier of radiotherapy in patients with primary squamous cell carcinoma of the head and neck (HNSCC): final outcome of the DAHANCA 10 trial. J Clin Oncol. 2009;27(155):15S. [Google Scholar]
- 74.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 75.Winkler F, Kozin SV, Tong RT, et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: role of oxygenation, angiopoitin-1, and matrix metalloproteinases. Cancer Cell. 2004;6(6):553–563. doi: 10.1016/j.ccr.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 76.Duda DG, Jain RK, Willett CG. Antiangiogenics: the potential role of integrating this novel treatment modality with chemoradiation for solid cancers. J Clin Oncol. 2007;25(26):4033–4042. doi: 10.1200/JCO.2007.11.3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kerbel RS. Antiangiogenic therapy: a universal chemosensitization strategy for cancer? Science. 2006;312(5777):1171–1175. doi: 10.1126/science.1125950. [DOI] [PubMed] [Google Scholar]
- 78.Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res. 2009;15(24):7471–7478. doi: 10.1158/1078-0432.CCR-09-1070. [DOI] [PubMed] [Google Scholar]
- 79.Hayden EC. Cutting off cancer's supply lines. Nature. 2009;458(7239):686–687. doi: 10.1038/458686b. [DOI] [PubMed] [Google Scholar]
- 80.Pennacchietti S, Michieli P, Galluzzo M, et al. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3(4):347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 81.Saito Y, Uchida N, Tanaka S, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275–280. doi: 10.1038/nbt.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mikkola HKA, Radu CG, Witte ON. Targeting leukemia stem cells. Nat Biotechnol. 2010;28(3):237–238. doi: 10.1038/nbt0310-237. [DOI] [PubMed] [Google Scholar]
- 83.Estey EH, Thall PF, Pierce S, et al. Randomized Phase II study of fludarabine + cytosine arabinoside + idarubicin ± all-trans retinoic acid ± granulocyte colony-stimulating factor in poor prognosis newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Blood. 1999;93(8):2478–2484. [PubMed] [Google Scholar]
- 84.Hensley ML, Hagerty KL, Kewalramani T, et al. American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. J Clin Oncol. 2009;27(1):127–145. doi: 10.1200/JCO.2008.17.2627. [DOI] [PubMed] [Google Scholar]
- 85.Ang KK, Peters LJ, Weber RS, et al. Concomitant boost radiotherapy schedules in the treatment of carcinoma of the oropharynx and nasopharynx. Int J Radiat Oncol Biol Phys. 1990;19(6):1339–1345. doi: 10.1016/0360-3016(90)90341-g. [DOI] [PubMed] [Google Scholar]
- 86.Fu KK, Pajak TF, Trotti A, et al. A Radiation Therapy Oncology Group (RTOG) Phase III randomized study to compare hyperfractionation and two variants of accelerated fractionation to standard fractionation radiotherapy for head and neck squamous cell carcinomas: first report of RTOG 9003. Int J Radiat Oncol Biol Phys. 2000;48(1):7–16. doi: 10.1016/s0360-3016(00)00663-5. [DOI] [PubMed] [Google Scholar]
- 87.Saunders M, Dische S, Barrett A, et al. Continuous, hyperfractionated, accelerated radiotherapy (CHART) versus conventional radiotherapy in non-small cell lung cancer: mature data from the randomized multicentre trial. Radiother Oncol. 1999;52(2):137–148. doi: 10.1016/s0167-8140(99)00087-0. [DOI] [PubMed] [Google Scholar]
- 88.Pignon JP, le Maitre A, Bourhis J, et al. Meta-analysis of chemotherapy in head and neck cancer meta-analysis of chemotherapy in head and neck cancer (MACH-NC): an update of 93 randomized trials and 17,346 patients. Radiother Oncol. 2009;92(1):4–14. doi: 10.1016/j.radonc.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 89.Mason KA, Hunter NR, Raju U, et al. Flavopiridol increases therapeutic ratio of radiotherapy by preferentially enhancing tumor radioresponse. Int J Radiat Oncol Biol Phys. 2004;59(4):1181–1189. doi: 10.1016/j.ijrobp.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 90.Raju U, Nakata E, Mason KA, et al. Flavopiridol, a cyclin-dependent kinase inhibitor, enhances radiosensitivity of ovarian carcinoma cells. Cancer Res. 2003;63(12):3263–3267. [PubMed] [Google Scholar]
- 91.Sims-Mourtada J, Izzo JG, Apisarnthanarax S, et al. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res. 2006;12(21):6565–6572. doi: 10.1158/1078-0432.CCR-06-0176. [DOI] [PubMed] [Google Scholar]
- 92.Revesz L. Effect of lethally damaged tumor cells upon the development of admixed viable cells. J Natl Cancer Inst. 1958;20(6):1157–1186. [PubMed] [Google Scholar]
- 93.Hewitt H, Blake E, Porter E. The effect of lethally irradiated cells on the transplantability of murine tumours. Br J Cancer. 1973;28(2):123–135. doi: 10.1038/bjc.1973.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Milas L, Furuta Y, Hunter N, et al. Dependence of indomethacin-induced potentiation of murine tumor radioresponse on tumor host immunocompetence. Cancer Res. 1990;50(15):4473–4477. [PubMed] [Google Scholar]
- 95.Choy H, Milas L. Enhancing radiotherapy with cyclooxygenase-2 enzyme inhibitors: a rational advance? J Natl Cancer Inst. 2003;95(19):1440–1452. doi: 10.1093/jnci/djg058. [DOI] [PubMed] [Google Scholar]
- 96.Milas L, Fang FM, Mason KA, et al. Importance of maintenance therapy in C225-induced enhancement of tumor control by fractionated radiation. Int J Radiat Oncol Biol Phys. 2007;67(2):568–572. doi: 10.1016/j.ijrobp.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 97.Riesterer O, Mason KA, Raju U, et al. Enhanced response to C225 of A431 tumor xenografts growing in irradiated tumor bed. Radiat Oncol. 2009;92(3):383–387. doi: 10.1016/j.radonc.2009.07.009. [DOI] [PubMed] [Google Scholar]; ▪ Demonstrates that cetuximab administration following definitive radiation treatment enhances tumor radiocurability by retarding tumor re-establishment in the irradiated tumor bed microenvironment.
- 98.Zips D, Eicheler W, Geyer P, et al. Enhanced susceptibility of irradiated tumor vessels to vascular endothelial growth factor receptor tyrosine kinase inhibition. Cancer Res. 2005;65(12):5374–5379. doi: 10.1158/0008-5472.CAN-04-3379. [DOI] [PubMed] [Google Scholar]
- 99.Moeller BJ, Dreher MR, Rabbani ZN, et al. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005;8(2):99–110. doi: 10.1016/j.ccr.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 100.Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10(8):858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 101.Kioi M, Vogel H, Schultz G, et al. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shaked Y, Kerbel RS. Antiangiogenic strategies on defense: on the possibility of blocking rebounds by the tumor vasculature after chemotherapy. Cancer Res. 2007;67(15):7055–7058. doi: 10.1158/0008-5472.CAN-07-0905. [DOI] [PubMed] [Google Scholar]
- 103.Yan M, Plowman GD. δ-like 4/Notch signaling and its therapeutic implications. Clin Cancer Res. 2007;13(24):7243–7246. doi: 10.1158/1078-0432.CCR-07-1393. [DOI] [PubMed] [Google Scholar]
- 104.Yan M, Callahan CA, Beyer JC, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature. 2010;463(7282):E6–E7. doi: 10.1038/nature08751. [DOI] [PubMed] [Google Scholar]
- 105.Neelapu SS, Gause BL, Harvey L, et al. A novel proteoliposomal vaccine induces antitumor immunity against follicular lymphoma. Blood. 2007;109(12):5160–5163. doi: 10.1182/blood-2006-12-063594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kantoff PW, Schuetz TJ, Blumenstein BA, et al. Overall survival analysis of a Phase II randomized controlled trial of a poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28(7):1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]