

Abstract
Periodic recurrence of painful vaso-occlusive crisis is the defining feature of sickle cell disease. Among multiple pathologies associated with this disease, sickle-red cell-endothelium interaction has been implicated as potential initiating mechanism in vaso-occlusive events. This review focuses on various interrelated mechanisms involved in human sickle red cell adhesion. We discuss in vitro and microcirculatory findings on sickle red cell adhesion, its potential role in vaso-occlusion, and the current understanding of receptor-ligand interactions involved in this pathological phenomenon. In addition, we discuss the contribution of other cellular interactions (leukocytes recruitment and leukocyte-red cell interaction) to vaso-occlusion as observed in transgenic sickle mouse models. Emphasis is given to recently discovered adhesion molecules that play a predominant role in mediating human sickle red cell adhesion. Finally, we analyze various therapeutic approaches for inhibiting sickle red cell adhesion by targeting adhesion molecules, and also consider therapeutic strategies that target stimuli involved in endothelial activation and initiation of adhesion.
INTRODUCTION
The primary defect in sickle cell disease is hemoglobin S (HbS) polymerization and red cell sickling under deoxygenated conditions. The unique features of this disease are recurring painful vaso-occlusive crisis and multiple organ damage. Although HbS polymerization and red cell sickling under deoxygenated conditions are central to the pathophysiology of this disease, emerging evidence indicates that initial events in vaso-occlusion may involve a complex interplay of multiple factors, both polymerization-dependent and polymerization-independent. The presence of low hematocrit, hemolysis, red cell sickling, intense oxidative stress and vascular endothelium activation are among the abnormalities that would continually affect the hemodynamics and physiological processes in this disease even in the crisis-free state resulting in an abnormal steady state. Among the multiple pathologies associated with this disease, sickle red cell-endothelial interaction has been implicated as one of the major potential initiating mechanisms in vaso-occlusion (27, 28, 41).
In this review, we will focus on in vitro and microcirculatory evidence of sickle red cell adhesion, its pathophysiological significance and the status of its underlying mechanisms. We will also draw attention to the role of red cell heterogeneity that is a salient feature of sickle cell disease, and enumerate the significant contribution of leukocytes to vaso-occlusion as observed in transgenic sickle mouse models under a unifying model of vaso-occlusion.
HEMOGLOBIN S (HbS) POLYMERIZATION
Under hypoxic conditions, HbS undergoes polymerization resulting in red cell sickling. An important feature of HbS polymerization is the delay time, i.e., time required for polymer formation upon deoxygenation (17). The delay time is an inverse function of HbS concentration from the 30th to the 50th power (11, 37). In other words, sickle red cells with high hemoglobin concentration will be most susceptible to HbS polymerization. Extensive studies of delay time have led to a more sophisticated understanding of the sickling phenomenon in vivo. In vivo, sickle red cells undergo reversible sickling (oxy-deoxy cycles) in blood circulation. However, oxy-deoxy cyles in vivo do not generally result in vaso-occlusion. This is due to the fact that the delay time for HbS polymerization is generally longer than the capillary transit times of red cells (<1 sec). Hence, most red cells escape sickling during the capillary transit. This explains why sickle patients, who have sufficient HbS in each red cell to sickle, are not sicker than they are. However, a prolonged transit time of red cells in the exchange compartment of the microcirculation would promote HbS polymerization and sickling resulting in the inability of sickled red cells to traverse narrow capillary diameters. Thus, factors that increase red cell transit times (e.g., red cell and/or leukocyte adhesion) may play a crucial role in the initiation of a vaso-occlusive episode.
SICKLE RED CELL HETEROGENEITY
Density gradient separation of blood from sickle patients reveals a marked heterogeneity of red cells. Red cell density is determined by mean corpuscular hemoglobin concentration (MCHC) expressed as g/dl. In a seminal study, published in 1982, Fabry and Nagel (19) showed a bimodal distribution of sickle red cells on density gradients. One major population comprised of discocytic red cells with MCHC similar to normal (AA) red cells (fraction 2); the other major population was very dense (fraction 4) sickle cells and contained mainly rigid, elongated, irreversibly sickled cells (ISCs). The red cells lighter than fraction 2 (fraction 1) contained a high percentage of reticulocytes. The fraction between 2 and 4 was characterized by the presence of very dense and somewhat irregular shaped discocytes (fraction 3) with high MCHC. The range of MCHC varied from ~30 g/dl for fraction 1 to >45 g/dl for fraction 4. Hence, at a given deoxygenation level, dense sickle cells will be the first to undergo HbS polymerization. Hemodymanic behavior of density-defined sickle red cell populations, examined in an ex vivo microcirculatory preparation, showed that deoxygenation caused maximal (~10-fold) increase in the vascular resistance to very dense sickle discocytes of fraction 3 over its oxygenated counterpart (45). In contrast, the bulk viscosity of each density gradient fraction showed a nearly uniform increase (~2-fold) increase upon deoxygenation. The hemodynamic results indicated that a profound vaso-occlusive behavior of dense sickle cells (fraction 3) with deoxygenation. Moreover, these studies showed that deoxygenation of fraction 1 and 2 cells resulted in the formation of typical sickle form, while deoxygenation had no effect on the morphology of high density discocytes (fraction 3) and ISCs (fraction 4) (45). The difference in the hemodynamic behavior suggested that heterogeneous sickle red cells would contribute differently to a vaso-occlusive event.
The generation of dense sickle cells is mainly attributed to cell dehydration caused by abnormal membrane transport (10, 40). Since the rate of HbS polymerization is extremely sensitive to intracellular hemoglobin concentration (i.e., MCHC), there is always a possibility of transient occlusion by dense red cells since these cells will be the first to undergo polymerization as they traverse capillaries. However, dense sickle red cells, present in varying proportions in the blood of sickle patients, may be more of a participant than a causative factor. This line of argument has been supported by the observations that patient to patient variation in the percentage of dense red cells shows no correlation with the disease severity (1, 5). In contrast, red cell deformability index is positively correlated with severity of painful crisis as patients with high deformability index experience more crises and crisis days (1). Moreover, results obtained on an animal model show that animals tolerate some degree of ischemia following the infusion of dense sickle cells. As suggested by Ferrone (21), transient occlusive events caused by intravascular sickling are not rare, but cannot be equated with a vaso-occlusive crisis. Nevertheless, dense sickle cells, because of their ability to polymerize rapidly, are likely to contribute importantly to the propagation of vaso-occlusion in which adhesion of deformable sickle red cells plays a predominant role.
SICKLE RED CELL ADHESION AND VASO-OCCLUSION
As noted above, sickling is necessary but not sufficient to initiate a vaso-occlusive episode. Abnormal adhesion of human sickle red cells to vascular endothelium has drawn intense attention as a potential initiating factor in vascular obstruction in sickle cell disease.
a) In vitro studies
The introduction of in vitro cell adhesion assays using cultured endothelial cells was instrumental in drawing attention to the involvement of vascular factors in the pathophysiology of this disease. This new perspective was evident in two landmark papers published independently and consecutively by Hoover et al. (39) and Hebbel et al. (35). Both these studies suggested that abnormal adhesion of sickle red cells to cultured human endothelium was a potential contributing factor to vaso-occlusion in sickle cell disease. Although Hebbel et al. found that adhesion of sickle red cells obtained from individual sickle patients showed a correlation with the disease severity, this relationship was apparently based on adhesion of dense red cell populations (32, 33). As discussed later, subsequent studies revealed that under shear flow conditions sickle cell adhesion to vascular endothelium was dependent on the differences in the deformability characteristics of heterogeneous sickle red cell populations, and that deformable sickle red cells showed a greater propensity for adhesion.
In another approach, Mohandas and Evans employed a micropipette technique to measure the amount of force required to detach a single sickle red cell from adhesive contact with cultured endothelium. In a paper published in 1984, these authors concluded “that alterations in membrane surface that accompany cellular dehydration may render sickle cells more adhesive.” However, in a follow-up study, Mohandas and Evans reported that the ability of ISCs (dense sickle red cells) to adhere was surprisingly poor, but found irregular deformable discocytes to be more adherent (59). They suggested that the irregular shape of ISCs prevented sufficient membrane contact area with the endothelium, thus inhibiting their ability to form attachments.
Development of the parallel plate flow chamber represented an important step forward for in vitro study of sickle red cell adhesion under defined shear stress. Endothelial cell monolayer on a glass slide formed the base of a modified Richardson flow chamber (3). A fundamental characteristic of the parallel plate flow chamber is that fluid behavior within it can be clearly defined. The straight, parallel streamlines of laminar flow ensure that the fluid velocity and shear stress conditions are known at the surface of the endothelial monolayer, where cell-cell adhesive interactions occur. The parallel plate flow chamber allowed control of a variable or a set of variables of interest (a specific cell, protein, shear stress, etc.). Such a device was first applied to the study of sickle red cell adhesion in sickle cell disease by Barabino, et al. (3, 4). These authors applied a controlled-flow rate to give a venular wall shear stress of 1 dyne/cm2. Pressure-driven flow of a dilute suspension of sickle red cells (1% v/v) at this shear stress resulted in markedly greater adhesion of these cells to HUVEC as compared to normal (AA) red cells. These studies revealed an inverse relationship between sickle red cell adhesion and shear stress. Sickle red cell adhesion to HUVEC was negligible above a shear rate of 200 sec−1 (corresponding to a shear stress of 2 dynes/cm2) in a parallel plate flow chamber, which is relevant to low shear regimes in the microcirculation.
Using the parallel plate flow chamber, Barabino et al. found that the greatest adhesion was exhibited by cells from the least dense fraction, the reticulocytes, and the least by the densest red cells (e.g., ISCs) (3). As demonstrated in Figure 1, adhesion of sickle red cells to HUVEC under flow was maximal for reticulocyte-rich fraction (top fraction) while it showed a marked decrease for ISC-rich dense sickle red cell fraction (bottom fraction).
Figure 1.

Adhesion of sickle red cells to HUVEC under flow conditions is maximal for reticulocyte-rich fraction (top fraction), while it shows a marked decrease for ISC-rich dense sickle red cell fraction (bottom fraction). Modified from Barabino et al. (3).
b) Intrvital studies
In vitro assay did not indicate whether sickle cell adhesion occurred in a living microcirculatory beds under shear flow conditions, and if so, whether there were arteriolar versus venular differences in adhesion. Microvascular sites of human sickle red cell adhesion and the relative contribution of heterogeneous sickle cell populations in adhesive and obstructive events were investigated under shear flow conditions using an ex vivo microvascular bed. Infusion of oxygenated sickle red cells resulted in adhesion of these cells exclusively in postcapillary venules. The adhesion showed an inverse correlation with the venular diameter with maximal adhesion occurring in small-diameter immediate postcapillary venules (44). Postcapillary venules are also the primary sites of adhesion in transgenic sickle mouse models, as well as in the nude mice infused with human sickle red cells (42, 47, 84). Moreover, the relationship between sickle red cell adhesion and the venular diameter has recently been corroborated by Telen and coworkers in vivo (84). Adherent sickle red cells are able to withstand the prevalent wall shear rates encountered in postcapillary venules (~200 to 600 sec−1) (42), indicating pathophysiological relevance of sickle cell adhesion. In fact, some studies have reported human sickle red cells adhering to endothelial cells at shear stress values ranging from 10 to 55 dyne/cm2 (24, 54, 67).
In ex vivo studies, selective fluorescent labeling of sickle red cell populations revealed unusually high concentration of dense sickle red cells in the areas of vaso-occlusion (44). In vessels with adherent sickle red cells but no obstruction, there was almost a total absence of dense cells. Based on these intravital microscopic observations, Kaul et al. presented a two-step model of adhesion-induced vaso-occlusion (Figure 2 A,B) in which preferential adhesion of deformable sickle red cells (reticulocytes and discocytes) in postcapillary venules is followed by selective trapping of dense sickle red cells (dense discocytes and ISCs) that can lead to vessel obstruction (43). The effect of deformability on the adhesive behavior of heterogeneous sickle red cell populations was essentially in accord with the studies of Barabino et al. (3). The results of the ex vivo studies (44) support the concept that preferential adhesion of deformable sickle red cells increases microvascular red cell transit times, induces hypoxia and facilitates entrapment of dense and sickled red cells in postcapillary venules bedecked with adherent deformable red cells (43). This view has been supported by microscopic examination of the vascular bed and by higher peripheral resistance caused by sickle red cell adhesion (50, 51). As pointed out later in this review, leukocyte recruitment may also contribute to the selective trapping of sickled and dense red cells (Figure 2C).
Figure 2.

Models of vaso-occlusion in sickle cell disease. (A) Initial adhesion of deformable sickle red cells in small-diameter postcapillary venules. (B) Adhesion of deformable sickle cells is followed by selective trapping of dense or sickled red cells among adherent red cells. (C) Alternatively, recruitment of leukocytes in inflamed venules may trigger selective trapping of sickled and dense red cells. Modified from Kaul et al. (43).
Human sickle red cell adhesion has been confirmed by a wide variety of assay systems involving both static and dynamic flow conditions. Transgenic sickle mouse models have provided an opportunity to examine in vivo red cells adhesion using intravital microscopy. Studies using the cremaster muscle microcirculation of transgenic sickle mice expressing HbS and HbS-Antilles (S+S-Antilles mice) showed frequent red cell adhesion exclusively in postcapillary venules (42). In contrast, red cell adhesion is less frequent in transgenic-knockout sickle (BERK) mice that express exclusively HbS. In fact, Turhan et al. (79) did not find any evidence of red cell adhesion in BERK bone marrow transplanted C57BL mice. Moreover, in contrast to S+S-Antilles mice, BERK mice show fewer dense red cells, and their thalassemic features (β-globin:α-globin ratio, 0.79) are associated with the presence of microcytic red cells with low mean corpuscular hemoglobin (MCH) (20). However, BERK mice show intravascular sickling, significant hemolysis and oxidative stress. These features render these mice more suitable for studying the role of hemolysis and inflammation (i.e., leukocyte recruitment) (48, 79) rather than evaluating the contribution of heterogeneous red cells (a salient feature of this disease) to adhesive and obstructive events. Also, the mouse red cells have much smaller mean diameter (~4.5-5.0 μm) than human red cells (~8.0 μm) (14), and may not express similar adhesion receptors as reported for human sickle red cells. Such differences should be kept in mind while applying findings in BERK mice to sickle cell disease.
c) Red cell deformability, adhesion and clinical severity
Although under flow conditions, deformable sickle red cells are most adhesive to vascular endothelium, the greater adhesion of dense sickle red cells in static assays has been attributed by Hebbel (26) to low affinity mechanisms, which could form stable contacts in static assay system, and plausibly in vessels of limiting diameters or under flow stagnation. In fact, the reported correlation between sickle red cell adhesiveness and vaso-occlusive severity in sickle patients was based on findings in a static assay system wherein dense sickle red cells showed greater adhesion to endothelium as compared with less dense deformable populations of sickle red cells (29, 31). On the contrary, under shear flow conditions, adhesion characteristics are determined by the ability of red cells to deform and establish sufficient areas of contact with endothelial cell surface, as well as by differences in membrane mechanical and surface characteristics (59). For instance, controlled dehydration of deformable sickle red cell population (using nystatin-sucrose to elevate MCHC to ~45 g/dl) resulted in reduced adhesion as compared to the unperturbed cells. However, the decrease in adhesion was less marked compared with native dense sickle red cells (MCHC, 44 to 46 g/dl), indicating that red cell density, shape and membrane characteristics are play an important role in adhesion of sickle red cells during flow conditions.
Thus, the role of adhesion in clinical severity of sickle cell disease needs to be reexamined using a dynamic system designed to evaluate the effect of red cell deformability.
LEUKOCYTES
Sickle cell patients show an elevated leukocyte count in the peripheral circulation, and infections are often followed by the occurrence of vaso-occlusive episodes (6, 62). Pro-inflammatory state in this disease can be ascribed to episodes of reperfusion injury caused by vaso-occlusive events. Reperfusion injury in sickle cell disease is evidenced by endothelial damage/activation, detachment of endothelial cells, oxidant generation, elevated cytokines and tissue factor (34, 68, 69). Under these conditions, increased recruitment of leukocytes and their potential interaction with sickle red cells in the microcirculation can lead to sluggish flow, increased transit times, hypoxia and sickling. Also, leukocyte-sickle red cells interactions have been reported in transgenic sickle mice (79) and under in vitro flow conditions (23). Since the two-step model, described above under intravital studies, was first proposed, it has become quite apparent that not only sickle red cell adhesion, but also leukocyte recruitment would influence in vivo microcirculatory flow. Studies in transgenic sickle mice have indicated that intermittent vaso-occlusive events (ischemia/reperfusion) in sickle cell disease may induce an inflammatory endothelial phenotype resulting in enhanced leukocyte-endothelium interactions (46). Thus, it is reasonable to propose that trapping of dense or sickled red cells is likely to occur in a similar manner in the event of red cell and/or leukocyte adhesion to postcapillary endothelium (Figure 2C). In fact, studies by Frenette and co-workers (79) in transgenic-knockout mice (expressing exclusively HbS) indicate that adherent leukocytes might similarly facilitate mechanical trapping of elongated sickled red cells.
In normoxic conditions, transgenic sickle mice show endothelial expression of P-selectin and increased leukocyte rolling (46, 82), indicating endothelial activation. Greater leukocyte recruitment caused by hypoxia/reoxygenation in transgenic sickle mice is ameliorated by antibodies to P-selectin (46). Similarly, P- and E-selectin double knockout mice transplanted with bone marrow from transgenic-knockout (BERK) mice show absence of inflammatory response (79). Frenette and co-workers have used the BERK bone marrow transplanted C57BL mice to further focus on the role of leukocytes in vaso-occlusive events in the recipient mice wherein intravenous human gamma globulin (IVIG) administration was followed by an inflammatory stimulus (i.e., TNF-α) (78). Pretreatment with IVIG caused a marked reduction in leukocyte recruitment and associated red cell trapping in the recipient mice. In a follow-up study, these authors pre-treated the BERK marrow recipient C57BL mice with TNF-α followed by IVIG administration and reported a reversal of vaso-occlusion induced by TNF-α (12). Possibly, immunoglobulin may express multiple epitopes to various adhesion molecules. Detailed mechanistic studies are needed before using IVIG in sickle patients although this approach may have a therapeutic potential. The above studies also reinforce the notion that BERK mice (or C57BL recipients of BERK bone marrow) are a suitable model for investigating the role of inflammation and leukocyte recruitment in vaso-occlusive processes.
ADHESION AND ENDOTHELIAL INJURY/ACTIVATION
It is well recognized that postcapillary venules are the primary sites of blood cell-endothelium interactions and inflammation. In sickle cell disease, red cell rheological abnormalities, presence of dense red cells and adhesive interactions between sickle red cells and endothelium are potential source of endothelial injury. Endothelial injury in this disease may lead to the reported sloughing off of the injured endothelial cells into the peripheral circulation, and apoptosis of the affected endothelium (60, 68). In addition, generation of oxygen radicals by sickle red cells will contribute to endothelial activation. Sickle red cells generate excessive amounts of reactive oxygen species due to the presence of unstable HbS and spontaneous autooxidation of iron in heme (25, 30). Enhanced sickle red cell adhesion induces oxidant stress in cultured endothelium as evidenced by increased peroxidation, activation of the transcription factor nuclear factor-κB (NF-κB), and increased expression of intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin (76). In vivo reversible sickling, red cell membrane loss and endothelial injury caused by adhesion are likely to release substances (e.g., hemoglobin and adenosine diphosphate [ADP]) into blood circulation that would promote platelet activation and aggregation. NO inactivation by cell-free plasma heme (a consequence of hemolysis) is accompanied by platelet activation (80). Also, damaged endothelium itself is known to release ADP (38), which is a potent activator of platelets and leads to their aggregation. Thus, it is likely that red cell adhesion itself can activate endothelium and lead to the activation and involvement of platelets and leukocytes in vaso-occlusive processes.
ADHESION MOLECULES
Previous studies have implicated multiple adhesion molecules in sickle red cell adhesion to endothelium, but little information is forthcoming on the up-regulation of specific adhesive molecules that initiate sickle red cell adhesion in vivo. Contrary to leukocyte-endothelium interactions in which rolling, firm adhesion and emigration of leukocytes occur via fairly well-defined sequence of receptor-ligand interactions, sickle red cell adhesion apparently follows no such paradigm (41). Recent reviews by Hebbel et al. (34), and Stuart and Nagel (73) have provided excellent accounts of various adhesion mechanisms involved in sickle red cell adhesion. Here, we will focus on the most pertinent adhesion molecules that predominantly determine these interactions, while briefly reviewing various other proposed adhesion mechanisms.
The adhesion molecules implicated in sickle red cell adhesion are broadly categorized as 1) Red cell receptors, 2) Adhesive bridging proteins, and 3) Endothelial receptors (Figure 3). Sickle red cell adhesion molecules/receptors include CD36 and integrin α4β1 expressed on stress sickle reticulocytes, as well as exposed sulfated glycolipids in the red cell memberane (2, 34). α4β1 integrin and CD36 expressed on stress reticulocytes can mediate adhesion of these cells by interacting, respectively, with thrombospondin (TSP) and vascular cell adhesion molecule-1 (VCAM-1) (8, 75, 77). Also, a new category of adhesion molecules on sickle red cells has been described that require activation via signal transduction; these include basal cell adhesion molecule-1/Lutheran (B-CAM-1/Lu), integrin-associated protein (IAP) and intercellular adhesion molecule-4 (ICAM-4) (9, 36, 50, 83). Other described mechanisms include exposed phosphatidyl serine (PS) on sickle red cells (66), but the tenacity of PS-mediated adhesion has not been evaluated.
Figure 3.

Potential mechanisms in sickle red cell adhesion to endothelium. IAP = integrin-associated protein; ICAM-4/LW = intercellular adhesion molecule-4/Landsteiner-Weiner protein; PS = phosphatidyl serine.
Adhesive bridging proteins include TSP and von Willebrand factor (vWf) that are synthesized in endothelium (and platelets) and can exist in soluble state in the plasma, as well as expressed on the endothelial surface (56, 81). Endothelial P-selectin is also implicated to play a role in sickle red cell adhesion (58). The integrin αVβ3 is the most prominent endothelial receptor (51, 83) (Figure 3).
Plausibly, depending on the wall shear rates both low and high affinity adhesion mechanisms may participate. For example, under low wall shear rates, P-selectin expressed on activated endothelium may facilitate a weak adhesion via interaction with red cell sialyl Lewis moieties (57). Embury and coworkers have shown that antibodies to P-selectin can partially inhibit sickle red cell adhesion to human endothelial cells in a flow system (58). In addition, these authors showed inhibition of P-selectin mediated sickle red cell adhesion by unfractionated heparin. Notably, heparin and other anionic polysaccharides are also known to inhibit TSP-mediated adhesion of sickle cells in the ex vivo mesocecum preparation, as well as to human endothelial cells under flow conditions (2), suggesting that inhibitory effect of anionic polysaccharides is not limited to P-selectin-mediated adhesion. Embury et al. (18) have shown that an agonist peptide for murine protease-activated receptor-1 (PAR-1), which selectively activates mouse endothelial cells, but not platelets, results in flow stoppage of infused BERK sickle red cells in the microcirculation, but not in mice lacking P-selectin. However, the contribution of leukocytes was not confirmed, and it was not readily apparent whether the adhesion of sickle red cells was indirectly through leukocyte adhesion. Also, endothelial activation by PAR-1 could concomitantly release vWF as both P-selectin and vWf are stored in endothelial Weibel-Palade bodies. Future studies will be needed to clarify the relative roles of P-selectin and extra-large forms of endothelial vWF in sickle red cell adhesion.
P-selectin-mediated transient interaction may affect local wall shear rates and followed by more tenacious adhesion via high affinity adhesion mechanisms. Recent evidence shows that αVβ3 integrin, expressed on activated endothelium, is likely to play a predominant role in stable sickle red cell adhesion. αVβ3 integrin is a receptor to both vWF and TSP (Figure 3). Antibodies to αVβ3 inhibit sickle RBC adhesion-induced vaso-occlusion in inflamed venules suggesting that αVβ3 may be an endothelial cell integrin functioning in vaso-occlusion (51, 52).
Recent evidence shows that αVβ3 integrin interacts directly with activated red cell ICAM-4 (Landsteiner-Weiner blood group glycoprotein) (Figure 3) (83). We have shown that peptides based on αV-binding domains of ICAM-4 markedly decrease sickle cell adhesion and vaso-occlusion in the ex vivo mesocecum vasculature under shear flow conditions (50). In these studies, we tested the effect of synthetic peptides, V(16)PFWVRMS (FWV) and T(19)RWATSRI (ATSR), based on αV-binding domains of ICAM-4 and capable of inhibiting ICAM-4 and αV-binding in vitro (55). Ex vivo mesocecum vasculature was perfused with platelet-activating factor (PAF), which induces endothelial oxidant generation (49, 74) and causes endothelial activation (53), both of which characterize sickle cell disease. PAF is elevated twofold in sickle patients, and it enhances sickle cell adhesion in the ex vivo mesocecum preparation. Infusion of sickle red cells in PAF-treated vasculature resulted in pronounced adhesion of these cells: small diameter vessels were sites of maximal adhesion and frequent blockage (Figure 4). Both FWV and ATSR markedly decreased adhesion, and no vessel blockage was observed with either of the peptides, resulting in improved hemodynamics (Figure 4). In marked contrast, control peptide A(76)WSSLAHC (AWSS) had minimal effect (Figure 4 and 5). ATSR also inhibited adhesion in unactivated vasculature. Although infused fluoresceinated ATSR colocalized with vascular endothelium, pretreatment with function-blocking antibody (7E3) to αVβ3 markedly inhibited this interaction (Figure 6). These studies showed that ICAM-4 on sickle red cells binds endothelium via αVβ3 and that this interaction contributes to vaso-occlusion. Thus, peptides or small molecules based on αV-binding domains of ICAM-4 may have therapeutic potential.
Figure 4.

Videomicrographs showing inhibition of PAF-induced sickle erythrocytes adhesion in the ex vivo mesocecum microvasculature in the presence of ICAM-4 peptide ATSR. (A-D) Ex vivo preparation treated with PAF. (A) Clear vessel lumen (a = arteriole, v = venule) during perfusion with Ringer-albumin solution; (B and C) sickle erythrocytes bolus (arrows in B indicate flow direction) is followed by adhesion of these cells exclusively in the venule but not in the arteriole; (D) Maximal adhesion is observed in small-diameter postcapillaty venules (arrow-heads), frequently resulting in occlusion. (E-H) Ex vivo preparation treated with PAF and an ICAM-4 F strand control peptide AWSS; (E and F) Infusion of sickle erythrocytes bolus results in adhesion in the venules (v), but not in the arteriole (b); (G) adherent sickle erythrocytes in venules; (H) adhesion results in frequent blockage of small-diameter venules (arrow-head). (I-L) Ex vivo preparation treated with PAF and ICAM-4 peptide ATSR; (I) clear vessel lumens during perfusion with Ringer-albumin; (J and K) after a bolus infusion of sickle erythrocytes, rapid flow is observed in both arteriole and venules (arrows indicate flow direction), resulting in no adhesion; (L) scanning of the vasculature revealed little or no adhesion in venules. From Kaul et al. (50).
Figure 5.
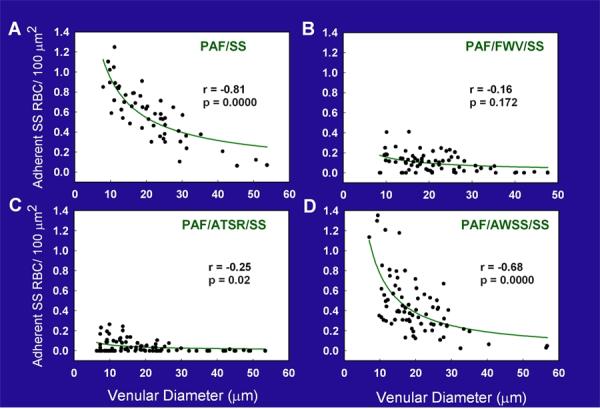
Regression plots for the number of adhered sickle erythrocytes (SS RBC)/100 μm2 relative to venular diameters in ex vivo preparations treated as follows: (A) PAF alone, (B) PAF and peptide FWV, (C) PAF and peptide ATSR and (D) PAF and control peptide AWSS. The regression lines represent the multiplicative equation of the form Y = aX-b for the best fit. In preparations treated with PAF alone, adhesion of sickle erythrocytes showed a strong correlation with the venular diameter. Preparations treated with peptide FWV or ATSR showed a marked inhibition of sickle erythrocyte adhesion in venules of all diameters, with ATSR having the maximal inhibitory effect, especially in small-diameter venules, the sites of frequent blockage. In contrast, in the presence of the control peptide AWSS, the resulting adhesion was essentially similar to that observed in PAF-treated preparations. Modified from Kaul et al. (50).
Figure 6.

Top panel: Colocalization of fluoresceinated ATSR peptide with vascular endothelium of the ex vivo preparation pretreated with PAF (A-C). (A) The presence of the fluorescent peptide is shown in green. (B) Blood vessels were identified by a polyclonal primary antibody to vWF and a secondary TRITC-conjugated antibody (red). (C) Merged image signals showed colocalization (yellow) of ATSR with the endothelial lining. No fluorescence staining was noted when the control peptide was infused (images not shown). Middle panel: The effect of a control antibody OC125 (D-F) on the colocalization of fluoresceinated ATSR peptide with vascular endothelium of the ex vivo preparation pretreated with PAF. (D) The presence of the fluorescent peptide is shown in green. (E) Blood vessels were identified by a polyclonal antibody to vWF as in B (red). (F) Merged image signals showed colocalization (yellow) of ATSR with the vessel wall. Bottom panel: The effect of 7E3 antibody to αVβ3 (G-I) on the colocalization of fluoresceinated ATSR peptide with vascular endothelium in PAF treated ex vivo preparation. (G) In the presence of 7E3 antibody, there was a marked decrease in ATSR localization with the vessel wall. ATSR infusion resulted in weak green staining likely attributable to autofluorescence or a low level of binding of ATSR peptide. (H) Vessel was identified by immunofluorescent staining for vWF (red). (I) No colocalization of ATSR with vessel wall in the presence of 7E3. From Kaul et al. (50).
ICAM-4 is unique in its expression on erythroid cells. ICAM-4 can bind diverse array of integrins including several αV integrins, β2 integrins (expressed on leukocytes), and α4β1 integrin, suggesting a multiple function of this adhesion molecule. Hence, future studies will be required to further explore the role of this molecule in sickle vaso-occlusion.
To further ascertain the role of αVβ3 integrin as a pivotal molecule in sickle cell adhesion, Finnegan et al. (22) tested the efficacy of two RGD-containing cyclic pentapetides, cRGDFV (EMD 66203) and cRGDF-ACHA (α-amino cyclohexyl carboxylic acid) (EMD 270179), based on their known ability to selectively bind αVβ3 (61;72). An inactive peptide cRβ-ADFV (EMD 135981) was used as control. Cyclization and the introduction of D-Phe (F) results in marked increase in the ability of cyclic peptides to selectively bind αVβ3 receptor. As shown in Figure 7, both EMD 66203 and EMD 270179 inhibited PAF-induced enhanced sickle red cell adhesion, and postcapillary blockage; the latter was evidenced by inhibition of adhesion in small-diameter venules. The inhibition of adhesion was accompanied by significantly improved hemodynamic behavior. Also, pretreatment of HUVEC with either αVβ3 antagonist resulted in a significant decrease in sickle red cell adhesion (Figure 8). Because of their metabolic stability (72), the use of these small-molecule cyclic pentapeptides may constitute an additional therapeutic approach to block sickle red cell adhesion and associated vaso-occlusion under flow conditions.
Figure 7.

Regression plots for the number of adherent SS RBC/100 :m2 relative to venular diameter in the ex vivo mesocecum treated as follows: (A) PAF alone, (B) PAF and peptide EMD 270179 (cRGDF-ACHA [α-amino cyclohexyl carboxylic acid]), (C) PAF and peptide EMD 66203 (cRGDFV), (D) PAF and control peptide EMD 135981 (cRβ-ADFV). The regression lines represent the multiplicative equation of the form Y = aX-b for the best fit. In preparations treated PAF alone, adhesion of SS RBCs showed a strong correlation with the venular diameter. Preparations treated with αVβ3 antagonist EMD 270179 or EMD 66203 showed a marked inhibition of SS RBC adhesion in venules of all diameters, with EMD 66203 having a greater inhibitory effect, especially in small-diameter venules, the site of frequent blockage. In contrast, in the presence of the control peptide EMD 135981, the resulting adhesion was essentially similar to that observed with PAF alone. Modified from Finnegan et al. (22).
Figure 8.
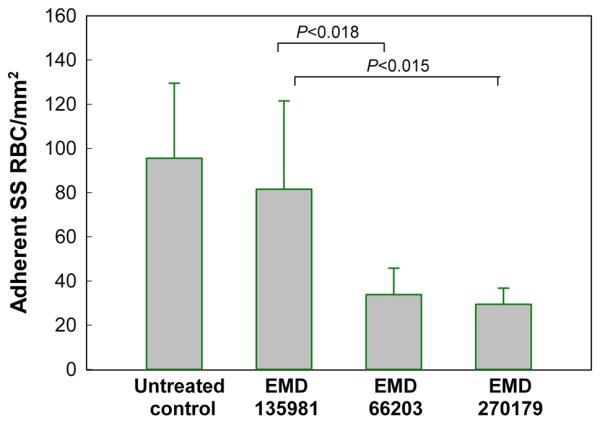
The effect of ∀V∃3 antagonists (EMD 66203 and EMD 270179) on SS RBC adhesion to HUVEC. The human endothelial monolyers were incubated with either peptide (100 :g/ml) for 30 min before the perfusion of the endothelialized flow chamber with SS RBC suspension (Hct 1%) at 1 dyne/cm2. The HUVEC treated with control peptide (n=7) showed no significant difference in adhesion of SS RBC/mm2 as compared to the baseline adhesion of these cells to untreated control HUVEC (n=6) (P>0.51). In contrast, both EMD 66203 (n=6) and EMD 270179 (n=4), caused marked inhibition of adhesion (i.e., 55% and 62%, respectively) of SS RBCs to HUVEC as compared with the control peptide group (P<0.018 and P<0.015, respectively). Modified from Finnegan et al. (22).
THE EFFECT OF HYDROXYUREA (HU) AND NITRIC OXIDE (NO)
Hydroxyurea (HU) is the only FDA approved drug for sickle patients to reduce the frequency of painful vaso-occlusive episodes. HU induces fetal hemoglobin (HbF) production in sickle cell disease. In the presence of HbF (α2γ2), polymer formation is efficiently prevented (20). HU may exert its therapeutic effect both by generation of NO (anti-inflammatory) and by increasing the level of antisickling fetal hemoglobin (HbF) (13, 16, 71). Setty et al. (65) have reported an inverse relationship between percent CD36 positive cells (reticulocytes) and percent F cells (HbF containing red cells) in sickle patients. These authors have shown that sickle patients with higher levels of F cells have a concomitant decrease in the number of reticulocytes expressing CD36 and α4β1 resulting in their reduced adherence. HU therapy also decreases sickle red cell adhesion and down-regulates endothelial adhesion molecules such as sVCAM-1 and sICAM-1 (7, 64). Recent studies of Schechter and co-workers (15, 16) have shown that NO donor properties of HU determine the induction of HbF by NO-dependent activation of soluble guanylate cyclase (cGMP). Notably, human sickle cell adhesion to TNF-α-activated human endothelial monolayers is markedly reduced by a NO donor DETA-NO (70). Future studies will be needed to differentiate the relative roles of HbF and NO in the therapeutic efficacy of HU.
FUTURE THERAPEUTIC CONSIDERATIONS
In addition to the above therapeutic approaches, it would be important to ascertain under in vivo conditions the predominant stimuli involved in the initiation of adhesion and vaso-occlusion. This would help design therapeutic strategies to inhibit sickle red cell adhesion and related vaso-occlusion. Because of anti-inflammatory properties of NO, NO supplementation may be of therapeutic value since NO is effectively inactivated by cell-free plasma heme and excessive oxidant generation in sickle cell disease (48, 63). The resulting NO deficiency may lead to inflammatory effects and up-regulation of endothelial adhesive molecules. Hence, therapies designed to reduce hemolysis and intravascular sickling (e.g., hydroxyurea and fetal hemoglobin elevation) may have beneficial effects. Finally, quenching endothelial oxidant generation results in amelioration of sickle red cell adhesion and vaso-occlusion in an ex vivo preparation (49). Thus, targeting the stimuli that cause endothelial activation, red cell adhesion and leukocyte recruitment will constitute novel therapeutic strategies to inhibit vaso-occlusion.
Acknowledgments
This work was supported by National Institutes of Health grants RO1HL070047, U54 070994 and HL071631.
REFERENCES
- 1.Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. Rheologic predictors of the severity of the painful sickle cell crisis. Blood. 1988;72:1216–1223. [PubMed] [Google Scholar]
- 2.Barabino GA, Liu XD, Ewenstein BM, Kaul DK. Anionic polysaccharides inhibit adhesion of sickle erythrocytes to the vascular endothelium and result in improved hemodynamic behavior. Blood. 1999;93:1422–1429. [PubMed] [Google Scholar]
- 3.Barabino GA, McIntire LV, Eskin SG, Sears DA, Udden M. Endothelial cell interactions with sickle cell, sickle trait, mechanically injured, and normal erythrocytes under controlled flow. Blood. 1987;70:152–157. [PubMed] [Google Scholar]
- 4.Barabino GA, McIntire LV, Eskin SG, Sears DA, Udden M. Rheological studies of erythrocyte-endothelial cell interactions in sickle cell disease. Progress in Clinical & Biological Research. 1987;240:113–127. [PubMed] [Google Scholar]
- 5.Billett HH, Kim K, Fabry ME, Nagel RL. The percentage of dense red cells does not predict incidence of sickle cell painful crisis. Blood. 1986;68:301–303. [PubMed] [Google Scholar]
- 6.Boggs DR, Hyde F, Srodes C. An unusual pattern of neutrophil kinetics in sickle cell anemia. Blood. 1973;41:59–65. [PubMed] [Google Scholar]
- 7.Bridges KR, Barabino GD, Brugnara C, Cho MR, Christoph GW, Dover G, Ewenstein BM, Golan DE, Guttmann CR, Hofrichter J, Mulkern RV, Zhang, Eaton WA. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88:4701–4710. [PubMed] [Google Scholar]
- 8.Brittain HA, Eckman JR, Swerlick RA, Howard RJ, Wick TM. Thrombospondin from activated platelets promotes sickle erythrocyte adherence to human microvascular endothelium under physiologic flow: a potential role for platelet activation in sickle cell vaso-occlusion. Blood. 1993;81:2137–2143. [PubMed] [Google Scholar]
- 9.Brittain JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV. Activation of sickle red blood cell adhesion via integrin-associated protein/CD47-induced signal transduction. J.Clin.Invest. 2001;107:1555–1562. doi: 10.1172/JCI10817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brugnara C, Bunn HF, Tosteson DC. Regulation of erythrocyte cation and water content in sickle cell anemia. Sceince. 1986;232:388–390. doi: 10.1126/science.3961486. [DOI] [PubMed] [Google Scholar]
- 11.Cao Z, Ferrone FA. A 50th order reaction predicted and observed for sickle hemoglobin nucleation. J.Mol.Biol. 1996;256:219–222. doi: 10.1006/jmbi.1996.0079. [DOI] [PubMed] [Google Scholar]
- 12.Chang J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood. 2008;111:915–923. doi: 10.1182/blood-2007-04-084061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charache S, Barton FB, Moore RD, Terrin ML, Steinberg MH, Dover GJ, Ballas SK, McMahon RP, Castro O, Orringer EP. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine. 1996;75:300–326. doi: 10.1097/00005792-199611000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Chen D, Kaul DK. Rheologic and hemodynamic characteristics of red cells of mouse, rat and human. Biorheology. 1994;31:103–113. doi: 10.3233/bir-1994-31109. [DOI] [PubMed] [Google Scholar]
- 15.Cokic VP, Andric SA, Stojilkovic SS, Noguchi CT, Schechter AN. Hydroxyurea nitrosylates and activates soluble guanylyl cyclase in human erythroid cells. Blood. 2008;111:1117–1123. doi: 10.1182/blood-2007-05-088732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cokic VP, Smith RD, Beleslin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, Schechter AN. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J.Clin.Invest. 2003;111:231–239. doi: 10.1172/JCI16672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eaton WA, Hofrichter J. Hemoglobin S gelation and sickle cell disease. Blood. 1987;70:1245–1266. [Review] [230 refs] [PubMed] [Google Scholar]
- 18.Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, Diwan BA, Mohandas N, Cheung AT. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood. 2004;104:3378–3385. doi: 10.1182/blood-2004-02-0713. [DOI] [PubMed] [Google Scholar]
- 19.Fabry ME, Nagel RL. Heterogeneity of red cells in the sickler: a characteristic with practical clinical and pathophysiological implications. Blood Cells. 1982;8:9–15. [PubMed] [Google Scholar]
- 20.Fabry ME, Suzuka SM, Weinberg RS, Lawrence C, Factor SM, Gilman JG, Costantini F, Nagel RL. Second generation knockout sickle mice: the effect of HbF. Blood. 2001;97:410–418. doi: 10.1182/blood.v97.2.410. [DOI] [PubMed] [Google Scholar]
- 21.Ferrone FA. Kinetic models and the pathophysiology of sickle cell disease. Ann.N.Y.Acad.Sci. 1989;565:63–74. doi: 10.1111/j.1749-6632.1989.tb24151.x. [DOI] [PubMed] [Google Scholar]
- 22.Finnegan EM, Barabino GA, Liu XD, Chang HY, Jonczyk A, Kaul DK. Small-Molecule Cyclic {alpha}V{beta}3 Antagonists Inhibit Sickle Red Cell Adhesion to Vascular Endothelium and Vaso-Occlusion. Am J Physiol Heart Circ Physiol. 2007;293:H1038–H1045. doi: 10.1152/ajpheart.01054.2006. [DOI] [PubMed] [Google Scholar]
- 23.Finnegan EM, Turhan A, Golan DE, Barabino GA. Adherent leukocytes capture sickle erythrocytes in an in vitro flow model of vaso-occlusion. Am.J.Hematol. 2007;82:266–275. doi: 10.1002/ajh.20819. [DOI] [PubMed] [Google Scholar]
- 24.Grabowski EF. Sickle erythrocytes adhere to endothelial cell monolayers (ECM’s) exposed to flowing blood. Progress in Clinical & Biological Research. 1987;240:167–179. [PubMed] [Google Scholar]
- 25.Hebbel RP. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood. 1991;77:214–237. [Review] [313 refs] [PubMed] [Google Scholar]
- 26.Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J.Clin.Invest. 1997;100:S83–S86. [Review] [17 refs] [PubMed] [Google Scholar]
- 27.Hebbel RP. Perspectives series: cell adhesion in vascular biology. Adhesive interactions of sickle erythrocytes with endothelium. J.Clin.Invest. 1997;99:2561–2564. doi: 10.1172/JCI119442. [Review] [17 refs] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hebbel RP. Blockade of adhesion of sickle cells to endothelium by monoclonal antibodies. N.Engl.J.Med. 2000;342:1910–1912. doi: 10.1056/NEJM200006223422512. [DOI] [PubMed] [Google Scholar]
- 29.Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N.Engl.J.Med. 1980;302:992–995. doi: 10.1056/NEJM198005013021803. [DOI] [PubMed] [Google Scholar]
- 30.Hebbel RP, Eaton JW, Balasingam M, Steinberg MH. Spontaneous oxygen radical generation by sickle erythrocytes. J.Clin.Invest. 1982;70:1253–1259. doi: 10.1172/JCI110724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions and the vasocclusive severity of sickle cell disease. Progress in Clinical & Biological Research. 1981;55:145–162. [PubMed] [Google Scholar]
- 32.Hebbel RP, Eaton JW, Steinberg MH, White JG. Erythrocyte/endothelial interactions in the pathogenesis of sickle-cell disease: a “real logical” assessment. Blood Cells. 1982;8:163–173. [PubMed] [Google Scholar]
- 33.Hebbel RP, Moldow CF, Steinberg MH. Modulation of erythrocyte-endothelial interactions and the vasocclusive severity of sickling disorders. Blood. 1981;58:947–952. [PubMed] [Google Scholar]
- 34.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- 35.Hebbel RP, Yamada O, Moldow CF, Jacob HS, White JG, Eaton JW. Abnormal adherence of sickle erythrocytes to cultured vascular endothelium: possible mechanism for microvascular occlusion in sickle cell disease. J.Clin.Invest. 1980;65:154–160. doi: 10.1172/JCI109646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hines PC, Zen Q, Burney SN, Shea DA, Ataga KI, Orringer EP, Telen MJ, Parise LV. Novel epinephrine and cyclic AMP-mediated activation of BCAM/Lu-dependent sickle (SS) RBC adhesion. Blood. 2003;101:3281–3287. doi: 10.1182/blood-2001-12-0289. [DOI] [PubMed] [Google Scholar]
- 37.Hofrichter J, Ross PD, Eaton WA. Supersaturation in sickle cell hemoglobin solutions. Proc.Natl.Acad.Sci.U.S.A. 1976;73:3035–3039. doi: 10.1073/pnas.73.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hollopeter G, Jantzen HM, Vincent D, Li G, England L, Ramakrishnan V, Yang RB, Nurden P, Nurden A, Julius D, Conley PB. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- 39.Hoover R, Rubin R, Wise G, Warren R. Adhesion of normal and sickle erythrocytes to endothelial monolayer cultures. Blood. 1979;54:872–876. [PubMed] [Google Scholar]
- 40.Joiner CH. Cation transport and volume regulation in sickle red blood cells. Am.J.Physiol. 1993;264:C251–C270. doi: 10.1152/ajpcell.1993.264.2.C251. [DOI] [PubMed] [Google Scholar]
- 41.Kaul DK, Fabry ME. In vivo studies of sickle red blood cells. Microcirculation. 2004;11:153–165. [PubMed] [Google Scholar]
- 42.Kaul DK, Fabry ME, Costantini F, Rubin EM, Nagel RL. In vivo demonstration of red cell-endothelial interaction, sickling and altered microvascular response to oxygen in the sickle transgenic mouse. J.Clin.Invest. 1995;96:2845–2853. doi: 10.1172/JCI118355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaul DK, Fabry ME, Nagel RL. Erythrocytic and vascular factors influencing the microcirculatory behavior of blood in sickle cell anemia. Ann.N.Y.Acad.Sci. 1989;565:316–326. doi: 10.1111/j.1749-6632.1989.tb24179.x. [DOI] [PubMed] [Google Scholar]
- 44.Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: pathophysiological implications. Proc.Natl.Acad.Sci.U.S.A. 1989;86:3356–3360. doi: 10.1073/pnas.86.9.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaul DK, Fabry ME, Windisch P, Baez S, Nagel RL. Erythrocytes in sickle cell anemia are heterogeneous in their rheological and hemodynamic characteristics. J.Clin.Invest. 1983;72:22–31. doi: 10.1172/JCI110960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J.Clin.Invest. 2000;106:411–420. doi: 10.1172/JCI9225. [see comments] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaul DK, Kollander R, Mahaseth H, Liu XD, Solovey A, Belcher J, Kelm RJ, Vercellotti GM, Hebbel RP. Robust vascular protective effect of hydroxamic Acid derivatives in a sickle mouse model of inflammation. Microcirculation. 2006;13:489–497. doi: 10.1080/10739680600778456. [DOI] [PubMed] [Google Scholar]
- 48.Kaul DK, Liu XD, Chang HY, Nagel RL, Fabry ME. Effect of fetal hemoglobin on microvascular regulation in sickle transgenic-knockout mice. J.Clin.Invest. 2004;114:1136–1145. doi: 10.1172/JCI21633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaul DK, Liu XD, Zhang X, Ma L, Hsia CJ, Nagel RL. Inhibition of sickle red cell adhesion and vasoocclusion in the microcirculation by antioxidants. Am J Physiol Heart Circ Physiol. 2006;291:H167–H175. doi: 10.1152/ajpheart.01096.2005. [DOI] [PubMed] [Google Scholar]
- 50.Kaul DK, Liu XD, Zhang X, Mankelow T, Parsons S, Spring F, An X, Mohandas N, Anstee D, Chasis JA. Peptides based on {alpha}V-binding domains of erythrocyte ICAM-4 inhibit sickle red cell-endothelial interactions and vaso-occlusion in the microcirculation. Am J Physiol Cell Physiol. 2006;291:C922–C930. doi: 10.1152/ajpcell.00639.2005. [DOI] [PubMed] [Google Scholar]
- 51.Kaul DK, Tsai HM, Liu XD, Nakada MT, Nagel RL, Coller BS. Monoclonal antibodies to alphaVbeta3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by platelet-activating factor. Blood. 2000;95:368–374. [see comments] [PubMed] [Google Scholar]
- 52.Kaul DK, Tsai HM, Nagel RL, Chen D. Platelet-activating factor enhances adhesion of sickle erythrocytes to vascular endothelium: the role of vascular integrin αvβ3 and von Willebrand factor. In: Beuzard Y, et al., editors. Sickle cell disease and thalassemias:New trends in therapy (INSERM Symposium) INSERM/John Libbey Eurotext; Montrouge, France: 1995. pp. 497–500. [Google Scholar]
- 53.Kurose I, Argenbright LW, Wolf R, Granger DN. Oxidative stress during platelet-activating factor-induced microvascular dysfunction. Microcirculation. 1996;3:401–410. doi: 10.3109/10739689609148313. [DOI] [PubMed] [Google Scholar]
- 54.La Celle PL. Alterations by leukocytes of erythrocyte flow in microchannels. Blood Cells. 1986;12:179–189. [PubMed] [Google Scholar]
- 55.Mankelow TJ, Spring FA, Parsons SF, Brady RL, Mohandas N, Chasis JA, Anstee DJ. Identification of critical amino-acid residues on the erythroid intercellular adhesion molecule-4 (ICAM-4) mediating adhesion to {alpha}V integrins. Blood. 2003;103:1503–1508. doi: 10.1182/blood-2003-08-2792. [DOI] [PubMed] [Google Scholar]
- 56.Manodori AB, Matsui NM, Chen JY, Embury SH. Enhanced adherence of sickle erythrocytes to thrombin-treated endothelial cells involves interendothelial cell gap formation. Blood. 1998;92:3445–3454. [PubMed] [Google Scholar]
- 57.Matsui NM, Borsig L, Rosen SD, Yaghmai M, Varki A, Embury SH. P-selectin mediates the adhesion of sickle erythrocytes to the endothelium. Blood. 2001;98:1955–1962. doi: 10.1182/blood.v98.6.1955. [DOI] [PubMed] [Google Scholar]
- 58.Matsui NM, Varki A, Embury SH. Heparin inhibits the flow adhesion of sickle red blood cells to P-selectin. Blood. 2002;100:3790–3796. doi: 10.1182/blood-2002-02-0626. [DOI] [PubMed] [Google Scholar]
- 59.Mohandas N, Evans E. Sickle erythrocyte adherence to vascular endothelium. Morphologic correlates and the requirement for divalent cations and collagen-binding plasma proteins. J.Clin.Invest. 1985;76:1605–1612. doi: 10.1172/JCI112144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ortiz A. Circulating endothelial cells in sickle cell anemia. N.Engl.J.Med. 1998;338:1162–1163. [PubMed] [Google Scholar]
- 61.Pfaff M, Tangemann K, Muller B, Gurrath M, Muller G, Kessler H, Timpl R, Engel J. Selective recognition of cyclic RGD peptides of NMR defined conformation by alpha IIb beta 3, alpha V beta 3, and alpha 5 beta 1 integrins. J Biol.Chem. 1994;269:20233–20238. [PubMed] [Google Scholar]
- 62.Platt OS. Sickle cell anemia as an inflammatory disease. J.Clin.Invest. 2000;106:337–338. doi: 10.1172/JCI10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, III, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat.Med. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- 64.Saleh AW, Duits AJ, Gerbers A, de Vries C, Hillen HF. Cytokines and soluble adhesion molecules in sickle cell anemia patients during hydroxyurea therapy. Acta Haematologica. 1998;100:26–31. doi: 10.1159/000040859. [DOI] [PubMed] [Google Scholar]
- 65.Setty BN, Kulkarni S, Dampier CD, Stuart MJ. Fetal hemoglobin in sickle cell anemia: relationship to erythrocyte adhesion markers and adhesion. Blood. 2001;97:2568–2573. doi: 10.1182/blood.v97.9.2568. [DOI] [PubMed] [Google Scholar]
- 66.Setty BN, Kulkarni S, Stuart MJ. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood. 2002;99:1564–1571. doi: 10.1182/blood.v99.5.1564. [DOI] [PubMed] [Google Scholar]
- 67.Smith BD, La Celle PL. Erythrocyte-endothelial cell adherence in sickle cell disorders. Blood. 1986;68:1050–1054. [PubMed] [Google Scholar]
- 68.Solovey A, Lin Y, Browne P, Choong S, Wayner E, Hebbel RP. Circulating activated endothelial cells in sickle cell anemia. N. Engl. J. Med. 1997;337:1584–1590. doi: 10.1056/NEJM199711273372203. [see comments] [DOI] [PubMed] [Google Scholar]
- 69.Sowemimo-Coker SO, Meiselman HJ, Francis RB., Jr. Increased circulating endothelial cells in sickle cell crisis. Am. J. Hematol. 1989;31:263–265. doi: 10.1002/ajh.2830310409. [DOI] [PubMed] [Google Scholar]
- 70.Space SL, Lane PA, Pickett CK, Weil JV. Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am J Hematol. 2000;63:200–204. doi: 10.1002/(sici)1096-8652(200004)63:4<200::aid-ajh7>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 71.Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood. 1997;89:1078–1088. [PubMed] [Google Scholar]
- 72.Storgard CM, Stupack DG, Jonczyk A, Goodman SL, Fox RI, Cheresh DA. Decreased angiogenesis and arthritic disease in rabbits treated with an alphavbeta3 antagonist. J.Clin.Invest. 1999;103:47–54. doi: 10.1172/JCI3756. [see comments] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–1360. doi: 10.1016/S0140-6736(04)17192-4. [DOI] [PubMed] [Google Scholar]
- 74.Suematsu M, Schmid-Schonbein GW, Chavez-Chavez RH, Yee TT, Tamatani T, Miyasaka M, DeLano FA, Zweifach BW. In vivo visualization of oxidative changes in microvessels during neutrophil activation. Am.J.Physiol. 1993;264:H881–H891. doi: 10.1152/ajpheart.1993.264.3.H881. [DOI] [PubMed] [Google Scholar]
- 75.Sugihara K, Sugihara T, Mohandas N, Hebbel RP. Thrombospondin mediates adherence of CD36+ sickle reticulocytes to endothelial cells. Blood. 1992;80:2634–2642. [PubMed] [Google Scholar]
- 76.Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood. 1998;92:3924–3935. [PubMed] [Google Scholar]
- 77.Swerlick RA, Eckman JR, Kumar A, Jeitler M, Wick TM. Alpha 4 beta 1-integrin expression on sickle reticulocytes: vascular cell adhesion molecule-1-dependent binding to endothelium. Blood. 1993;82:1891–1899. [PubMed] [Google Scholar]
- 78.Turhan A, Jenab P, Bruhns P, Ravetch JV, Coller BS, Frenette PS. Intravenous immune globulin prevents venular vaso-occlusion in sickle cell mice by inhibiting leukocyte adhesion and the interactions between sickle erythrocytes and adherent leukocytes. Blood. 2004;103:2397–2400. doi: 10.1182/blood-2003-07-2209. [DOI] [PubMed] [Google Scholar]
- 79.Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc.Natl.Acad.Sci.U.S.A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Villagra J, Shiva S, Hunter LA, Machado RF, Gladwin MT, Kato GJ. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension and nitric oxide scavenging by cell-free hemoglobin. Blood. 2007;110:2166–2172. doi: 10.1182/blood-2006-12-061697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wick TM, Moake JL, Udden MM, Eskin SG, Sears DA, McIntire LV. Unusually large von Willebrand factor multimers increase adhesion of sickle erythrocytes to human endothelial cells under controlled flow. J.Clin.Invest. 1987;80:905–910. doi: 10.1172/JCI113151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am.J.Physiol Heart Circ.Physiol. 2004;286:H1608–H1614. doi: 10.1152/ajpheart.01056.2003. [DOI] [PubMed] [Google Scholar]
- 83.Zennadi R, Hines PC, De Castro LM, Cartron JP, Parise LV, Telen MJ. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood. 2004;104:3774–3781. doi: 10.1182/blood-2004-01-0042. [DOI] [PubMed] [Google Scholar]
- 84.Zennadi R, Moeller BJ, Whalen EJ, Batchvarova M, Xu K, Shan S, Delahunty M, Dewhirst MW, Telen MJ. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood. 2007;110:2708–2717. doi: 10.1182/blood-2006-11-056101. [DOI] [PMC free article] [PubMed] [Google Scholar]