

Abstract
Overexpression of anti-apoptotic Bcl-2 family proteins is commonly related with tumor maintenance, progression, and chemoresistance. Inhibition of these anti-apoptotic proteins is an attractive approach for cancer therapy. Guided by nuclear magnetic resonance (NMR) binding assays, a series of 5, 5′ substituted compound 6a (Apogossypolone) derivatives was synthesized and identified pan-active antagonists of anti-apoptotic Bcl-2 family proteins, with binding potency in the low micromolar to nanomolar range. Compound 6f inhibits the binding of BH3 peptides to Bcl-XL, Bcl-2 and Mcl-1 with IC50 values of 3.10, 3.12 and 2.05 μM, respectively. In a cellular assay, 6f potently inhibits cell growth in several human cancer cell lines in a dose-dependent manner. Compound 6f further displays in vivo efficacy in transgenic mice and demonstrated superior single-agent antitumor efficacy in a PPC-1 mouse xenograft model. Together with its negligible toxicity, compound 6f represents a promising drug lead for the development of novel apoptosis-based therapies for cancer.
Introduction
For the maintenance of normal tissue homeostasis, ensuring a proper balance of cell production and cell loss, cells undergo a process known as apoptosis or programmed cell-death.1, 2 Defective apoptosis contributes to tumorgenesis and chemoresistance.3, 4 Central regulators of this process are the Bcl-2 (B-cell lymphoma/leukemia-2) family proteins.5–7 To date, six anti-apoptotic members of the Bcl-2 family have been identified and characterized in human, including Bcl-2, Bcl-XL, Mcl-1, Bfl-1, Bcl-W and Bcl-B. Both X-ray crystallography and NMR spectroscopy structural characterizations of several of these proteins have identified a heterodimerization interface comprised of an hydrophobic crevice on the surface of anti-apoptotic Bcl-2 family proteins and the BH3 dimerization domain of pro-apoptotic family members.8 Hence, small molecues molecules that bind in the hydrophobic crevide of anto-apoptotic Bcl-2 protein would mimic the BH3 domain of pro-apoptotic proteins, presumably inducing apoptosis and/or abrogating the ability of anti-apoptotic Bcl-2 proteins to inhibit cancer cell death.8–11
We and others have reported that the natural product 1 (Gossypol) (Figure 1A) is a potent inhibitor of Bcl-2, Bcl-XL and Mcl-1, functioning as a BH3 mimic.12–16 The (−) atropisomer of compound 1 (AT101, Ascenta Pharmaceuticals) is currently in phase II clinical trials, displaying single-agent antitumor activity in patients with advanced malignancies.15–17 Given that compound 1 may target other proteins due to two reactive aldehyde groups, we designed compound 2a (Apogossypol) (Figure 1A), a compound that lacks these aldehydes, but retains activity against anti-apoptotic Bcl-2 family proteins in vitro.18 Recently, we further compared the efficacy and toxicity in mice of compounds 1 and 2a and our preclinical in vivo data show that compound 2a has superior efficacy and markedly reduced toxicity compared to 1.19 Moreover, we evaluated of the single-dose pharmacokinetic characteristics of 2a in mice and compound 2a displayed superior blood concentrations over time compared to compound 1, due to slower clearance.20 Recently, we reported the separation and characterization of atropoisomers of 2a 21 and these studies revealed that the racemic 2a is as effective as its individual isomers.21 We further reported the synthesis and evaluation of 5, 5′ alkyl, ketone and amide substituted 2a derivatives, with the best compounds 3a (BI79D10) 22 and 4a (8r) 23 displaying improved in vitro and in vivo efficacy compared to 2a (Figure 1A), and of the optically pure compound 9 (BI97C1) (Supplementary Figure 1A),24 with marked efficacy in vivo.24
Figure 1.

(A) Structure of compounds 1, 2a, 3a and 4a. (B) Structure of compounds 5, 6a, 7 and 8a. Molecular docking studies. Docked structures of (C) compound 6a and (D) compound 6f into Bcl-2 (PDB ID:1YSW).
Compound 5 (Gossypolone), a major metabolite of compound 1 formed by oxidation, displayed similar cytotoxic effects as compound 1 on several cancer cell lines (Figure 1B) and has been recently proposed for treatment of cancer.25, 26 Compound 6a (Apogossypolone) a derivative of 5, has as well been reported as a potent inhibitor of Mcl-1 and Bcl-2 proteins.27–32 Compound 6a blocks binding of Bim and Bcl-2 and induced apoptosis in a number of human cancer cell lines (Figure 1B).27–32 Compound 6a also induced regression in several tumor xenograft models and its maximum tolerated dose (MTD) when administered orally is above 240 mg/kg while the MTD of (−) 1 is 50 mg/kg.27, 31–32 It is therefore attractive to further explore whether 6a derivatives displayed similar or improved biological activities compared to 6a. In fact, we have previously demonstrated that certain 5,5′ substituted 2a derivatives (3a and 4a) displayed improved in vitro and in vivo activities compared to 2a.22, 23 Therefore, we envision that 5,5′ substitution of 6a could result in compounds with improved biological activities. Hence, we report on the synthesis and biological evaluation of novel 5, 5′ substituted 6a derivatives (6–8) which replace the isopropyl groups of 6a with various alkyl (6), ketone (7) and amide (8) groups at 5, 5′ positions (Figure 1B).
Results and Discussion
Compound 6a has recently been reported as an inhibitor of Bcl-XL, Bcl-2 and Mcl-1.26–28 Molecular docking studies of compound 6a into the BH3 binding groove in Bcl-2 33,34(Figure 1C) suggest that 6a forms two hydrogen bonds with residues Arg 143 and Tyr 199 in Bcl-2 through the 1′ oxygen and 6′ hydroxyl groups, respectively. The isopropyl group on the left naphthalene ring inserts into the first hydrophobic pocket (P1) in Bcl-2 (Figure 1C), while the isopropyl group on the right naphthalene ring inserts into the hydrophobic pocket (P2) (Figure 1C). Analysis of the predicted binding models indicates that while the overall core structure of compound 6a fits very well into BH3 binding groove of Bcl-2, the two isopropyl groups do not fully occupy the hydrophobic pockets P1 and P2. Therefore, a library of 5, 5′ substituted 6a derivatives (Figure 1B) that replace the isopropyl groups with suitable substituents was designed with the aim of deriving novel molecules that could occupy the hydrophobic pockets on Bcl-2 and other members of this protein family more efficiently.
Synthetic routes (Scheme 1–2) were developed to install a variety of groups at 5, 5′ position and synthesis of 6a and its 5, 5′ amide substituted derivatives are outlined in Scheme 1. Compound 1 was treated with NaOH solution at 90 °C to provide compound 2a, which was readily methylated in the presence of potassium carbonate to afford compound 9 (Scheme 1). Compound 9 was oxidized to compound 10 using periodic acid.35 Subsequent demethylation of the compound 10 using boron tribromide afforded compound 6a. Reaction of compound 9 with TiCl4 followed by dichloromethyl methyl ether resulted in loss of isopropyl groups and simultaneous bisformylation to give aldehyde compound 11.36 The aldehyde groups of compound 11 were oxidized and coupled with a variety of commercially available amines in the presence of 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide (EDCI) to afford amide compound 12. Subsequent demethylation of compound 12 afford compound 4.37, 38 Several oxidation reagents such as [bis(trifluoroacetoxy)iodo]benzene,39 potassium nitrosodisulfonate40 and ferric chloride41 were used to converted phenol 4 to quinone 8 and the ferric chloride is the most efficient oxidation reagent for this conversion in our hand (Scheme 1).
Scheme 1. Reagents and conditions.

(a) NaOH, H2O, reflux; (b) H2SO4; (c) DMS, K2CO3; (d) H5IO6, 95°C; (e) BBr3, CH2Cl2; (f) HCl, H2O; (g) TiCl4, rt; (h) Cl2CHOCH3, rt; (i) HCl, H2O; (j) NaClO2, H2O2, KH2PO4, CH3CN, rt; (k) EDCI, NH2R, HOBT, rt; (l) FeCl3, CH3COOH, 60°C; (m) 20% H2SO4.
Scheme 2. Reagents and conditions.

(a) RMgBr or RLi, rt; (b) NH4Cl, H2O; (c) Pyridinium chlorochromate, CH2Cl2, rt; (d) BBr3; (e) HCl, H2O; (f) Et3SiH, TFA; (g) Pd/C, H2, CH3COOH; (h) FeCl3, CH3COOH, 60°C; (i) 20% H2SO4
The synthesis of 5, 5′ alkyl substituted 6a derivatives were outlined in Scheme 2. Compound 11 was treated with different Grignard or lithium reagents to afford a secondary alcohol 13, which was oxidized to give the phenone 14 by pyridinium chlorochromate. Alcohol 13 and phenone 14 were readily reduced using triethylsilane to afford alkyl compound 15.42 Compound 15 was then demethylated using boron tribromide to afford compound 2 (Scheme 2). Oxidation of compound 2 using ferric chloride gave 6 as 5,5′ alkyl substituted 6a derivatives. The reduction of ketone 3 using H2 in the presence of Pd/C also affored compound 2.43 Demethylation of compound 1422 followed by ferric chloride oxidation afforded compound 7 as a 5,5′ ketone substituted 6a derivative (Scheme 2).
The synthesized 5, 5′ substituted 6a derivatives were first screened by one-dimensional 1H nuclear magnetic resonance spectroscopy (1D 1H NMR) binding assays against Bcl-XL, as we reported previously (Table 1 and supplementary Figure 1B).44 A group of compounds (6a, 6b, 6f, 6i, 6l, 6m, 7, 8a–c) induced chemical shift changes in active site methyl groups (region between −0.38 and 0.42 ppm) in the one-dimensional 1H-NMR spectra of Bcl-XL (Table 1). To confirm the result from the 1D 1H NMR binding assay, we also produced uniformly 15N-labeled Bcl-XL protein and measured 2D [15N, 1H]-HSQC correlation spectra in absence and presence of selected compounds. Consistent with 1D 1H NMR binding assays, compounds 6f, 6i and 8a displayed strong binding to Bcl-XL, as qualitatively evaluated by the nature of the shifts at the ligand/protein ratio of 1:1 (Supplementary Figure 1C–D). To confirm the results of the NMR binding data, we further evaluated the binding affinity of selected compounds for Bcl-XL using FP assay (Table 2). In agreement with NMR binding, compound 6f displayed potent binding affinity to Bcl-XL with an IC50 value of 3.1 μM in FP assay (Table 2 and Figure 2A). A group of compounds (6a, 6b, 6i, 6l, 6m, 7, 8a, 8c) were 5–19 times more potent than 6f, with IC50 values ranging from 0.16 to 0.63 μM in same assay (Table 2 and Supplementary Figure 2A–B).
Table 1.
Evaluation of 5, 5′ substituted 6a derivatives using a combination of 1D 1H-NMR binding assays and cell viability assays.
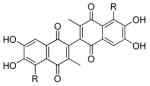 |
EC50 (μM) | ||||||
|---|---|---|---|---|---|---|---|
| Compound | R = | 1D-1H NMR a* | PC3 b* (μM) | H460 b* (μM) | H1299 b* (μM) | BP3 c* (μM) | RS4;11 c* (μM) |
| 6a | 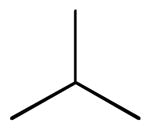 |
+++ | 1.46 ± 0.33 | 0.40 ± 0.07 | 2.76 ± 0.72 | 11.73 ± 2.15 | 7.47 ± 2.78 |
| 8a | 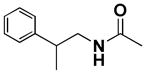 |
+++ | 7.6 ± 1.41 | 5.75 ± 1.34 | >30 | 12.9 ± 1.91 | >30 |
| 8b | 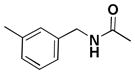 |
+++ | 5.44 ± 0.34 | 6.16 ± 1.48 | >30 | 21.64 ± 0.85 | >30 |
| 8c | 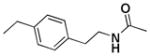 |
+++ | 7.70 ± 0.64 | 4.40 ± 0.70 | 9.10 ± 5.20 | 22.23 ± 1.43 | >30 |
| 7 | 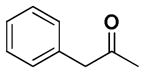 |
+++ | 5.44 ± 0.24 | 7.38 ± 1.07 | >30 | 9.58 ± 4.16 | >30 |
| 6b | 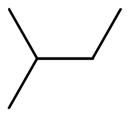 |
+++ | 8.50 ± 2.89 | 0.73 ± 0.29 | 10.64 ± 5.02 | 10.93 ± 0.43 | 11.72 ± 0.68 |
| 6c | 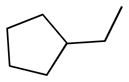 |
+ | 5.50 ± 0.74 | 0.70 ± 0.34 | 2.32 ± 0.51 | 12.84 ± 1.16 | 9.83 ± 0.26 |
| 6d | 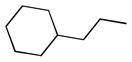 |
+ | 2.40 ± 0.24 | 2.20 ± 0.76 | 1.32 ± 0.30 | 15.61 ± 0.10 | 15.43 ± 1.86 |
| 6e | 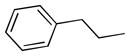 |
++ | 1.40 ± 0.14 | 1.04 ± 0.13 | 0.85 ± 0.36 | 8.72 ± 2.53 | 4.75 ± 0.01 |
| 6f | 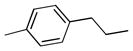 |
++ | 1.10 ± 0.08 | 0.59 ± 0.06 | 1.56 ± 0.17 | 4.18 ± 0.50 | 3.08 ± 0.59 |
| 6g | 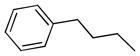 |
++ | 1.30 ± 0.18 | 0.92 ± 0.09 | 1.53 ± 0.41 | 6.0 ± 0.10 | 3.83 ± 0.70 |
| 6h | 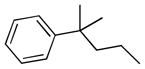 |
++ | 2.00 ± 0.17 | 1.62 ± 0.16 | 2.16 ± 0.27 | 30.0 ± 2.40 | 15.0 ± 0.24 |
| 6i | 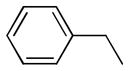 |
+++ | 0.59 ± 0.22 | 0.13 ± 0.08 | 0.31 ± 0.19 | 10.1 ± 0.04 | 6.9 ± 1.76 |
| 6j | + | 8.99 ± 2.49 | 1.44 ± 0.20 | 2.38 ± 0.21 | 16.12 ± 0.07 | 7.32 ± 0.90 | |
| 6k | 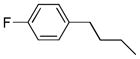 |
+ | 2.62 ± 0.31 | 1.48 ± 0.02 | 1.91 ± 0.17 | 14.61 ± 0.06 | 11.25 ± 0.54 |
| 6l | 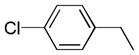 |
+++ | 0.21 ± 0.05 | 0.19 ± 0.04 | 2.99 ± 1.21 | 0.48 ± 0.01 | 1.13 ± 0.60 |
| 6m | +++ | 2.97 ± 0.98 | 0.98 ± 0.15 | 10.30 ± 3.84 | 0.81 ± 0.06 | 1.74 ± 0.11 | |
4-point-rating scale: +++: Very Active; ++: Active; +: Mild; −: Weak
Compounds against cell line using ATP-LITE assay
Compounds against cell line using Annexin V-FITC and propidium iodide assay
Table 2.
Cross-activity of selected 5, 5′ substituted 6a derivatives against Bcl-XL, Bcl-2, Mcl-1 and Bfl-1.
| Compound | IC50 (μM) FPA | Kd (μM) ITC | |||
|---|---|---|---|---|---|
| Bcl-XL | Bcl-2 | Bfl-1 | Mcl-1 | Bcl-XL | |
| 6a | 0.63 ± 0.02 | 0.37 ± 0.02 | 2.17 ± 0.35 | 0.54 ± 0.03 | 2.80 ± 0.60 |
| 6b | 0.55 ± 0.06 | 0.25 ± 0.02 | 1.41 ± 0.11 | 0.47 ± 0.03 | 1.50 ± 0.80 |
| 6f | 3.10 ± 0.28 | 3.12 ± 0.15 | 14.7 ± 6.63 | 2.05 ± 0.15 | 2.50 ± 2.20 |
| 6i | 0.34 ± 0.03 | 0.29 ± 0.01 | 0.65 ± 0.05 | 0.24 ± 0.02 | 0.45 ± 0.26 |
| 6c | 2.99 ± 0.16 | 2.27 ± 0.15 | ND a* | 3.08 ± 0.17 | ND |
| 6d | 12.65 ± 4.34 | 6.73 ± 2.24 | ND | 5.90 ± 0.54 | ND |
| 6e | 1.79 ± 0.14 | 2.57 ± 0.12 | 9.72 ± 1.38 | 1.29 ± 0.05 | ND |
| 6g | 1.44 ± 0.06 | 2.17 ± 0.14 | 5.27 ± 0.76 | 0.67 ± 0.03 | ND |
| 6l | 0.15 ± 0.06 | 0.34 ± 0.06 | 0.70 ± 0.07 | 0.40 ± 0.05 | ND |
| 7 | 0.34 ± 0.02 | 0.22 ± 0.02 | 0.69 ± 0.03 | 0.35 ± 0.02 | ND |
| 8a | 0.32 ± 0.01 | 0.23 ± 0.01 | 0.71 ± 0.06 | 0.47 ± 0.03 | ND |
| 8c | 0.24 ± 0.02 | 0.21 ± 0.01 | 1.25 ± 0.09 | 0.32 ± 0.02 | ND |
NDa* = Not determined
Figure 2.

(A) Fluorescence polarization-based competitive curves of 6f using Bcl-XL (red square), Bcl-2 (blue dot) and Mcl-1 (green down triangle). (B) Inhibition of cell growth by compounds 1 (dark dot), 6a (blue square), 6i (red down triangle), 8a (purple diamond) and 6f (green up triangle) in the PC-3 human prostate cancer cell line. Cells were treated for 3 days and cell viability was evaluated using ATP-LITE assay. (C) Inhibition of cell growth by compounds 6a (red dot), 6b (green square), 6i (blue up triangle) and 6f (purple down triangle) in the H460 human lung cancer cell line. Cells were treated for 3 days and cell viability was evaluated using ATP-LITE assay. (D) Inhibition of cell growth by compounds 6a (dark square), 6f (red diamond) and 6i (green cycle) in the human primary CLL cells. Cells were treated for 1 days and cell viability was evaluated using Annexin-V apoptosis assay.
To further confirm results of the NMR binding data and the FP assays, we also evaluated the binding affinity of selected compounds (6a, 6b, 6f, 6i) for Bcl-XL using isothermal titration calorimetry (ITC) (Table 2). In agreement with NMR binding and FPA data, compound 6f displayed high binding affinity to Bcl-XL with a Kd value of 2.5 μM by ITC and compound 6i showed increased binding affinity with a Kd value of 0.45 μM in same assay. Compound 6a showed similar binding affinity with 6f with a Kd value of 2.80 μM by ITC. Consistent with the NMR binding, FPA and ITC data, compounds 6a, 6d–6g, 6i and 6l displayed potent efficacy in inhibiting cell growth in a 3 day ATP-Lite assay in the PC3 cell line, which expresses high levels of Bcl-XL (Table 1 and Figure 2B). The average EC50 value of 6i and 6l is 0.40 μM, hence 3.5-fold more potent than 6a (EC50 = 1.5 μM) (Figure 2B). Compound 6f (EC50 = 1.1 μM) displayed similar efficacy in inhibiting PC3 cell growth as the potent compound 6a in same assay (Table 1). However, although compounds 8a and 8c displayed strong binding affinity to Bcl-XL in the NMR binding assay and FP assay, they showed relative weaker efficacy in inhibiting growth of PC3 cells with EC50 values around 7.6 μM (Table 1). The discrepancy is likely due to high hydrophilicity and molecular weight of 8a and 8c which may result in low cell permeability. Cell permeability of selected compounds was therefore evaluated using the parallel artificial membrane permeability assay (PAMPA) (Table 3). As anticipated, compounds 8a has a lower LogPe value of −7.9 indicating poor cell membrane permeability while the LogPe value of 6f is −5.6 indicating excellent cell membrane permeability. Compared to 6f, compound 6a also has relatively lower cell membrane permeability (LogPe = −5.9).
Table 3.
Plasma stability, microsomal stability and membrane permeability of selected 5, 5′ substituted 6a derivatives.
| Compounds | Plasma Stability (T = 40 mins) | Microsomal Stability (T = 40 mins) | Membrane Permeability (PAMPA, LogPe) |
|---|---|---|---|
| 6a | 77% | 47% | −5.94 ± 0.09 |
| 8a | 91% | 71% | −7.88 ± 0.08 |
| 6b | 98% | 69% | −6.17 ± 0.02 |
| 6d | 74% | 98% | −5.82 ± 0.20 |
| 6e | 81% | 89% | −6.58 ± 0.05 |
| 6f | 86% | 63% | −5.59 ± 0.08 |
| 6g | 87% | 88% | −6.65 ± 0.05 |
| 6i | 95% | 91% | −7.11 ± 0.04 |
In addition to Bcl-XL, other members of the Bcl-2 family are known to play critical roles in tumor survival.45, 46 Therefore, we further evaluated the binding properties and specificity of selected 5, 5′ substituted 6a derivatives against Bcl-2, Mcl-1 and Bfl-1 using FP assays (Table 2 and Supplementary Figure 2B). Compound 6f displayed potent affinity against Bcl-2 (IC50 = 3.12 μM), Mcl-1 (IC50 = 2.05 μM) and relative lower affinity against Bfl-1 (IC50 = 14.0 μM) in FP assays (Table 2). Compound 6f was further evaluated against H460 and H1299 cancer cell lines, which express high levels of Bcl-2 and Mcl-1, respectively (Table 1).46–48 Consistent with FPA data, compound 6f displayed potent efficacy in inhibiting cell growth in H460 and H1299 cell lines in a 3 day ATP-Lite assay, with EC50 values of 0.59 μM and 1.5 μM, respectively, which is comparable with 6a (Table 1 and Figure 2C). Molecular docking studies of compound 6f demonstrated that 1-methyl-4-propylbenzene groups at 5, 5′ positions inserted deeper into hydrophobic pockets (P1 and P2) in Bcl-2 (Figure 1D). Based on the docking models, compound 6f also forms two hydrogen bonds with residues Arg 143 and Tyr 199 in Bcl-2 through the 1′ oxygen and 6′ hydroxyl groups, respectively. In addition, the 7′ hydroxyl group on the right naphthalene ring also formed an additional hydrogen bond with residue Tyr141 (Figure 1D). Other 5, 5′ substituted 6a derivatives, such as 6b, 6i and 6l also displayed strong pan-active inhibitory properties against Bcl-2, Mcl-1 and Bfl-1. The most potent compound 6i displaces BH3 binding to Bcl-2, Mcl-1 and Bfl-1 with IC50 values of 0.29, 0.24 and 0.65 μM (Table 2 and Supplementary Figure 2B), respectively, in FP assays. In agreement with these FPA results, the compound 6i showed potent cell growth inhibitory activity against the H460 and H1299 cell lines in a 3 day ATP-Lite assay, with IC50 values of 0.13 and 0.31 μM, respectively (Table 1 and Figure 2C). The H460 cell line has been studied by several groups with respect to sensitivity to Bcl-2 antagonists.47–49 However, although compounds 7, 8a and 8c display potent binding affinity to Bcl-2 and Mcl-1 with average IC50 values of 0.22 μM and 0.38 μM, respectively, in FP assays, they showed relative weak efficacy in inhibiting growth of H460 and H1299 cells with average EC50 values of around 5.8 μM and 17 μM, respectively (Table 1). This discrepancy is partially due to high hydrophilicity and molecular weight of 5,5′ substituted ketone and amide 6a derivatives, resulting in reduced cell permeability.
We evaluated the ability of 5, 5′ substituted 6a derivatives to induce apoptosis of the human leukemia RS4;11 cell line (which expresses high levels of Bcl-2 and Bcl-XL) and human lymphoma BP3 cell line (which express high levels of Bfl-1 and Mcl-1) in one day Annexin-V apoptosis assay. For this assay, we used Annexin V-FITC and propidium iodide (PI) double staining, followed by flow-cytometry analysis (Table 1). The pan-Bcl-2 family inhibitor 6f effectively induced apoptosis of the RS4;11 and BP3 cell lines in a dose-dependent manner (Table 1 and Supplementary Figure 2C) with EC50 values of 3.5 and 3.0 μM, respectively, which are 2–3 times more potent than 6a (EC50 values of 7.4 and 9.2 μM, respectively) in same assays (Table 1 and Supplementary Figure 2D). By comparison, the potent Bcl-XL and Bcl-2 antagonist compound 10 (ABT-737) 33 displayed no cytotoxic activity against BP3 cell lines presumably because compound 10 is not effective against Mcl-1 and Bfl-1.23, 33, 45, 50 Consistent with previous results obtained for human PC3, H460 and H1299 cancer cell lines, most of synthesized 6a derivatives induced apoptosis of the RS4;11 and BP3 cell lines in a dose-dependent manner (Table 1 and Supplementary Figure 2D).
To further explore the anticancer activities of selected 5, 5′ substituted 6a derivatives, we tested their ability to induce apoptosis of primary lymphocytic leukemia cells freshly isolated from different patients affected by chronic lymphocytic leukemia (CLL) in a one day Annexin-V apoptosis assay (Figure 2D and Supplementary Figure 3A–B). Consistent with previous results obtained for human RS4;11 and BP3 cell lines, the most potent compound 6f effectively induced apoptosis of two primary CLL samples (Figure 2D and Supplementary 3B) in a dose-dependent manner with LD50 values of 10 μM and 15 μM, respectively. By comparison, compound 6a display weak activities in these two primary cells (LD50 > 30 μM). Compound 6f was further tested against primary leukemic cells freshly isolated from different six patients affected by CLL using the same assay. In agreement with previous CLL results, compound 6f effectively induced apoptosis of all six CLL samples with LD50 values ranging from 1.0–16.9 μM (Supplementary table 1). Compound 6c also effectively induced apoptosis of primary CLL samples, with a LD50 value of 6.5 μM while 6a is less effective (LD50 > 30 μM) (Supplementary Figure 3A).
To test the pharmacological properties of 5, 5′ substituted compound 6a derivatives, we determined their in vitro plasma stability, microsomal stability, and cell membrane permeability (Table 3). From these studies, we could conclude that most of our synthesized compounds displayed superior plasma and microsomal stability compared to 6a (Table 3). Compounds 6f and 6i degraded 37% and 9%, respectively, after 1 h incubation in rat plasma while 6a degraded 53% under the same conditions. In addition, compounds 6f and 6i also displayed better plasma stability and only degraded by 14% and 5%, respectively, after 1 h incubation in rat plasma preparations while 6a degraded 23% under the same conditions.
Hence, using a combination of 1D 1H-NMR binding assays, FP assays, ITC, cytotoxicity assays and preliminary in vitro ADME data, we selected compounds for in vivo studies using a B6-Bcl-2 transgenic mouse model. B-cells of the B6 transgenic mice overexpress human Bcl-2 and accumulate in the spleen of mice. The spleen weight is used as an end-point for assessing in vivo activity as we have determined that the spleen weight is highly consistent in age- and sex-matched Bcl-2-transgenic mice, varying by only ±2% among control Bcl2 mice.19 We first screened the in vivo activities of compounds such as 6b and 6f side by side with 6a in a single Bcl-2 transgenic mouse with a single intraperitoneal (ip) injection at 60 and 120 μmol/kg, respectively. In agreement with all in vitro data, tested 5, 5′ substituted 6a derivatives induce significant spleen weight reduction of mice in a dose-dependent manner. Compounds 6b, 6d and 6f displayed superior in vivo activity compared to 6a at dose of 60 μmol/kg (Figure 3A). In particular, compounds 6b and 6f induced more than 30–40% spleen weight reduction compared to the 20% induced by 6a. Since the maximum spleen shrinkage would be no more than 50% in this experimental model,19 these compounds induced near maximal (60–80%) biological activity, while 6a induced only 40% of maximum reduction in spleen weight at the same dose. Mice treated with 6b, 6d and 6f tolerated treatment well with no observed toxicity. However, mice treated with compounds 6i, 6l and 8a at 60 μmol/kg i.p., died. Nevertheless, compounds 6i and 6l are well tolerated when adimistested i.p. at 30 μmol/kg resulting in significant maximal reduction of speen size of 86% ± 8.0% and 76% ± 14.0%, respectively.
Figure 3.

Characterization of compounds in vivo. (A) Effects of 5, 5′ substituted 6a derivatives on shrinkage of Bcl-2 mouse spleen at a single intraperitoneal injection dose of 60 μmmol/kg and 120 μmmol/kg, respectively. All shrinkage data are percentage of maximum reduction of mice spleen size. (B) Effects of compound 6f at 60 μmmol/kg on reduction of spleen weight of six Bcl-2 mice treatment with a single intraperitoneal injection. Data shown as means ± S.E. (n =7). P < 0.0002. (C) Effect of i.p. 6f and 6a at 50 mg/kg on the growth of PCC-1 tumors in nude mice. Compound 6f significantly inhibited tumor growth compared to vehicle control determined with Anova statistics (P<0.001). Tumor growth inhibition ratios (T/C %) were calculated by dividing the average tumor volume in the treatment group by the average tumor volume in the control group. Dark down arrow “↓” represents the date mice were treated with compounds (D) Average body weight changes during treatment.
To further confirm results of the single transgenic mouse experiment, we next evaluated the in vivo activity of compound 6f in groups of seven B6-Bcl-2 transgenic mice each at a dose of 60 μmol/kg. Consistent with the single mouse experiment, compound 6f treatment resulted in a significant (~60%) reduction of spleen weight (P<0.0002) compared to the control group of seven mice (Figure 3B). All mice tolerated the treatment well, with no evident signs of toxicity. The average weight loss of mice was ~ 5.0% during the course of this study with compound 6f.
To examine the therapeutic potential of compound 6a and its derivatives (6f and 6i) as a single agent against prostate cancer, the in vivo efficacy of these compounds were investigated side by side with compound 1 on the growth of PPC-1 xenograft tumors (Figure 3C). When dosed i.p. three times in first week at 50 mg/kg, compound 6f and 6a induced strong tumor regression compared with the control group. Mice treated with 6f and 6a tolerate the treatment well in first week with modest (~5%) weight loss. However, mice treated with 6i (50 mg/kg, i.p.) and 1 (25 mg/kg, i.p.) died in the first week of this experiment. Mice were treated with 6f and 6a twice in the second week and once in third week at 50 mg/kg. Overall, compound 6f displayed significant antitumor activity compared to control group, with T/C% ratios of 33% (P < 0.001) in PPC-1 xenograft-bearing nude mice (Figure 3C). Compound 6a showed weaker antitumor activity compared to 6f, with T/C% ratios of 65% in same xenograft model (Figure 3C). Mice treated with 6f tolerated the treatment were well with no observable signs of toxicity (Figure 3D). Average body weight losses during the treatment are 6.8%, 7.1% and 9.3% for 6f, control and 6a group, respectively.
Conclusions
In summary, a series of 5, 5′ substituted 6a derivatives were synthesized and evaluated in a variety of in vitro and in vivo assays. The compound 6f was found to bind to Bcl-XL, Bcl-2 and Mcl-1 with IC50 values of 3.10 μM, 3.12 μM and 2.05 μM, respectively. In a cellular assay 6f potently inhibited growth in cultures of the PC3, H460, H1299 and BP3 cancer cell lines, which express Bcl-XL, Bcl-2, Mcl-1 and Bfl-1, respectively, with EC50 values in the single digit micromolar to nanomolar range. Compound 6f effectively induced apoptosis of the RS4;11 human lymphoma cell line and primary human chronic lymphocytic leukemia cells in a dose-dependent manner. Compound 6f also displays in vivo efficacy in transgenic mice in which Bcl-2 is overexpressed in splenic B-cells. Finally, compound 6f showed favorable in vitro ADME properties and superior in vivo efficacy as a single agent in a PPC-1 nude mouse xenograft model relative to 6a. Considering the critical roles of anti-apoptotic Bcl-2 family proteins in tumorgenesis, chemoresistance, and the potent inhibitory activity of 6f against anti-apoptotic Bcl-2 family proteins, compound 6f represents a viable drug candidate for the development of novel apoptosis-based cancer therapies.
Experimental Section
General Synthetic Procedures
Unless otherwise indicated, all reagents and anhydrous solvents (CH2Cl2, THF, diethyl ether, etc) were obtained from commercial sources and used without purification. All reactions were performed in oven-dried glassware. All reactions involving air or moisture sensitive reagents were performed under a nitrogen atmosphere. Silica gel or reverse phase chromatography was performed using prepacked silica gel or C-18 cartridges (RediSep), respectively. All final compounds were purified to > 95% purity, as determined by a HPLC Breeze from Waters Co. using an Atlantis T3 3 μM 4.6 mm × 150 mm reverse phase column. Method A: The eluant was a linear gradient with a flow rate of 1 mL/min from 50% A and 50% B to 5% A and 95% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: ACN with 0.1% TFA). Compounds were detected at λ = 254 nm. Method B: The eluant was a linear gradient with a flow rate of 1 mL/min from 20% A and 80% B to 100% B in 15 min followed by 5 min at 100% B (Solvent A: H2O with 0.1% TFA; Solvent B: ACN with 0.1% TFA). Compounds were detected at λ = 254 nm. 1H NMR spectra were recorded on Varian 300 or Bruker 600 MHz instruments. Chemical shifts are reported in ppm (δ) relative to 1H (Me4Si at 0.00 ppm). Coupling constant 11 are reported in Hz throughout. Mass spectral data were acquired on Shimadzu LCMS-2010EV for low resolution, and on an Agilent ESI-TOF for high resolution.
1,1′,6,6′,7,7′-Hexahydroxy-5,5′-diisopropyl-3,3′-dimethyl-2,2′-binaphthyl-8,8′-dicarboxaldehyde (1)
Compound 1 (Gossypol) is commercially available from Yixin Pharmaceutical Co. HPLC purity 99.0%, tR = 12.50 min (Method A).
5,5′-Diisopropyl-1,1′,6,6′,7,7′-hexamethoxy-3,3′-dimethyl-2,2′-binaphthalene (9)
The compound 1 (5 g, 8.65 mmol) in 50 mL of 40% NaOH was heated under nitrogen at 90 °C for 3.5 h in the dark. The reaction mixture was cooled and poured slowly onto ice (300 mL) and concentrated H2SO4 (35 mL) mixture to form white precipitate. The precipitate was filtered, washed with water and dried to afford 3.8 g of compound 2a (95%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.61 (s, 2H), 7.50 (s, 2H), 5.93 (s, 2H), 5.27 (s, 2H), 5.13 (s, 2H), 3.88 (m, 2H), 2.12 (s, 6H), 1.55 (d, J = 5.5 Hz, 12H). HPLC purity 99.2%, tR = 13.12 min. HRMS calcd for C28H30O6 463.2115 (M + H), found 463.2108. The compound 2a (3.8 g, 8.21 mmol) was dissolved into acetone (200 mL). K2CO3 (23.9 g, 206.7 mmol) and dimethyl sulfate (16.3 mL, 206.7 mmol) were added and the reaction mixture was refluxed under nitrogen for 24 h. The solid was collected by filtration and washed using acetone and water and dried to yield 4.2 g of compound 9 as white solid (93%). 1H NMR (300 MHz, CDCl3) 7.83 (s, 2H), 7.43 (s, 2H), 3.98 (m, 8H), 3.94 (s, 6H), 3.57 (s, 6H), 2.20 (s, 6H), 1.56 (s, 12H).
5,5′-Diisopropyl-6,6′,7,7′-tetramethoxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (10)
Periodic acid (10 g, 43.8 mmol) was added to a solution of compound 9 (0.62 g, 1.12 mmol) in 20 mL of dioxane and the reaction mixture was stirred at 95 °C for 15 min. Crushed ice was added to quench the reaction. The solution was extracted twice with ethyl acetate and the organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified by flash silica column chromatography to give 142 mg of compound 10 (23%) as yellow solid. 1H NMR (600 MHz, CD3OD) δ 7.56 (s, 2H), 4.31 (m, 2H), 3.97 (s, 6H), 3.94 (s, 6H), 2.03 (s, 6H), 1.40 (d, J = 1.8 Hz, 6H), 1.39 (d, J = 1.8 Hz, 6H).
6,6′,7,7′-tetrahydroxy-5,5′-diisopropyl-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6a)
0.54 mL of BBr3 (1.43 g, 5.71 mmol) was added dropwise into a solution of compound 10 (260 mg, 0.48 mmol) in 10 mL of anhydrous CH2Cl2 at −78 °C. Stirring was continued at −78 °C for 1 h, 0 °C for 1 h, and ambient temperature for 1 h. 50 grams of ice containing 5 mL of 6M HCl was added to the mixture and stirred for 1 h at room temperature. The aqueous layer was extracted with dichloromethane (3 × 50 mL). The combined organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified using C-18 column chromatography (H2O/Acetonitrile) followed by recrystallization from ethyl acetate/hexane to give 163 mg of compound 6a (70%) as brown-yellow solid. 1H NMR (600 MHz, CD3OD) δ 7.31 (s, 2H), 4.32 (m, 2H), 1.88 (s, 6H), 1.42 (s, 6H), 1.40 (s, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 187.10, 182.51, 150.92, 149.54, 147.60, 137.78, 137.10, 126.24, 125.00, 111.03, 27.07, 20.50, 20.35, 15.00. HPLC purity 99.5%, tR = 11.60 min (Method A). HRMS calcd for C28H26O8 491.1700 (M + H), found 491.1696.
The syntheses of compounds 3 and 4 have been previously described.22, 23
6,6′,7,7′-Tetrahydroxy-3,3′-dimethyl-1,1′,4,4′-tetraoxo-N5,N5′-bis(2-phenylpropyl)-1,1′,4,4′-tetrahydro-2,2′-binaphthyl-5,5′-dicarboxamide (8a)
A solution of compound 4a (290 mg, 0.414 mmol) in 12 mL of acetone and 23 mL of acetic acid was heated on an oil bath (60–67°C) during the addition of 18 mL of a 10% aqueous solution of ferric chloride (6.64 mmol) and for several minutes longer. The solution was cooled and 30 mL of water was added followed by 20 mL of aqueous 20% sulfuric acid. The solution was extracted twice with diethyl ether and the organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified by C-18 column chromatography (H2O/Acetonitrile) to give 60 mg of compound 8a (45%) as yellow solid. 1H NMR (600 MHz, CD3OD) δ 7.42 (s, 2H), 7.34 (d, J = 7.2 Hz, 4H), 7.30 (t, J1 = J2 = 7.2 Hz, 4H), 7.18 (t, J1 = J2 = 7.2 Hz, 4H), 3.54 (d, J = 7.2 Hz, 4H), 3.22 (m, 2H), 1.91 (s, 6H), 1.39 (s, 6H), 1.38 (d, J = 6.6 Hz, 6H). 13C NMR (600 MHz, CD3OD) δ 184.50, 183.68, 170.44, 151.92, 150.19, 146.99, 146.55, 140.68, 129.63, 128.54, 127.53, 127.23, 126.13, 124.30, 113.14, 48.34, 40.74, 19.97, 14.50. HPLC purity 98.3%, tR = 5.82 min (Method A). HRMS calcd for C42H36N2O10 729.2443 (M + H), found 729.2441.
Following above mentioned procedure and the appropriate starting materials and reagents used; compounds 7 and 8b–8c were synthesized.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-N5,N5′-bis(3-methylbenzyl)-1,1′,4,4′-tetraoxo-1,1′,4,4′-tetrahydro-2,2′-binaphthyl-5,5′-dicarboxamide (8b)
Yield, 50%; 1H NMR (600 MHz, CD3OD) δ 7.44 (s, 2H), 7.39 (s, 2H), 7.29 (d, J = 7.2 Hz, 2H), 7.20 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 4H), 7.06 (d, J = 7.8 Hz, 2H), 4.61 (dd, J1 = 15 Hz, J2 = 4.8 Hz, 4H), 2.35 (s, 6H), 1.91 (s, 6H). 13C NMR (600 MHz, CD3OD) δ 184.57, 183.70, 170.33, 152.01, 150.28, 147.04, 139.71, 139.24, 129.68, 129.41, 128.87, 127.33, 126.04, 126.00, 124.32, 113.21, 44.65, 21.66, 14.49. HPLC purity 99.0%, tR = 5.53 min (Method A). HRMS calcd for C40H32N2O10 701.2130 (M + H), found 701.2128.
N5,N5′-Bis(4-ethylphenethyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-1,1′,4,4′-tetraoxo-1,1′,4,4′-tetrahydro-2,2′-binaphthyl-5,5′-dicarboxamide (8c)
Yield, 52%; 1H NMR (600 MHz, CD3OD) δ 7.43 (s, 2H), 7.23 (d, J = 6.6 Hz, 4H), 7.12 (d, J = 6.6 Hz, 4H), 3.60 (m, 4H), 2.96 (t, J1 = J2 = 6.6 Hz, 4H), 2.50 (q, J1 = J2 = 6.6 Hz, 4H), 1.93 (s, 6H), 1.20 (t, J1 = J2 = 6.6 Hz, 6H). 13C NMR (600 MHz, CD3OD) δ 184.55, 183.68, 170.37, 151.99, 150.18, 147.01, 143.51, 140.73, 138.24, 130.09, 129.09, 127.38, 126.09, 124.31, 113.18, 43.03, 36.03, 29.68, 16.49, 14.51. HPLC purity 97.6%, tR = 6.99 min (Method A). HRMS calcd for C44H40N2O10 757.2756 (M + H), found 757.2745.
6,6′,7,7′-Tetrahydroxy-3,3′-dimethyl-5,5′-bis(2-phenylacetyl)-2,2′-binaphthyl-1,1′,4,4′-tetraone (7)
Yield, 49%; 1H NMR (600 MHz, CD3OD) δ 7.32 (s, 2H), 7.28 (d, J = 6.0 Hz, 4H), 7.22 (t, J1 = J2 = 6.0 Hz, 4H), 7.15 (t, J1 = J2 = 6.0 Hz, 2H), 4.13 (m, 4H), 1.93 (s, 6H). 13C NMR (600 MHz, CD3OD) δ 204.02, 183.63, 181.97, 150.74, 147.30, 145.10, 139.75, 134.05, 130.14, 129.78, 127.70, 126.40, 125.69, 122.90, 111.65, 49.31, 12.84. HPLC purity 99.0%, tR = 9.44 min (Method A). HRMS calcd for C38H26O10 643.1599 (M + H), found 643.1601.
1,1′,6,6′,7,7′-hexamethoxy-3,3′-dimethyl-5,5′-bis(4-methylphenethyl)-2,2′-binaphthyl (15f)
To a freshly prepared 4-methylbenzylmagnesium chloride (30.85 mmol) solution at room temperature was added a solution of 11 (2.0 g, 3.86 mmol) in anhydrous tetrahydrofuran (30 mL) and the reaction mixture was heated at 30°C for 18 h. The reaction mixture was poured onto saturated ammonium chloride solution and the aqueous layer was extracted twice with diethyl ether, washed with brine and dried over MgSO4. Filtration followed by evaporation of the ether gave yellow oil 13. To a solution of the yellow oil 13 (1.4 g, 1.929 mmol) in 25 mL TFA was added 3.1 mL of triethylsilane dropwise. The solution was heated at 75°C for 1 h followed by stirred at room temperature for 18 h. The solution was concentrated in vacuo followed by silica gel column chromatography to give 660 mg compound 15f as colorless oil (50% from 11). 1H NMR (600 MHz, CDCl3) δ 7.64 (s, 2H), 7.44 (s, 2H), 7.26 (d, J = 7.8 Hz, 4H), 7.15 (d, J = 7.8 Hz, 4H), 3.99 (s, 6H), 3.94 (s, 6H), 3.60 (s, 6H), 3.37 (t, J1 = J2 = 8.40 Hz, 4H), 2.98 (t, J1 = J2 = 8.4 Hz, 4H), 2.35 (s, 6H), 2.20 (s, 6H).
3,3′-dimethyl-5,5′-bis(4-methylphenethyl)-2,2′-binaphthyl-1,1′,6,6′,7,7′-hexaol (2f)
2.1 mL of BBr3 solution (5.56 g, 22.2 mmol) was added dropwise into a solution of 15f (1.23 g, 1.76 mmol) in 60 mL of anhydrous CH2Cl2 at −78 °C. Stirring was continued at −78 °C for 1 h, 0 °C for 1 h, and ambient temperature for 1 h, respectively. 300 grams of ice containing 30 mL of 6M HCl was added to the mixture and stirred for 0.5 h at room temperature. The aqueous layer was extracted with dichloromethane (3 × 100 mL). The combined organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified by C-18 column chromatography (H2O/Acetonitrile) to give 1.1 g of compound 2f (90%) as white solid. Yield, 45%; 1H NMR (600 MHz, CD3OD) δ 7.45 (s, 2H), 7.34 (s, 2H), 7.20 (d, J = 7.2 Hz, 4H), 7.08 (d, J = 7.2 Hz, 4H), 3.27 (m, 4H), 2.87 (m, 4H), 2.31 (s, 6H), 2.03 (s, 6H). HPLC purity 96.6%, tR = 17.00 min (Method A). HRMS calcd for C40H38O6 615.2741 (M + H), found 615.2720.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-5,5′-bis(4-methylphenethyl)-2,2′-binaphthyl-1,1′,4,4′-tetraone (6f)
A solution of compound 2f (1.0 g, 1.55 mmol) in 50 mL of acetone and 80 mL of acetic acid was heated on an oil bath (60–67 °C) during the addition of 68 mL of a 10% aqueous solution of ferric chloride and for several minutes longer. The solution was cooled and 50 mL of water was added followed by 30 mL of aqueous 20% sulfuric acid. The solution was extracted twice with diethyl ether and the organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified by C-18 column chromatography (H2O/Acetonitrile) to give 350 mg of compound 6f (35%) as yellow solid. 1H NMR (600 MHz, CD3OD) δ 7.40 (s, 2H), 7.22 (d, J = 7.8 Hz, 4H), 7.08 (d, J = 7.2 Hz, 4H), 3.45 (m, 4H), 2.78 (t, J1 = 8.4 Hz, J2 = 7.8 Hz, 4H), 2.30 (s, 6H), 1.93 (s, 6H). 13C NMR (600 MHz, CD3OD) δ 185.65, 182.87, 149.35, 148.72, 146.61, 139.47, 138.20, 134.62, 131.79, 128.32, 128.14, 126.47, 123.95, 110.50, 34.44, 28.68, 19.69, 13.36. HPLC purity 99.0%, tR = 17.53 min (Method A). HRMS calcd for C40H34O8 643.2326 (M + H), found 643.2326.
Following above mentioned procedure and the appropriate starting materials and reagents used; compounds 6b-I, 6l and 6m were synthesized.
6,6′,7,7′-tetrahydroxy-5,5′-diisobutyl-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6b)
Yield, 50%; 1H NMR (600 MHz, CD3OD) δ 7.39 (s, 2H), 3.18 (m, 4H), 1.94 (m, 2H), 1.93 (s, 6H), 0.96 (d, J = 6.0 Hz, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.67, 182.75, 149.97, 149.53, 146.67, 138.38, 132.10, 126.65, 123.60, 111.39, 34.43, 29.18, 23.13, 23.11, 14.96. HPLC purity 96.7%, tR = 13.68 min (Method A). HRMS calcd for C30H30O8 519.2013 (M + H), found 519.2012.
5,5′-bis(cyclopentylmethyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6c)
Yield, 40%; 1H NMR (600 MHz, (CD3)2SO) δ 10.99 (s, br, 2H), 9.54 (s, br, 2H), 7.34 (s, 2H), 3.23 (dd, J1 = 7.2 Hz, J2 = 4.8 Hz, 2H), 3.15 (dd, J1 = 7.2 Hz, J2 = 4.8 Hz, 2H), 2.10 (m, 2H), 1.87 (s, 6H), 1.61 (m, 8H), 1.45 (m, 4H), 1.26 (m, 4H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.25, 182.29, 149.36, 149.00, 146.23, 137.95, 132.26, 126.19, 123.05, 110.84, 40.26, 32.00, 30.65, 24.50, 14.52. HPLC purity 99.0%, tR = 16.80 min (Method A). HRMS calcd for C34H34O8 571.2326 (M + H), found 571.2323.
5,5′-bis(2-cyclohexylethyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6d)
Yield, 50%; 1H NMR (600 MHz, (CD3)2SO) δ 10.88 (s, br, 2H), 9.51 (s, br, 2H), 7.30 (s, 2H), 3.08 (m, 4H), 1.85 (s, 6H), 1.80 (d, J = 12.0 Hz, 4H), 1.68 (d, J = 12.6 Hz, 4H), 1.61 (d, J = 11.4 Hz, 2H), 1.35 (m, 6H), 1.23 (q, J1 = 24.6 Hz, J2 = 12.6 Hz, 4H), 1.16 (m, 2H), 0.96 (m, 4H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.20, 182.22, 148.95, 148.85, 146.16, 138.00, 133.20, 125.96, 123.03, 110.72, 38.02, 36.26, 32.87, 26.32, 25.91, 23.93, 14.44. HPLC purity 98.5%, tR = 14.76 min (Method B). HRMS calcd for C38H42O8 627.2952 (M + H), found 627.2952.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-5,5′-diphenethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6e)
Yield, 38%; 1H NMR (600 MHz, CD3OD) δ 7.41 (s, 2H), 7.36 (d, J = 7.8 Hz, 4H), 7.28 (t, J1 = 7.8Hz, J2 = 7.2 Hz, 4H), 7.16 (t, J1 = 7.8 Hz, J2 = 7.2 Hz, 2H), 3.47 (m, 4H), 2.84 (t, J1 = 8.4 Hz, J2 = 7.8 Hz, 4H), 1.95 (s, 6H). HPLC purity 99.0%, tR = 15.48 min (Method A). HRMS calcd for C38H30O8 615.2013 (M + H), found 615.2015.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-5,5′-bis(3-phenylpropyl)-2,2′-binaphthyl-1,1′,4,4′-tetraone (6g)
Yield, 42%; 1H NMR (600 MHz, CD3OD) δ 7.39 (s, 2H), 7.26 (m, 8H), 7.14 (m, 2H), 3.27 (m, 4H), 2.81 (t, J1 = J2 = 7.8 Hz, 4H), 1.97 (s, 6H), 1.90 (p, J1 = J2 = 7.8 Hz, 4H). 13C NMR (600 MHz, CD3OD) δ 185.90, 183.4, 149.40, 148.80, 146.70, 142.80, 138.22, 132.54, 127.96, 127.75, 126.61, 125.11, 123.67, 110.42, 36.15, 30.73, 26.21. HPLC purity 98.0%, tR = 16.20 min (Method A). HRMS calcd for C40H34O8 643.2326 (M + H), found 643.2334.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-5,5′-bis(3-methyl-3-phenylbutyl)-2,2′-binaphthyl-1,1′,4,4′-tetraone (6h)
Yield, 50%; 1H NMR (600 MHz, CD3OD) δ 7.52 (d, J = 7.8 Hz, 4H), 7.36 (s, 2H), 7.32 (t, J1 = J2 = 7.8 Hz, 4H), 7.16 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 2H), 3.04 (t, J1 = 7.8 Hz, J2 = 8.4 Hz, 4H), 1.96 (s, 6H), 1.91 (m, 4H), 1.47 (s, 12H). HPLC purity 98.0%, tR = 13.5 min (Method B). 13C NMR (600 MHz, CD3OD) δ 185.36, 182.93, 149.38, 148.47, 146.58, 138.18, 133.24, 131.96, 127.47, 126.47, 125.66, 124.84, 123.69, 110.29, 42.15, 37.51, 35.48, 28.19, 22.01. HRMS calcd for C44H42O8 699.2952 (M + H), found 699.2964.
5,5′-dibenzyl-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6i)
Yield, 55%; 1H NMR (600 MHz, CD3OD) δ 7.44 (s, 2H), 7.22 (d, J = 7.2 Hz, 4H), 7.17 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 4H), 7.17 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 2H), 4.63 (q, J1 = 11.4 Hz, J2 = 14.4 Hz, 4H), 1.86 (s, 6H). 13C NMR (600 MHz, (CD3)2SO)) δ 185.38, 182.54, 149.94, 149.77, 146.53, 140.89, 138.49, 130.01, 128.60, 128.33, 126.45, 125.76, 123.69, 111.68, 31.68, 14.77. HPLC purity 99.6%, tR = 12.12 min (Method A). HRMS calcd for C36H26O8 587.1700 (M + H), found 587.1710.
5,5′-bis(4-chlorobenzyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6l)
Yield, 60%; 1H NMR (600 MHz, CD3OD) δ 7.45 (s, 2H), 7.22 (d, J = 6.4 Hz, 4H), 7.18 (d, J = 6.4 Hz, 4H), 4.59 (dd, J1 = 13.8 Hz, J2 = 26.4 Hz, 4H), 1.85 (s, 6H). HPLC purity 99.0%, tR = 14.88 min (Method A). HRMS calcd for C40H32F2O8 655.0921 (M + H), found 655.0931.
5,5′-bis(biphenyl-4-ylmethyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6m)
Yield, 52%; 1H NMR (600 MHz, CD3OD) δ 7.55 (d, J = 7.2 Hz, 4H), 7.44 (d, J = 7.8 Hz, 6H), 7.38 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 4H), 7.30 (d, J = 7.8 Hz, 4H), 7.27 (t, J1 = 7.2 Hz, J2 = 6.6 Hz, 2H), 4.59 (dd, J1 = 13.8 Hz, J2 = 27.0 Hz, 4H), 1.88 (s, 6H). HPLC purity 99.0%, tR = 16.96 min (Method A). HRMS calcd for C40H32F2O8 739.2326 (M + H), found 739.2329.
6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-5,5′-bis(4-(trifluoromethoxy)phenethyl)-2,2′-binaphthyl-1,1′,4,4′-tetraone (6j)
To a solution of 3j (100 mg, 0.13 mmol) in 25 mL of ethonal and 1mL acetic acid at room temperature under H2, 10% palladium on carbon (0.10g) was added and stirred overnight. The solution was extracted twice with diethyl ether and the organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the crude residue (2j) was dissolved in 5 mL of acetone and 8 mL of acetic acid was heated on an oil bath (60–67 °C) during the addition of 7 ml of a 10% aqueous solution of ferric chloride and for several minutes longer. The solution was cooled and 5 mL of water was added followed by 3 mL of aqueous 20% sulfuric acid. The solution was extracted twice with diethyl ether and the organic layer was washed with water, brine and dried over MgSO4. The solvent was concentrated in vacuo and the residue was purified by C-18 column chromatography (H2O/Acetonitrile) to give 30 mg of compound 6j (30%) as yellow-brown solid. 1H NMR (600 MHz, CD3OD) δ 7.48 (d, J = 7.8 Hz, 4H), 7.42 (s, 2H), 7.20 (d, J = 7.8 Hz, 4H), 3.48 (m, 4H), 2.88 (t, J1 = 8.4 Hz, J2 = 7.2 Hz, 4H), 1.95 (s, 6H). HPLC purity 97.0%, tR = 11.67 min (Method B). HRMS calcd for C40H28F6O10 783.1659 (M + H), found 783.1659.
Following above mentioned procedure and the appropriate starting materials and reagents used; compound 6k was synthesized.
5,5′-bis(3-(4-fluorophenyl)propyl)-6,6′,7,7′-tetrahydroxy-3,3′-dimethyl-2,2′-binaphthyl-1,1′,4,4′-tetraone (6k)
Yield, 45%; 1H NMR (600 MHz, CD3OD) δ 7.37 (s, 2H), 7.24 (m, 4H), 6.97 (m, 4H), 3.26 (m, 4H), 2.78 (t, J1 = 7.2 Hz, J2 = 7.8 Hz, 4H), 1.94 (s, 6H), 1.90 (m, 4H). HPLC purity 96.5%, tR = 8.76 min (Method B). HRMS calcd for C40H32F2O8 679.2138 (M + H), found 679.2150.
Molecular Modeling
Molecular modeling studies were conducted on a Linux workstation and a 64 3.2-GHz CPUs Linux cluster. Docking studies were performed using the crystal structure of Bcl-2 in complex with a benzothiazole BH3 mimetic ligand (Protein Data Bank code 1YSW) 33, 34 The ligand was extracted from the protein structure and was used to define the binding site for small molecules. Compound 6a and its derivatives were docked into the Bcl-2 protein by the GOLD 51 docking program using ChemScore 52 as the scoring function. The active site radius was set at 10 Å and 10 GA solutions were generated for each molecule. The GA docking procedure in GOLD 51 allowed the small molecules to flexibly explore the best binding conformations whereas the protein structure was static. The protein surface was prepared with the program MOLCAD 53 as implemented in Sybyl (Tripos, St. Louis) and was used to analyze the binding poses for studied small molecules.
Fluorescence Polarization Assays (FPAs)
A Bak BH3 peptide (F-BakBH3) (GQVGRQLAIIGDDINR) was labeled at the N-terminus with fluorescein isothiocyanate (FITC) (Molecular Probes) and purified by HPLC. For competitive binding assays, 100 nM GST–Bcl-XL ΔTM protein was preincubated with the tested compound at varying concentrations in 47.5 μL PBS (pH=7.4) in 96-well black plates at room temperature for 10 min, then 2.5 μL of 100 nM FITC-labeled Bak BH3 peptide was added to produce a final volume of 50 μL. The wild-type and mutant Bak BH3 peptides were included in each assay plate as positive and negative controls, respectively. After 30 min incubation at room temperature, the polarization values in millipolarization units 54 were measured at excitation/emission wavelengths of 480/535 nm with a multilabel plate reader (PerkinElmer). IC50 was determined by fitting the experimental data to a sigmoidal dose-response nonlinear regression model (SigmaPlot 10.0.1, Systat Software, Inc., San Jose, CA, USA). Data reported are mean of three independent experiments ± standard error (SE). Performance of Bcl-2 and Mcl-1 FPA are similar. Briefly, 50 nM of GST-Bcl-2 or -Mcl-1 were incubated with various concentrations of compound 6a, or its 5, 5′ substituted derivatives for 2 min, then 15 nM FITC-conjugated-Bim BH3 peptide 55 was added in PBS buffer. Fluorescence polarization was measured after 10 min.
Cell Viability and Apoptosis Assays
The activity of the compounds against human cancer cell lines (PC3, H460, H1299) were assessed by using the ATP-LITE assay (PerkinElmer). All cells were seeded in either 12F2 or RPMI1640 medium with 5 mM L-glutamine supplemented with 5% fetal bovine serum (Mediatech Inc.), penicillin and streptomycin (Omega). For maintenance, cells were cultured in 5% FBS. Cells plated into 96 well plates at varying initial densities depending on doubling time. H460 and H1299 plated at 2000 cells/well and PC3 at 3000 cells/well. Compounds were diluted to final concentrations with 0.1% DMSO. Prior to dispensing compounds onto cells, fresh 5% media was placed into wells. Administration of compounds occurred 24 hours after seeding into the fresh media. Cell viability was evaluated using ATP-LITE reagent (PerkinElmer) after 72 hours of treatment. Data were normalized to the DMSO control-treated cells using Prism version 5.01 (Graphpad Software).
The apoptotic activity of the compounds against RS4;11, BP3 and primary CLL cells was assessed by staining with Annexin V-FITC and propidium iodide (PI). Cells were cultured in RPMI 1640 medium (Mediatech Inc., Herndon, VA 20171) containing 10% fetal bovine serum (Mediatech Inc., Herndon, VA 20171) and Penicillin/Streptomycin (Mediatech Inc., Herndon, VA 20171). Cells were cultured with various concentrations of 5, 5′ substituted 6a derivatives for 1 day. The percentage of viable cells was determined by FITC-Annexin V- and propidium iodide (PI)-labeling, using an Apoptosis Detection kit (BioVision Inc.), and analyzing stained cells by flow cytometry (FACSort; Bectin-Dickinson, Inc.; Mountain View, CA). Cells that were annexin-V-negative and PI-negative were considered viable.
Bcl-2 Transgenic Mice Studies
Transgenic mice expressing Bcl-2 have been described as the B6 line.56 The BCL-2 transgene represents a minigene version of a t(14;18) translocation in which the human BCL-2 gene is fused with the immunoglobulin heavy-chain (IgH) locus and associated IgH enhancer. The transgene was propagated on the Balb/c background. These mice develop polyclonal B-cell hyperplasia with asynchronous transformation to monoclonal aggressive lymphomas beginning at approximately 6 months of age, with approximately 90% of mice undergoing transformation by the age of 12 to 24 months. All animals used here had not yet developed aggressive lymphoma. Compounds dissolved in 500 μL of solution (Ethanol: Cremophor EL: Saline = 10: 10: 80) were injected intraperitoneally to age- and sex-matched B6Bcl2 mouse, while control-mice were injected intraperitoneally with 500 μL of the same formulation without compound. After 24 hours, B6Bcl2 mice were sacrificed by intraperitoneal injection of lethal dose of Avertin. Spleen was removed and weighed. The spleen weight of mice is used as an end-point for assessing activity as we determined that spleen weight is highly consistent in age- and sex-matched Bcl-2-transgenic mice in preliminary studies.20 Variability of spleen weight was within ±2% among control-treated age-matched, sex-matched B6Bcl2 mice.
Tumor Xenograft Studies
Female 6-week-old nude mice were purchased from Charles River Laboratories. 5 ×106 PCC-1 cells suspended in 0.2 ml PBS were injected subcutaneously into a flank region of each nude mouse. Tumor bearing mice were size matched (200 mm3) into treatment and control group, ear tagged, and monitored individually. Tumor volume was measured two to three times weekly by digital calipers (volume = length × width2/2). All studies use 6 mice per group. Compounds dissolved in 500 μL of solvent (Ethanol:Cremophor EL/saline=10:10:80) were injected intraperitoneally (i.p.) into tumor-beraing mice. Control mice received saline. The injections were given three times in first week, twice in second week and once in third week and a total of six injections were administered during the experiment. When all tumors of the control group exceed 2000 mm3 in volume, the animal experiment was terminated. Tumor growth inhibition ratios (T/C %) were calculated by dividing the average tumor volume in the treatment group by the average tumor volume in the control group.
Supplementary Material
Acknowledgments
We thank NIH (Grants CA113318 and CA149668 to MP and JCR) for financial support.
Abbreviations list
- Bcl-2
B-cell lymphoma/leukemia-2
- EDCI
1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide
- 1D-1H NMR
one-dimensional 1H nuclear magnetic resonance spectroscopy
- SAR
Structure-activity relationship
- FPA
Fluorescence Polarization Assays
- ITC
Isothermal Titration Calorimetry
- CLL
Chronic lymphocytic leukemia
- WT
Wild type
- MEFs
Mouse embryonic fibroblast cells
- DKO
Bax/Bak Double knockout
- DKO/MEFs
Bax/Bak Double knockout mouse embryonic fibroblast cells
- ACN
Acetonitrile
- LC-MS
Liquid chromatography and tandem mass spectrometry
- HPLC
High-performance liquid chromatography
- TROSY
Transverse Relaxation-Optimized Spectroscopy
- ADME
Absorption, Distribution, Metabolism, and Excretion
- DMSO
Dimethyl sulphoxide
- PPC-1
Human Prostatic cancer cell line
- PAMPA
Parallel artificial membrane permeation assay
- FITC
Fluorescein isothiocyanate
- GST
Glutathione-S-transferase
- PBS
Phosphate-buffered saline
- SE
Standard error
- PI
Propidium iodide
- NADPH
Nicotinamide adenine dinucleotide phosphate
- Rpm
Rotations Per Minute
- AUC
Area under the curve
Footnotes
Supporting Information Available: An experimental section including information on chemical data for compounds 15b–i and 2b–2m, NMR experiments, isothermal titration calorimetry assays, in vitro ADME studies, and in vivo mice studies. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 2.Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941–2953. doi: 10.1200/JCO.1999.17.9.2941. [DOI] [PubMed] [Google Scholar]
- 3.Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 4.Reed JC. Apoptosis-based therapies. Nature reviews Drug discovery. 2002;1:111–121. doi: 10.1038/nrd726. [DOI] [PubMed] [Google Scholar]
- 5.Reed JC. Molecular biology of chronic lymphocytic leukemia: implications for therapy. Seminars in hematology. 1998;35:3–13. [PubMed] [Google Scholar]
- 6.Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science (New York, N Y ) 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 7.Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes & development. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- 8.Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- 9.Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES, Huang Z. Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7124–7129. doi: 10.1073/pnas.97.13.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T, Yuan J. Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat Cell Biol. 2001;3:173–182. doi: 10.1038/35055085. [DOI] [PubMed] [Google Scholar]
- 11.Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Advances in pharmacology (San Diego, Calif ) 1997;41:501–532. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- 12.Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. Journal of medicinal chemistry. 2003;46:4259–4264. doi: 10.1021/jm030190z. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M, Liu H, Guo R, Ling Y, Wu X, Li B, Roller PP, Wang S, Yang D. Molecular mechanism of gossypol-induced cell growth inhibition and cell death of HT-29 human colon carcinoma cells. Biochemical pharmacology. 2003;66:93–103. doi: 10.1016/s0006-2952(03)00248-x. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, Yang D. Small Molecular Antagonists of Bcl-2 family proteins. 2003008924. US patent applications series. 2002
- 15.Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, Shangary S, Qiu S, Gao W, Yang D, Meagher J, Stuckey J, Krajewski K, Jiang S, Roller PP, Abaan HO, Tomita Y, Wang S. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. Journal of medicinal chemistry. 2006;49:6139–6142. doi: 10.1021/jm060460o. [DOI] [PubMed] [Google Scholar]
- 16.Mohammad RM, Wang S, Aboukameel A, Chen B, Wu X, Chen J, Al-Katib A. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-X(L) [(−)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther. 2005;4:13–21. [PubMed] [Google Scholar]
- 17.Meng Y, Tang W, Dai Y, Wu X, Liu M, Ji Q, Ji M, Pienta K, Lawrence T, Xu L. Natural BH3 mimetic (−)-gossypol chemosensitizes human prostate cancer via Bcl-xL inhibition accompanied by increase of Puma and Noxa. Mol Cancer Ther. 2008;7:2192–2202. doi: 10.1158/1535-7163.MCT-08-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, Kipps TJ, Reed JC, Pellecchia M. Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L) Chemistry & biology. 2004;11:389–395. doi: 10.1016/j.chembiol.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 19.Kitada S, Kress CL, Krajewska M, Jia L, Pellecchia M, Reed JC. Bcl-2 antagonist apogossypol (NSC736630) displays single-agent activity in Bcl-2-transge3nic mice and has superior efficacy with less toxicity compared with gossypol (NSC19048) Blood. 2008;111:3211–3219. doi: 10.1182/blood-2007-09-113647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coward L, Gorman G, Noker P, Kerstner-Wood C, Pellecchia M, Reed JC, Jia L. Quantitative determination of apogossypol, a pro-apoptotic analog of gossypol, in mouse plasma using LC/MS/MS. Journal of pharmaceutical and biomedical analysis. 2006;42:581–586. doi: 10.1016/j.jpba.2006.05.020. [DOI] [PubMed] [Google Scholar]
- 21.Wei J, Rega MF, Kitada S, Yuan H, Zhai D, Risbood P, Seltzman HH, Twine CE, Reed JC, Pellecchia M. Synthesis and evaluation of Apogossypol atropisomers as potential Bcl-xL antagonists. Cancer Lett. 2009;273:107–113. doi: 10.1016/j.canlet.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wei J, Kitada S, Rega MF, Emdadi A, Yuan H, Cellitti J, Stebbins JL, Zhai D, Sun J, Yang L, Dahl R, Zhang Z, Wu B, Wang S, Reed TA, Lawrence N, Sebti S, Reed JC, Pellecchia M. Apogossypol derivatives as antagonists of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2009;8:904–913. doi: 10.1158/1535-7163.MCT-08-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei J, Kitada S, Rega MF, Stebbins JL, Zhai D, Cellitti J, Yuan H, Emdadi A, Dahl R, Zhang Z, Yang L, Reed JC, Pellecchia M. Apogossypol derivatives as pan-active inhibitors of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. Journal of medicinal chemistry. 2009;52:4511–4523. doi: 10.1021/jm900472s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei J, Stebbins JL, Kitada S, Dash R, Placzek W, Rega MF, Wu B, Cellitti J, Zhai D, Yang L, Dahl R, Fisher PB, Reed JC, Pellecchia M. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl- 2) family proteins. J Med Chem. 2010;53:4166–4176. doi: 10.1021/jm1001265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shelley MD, Hartley L, Fish RG, Groundwater P, Morgan JJ, Mort D, Mason M, Evans A. Stereo-specific cytotoxic effects of gossypol enantiomers and gossypolone in tumour cell lines. Cancer letters. 1999;135:171–180. doi: 10.1016/s0304-3835(98)00302-4. [DOI] [PubMed] [Google Scholar]
- 26.Flack MR, Knazek R, Reidenberg M. Gossypolone for the treatment of cancer. 40862. US patent. 2009
- 27.Wang S, Nikolovska-Coleska Z, Yang C-Y, Chen J. Apogossypolone and the uses thereof. WO 2006050447 A2 20060511. PCT Int Appl. 2006
- 28.Sun J, Li ZM, Hu ZY, Zeng ZL, Yang DJ, Jiang WQ. Apogossypolone inhibits cell growth by inducing cell cycle arrest in U937 cells. Oncology reports. 2009;22:193–198. doi: 10.3892/or_00000424. [DOI] [PubMed] [Google Scholar]
- 29.Lin J, Wu YJ, Yang DJ, Zhao Y-Q. Effect of apogossypolone on induction apoptosis in multiple myeloma cells and its mechanisms. Zhongguo shi yan xue ye xue za zhi/Zhongguo bing li sheng li xue hui = Journal of experimental hematology/Chinese Association of Pathophysiology. 2009;17:92–98. [PubMed] [Google Scholar]
- 30.Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, Mohammad RM. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Molecular cancer. 2008;7:20. doi: 10.1186/1476-4598-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mi JX, Wang GF, Wang HB, Sun XQ, Ni XY, Zhang XW, Tang JM, Yang DJ. Synergistic antitumoral activity and induction of apoptosis by novel pan Bcl-2 proteins inhibitor apogossypolone with adriamycin in human hepatocellular carcinoma. Acta Pharmacol Sin. 2008;29:1467–1477. doi: 10.1111/j.1745-7254.2008.00901.x. [DOI] [PubMed] [Google Scholar]
- 32.Hu ZY, Zhu XF, Zhong ZD, Sun J, Wang J, Yang D, Zeng YX. ApoG2, a novel inhibitor of antiapoptotic Bcl-2 family proteins, induces apoptosis and suppresses tumor growth in nasopharyngeal carcinoma xenografts. Int J Cancer. 2008;123:2418–2429. doi: 10.1002/ijc.23752. [DOI] [PubMed] [Google Scholar]
- 33.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 34.Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW. Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem. 2007;50:641–662. doi: 10.1021/jm061152t. [DOI] [PubMed] [Google Scholar]
- 35.Adams R, Butterbaugh DJ. Structure of Gossypol. X. Apogossypol and its Degradation Products. J Am Chem SOC. 1938;60:2174–2180. [Google Scholar]
- 36.Meltzer PC, Bickford HP, Lambert GJ. A Regioselective Route to Gossypol Analogues: The Synthesis of Gossypol and 5,5′-Didesisopropyl-5,5′-diethylgossypol. J Org Chem. 1985;50:3121–3124. [Google Scholar]
- 37.Yamanoi Y, Nishihara H. Direct and selective arylation of tertiary silanes with rhodium catalyst. J Org Chem. 2008;73:6671–6678. doi: 10.1021/jo8008148. [DOI] [PubMed] [Google Scholar]
- 38.Royer RE, Deck LM, Vander Jagt TJ, Martinez FJ, Mills RG, Young SA, Vander Jagt DL. Synthesis and anti-HIV activity of 1,1′-dideoxygossypol and related compounds. J Med Chem. 1995;38:2427–2432. doi: 10.1021/jm00013a018. [DOI] [PubMed] [Google Scholar]
- 39.Du Y, Liu R, Linn G, Zhao K. Synthesis of N-substituted indole derivatives via PIFAmediated intramolecular cyclization. Org Lett. 2006;8:5919–5922. doi: 10.1021/ol062288o. [DOI] [PubMed] [Google Scholar]
- 40.Islam I, Skibo EB, Dorr RT, Alberts DS. Structure-activity studies of antitumor agents based on pyrrolo[1,2-a]benzimidazoles: new reductive alkylating DNA cleaving agents. J Med Chem. 1991;34:2954–2961. doi: 10.1021/jm00114a003. [DOI] [PubMed] [Google Scholar]
- 41.Dao VT, Gaspard C, Mayer M, Werner GH, Nguyen SN, Michelot RJ. Synthesis and cytotoxicity of gossypol related compounds. Eur J Med Chem. 2000;35:805–813. doi: 10.1016/s0223-5234(00)00165-3. [DOI] [PubMed] [Google Scholar]
- 42.Tang G, Ding K, Nikolovska-Coleska Z, Yang CY, Qiu S, Shangary S, Wang R, Guo J, Gao W, Meagher J, Stuckey J, Krajewski K, Jiang S, Roller PP, Wang S. Structure-based design of flavonoid compounds as a new class of small-molecule inhibitors of the anti-apoptotic Bcl-2 proteins. J Med Chem. 2007;50:3163–3166. doi: 10.1021/jm070383c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakamoto K, Miyoshi H, Takegami K, Mogi T, Anraku Y, Iwamura H. Probing substrate binding site of the Escherichia coli quinol oxidases using synthetic ubiquinol analogues. J Biol Chem. 1996;271:29897–29902. doi: 10.1074/jbc.271.47.29897. [DOI] [PubMed] [Google Scholar]
- 44.Rega MF, Leone M, Jung D, Cotton NJ, Stebbins JL, Pellecchia M. Structurebased discovery of a new class of Bcl-xL antagonists. Bioorg Chem. 2007;35:344–353. doi: 10.1016/j.bioorg.2007.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wesarg E, Hoffarth S, Wiewrodt R, Kroll M, Biesterfeld S, Huber C, Schuler M. Targeting BCL-2 family proteins to overcome drug resistance in non-small cell lung cancer. Int J Cancer. 2007;121:2387–2394. doi: 10.1002/ijc.22977. [DOI] [PubMed] [Google Scholar]
- 46.Brien G, Trescol-Biemont MC, Bonnefoy-Berard N. Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene. 2007;26:5828–5832. doi: 10.1038/sj.onc.1210363. [DOI] [PubMed] [Google Scholar]
- 47.Li J, Viallet J, Haura EB. A small molecule pan-Bcl-2 family inhibitor, GX15-070, induces apoptosis and enhances cisplatin-induced apoptosis in non-small cell lung cancer cells. Cancer Chemother Pharmacol. 2008;61:525–534. doi: 10.1007/s00280-007-0499-3. [DOI] [PubMed] [Google Scholar]
- 48.Voortman J, Checinska A, Giaccone G, Rodriguez JA, Kruyt FA. Bortezomib, but not cisplatin, induces mitochondria-dependent apoptosis accompanied by up-regulation of noxa in the non-small cell lung cancer cell line NCI-H460. Mol Cancer Ther. 2007;6:1046–1053. doi: 10.1158/1535-7163.MCT-06-0577. [DOI] [PubMed] [Google Scholar]
- 49.Ferreira CG, Span SW, Peters GJ, Kruyt FA, Giaccone G. Chemotherapy triggers apoptosis in a caspase-8-dependent and mitochondria-controlled manner in the non-small cell lung cancer cell line NCI-H460. Cancer Res. 2000;60:7133–7141. [PubMed] [Google Scholar]
- 50.Cory S, Adams JM. Killing cancer cells by flipping the Bcl-2/Bax switch. Cancer cell. 2005;8:5–6. doi: 10.1016/j.ccr.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. Journal of molecular biology. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 52.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J Comput Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 53.Teschner M, Henn C, Vollhardt H, Reiling S, Brickmann J. Texture mapping: a new tool for molecular graphics. J Mol Graph. 1994;12:98–105. doi: 10.1016/0263-7855(94)80074-x. [DOI] [PubMed] [Google Scholar]
- 54.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of BclxL- Bak peptide complex: recognition between regulators of apoptosis. Science (New York, N Y) 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 55.Ramjaun AR, Tomlinson S, Eddaoudi A, Downward J. Upregulation of two BH3-only proteins, Bmf and Bim, during TGF beta-induced apoptosis. Oncogene. 2007;26:970–981. doi: 10.1038/sj.onc.1209852. [DOI] [PubMed] [Google Scholar]
- 56.Katsumata M, Siegel RM, Louie DC, Miyashita T, Tsujimoto Y, Nowell PC, Greene MI, Reed JC. Differential effects of Bcl-2 on T and B cells in transgenic mice. Proc Natl Acad Sci U S A. 1992;89:11376–11380. doi: 10.1073/pnas.89.23.11376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.