

Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder predominantly occurring in females with an incidence of 1:10,000 births and caused by sporadic mutations in the MECP2 gene, which encodes methyl-CpG-binding protein-2, an epigenetic transcription factor that binds methylated DNA. The clinical hallmarks include a period of apparently normal early development followed by a plateau and then subsequent frank regression. Impaired visual and aural contact often leads to an initial diagnosis of autism. The characterization of experimental models based on the loss-of-function of the mouse Mecp2 gene revealed that subtle changes in the morphology and function of brain cells and synapses have profound consequences on network activities that underlie critical brain functions. Furthermore, these experimental models have been used for successful reversals of RTT-like symptoms by genetic, pharmacological and environmental manipulations, raising hope for novel therapeutic strategies to improve the quality of life of RTT individuals.
1. INTRODUCTION
Rett syndrome (RTT; Online Mendelian Inheritance in Man #312750; http://www.ncbi.nlm.nih.gov/omim/), first recognized and described in German by Andreas Rett (1), is a neurodevelopmental disorder predominantly occurring in females. Almost 20 years later, Hagberg and colleagues presented the first description of RTT in the English language, leading to worldwide diagnosis in all ethnic and racial groups (2). Currently, the incidence of RTT is estimated to be approximately 1:10,000 female births (3, 4). Early studies proposing a genetic basis for RTT were later confirmed by Zoghbi and colleagues with the identification of mutations in the MECP2 gene located at chromosome Xq28 (5). MECP2 encodes methyl-CpG-binding protein-2, a transcription factor that binds methylated DNA and is ubiquitously expressed in mammalian tissues (6). Following the identification of loss-of-function mutations in the MECP2 gene in RTT individuals, research efforts expanded rapidly on an international scale with comprehensive clinical investigations into the complex array of medical and behavioral issues. In addition, intensive laboratory-based studies have been spurred by the availability of human autopsy tissue and experimental animal models, such as deletions of the endogenous Mecp2 gene (knockout), insertions of premature STOP codons or RTT-associated mutations (knock-in) common in the human MECP2 gene, and overexpression of Mecp2 to model the newly identified MECP2 duplication syndrome (7–9).
RTT is a sporadic condition in >99% of the cases, with the risk of familial recurrence being extremely low. Indeed, MECP2 mutations appear to be spontaneous transitions occurring in the paternal germline (10, 11), explaining in part the paucity of males with RTT or carrying MECP2 mutations. The clinical hallmarks of RTT include a period of apparently normal early development followed by a plateau or stagnation in development and a subsequent frank regression. It is during this period that both visual and aural contact is impaired leading to an initial diagnosis of autism. The convergence of clinical presentations in RTT and autism in association with MECP2 mutations represents an intriguing link between these disorders and other neurodevelopmental conditions with an established genetic basis and clinical features consistent with autism spectrum disorders, such as fragile X syndrome and Down syndrome (12). Here, we briefly review the features of RTT and its genetic bases. We then provide a detailed description of the existing mouse models of RTT based on MeCP2 dysfunction, and their experimental use to test novel therapeutic approaches for the reversal of established neurological deficits.
2. RTT NEUROPATHOLOGY
Gross anatomy and cellular morphology in autopsy brains from RTT individuals reveals very consistent and distinct features (13). First and foremost, the absence of any recognizable pattern of neuronal or glial cell atrophy, degeneration or death, gliosis, demyelination or neuronal migration defects, as well as the lack of disease progression is critical to differentiate RTT from a neurodegenerative disorder (14, 15).
Reduced brain and neuronal size with increased cell density is consistently observed in several brain regions, including cerebral cortex, hypothalamus and the hippocampal formation (16, 17). In addition, biopsies of nasal epithelium revealed far fewer terminally differentiated olfactory receptor neurons and significantly greater numbers of immature neurons in RTT individuals compared to unaffected controls (18). Furthermore, the size and complexity of dendritic trees was reduced in pyramidal cells of the frontal and motor cortices and of the subiculum (19, 20), while levels of microtubule-associated protein-2 (MAP-2), a protein involved in microtubule stabilization, were lower throughout the neocortex of RTT autopsy material (21, 22). In addition, autoradiography in frontal cortex and basal ganglia of autopsy RTT brains revealed complex, age-related abnormalities in the density of neurotransmitter receptors, such as excitatory NMDA-(N-methyl-d-aspartate), AMPA-(α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), kainate- and metabotropic-type glutamate receptors (GluRs) as well as inhibitory GABA receptors (23, 24). Furthermore, 1H spectroscopy at 4.1 Tesla revealed that the ratio of glutamate to N-acetylaspartate was elevated in gray matter of RTT individuals compared to their unaffected siblings, while unchanged in white matter (25). Finally, the density of dendritic spines is reduced in pyramidal neurons of the frontal cortex (14, 26) and of the CA1 region of the hippocampal formation (Fig. 1) (27), which is consistent with a reduced expression of cyclooxygenase, a protein enriched in dendritic spines (28). This so-called spine dysgenesis phenotype (29) is common to other neurodevelopmental disorders, including Down syndrome, autism, Angelman syndrome and fragile X syndrome (12, 20, 30). Such striking commonality across a spectrum of disorders, most with distinct molecular mechanisms, suggests a fundamental linkage through common pathways of neurobiological development responsible for cognitive performance (31).
FIGURE 1. Dendritic Spines of Pyramidal Neurons in Rat Hippocampal Slice Cultures and from Human Hippocampus.
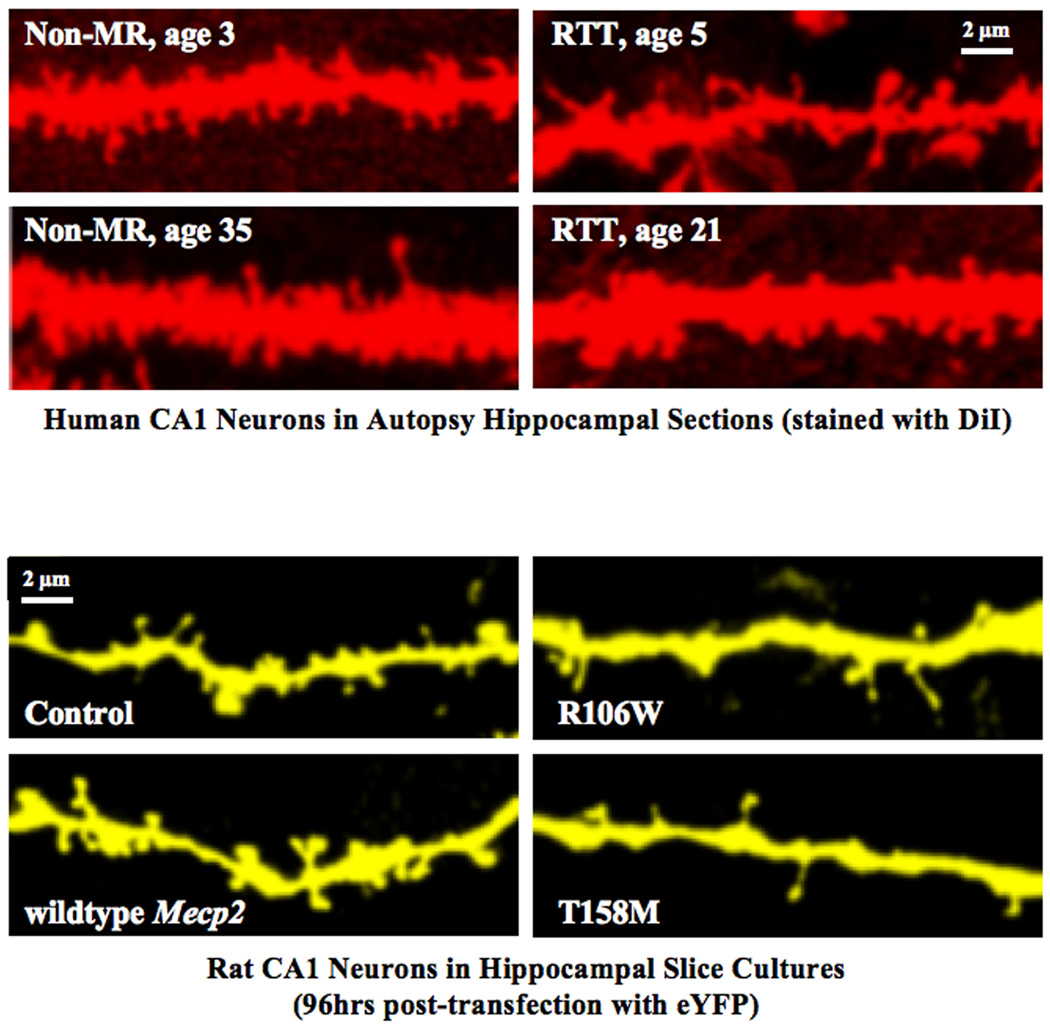
Upper panels: CA1 pyramidal neurons from human hippocampus stained with DiI by "diolistics" showing marked reduction and aberrant morphology of dendritic spines in samples from child (age 5) and adult (age 21) with RTT, compared to non-MR controls, child (age 3) and adult (age 35), respectively.
Lower panels: Rat CA1 neurons (96 hours post-transfection with enhanced yellow fluorescent protein, eYFP) in slice cultures showing an increase in spine density (wildtype MECP2 over-expression) and decrease in spine density (mutant R106W and T158M MECP2 over-expression).
Modified with permission from (27).
3. GENE FUNCTION: MECP2
MeCP2 is a member of a family of proteins that bind regions of DNA enriched with methylated CpG regions, i.e. cytosine and guanine nucleosides separated by a phosphate group (32). Two major functional domains characterize this family of proteins: the methyl-CpG binding domain (MBD) and the transcriptional repression domain (TRD) (33, 34). The best and first characterized function of Mecp2 is to repress gene transcription by recruiting co-repressor and the mSin3a/histone deacetylase complex (HDACs) and altering the structure of genomic DNA (35). This classical view of Mecp2 as a global transcriptional repressor exclusively has been questioned by the recent realization that out of all the genes misregulated in the hypothalamus of both MECP2 overexpressing and Mecp2 knockout mice (see below), the majority (~85%) was activated in the overexpressing mice and down-regulated in the knockout mice, which indicates that Mecp2 has a broader gene transcription role than originally thought (36). In addition, Mecp2 interacts with the RNA-binding protein Y box-binding protein 1 and regulates splicing of reporter minigenes, which may explain the aberrant alternative splicing patterns observed in Mecp2308/Y mice (37), which carry a premature STOP codon and express a truncated non-functional Mecp2 protein (see below) (38).
The human MECP2 gene has four exons (39), from which two protein isoforms differing in their N-termini are expressed by alternative splicing: MECP2-e1 (previously identified as MECP2B/MECP2α), the most abundant isoform; and MECP2-e2 (previously identified as MECP2A/MECP2β) (40, 41). Total protein and mRNA expression from the mouse Mecp2 gene (without differentiating between isoforms) is widely distributed throughout the developing and adult brain (38, 42, 43). More recently, brain-region specific splicing of the Mecp2 gene was observed during mouse brain development: Mecp2-e2 mRNA was enriched in the dorsal thalamus and layer V of the cerebral cortex, while more Mecp2-e1 transcript was detected in the hypothalamus than in the thalamus between postnatal days 1 and 21 (44).
So far, more than 250 different MECP2 mutations have been identified in individuals with RTT. However, 8 common point mutations (R106W, R133C, T158M, R168X, R255X, R270X, R294X, R306C) account for about 65% of those with RTT, while large deletions involving one or more exons, and 3’ deletions account for another 15–18% of RTT individuals (RettBASE: IRSF MECP2 Variation Database; http://mecp2.chw.edu.au/). While the vast majority of individuals expressing a MECP2 mutation do fulfill the diagnostic criteria for typical RTT, significant phenotypic variation is associated with such mutations (45, 46). These include individuals meeting variant atypical RTT criteria, female carriers who may be normal or have mild learning or cognitive disabilities, and more significantly include individuals with prominent behavioral phenotypes, such as autism and obsessive-compulsive and aggressive behaviors in associations with moderate to severe cognitive delay. Individuals meeting the RTT variant criteria, such as preserved speech but with a significant delayed onset (up to 10 years of age or more) are noted in association with milder involvement, whereas early onset seizures and congenital onset are noted with more severe clinical features (45, 47). Inasmuch as RTT is a sporadic condition with extremely low risk of familial recurrence, female carriers represent less than 3% of the total participants in the RTT natural history study (48). However, this group is likely to be under-represented because they would not be recognized if it not were for an affected child or sibling.
A possible explanation for the wide phenotypic variability in clinical presentations is skewing of the normally expected random X chromosome inactivation (XCI), whereby the inactivation of one of the X chromosomes in every female cell typically permits the expression of genes from the active X chromosome in the adult. As such, individuals with normal function or only mild involvement would represent cases of wildtype MECP2 expression in the majority of cells. However, it is important to mention that this explanation may account for only ~20% of RTT cases related to variances in severity (49).
Among males, the most common MECP2 dysfunction is associated with duplications of the Xq28 region, which include variable numbers of other genes. However, the principal clinical features in these male individuals appear to relate to the overexpression of MECP2, because overexpressing the human MECP2 gene in mice caused significant neurological deficits (see below) (50). Thus, a MECP2 duplication syndrome has been defined (7–9). Male individuals carrying MECP2 mutations generally have non-specific cognitive delay with or without progressive motor problems or a severe early infantile encephalopathy (51), as well as clear autistic features (52). As with carrier females, these males are likely under-represented, unless a sibling has been identified with RTT or a progressive encephalopathy. In addition, typical RTT features have been described in a small number of males with either somatic mosaicism or in combination with Klinefelter syndrome (47XXY), because in both instances, two cell populations of X chromosomes would exist, similar to females with RTT.
4. MODEL SYSTEMS: Mecp2 KNOCKOUT, Mecp2 MUTANT AND MECP2 KNOCK-IN MICE
To advance the basic knowledge of the role of Mecp2 on brain development and function, as well as to understand the molecular and cellular bases of RTT pathophysiology, several different mouse models were generated based on targeted manipulations of the endogenous Mecp2 gene or targeted introduction of the human MECP2 gene, either wildtype or carrying RTT-associated mutations. The following mouse models have been generated, and their similarities to the clinical presentation in RTT individuals will be discussed in the next section.
(1) Mecp2 knockout mice
The first report of constitutive Mecp2 deletion in mice described embryonic lethality (53), which led to the generation of mice using the Cre recombinase-loxP system of conditional deletion in selected tissues at desired times (54). Embryonic lethality was prevented by breeding mice that have Mecp2 exons 3 and 4 flanked by loxP sites (i.e. “floxed” or Mecp22lox) with “Cre deleter” mice that ubiquitously express a Cre transgene in the X chromosome (55). The resulting progeny (Mecp2tm1.1Bird, “Bird strain”) carry deletions spanning Mecp2 exons 3 and 4 starting in early embryonic development (56). These mice showed no signs of Mecp2 expression using antibodies directed against either N- or C-termini of the protein (56, 57). In addition, brain-specific targeting to the neuron and glial lineage was achieved by breeding floxed Mecp2 mice with mice expressing a Nestin-Cre transgene, which is highly expressed in neural precursor cells beginning at E12 (58). However, it should be noted that the mice with Mecp2 exons flanked by loxP sites used in the generation of these knockouts (Bird Mecp22lox/Y) already show ~50% lower levels of Mecp2 protein than wildtype controls (without Cre-mediated recombination), and thus should be considered “Mecp2 hypomorphs” (see below) (59).
Another strain of Mecp2 knockout mice was generated by removal of the coding methyl-CpG-binding domain (MBD) sequence (entire exon 3 and part of exon 4) and introduction of a non-functional splicing site at the 5' end of the gene sequence encoding the transcriptional repression domain (TRD), preventing splicing and transcription of downstream Mecp2 sequence for the TRD, the C-terminal domain and the 3' UTR (Mecp2tm1Tam) (60). These mice showed no signs of Mecp2 protein expression using antibodies directed against either N- or C-termini, and developed a postnatal behavioral phenotype that resembles RTT.
(2) Mecp2 mutant mice expressing a truncated protein
Brain-specific deletions of Mecp2 exon 3, which encodes 116 amino acids including most of the MBD, led to the expression of a truncated protein in the neuron and astrocyte lineage beginning at E12 using a Nestin-Cre transgene (61) (Mecp22lox/X; Nestin-Cre). Because these mice express a truncated Mecp2 protein lacking the MBD but with an intact C-terminus – which includes the nuclear localization signal and potentially the TRD and other downstream domains (57, 61, 62) – they should be considered mutant mice rather than “knockout” or “null” mice. Therefore, the consequences of the expression of a mutant protein of unknown function should be taken into account. In addition, it is unknown whether the introduction of the loxP sites flanking Mecp2 exon 3 in Jaenisch Mecp22lox mice (before Cre-mediated loxP deletion) had consequences on Mecp2 protein expression similar to that detected in Bird Mecp22lox/Y mice (59). It should be noted that most studies of this so-called “Jaenisch strain” use mice derived by germline recombination (Mecp21lox) where the mutations in Mecp2 are not longer brain-specific (61).
The introduction of a premature STOP codon in the mouse Mecp2 gene led to the expression of a protein truncated at amino-acid 308 (Mecp2308) (38). These mice express a truncated protein with the MBD and a portion of the TRD elements still intact, suggesting residual protein function, as thought to be the case in RTT patients presenting with milder disease features.
(3) Cell-type specific Mecp2 deletions or mutations
The Cre/loxP recombination system has been used to generate the following conditional Mecp2 knockout or mutant mice:
Breeding Jaenisch Mecp22lox mice with mice expressing Cre under control of the CamkII promoter (CamkII-Cre93 line) (63) allowed the Mecp2 mutations to be selectively expressed in forebrain excitatory neurons after approximately postnatal-day 20 (61). It should be noted that these mice express a milder behavioral phenotype with a delayed onset compared to that caused by more widespread deletion (56) or truncation (61) of Mecp2 using the Nestin-Cre transgene.
Breeding Bird Mecp22lox/Y mice with mice expressing Cre under control of the Sim1 promoter (64) yielded cell-type specific Mecp2 deletions in hypothalamic neurons (65).
Breeding Bird Mecp22lox/Y mice with mice expressing Cre in tyrosine hydroxylase (TH) neurons (66) generated another cell-type specific Mecp2 deletion (67).
Breeding Bird Mecp22lox/Y mice with mice expressing Cre in PC12 ets factor 1 (PET1)-expressing neurons (68) generated another cell-type specific Mecp2 deletion (67).
Lastly, infusions of Cre-expressing lentiviruses into specific brain areas of Jaenisch Mecp22lox mice yielded useful mouse models of RTT amenable for behavioral studies without confounding issues of brain development (69).
(4) Mice expressing reduced levels of Mecp2
The introduction of loxP sites flanking Mecp2 exons 3 and 4 for the generation of conditional knockout mice of the Bird knockout mouse line (56) may be the reason for a ~ 50% reduction in the expression of Mecp2 protein in male Mecp22lox/Y compared to wildtype littermates (59). Interestingly, these mice express a delayed and milder behavioral phenotype similar to other Mecp2-deficient mice, which may originate from reduced expression levels of wildtype Mecp2 protein.
An alternative approach to reduce the expression of endogenous Mecp2 is by knock-down with small interference RNAs (70, 71). Intraventricular injections of lentiviruses that express a Mecp2-targeted short hairpin RNA (shRNA) in 1 day-old rat pups reduced Mecp2 mRNA levels and caused subtle but transient sensory-motor impairments (72). So far, the in utero transfection of shRNA constructs to knock-down Mecp2 expression in sparsely distributed cortical pyramidal neurons in mice has been used to map intracortical connectivity by glutamate uncaging and laser scanning photostimulation in brain slices (73).
(5) Mice overexpressing wildtype full-length MeCP2
The introduction of the human MECP2 gene under control of its entire regulatory promoter using the P1-derived artificial chromosome (PAC) led to a ~2-fold increase in expression levels compared to the endogenous mouse Mecp2 (MeCP2Tg1 mice) (50). Another mouse line overexpressed a Tau-MeCP2 fusion protein selectively in postmitotic neurons from the Tau locus in homozygous Tau knockout mice (62).
(6) Knock-in mice carrying RTT-associated MECP2 mutations
So far, only 2 mouse strains carrying RTT-associated mutations have been generated: the R168X mouse (Mecp2R168X) (74), and the MeCP2 A140V mouse (75). In theory, female heterozygous mice with mosaic expression of RTT-associated mutations in MECP2 would represent the closest experimental animal model of the human disease. The next closest experimental model would be human neurons derived from induced pluripotent stem (iPS) cells obtained from individual RTT patients (e.g. (76), with the obvious limitation of being a dissociated culture system allowing only studies at the molecular and cellular level, and not network or behavioral studies.
The most important feature of all these genetically manipulated mice is that all present some behavioral features that correlate well with specific clinical symptoms observed in RTT individuals, although no single mouse line truly mimics the human disease (reviewed by 77, 78).
It should be stressed that most studies use male hemizygous mice (i.e. Mecp2−/y) because they consistently develop a severe and characteristic behavioral phenotype much earlier than female heterozygous mice (i.e. Mecp2−/+), which express a mosaic pattern of wildtype and mutant cells due to XCI. However, XCI is not uniform in female heterozygous mice from the Mecp2tm1.1Jae and Mecp2308/X mutant lines, or from the Mecp2tm1.1Bird knockout strains, being skewed towards the wildtype Mecp2 allele (57, 79). Intriguingly, wildtype cells in Mecp2tm1.1Jae mutant and Mecp2tm1.1Bird knockout mice express lower levels of Mecp2 protein than in wildtype mice (57). Therefore, the delayed appearance and variability of the behavioral phenotypes observed in Mecp2−/+ heterozygous female mice could be caused by the combination of mosaic expression of mutant Mecp2, the degree of XCI unbalance, as well as reduced Mecp2 levels in wildtype cells. To simplify the analyses by reducing the contribution of these confounding factors, Mecp2−/y male hemizygous mice are used as a more homogenous population, which seem more amenable for experimental work in the laboratory. There are however, a number of studies that compare RTT-like phenotypes between Mecp2−/+ heterozygous female and Mecp2−/y hemizygous male mice and their wildtype littermates, and all agree that female heterozygous mice display a delayed onset, milder phenotype than male hemizygous mice (e.g. 80–90).
Another critical point is that only one study characterized the consequences of the complete deletion of the Mecp2 protein without potential deleterious effects of genetic engineering (e.g. Neo cassettes) (60), since at least one of the mouse lines commonly called “Mecp2 null” or “Mecp2 knockout” in fact expresses an internally deleted protein detectable by western blotting and immunohistochemstry (57, 61, 62). Despite these limitations, Mecp2-based mouse models represent useful experimental models of RTT in which to test novel therapeutic approaches before moving to the clinic.
4.1 General features of mouse models based on Mecp2 dysfunction
Male hemizygous mice of the Mecp2 knockout strains (Mecp2tm1.1Bird and Mecp2tm1Tam) (56, 60), and of the Mecp2 mutant line (Mecp2tm1.1Jae) (61) appeared normal until the 4th – 5th week of life when a consistent behavioral phenotype appears, which include unusual gait with splaying hind limbs, clasping of hind limb when suspended by the tail, dishevelled fur and erected whiskers, labored breathing, tremors and episodes of seizures. This phenotype progresses in severity, and also includes varied neurological features of sensory-motor or cognitive origin, such as increased anxiety-related behaviors and reduced social interactions with novel mice, learning and memory deficits. From the onset of these symptoms, marked weight loss, kyphosis and irregular breathing become more severe, until these mice die at ~10 weeks of life. Considering that a conditional deletion in neural precursor cells starting at ~E12 using Nestin-Cre mice (56, 61) gave similar phenotypes to widespread deletions using X chromosome-Cre transgenic mice (56), the complex phenotype seems to arise from impaired Mecp2 expression in the CNS. It should be noted that more restricted deletions in forebrain excitatory neurons starting at ~P20 using CamkII-Cre deleter mice cause a delayed and milder phenotype (61), suggesting that proper Mecp2 expression in both neurons and glial cells is critical for CNS development (see below). As mentioned above, this symptomatology is highly reminiscent of the complex clinical manifestations observed in RTT individuals used for inclusion criteria, with the exception of the progressive process (2, 48, 91).
Similar to the Mecp2 knockout and mutant strains, male hemizygous of the Mecp2308/Y strain appeared normal until the 6th week of life when they develop a similar but milder neurological phenotype. The features of that phenotype included tremors, motor impairments, hypoactivity, increased anxiety-related behavior, seizures, kyphosis, and stereotypic forelimb motions (38), as well as deficits in hippocampus-dependent spatial memory, contextual fear memory, and social memory (92).
Consistent with XCI of the mutant Mecp2 allele in a fraction of cells, female heterozygous mice of the Mecp2 knockout, mutant and truncated Mecp2308 strains develop a milder behavioral phenotype than the hemizygous male littermates, but with a delayed onset. In addition, the features of the phenotype in heterozygous females are much more variable than in hemizygous males.
Forebrain neuron-specific conditional Mecp2 mutant mice generated by crossing CamkII-Cre93 mice (63) with Jaenisch floxed mice (Mecp22lox) showed behavioral abnormalities similar to RTT phenotypes, including hind limb clasping, impaired motor coordination, increased anxiety, and abnormal congener social behavior (61). However, locomotor activity and context-dependent fear conditioning were unaffected in these mice (93).
Hypothalamus-specific conditional Mecp2 knockout mice generated by crossing Sim1-Cre BAC transgenic mice (64) with Bird floxed mice (Mecp22lox/Y) lacked Mecp2 protein during embryonic development and after birth in the paraventricular, supraoptic, and posterior hypothalamic nuclei, as well as in the nucleus of the lateral olfactory tract of the amygdala (65). These mice recapitulated the abnormal physiological stress response that is seen upon expression of a truncated non-functional Mecp2 protein in the entire brain (38). Hypothalamus-specific Mecp2 conditional knockout mice were also aggressive, hyperphagic, and obese, consistent with a role for Mecp2 in the regulation of social and feeding behaviors. It should be noted that reduced Mecp2 protein levels in Bird Mecp22lox/Y mice were taken in consideration when comparing different genotypes in this study (65).
Deletion of Mecp2 in TH-positive dopaminergic and noradrenergic neurons by breeding TH-Cre transgenic mouse line (66) with Bird Mecp22lox/Y mice caused a specific alteration in locomotor activity (i.e. decreased total distance and vertical activity in the open field, poor performance on the dowel walking task), without any impairments in motor learning, anxiety, social interaction, breathing patterns and learning and memory (67). In this study as well, reduced Mecp2 protein levels in Bird Mecp22lox/Y mice were taken in consideration.
Deletion of Mecp2 in serotonergic neurons of the dorsal and medial raphe nuclei by breeding PET1-Cre transgenic mouse line (68) with Bird Mecp22lox/Y mice caused an increased aggressive behavior (i.e. congener resident intruder test), without affecting motor function, anxiety, self-grooming or repetitive behaviors, breathing patterns, motor learning or learning and memory. Also here, reduced Mecp2 protein levels in Bird Mecp22lox/Y mice were taken in consideration (67).
Bilateral injections of adeno-associated viruses that express GFP-tagged Cre recombinase in the basolateral amygdala of Jaenisch Mecp22lox mice caused elevated anxiety-like behavior with normal locomotion, motor coordination and social interaction behavior (69). In addition, these mice have impaired cue-dependent fear conditioning. However, it is unknown whether Jaenisch Mecp22lox mice show a similar reduction in Mecp2 protein like that detected in Bird Mecp22lox/Y mice.
Supporting the notion that proper and tightly controlled dosage of MeCP2 protein are critical for brain development and function, mice overexpressing human MeCP2 at approximately twice the endogenous levels (MeCP2Tg1) exhibited a delayed (10th week) neurological syndrome that included enhanced motor and contextual learning and enhanced synaptic plasticity in the hippocampus. After 20 weeks of age, these mice developed seizures, became hypoactive and approximately 30% of them died by 1 year of age (50). Without exception, higher levels of MeCP2 caused a more severe neurological syndrome (50), even when expressed exclusively in postmitotic neurons (62).
Consistent with this model of tightly controlled dosage of MeCP2 protein, a ~50% reduction in expression of the wildtype Mecp2 protein led to a neurological phenotype in male Bird Mecp22lox/Y mice, before crossing with any Cre deleter line. These mice showed learning and motor deficits, decreased anxiety, altered social behavior and nest building, decreased pain recognition and disrupted breathing patterns (59). Although no typical RTT-like symptoms were observed in rat pups injected with lentiviruses expressing Mecp2-targeted sh/RNAs, they displayed some transiently altered sensory-motor reflexes and neurobehavioral abnormalities during early development (72).
The introduction of human MECP2 carrying RTT-specific mutations also caused neurological symptoms related to the human disease. The R168X mice (Mecp2R168X) carry a premature STOP codon in position 168, and thus do not express full-length wildtype Mecp2 protein. The truncated protein contains an intact MBD but lacks the TRD and C-terminal domains including the nuclear localization signal. As in the other Mecp2 mouse lines, male hemizygotes are more severely affected than the female heterozygotes, with a shortened lifespan and neurological features starting by the 5th – 6th week of life, which include forelimb stereotypies, hindlimb clasping and atrophy, hypoactivity, and breathing irregularities; female heterozygotes showed similar signs only after 6 month of age (74). It should be noted that, during the cloning of the mutant constructs, additional changes were introduced between codons 167 and 172. Since the STOP codon is at codon 168, these changes are not represented in the mutant Mecp2 protein; however, these mice are not useful to test the effectiveness of read-through compounds that skip premature STOP codon mutations (94).
The A140V MeCP2 mutation is noted only in ~0.6% of individuals, the majority being males with X-linked mental retardation, and including some with manic-depressive or schizophrenic behaviors (RettBASE: IRSF MECP2 Variation Database; http://mecp2.chw.edu.au/). Of the 7 females carrying this MECP2 mutation listed in the RettBASE, most were unaffected siblings and none had RTT. In addition, The A140V MeCP2 mutation has been described elsewhere in familial X-linked mental retardation, and in Parkinsonism/Pyramidal signs/Macroorchidism/X-linked mental retardation (95). The A140V mutant MeCP2 protein has been reported to preserve the methyl-CpG binding while compromising its ability to bind to the mental retardation associated protein ATRX (96). In contrast to the other mouse models, male hemizygous mice carrying the A140V Mecp2 mutation have an apparently normal lifespan and normal weight gain patterns without obvious seizures, tremors, breathing difficulties or kyphosis. However, they show some typical features of autopsy brains from RTT individuals, such as increased cell packing density and reduced complexity of dendritic branching (75).
4.2 On the morphology of neurons and synapses in Mecp2-based mouse models of RTT
The most prototypical morphological features of autopsy brains from RTT individuals at the anatomical, histological and cytological levels are present in brains from Bird Mecp2 knockout and Jaenisch Mecp2 mutant mice, which are the most widely studied mouse models of RTT. For example, these mice have smaller cortical neurons packed at a higher density than their wildtype littermates (43, 61, 81). In addition, pyramidal neurons in the cortex (43) and hippocampal CA3 region, as well as granule cells of the dentate gyrus show reduced dendritic complexity (97). In addition, Jaenisch Mecp2 mutant mice show a disorganized olfactory neuroepithelium indicative of delayed terminal differentiation, in addition to impaired turnover of olfactory neurons (98). In MeCP2 A140V mice, pyramidal neurons in layer II/III of the somatosensory cortex also have decreased dendritic complexity compared to wildtype controls (75). However, dendritic branching in layer III and V pyramidal neurons of the frontal cortex of male Mecp2308/Y mice was comparable to that in wildtype controls (92).
In addition to smaller neurons with fewer dendritic branches, RTT mouse models exhibit the so-called spine dysgenesis phenotype (29), which is common to other neurodevelopmental disorders, including Down syndrome, autism, Angelman syndrome and fragile X syndrome (12, 20, 30, 31). Pyramidal neurons in the somatosensory cortex of 6 week-old Bird Mecp2 knockout mice have lower spine densities compared to wildtype controls (99). Newly generated granule cells in the dentate gyrus of 8 week-old Jaenisch Mecp2 mutant mice also show impaired dendritic spine density and distribution (100). Dendritic spine density is also lower in pyramidal neurons of layers V-VI of motor cortex from male hemizygous and female heterozygous Bird Mecp2 knockout mice (86). The onset of this dendritic spine phenotype is delayed in female heterozygous mice, and it seemed more severe in Mecp2-expressing neurons than in Mecp2-lacking neurons, suggesting both cell autonomous and non-cell autonomous consequences of Mecp2 deletion (86). In addition to similar lower spine densities, the intensity of immunolabeling for PSD-95 (a postsynaptic marker of mature excitatory synapses) was lower in layer V pyramidal neurons of motor cortex of Jaenisch Mecp2 mutant mice (101). Furthermore, pyramidal neurons in CA1 region of the hippocampus from Bird Mecp2 knockout mice and Jaenisch Mecp2 mutant mice also show lower spine densities, in addition to dendritic swelling, reduced diameter of dendritic spine heads and elongation of their necks (102). Intriguingly, this study described significant differences between the 2 mouse strains examined, suggesting different consequences of a deletion (56) and expression of a truncated Mecp2 protein (61).
It should be noted that the lower dendritic spine density observed in RTT mouse models is not a consistent phenotype; for example Kishi and Macklis (2004) described comparable spine densities in cortical layers II/III of either Bird Mecp2 knockout or Jaenisch Mecp2 mutant mice and their wildtype controls at 8 weeks of age (43). Similarly, neither dendritic spine density in layer III and V pyramidal neurons of the frontal cortex nor asymmetric spine synapse density in area CA1 were affected in male Mecp2308/Y mice, despite significant impairments in hippocampal-dependent learning and memory, as well as hippocampal synaptic transmission and plasticity (see below); however, the length of individual postsynaptic densities (PSDs) was shorter in the mutant mice (92).
With regards to the morphology of axons and presynaptic terminals, the motor cortex of Bird Mecp2 knockout mice show defects in axonal fasciculation (102). Similarly, Jaenisch Mecp2 mutant mice show impairments in olfactory axonal fasciculation, guidance and targeting before synaptogenesis (103). In addition, the immunolabeling intensity of VGLUT1 (vesicular glutamate transporter) – a presynaptic marker of mature excitatory synapses – is lower in the dendritic region of the hippocampal CA1 region from Bird Mecp2 knockout compared to wildtype controls, but only at 2 weeks of age (104). Since the immunolabeling intensity of the postsynaptic dendritic marker MAP2 was not different, the authors interpreted that Mecp2 deletion caused a reduction in the number of mature synapses in area CA1, consistent with their results from autaptic neuronal cultures (see below) (104). On the other hand, the number of docked and total synaptic vesicles within presynaptic terminals was unaffected in area CA1 of Mecp2308 (92).
The consequences of Mecp2 deletion on synaptic circuits are different across the CNS, even for specific neuronal subtypes such as inhibitory GABAergic interneurons. The cerebellum of Jaenisch Mecp2 mutant mice shows more GABA positive presynaptic terminals from stellate and basket cells onto Purkinje cells than in wildtype controls, while the density of GABA-positive presynaptic terminals was not affected in layer III of the somatosensory cortex (88). These differences may even occur within individual brain regions. In the thalamus of Bird Mecp2 knockout mice for example, the ventral basal complex shows fewer puncta immunopositive for VGAT (vesicular GABA transporter, a presynaptic marker of mature inhibitory synapses), while the reticular thalamic nucleus shows more VGAT-positive puncta (105).
4.3 On the physiology of neurons and synapses in Mecp2-based mouse models of RTT
Intrinsic properties
Despite the consistent observations of smaller neuronal somata and dendritic arborizations, most electrophysiological studies in brain slices describe that intrinsic membrane properties are not affected in Mecp2 knockout or Mecp2 mutant mice. It should be noted that the majority of such studies were performed in brain slices from young mice before the appearance of RTT-like symptoms, and thus it is unknown whether these features deteriorate later in symptomatic mice. For example, layer V pyramidal neurons in slices of primary somatosensory cortex from Jaenisch Mecp2 mutant mice have similar resting membrane potential (RMP), input resistance (Ri), and intrinsic excitability (i.e. spike rate vs. current injection relationships) to their wildtype controls (106). Pyramidal neurons layer II/III of the frontal cortex of Bird Mecp2 knockout mice also show input resistances comparable to those in wildtype, although the effect ouabain, a Na+,K+-ATPase inhibitor, was smaller in knockout neurons. The reduced basal activity of this electrogenic pump in Mecp2 null neurons seems to originate from increased expression levels of FXYD, a small, single-spanning membrane protein that controls cell excitability by modulating Na+,K+-ATPase activity (107).
Similarly, CA3 and CA1 pyramidal neurons in hippocampal slices from Bird Mecp2 knockout mice show unaffected RMP, Ri and action potential amplitude and threshold (108). Intrinsic membrane properties – such as RMP, Ri, membrane capacitance (Cm), action potential threshold and firing patterns – were also unaffected in inhibitory neurons of the reticular thalamic nucleus (105). Relay neurons in the nucleus tractus solitarius of the brainstem from Jaenisch Mecp2 mutant mice also showed RMP, Ri, action potential amplitude and threshold, afterhyperpolarizations and intrinsic excitability (i.e. spike rate vs. current injection relationship) comparable to those in wildtype controls (109). In addition, expression of a truncated non-functional protein did not affect Ri and Cm in principal neurons of the lateral nucleus of the amygdala from Mecp2308/Y mice (110). Finally, Mecp2 knockdown in cortical layer II/III pyramidal neurons by in utero electroporation did not affect RMP, Ri, membrane time constant, voltage-current relationship, rheobase (i.e. lowest-amplitude current step evoking at least one action potential), action potential half-width, spike frequency adaptation, or firing-current relationships (73).
On the other hand, TH-positive neurons in the locus ceruleus of Jaenisch Mecp2 mutant mice have lower membrane conductance, Cm, slow afterhyperpolarizations and spike frequency adaptation than wildtype controls (all consistent with significantly smaller neuronal somata), all of which could contribute to their enhanced intrinsic excitability (i.e. spike rate vs. current injection relationship) (111).
Synaptic properties
As with morphological features, the consequences of Mecp2 dysfunction on the physiology of synapses vary across different brain regions and with neurotransmitter types. In slices of primary somatosensory cortex from Jaenisch Mecp2 mutant mice, the cumulative charge (Q) of spontaneous excitatory postsynaptic currents (EPSCs) recorded in layer V pyramidal neurons was smaller, while the charge of spontaneous inhibitory postsynaptic currents (IPSCs) was larger than wildtype controls, which may contribute to the lower spontaneous firing rate of Mecp2 mutant pyramidal neurons (106). These differences seem to arise from impaired quantal transmitter release from excitatory synapses because action potential-independent miniature EPSCs (mEPSCs) were smaller in Mecp2 mutant neurons (without changes in their frequency), whereas miniature IPSCs (mIPSCs) were unaffected (106). In addition, studies of monosynaptic connections between layer V pyramidal neurons in cortical slices revealed reduced connectivity and weaker individual connections (112), which likely contribute to reduced network activity that is reflected in the lower frequency of spontaneous action potentials recorded in Mecp2 mutant neurons (106, 113). On the other hand, the input-output relationship of extracellularly recorded fEPSPs evoked in layer II/III of primary somatosensory cortex by stimulation in white matter/layer IV was comparable between Jaenisch Mecp2 mutant mice and wildtype controls (88). Similarly, input-output relationships of fEPSPs in layers II/III of slices from primary motor or primary sensory cortices were unaffected by expression of truncated Mecp2 in Mecp2308/Y mice (92).
The consequences of embryonic Mecp2 knockdown on the degree of synaptic connectivity in postnatal cortical slices was also evaluated by comparing input maps generated by glutamate uncaging onto layer II/III pyramidal neurons using laser-scanning photostimulation. In utero electroporation of Mecp2-specific shRNA constructs caused a significant reduction in local excitatory input pathways in Mecp2-knockdown neurons compared to untransfected neighbors, without changes in inhibitory synaptic inputs (73).
The brainstem of Mecp2 knockout and Mecp2 mutant mice also exhibits altered synaptic transmission. The amplitude and frequency of spontaneous EPSCs recorded from neurons in the rostroventrolateral medulla (which included the pre-Bötzinger complex) were higher, while the amplitude and frequency of spontaneous IPSCs were lower in brainstem slices from 1 week-old Mecp2 knockout mice compared to wildtype controls, well before any RTT-like symptoms are observed (114). Such consequence on GABAergic transmission reflects differences in presynaptic function because the frequency of both spontaneous mIPSCs and sucrose-evoked postsynaptic currents was lower in Mecp2 mutant neurons; however, postsynaptic GABAA receptors were also impaired, as reflected by smaller agonist-induced currents in Mecp2 knockout slices (114). Similarly, evoked EPSCs and spontaneous mEPSCs were larger in relay neurons of the nucleus tractus solitarius in the brainstem of Mecp2 mutant mice than in wildtype controls, which led to a higher probability of action potential firing in response to afferent stimulation (109). These studies demonstrate an imbalance of excitatory and inhibitory synaptic input to brainstem circuits responsible for the respiratory rhythm, and thus strongly implicate this synaptic dysfunction in the breathing irregularities experienced by RTT individuals. Raising hopes for novel therapeutic approaches for RTT, breathing irregularities in Mecp2 mutant mice were reverted by treatment with an AMPAkine drug that facilitates activation of glutamatergic AMPA receptors and increases the expression of brain-derived neurotrophic factor (BDNF) (115), consistent with the reversal of synaptic dysfunction by direct application of recombinant BDNF to brainstem slices (109).
Altered synaptic properties were also found in the amygdala of Mecp2308/Y mice that express a truncated non-functional protein, albeit restricted to its cortical inputs. Intracellular whole-cell recordings from principal neurons of the lateral nucleus of the amygdala revealed that both synaptic maturation and elimination during development were enhanced in slices from Mecp2308/Y mice, but only on those synapses made by cortical afferents and not by projections from the thalamus (110). Whether the probability of neurotransmitter release (Pr) at these cortico-amygdalar synapses is affected in Mecp2308/Y slices is unclear, because the paired-pulse ratio is smaller (consistent with higher Pr, see (116) but the maximal amplitude of evoked EPSCs, the amplitude of EPSCs evoked by minimal stimulation, and the frequency of spontaneous mEPSCs are all smaller than in wildtype slices (110), which is consistent with lower Pr.
On the other hand, studies of the consequences of Mecp2 dysfunction on synaptic transmission in the hippocampus have yielded more conflicting results. Extracellular recordings of field excitatory postsynaptic potentials (fEPSPs) evoked in area CA1 by stimulation of Schaffer collaterals (the axons of CA3 pyramidal neurons) revealed that basal excitatory synaptic transmission is not affected by either Mecp2 deletion in Bird knockout mice or expression of a mutant protein in Jaenisch mutant mice (117). However, paired-pulse facilitation of fEPSPs at these synapses was smaller in Mecp2 deficient slices, which could be interpreted as a higher release probability (Pr) (see (116); it is unclear though, why this potential difference in Pr was not reflected in the amplitude of post-tetanic potentiation or in the slope of input-output relationships (117). On the other hand, similar recordings performed in area CA1 of hippocampal slices from Mecp2308/Y mice expressing a truncated Mecp2 protein showed that excitatory synaptic transmission is enhanced compared to wildtype controls, as reflected in steeper input-output relationships of fEPSPs and reduced paired-pulse facilitation (at the shortest inter-stimulus intervals, 10ms and 20ms) (92).
The CA3 region of the hippocampus seems to exhibit pronounced changes in Mecp2 knockout mice. Intracellular whole-cell recordings from CA3 pyramidal neurons demonstrated that spontaneous IPSCs were larger in hippocampal slices from Bird knockout mice, suggesting that the activity of GABAergic interneurons is more synchronized than in wildtype slices (108). In addition, the cumulative charge transfer and the amplitude of spontaneous EPSCs in CA3 pyramidal was smaller than in wildtype slices, which may contribute to the slower frequency of spontaneous rhythmic field potentials exhibited by the Mecp2 knockout slices. Paradoxically, this combination leads to a hippocampal circuitry more prone to hyperexcitability, as reflected by the repetitive sharp wave-like discharges induced in area CA3 by a brief high-frequency stimulation to the alveus and CA3 stratum oriens, which contain recurrent collaterals of CA3 pyramidal neurons (108).
Consistent with the notion that tightly controlled MeCP2 levels are critical for brain development and function, fEPSPs evoked in area CA1 of slices from MECP2 overexpressing (MeCP2Tg1) showed larger paired-pulse facilitation than in wildtype slices. Since the input-output relationships of fEPSPs was unaffected by MECP2 overexpression, those results were interpreted as presynaptic differences in release probability (50).
Long-term synaptic plasticity
Despite the various discrepancies mentioned above regarding the consequences of Mecp2 dysfunction on synaptic transmission and short-term plasticity at excitatory synapses, more consistent results have been obtained with respect to long-term synaptic plasticity (118). Several studies in Mecp2 deficient mice have documented impairments in long-term potentiation (LTP) and depression (LTD), two widely accepted forms of synaptic plasticity thought to underlie experience-dependent modifications of brain function, including learning and memory (119).
When recorded with extracellular electrodes, the strength of excitatory synapses within layer II/III of slices from primary somatosensory cortex of Jaenisch Mecp2 mutant mice displayed smaller LTP after theta-burst stimulation (TBS) of white matter/layer VI inputs than that recorded in wildtype controls, an impairment reversed by environmental enrichment (see below) (88). Extracellular fEPSPs evoked in layer II/III of slices from primary motor or primary sensory cortices of Mecp2308/Y mice expressing truncated Mecp2 also showed smaller TBS-induced LTP than that shown by wildtype controls (92). However, an intracellular study of monosynaptic connections between pyramidal neurons in layer V of primary somatosensory cortex of Jaenisch Mecp2 mutant mice demonstrated that LTP was intact at these synapses, provided that sufficient postsynaptic depolarization was achieved by step depolarization or by evoked action potentials during the induction of spike-timing dependent plasticity (112). Thus, the sparser connectivity with weaker synapses could be the reason of reduced LTP amplitude when induced without control of postsynaptic depolarization (as with extracellular recordings of fEPSPs). Alternatively, differences in GABAergic inhibition and the overall excitation/inhibition balance could also contribute to impaired LTP induction in Mecp2 deficient slices. Finally, a potential “ceiling” effect on LTP induction has not been considered to explain its reduction or absence in Mecp2 deficient slices (i.e. saturation of LTP induction).
Similar to the studies in cortical slices, extracellular field EPSPs evoked in CA1 by CA3 afferent stimulation showed smaller LTP in hippocampal slices from either Bird Mecp2 knockout or Jaenisch Mecp2 mutant male mice compared to that induced in wildtype slices, a difference that occurred only after the appearance of RTT-like symptoms (117). Such impaired LTP was observed using either high frequency afferent stimulation or a more physiological pattern of afferent stimulation with shorter trains at theta frequency (5Hz, i.e. TBS). Furthermore, high frequency- and TBS-induced LTP was impaired in area CA1 of slices from mice where the endogenous Mecp2 gene is silenced by insertion of a lox-Stop cassette (which can be conditionally activated under the control of its own promoter and regulatory elements by cassette deletion; see below) (120). In addition, LTP induced by either high frequency or TBS stimulation to CA1 afferents was smaller in slices from Mecp2308/Y mice expressing a truncated Mecp2 protein than that in wildtype slices (92).
Again consistent with the critical role of tightly regulated MeCP2 levels, LTP of field EPSPs in area CA1 was enhanced in MeCP2Tg1 overexpressing mice (50). The enhanced synaptic plasticity in young MeCP2Tg1 mice might have contributed to their better behavioral performance in motor and contextual learning paradigms, but it also could have caused their progressive neurological symptoms (e.g. forepaw clasping, aggressiveness, kyphosis and hypoactivity) and premature death (50).
Regarding LTD, the other form of long-term synaptic plasticity, low frequency afferent stimulation (15min at 1Hz) induced smaller LTD in area CA1 of slices from either Bird Mecp2 knockout or Jaenisch Mecp2 mutant mice, but only in symptomatic mice (117). Similarly, LTD induced in area CA1 by paired-pulse low frequency stimulation of its afferent fibers was impaired in slices from Mecp2308/Y mice (92).
4.4 Morphological and physiological phenotypes are expressed in vitro by cultured neurons from Mecp2-deficient mice
A few studies evaluated whether neurons dissociated from either embryonic or early postnatal Mecp2-deficient mice, and cultured in vitro for several weeks display morphological and physiological characteristics resembling those described in more intact preparations from older presymptomatic and symptomatic animals (i.e. brain slices). As we summarize below, somewhat contradicting results have been obtained so far, which may be due to very different cell culture conditions.
On one hand, dendritic length and branching were significantly affected in conventional high-density cultures of hippocampal neurons from Mecp2 knockout or Mecp2 mutant mice (97, 121). Consistently, rat hippocampal neurons transfected with a Mecp2-specific shRNA showed shorter dendrites than control cells in mass cultures (122). On the contrary, the dendritic length and branch points of hippocampal neurons cultured at low density on glial micro-islands were not affected by Mecp2 deletion or MECP2 overexpression (104). This type of neuronal cultures is often called “autaptic” because individual neurons make synapses onto themselves, i.e. autapses (123). It is unclear whether differences in the morphological consequences of Mecp2 deletion between “mass” high-density cultures and autaptic cultures are due to their widely different cell densities and the ensuing neuronal activity (see below).
In addition, conventional mass cultures of mouse hippocampal neurons transfected with full-length Mecp2 displayed enhanced dendritic and axonal length and branching, a feature absent in cells expressing mutant Mecp2 coding for a truncated protein that retains the MBD and a putative nuclear localization signal, but terminates within the TRD (MeCP2293) (124). Similarly, rat hippocampal neurons transfected with wildtype human MECP2 showed enhanced dendritic and axonal length and branching (122). Consistent with the human neuropathology, two different MECP2 mutations common in RTT individuals, T158M and R106W, caused dendritic and axonal shortening, an effect prevented by overexpression of Bdnf (122), one of the gene targets of MeCP2 (see below).
This role of MeCP2 in dendritic complexity is also evident in more mature preparations, such as organotypic cultures of postnatal hippocampal slices, where pyramidal neurons have decreased dendritic branching after shRNA-mediated Mecp2 knockdown or overexpression of wildtype Mecp2, while expression of a phosphorylation mutant (S421A) had no effect on dendritic morphology (125). In addition, dendritic spines of neurons overexpressing wildtype Mecp2 were longer, thinner, and more filopodia-like (i.e. resembling immature spines) than those in control neurons or neurons transfected with the S421A mutant MeCP2, while shRNA Mecp2 had no effect (125). It should be noted that Mecp2 protein levels were up to 4-fold higher than control values in these experiments, and that slice cultures were co-transfected with the antiapoptotic protein Bcl-XL. Incidentally, Bcl-XL promotes dendritic spine formation in cultured hippocampal neurons (126).
On the other hand, similar experiments in hippocampal slice cultures but with lower MeCP2 expression levels (~2-fold endogenous levels), revealed that the RTT-associated T158M mutation caused only a small and transient increase in dendritic complexity in CA3 pyramidal neurons, while the wildtype protein only transiently reduced dendritic branch points (27). In addition, shRNA-mediated Mecp2 knockdown caused a reduction of dendritic length and branch points. In regards to dendritic spines, the effects of the two different RTT-associated MECP2 mutations were much more pronounced than the gain or loss of the intact protein. CA3 and CA1 pyramidal neurons expressing T158M and R106W showed fewer and more immature dendritic spines than controls cells, while overexpression of wildtype MECP2 only transiently reduced spine density (Fig. 1). Similarly, shRNA-mediated Mecp2 knockdown only shifted the proportion of morphological spine types promoting the immature ones, but not their density (27). Considering that the T158M and R106W mutations cause single amino acid substitutions in the MBD that alter MeCP2 binding to methylated DNA, and subsequent DNA transcription (127–130), the consequences of their expression on dendritic spine density and morphology may reflect a “toxic” gain-of-function of the mutant protein.
At the physiological level, hippocampal neurons from postnatal day-1 Bird Mecp2 knockout mice maintained in conventional mass cultures for 11–14 days in vitro showed a lower frequency of spontaneous action potential-independent mEPSCs than that recorded in neurons from wildtype mice, without differences in their amplitude or kinetics (131). Given that neither (1) the number of synapsin-positive presynaptic terminals in contact with MAP2-positive dendrites; (2) the size and destaining kinetics of the total recycling pool of vesicles labeled with FM1–43 and high K+ solutions; or (3) the size of the readily-releasable pool of vesicles estimated by the amplitude of membrane currents evoked by hypertonic sucrose, were affected by Mecp2 deletion, the lower mEPSC frequency was interpreted to reflect differences in the probability of vesicular neurotransmitter release (Pr). Indeed, the kinetics of depression of evoked EPSCs during 10Hz stimulation (reflecting vesicle depletion) and the time-course of their recovery were both enhanced in Mecp2-deficient neurons, suggesting higher Pr. Consistently, the responses to paired pulses were reduced in Mecp2 knockout neurons, but only at the shortest intervals (i.e. higher frequencies). A similar synaptic phenotype was observed after acute deletion in cultures from Mecp22lox mice transfected with Cre-expressing lentiviruses. On the contrary, the frequency and amplitude of mIPSCs were not affected, suggesting a selective role of Mecp2 at excitatory synapses (131).
Conversely, a study of autaptic connections between isolated hippocampal neurons grown in micro-island cultures concluded that MeCP2 controls the number of synapses, but not their individual properties. In this study, glutamatergic hippocampal neurons from Mecp2-deficient mice showed smaller membrane currents evoked by a single action potential fired by the cell under recording (autaptic EPSCs), while neurons from MeCP2Tg1 mice had larger autaptic EPSCs. The authors concluded that those effects were likely due to differences in the number of glutamatergic synapses per neuron because (1) the frequency of mEPSCs was reduced in the Mecp2-deficient neurons and increased in overexpressing cells; (2) the size of the ready releasable pool (RRP) of synaptic vesicles (as estimated by integrating the membrane currents evoked by hypertonic sucrose applications; (132) was smaller in Mecp2-deficient neurons and larger in overexpressing cells; and (3) neither release probability per vesicle (estimated from the RRP) or per terminal (estimated by the MK-801 block method; (133) nor short-term plasticity were affected by loss or doubling of MeCP2 (104). Whether differences in these physiological consequences of MeCP2 manipulations are due to the widely different cell densities of micro-islands vs. mass cultures, or more importantly, different patterns or magnitude of neuronal activity induced by autapses vs. synapses, is unclear at this time. For example, the consequences of synaptotagmin-I deletion on evoked synaptic transmission are different when evaluated at autapses in micro-island cultures than when tested at synapses between neurons maintained at higher density in mass cultures (134).
The studies summarized so far strongly suggest that the pathology observed in RTT individuals and Mecp2-based mouse models results from Mecp2 protein dysfunction in neurons, partly because the initial studies of Mecp2 immunolocalization indicated an exclusive expression in neurons (38, 42, 43), and also because transgenic Mecp2 expression (62, 83) or reactivation of endogenous Mecp2 (120, 135) in postmitotic neurons reverted some phenotypes of the Mecp2-deficient mice (see below). However, postnatal Cre-mediated Mecp2 deletion in forebrain neurons using the CamkII promoter resulted in a delayed and milder phenotype than that caused by germline or Nestin-Cre deletions (61), while transgenic Mecp2 expression in Mecp2 mutant mice using strictly neuronal promoters did not improve the RTT-like phenotype, unlike the widespread re-expression of endogenous Mecp2 by removal of a conditional STOP codon (see below) (120). In addition, Mecp2 protein has been recently detected in all types of glial cells, including astrocytes, oligodendrocyte progenitor cells, and mature oligodendrocytes (97, 121), as well as in microglia (136). Moreover, astrocytes and microglia from either Bird Mecp2 knockout or Jaenisch Mecp2 mutant mice caused impaired dendritic branching in co-cultured neurons from either wildtype or mutant mice (97, 121, 136). The pathogenic effect of Mecp2-deficient glia is reproduced by their conditioned media, suggesting that aberrant secretion of soluble factors (e.g. glutamate (136) may cause non-cell autonomous effects on neuronal morphology (e.g. (86).
5. REVERSAL OF BEHAVIORAL AND CELLULAR IMPAIRMENTS IN MeCP2-BASED MOUSE MODELS OF RTT
The experimental reversal of behavioral and synaptic impairments in several models of neurodevelopmental disorders by pharmacological approaches in adult animals has raised hope for similar interventions in humans after the onset of neurological symptoms (137). For example, the behavioral impairment in a mouse model of Down syndrome (Ts65Dn) caused by an excitatory/inhibitory imbalance of synaptic function in the hippocampus can be reverted by GABAAR antagonists in symptomatic animals (138, 139). A mouse model of neurofibromatosis-1 (Nf1+/−) also has higher levels of inhibition than their wildtype littermates (but comparable excitation), which can be reversed by decreasing Ras/MAPK signaling (140, 141). Also, mice that model tuberous sclerosis (Tsc2+/−) improve after treatment with inhibitors of the mTOR/Akt signaling cascade (142–144). In addition, a mouse model of Rubinstein-Taybi syndrome (CBP+/−) improves after treatment with inhibitors of either phosphodiesterase 4 (145) or histone deacetylases (146). Using a genetic manipulation, most neurological deficits in a mouse model of Fragile X syndrome (Fmr1−/−) are prevented after breeding them with heterozygous mGluR5 mice (147). Likewise, reducing αCaMKII inhibitory phosphorylation in a mouse model of Angelman syndrome (Ube3a mutants) by crossing them with mice harboring a targeted αCaMKII mutation (T305V/T306A) prevents the development of Angelman syndrome-like behavioral deficits (148). As we reviewed in the preceding sections, the neuropathology in RTT individuals and Mecp2-deficient mice is subtle, including reduced neuronal complexity and dendritic spine density rather than severe neuronal degeneration (26, 27, 43, 86, 102, 149, 150), thus raising the possibility that some specific deficits in RTT individuals may be reversible (151).
Several experimental approaches have been tested for the reversal of behavioral impairments in symptomatic Mecp2 deficient mice, 4 based on gene expression manipulations, two on pharmacological treatments, in addition to a dietary supplementation and behavioral interventions in the form of rearing in enriched environments.
To selectively increase the expression of full-length wildtype Mecp2 in postmitotic neurons of Mecp2 mutant mice, they were crossed with a mouse line overexpressing a Tau-MeCP2 fusion protein from the Tau locus in homozygous Tau knockout mice. The resulting offspring showed improved body and brain weights, as well as locomotor activity and fertility compared to Mecp2 mutants, and seemed indistinguishable from their wildtype littermates (62).
Inducible and neuron-specific expression of human MECP2 in either Mecp2 knockout mice or Mecp2308/Y mice (which express a truncated non-functional protein) was achieved by using tetracycline-inducible MECP2 under control of either the CamkII or the Eno2 promoters. Despite the presence of specific patterns of transgene expression, most behavioral impairments in Mecp2308/Y mice (i.e. dowel test, suspended wire, and accelerating rotarod) were not improved by neuron-specific MECP2 transgene expression. Similarly, neuron-specific MECP2 transgene expression failed to extend the longevity or prevent the tremors and breathing irregularities of Mecp2 knockout mice, with a subtle effect on locomotor activity in their home cages (152). The conclusion of these studies was that either the levels of MeCP2 achieved were insufficient or not in the relevant brain regions. Alternatively, these results may reflect the critical role of proper Mecp2 expression in glial cells and its non-cell autonomous consequences on neuronal structure and function (see preceding section).
Mecp2 overexpression in postmitotic neurons of Mecp2 knockout mice was achieved by using tetracycline-inducible Mecp2-e2 cDNA under control of the CamkII promoter. Females of this rescue line showed improved rearing activity, overall mobility and rotarod performance compared to female Mecp2−/+ mice, reaching levels of performance comparable to wildtype littermates (83).
The reactivation of the endogenous Mecp2 gene under control of its own promoter and regulatory elements was achieved by silencing it with a lox-Stop cassette, which can be removed by transgene expression of a fusion protein between Cre recombinase and a modified estrogen receptor (Cre-ER). The Cre-ER protein remains in the cytoplasm unless exposed to the estrogen analog tamoxifen, which induces its nuclear translocation. Male mice of this strain (Mecp2lox-Stop/y) developed RTT-like symptoms at 16 weeks of age and survived for ~11 weeks, being comparable to Mecp2 knockout mice. Tamoxifen injections in Mecp2lox-Stop/y mice with advanced symptoms (between 7–17 weeks of age) led to milder symptoms and extended lifespan, demonstrating that reactivation of the endogenous Mecp2 gene reverses established symptoms. Similar behavioral results were obtained in female Mecp2lox-Stop/+ mice after treatment with tamoxifen. Furthermore, the impairment in high frequency- and TBS-induced LTP in area CA1 of slices from symptomatic female Mecp2lox-Stop/+ was completely prevented by tamoxifen treatment (120).
A conditional Mecp2 transgene that can be activated by Cre-mediated deletion of a loxP-STOP-loxP cassette was used in Mecp2 mutant mice to show that increasing Mecp2 levels as late as 2–4 weeks of age prevented the onset of RTT-like symptoms. The mouse Mecp2e2 cDNA was placed downstream of a loxP-flanked STOP cassette, which in turn was downstream of the synthetic CAGGS promoter/enhancer/intron. Mice carrying this rescue transgene were crossed with Mecp2 mutant mice, and the resulting offspring bred with Cre deleter mice. When Cre was driven by the Nestin promoter (E12 in neural precursors of neurons and glia), or by Tau promoter (postmitotic neurons), the lifespan of the rescue mice increased to more than 8 months (compared to 10–12 weeks in Mecp2 mutants). Activation of the Mecp2 transgene by the CamkII promoter also extended the life span, but to a lesser extent. Nocturnal locomotor activity was also improved in all the lines of rescue mice, albeit more efficiently in those where Cre expression was driven earlier and in most neurons, i.e. Nest-Cre and Tau-Cre. Finally, rescue mice lacked the decrease in brain weight and neuronal soma size in hippocampus and cerebral cortex characteristic of Mecp2 mutant mice (135).
-
Bdnf, the gene coding for brain-derived neurotrophic factor, was one of the first Mecp2 targets to be identified, binding to its promoter region (153, 154). The initial interpretation of Mecp2 as a transcriptional repressor of Bdnf was later confronted with the observations that Mecp2 mutant mice express lower levels of BDNF mRNA and protein in the cerebral cortex and cerebellum than wildtype controls, and that conditional postnatal deletion of Bdnf in the forebrain, parts of midbrain and hindbrain of Mecp2 mutant mice exacerbated the onset of their locomotor dysfunction and shortened their longevity, two consistent RTT-associated impairments in these mice (113). Furthermore, a microarray study comparing hypothalamic samples from Mecp2 deficient and MeCP2Tg1 overexpressing mice found that BDNF mRNA levels were lower in the absence of Mecp2 and higher when MeCP2 levels were doubled (36). Considering the well-established role of BDNF on synaptic transmission and plasticity (31, 155–158), restoring proper levels of BDNF in Mecp2-deficient brains is an attractive therapeutic strategy.
To selectively overexpress BDNF in postmitotic forebrain neurons of Mecp2 mutant mice, they were first bred with a mouse line that expresses Cre recombinase under control of the CamkII promoter (cre93 transgenic line) (159). The resulting mice (Mecp2+/−; cre93) were then crossed with mice carrying a human BDNF transgene under regulation of the synthetic CAGGS promoter/enhancer/intron followed by a loxP-STOP-loxP cassette. In the presence of Cre in postmitotic forebrain neurons, the STOP cassette is removed resulting in the activation of the BDNF transgene. The resulting overexpression of BDNF in postnatal forebrain neurons in a Mecp2-deficient background extended the lifespan, and prevented a locomotor defect (hypoactivity) as well as an electrophysiological deficit (low spike firing frequency in cortical layer V pyramidal neurons) consistently observed in Mecp2 mutant mice (113). In support of these observations, an in vitro study showed that overexpression of Bdnf in primary hippocampal cultures rescued the dendritic phenotype caused by either shRNA-mediated Mecp2 knockdown or expression of RTT-associated MECP2 mutations (122). Altogether, these studies indicate that BDNF levels can be targeted for therapeutic interventions to alleviate RTT symptoms, and are the bases of two pharmacological approaches and the beneficial effect of environmental enrichment (see below).
The inability of BDNF to cross the blood-brain barrier has hampered its use as a therapeutic agent in several neurological disorders. AMPAkines are a family of allosteric modulators of AMPA-type glutamate receptors known to enhance BDNF mRNA and protein levels (160, 161). In support of their use to ameliorate RTT symptoms by elevating BDNF levels, the breathing pattern irregularities in Mecp2 mutant mice are alleviated by a 10-day treatment with the AMPAkine CX546 (115). Consistently, direct application of recombinant BDNF to brainstem slices from Mecp2 mutant mice reversed their synaptic dysfunction phenotype (109). Intriguingly, cultured neurons from Mecp2 knockout mice are able to release more BDNF than wildtype cells, despite showing lower BDNF expression levels. Such hypersecretion phenotype was also observed for catecholamine release from chromaffin cells (162). The parsimonious interpretation of these observations is that Mecp2 null neurons may eventually exhaust their pool of releasable BDNF.
Supporting the potential use of “BDNF-mimetic” trophic factors to reverse the RTT-like impairments in Jaenisch Mecp2 mutant mice, a 2-week treatment with an active tri-peptide fragment of Insulin-like Growth Factor 1 (IGF-1) extended the lifespan, improved locomotor function, ameliorated breathing patterns, reduced heart rate irregularity, and increased brain weight. Indeed, the IGF-1 receptor activates intracellular pathways common to those induced by BDNF signaling through its TrkB receptor (i.e. PI3K/Akt and MAPK) (163). Furthermore, IGF-1 partially restored dendritic spine density in pyramidal neurons of layer V in motor cortex, the amplitude of spontaneous EPSCs in pyramidal neurons of sensorimotor cortex, the cortical expression of the synaptic scaffolding protein PSD-95, and stabilized cortical plasticity in Mecp2 mutant mice to wild-type levels (101).
Daily injections of desipramine, a selective inhibitor of the norepinephrine transporter used to increase extracellular levels of this neurotransmitter, improved respiratory rhythm, the number of tyrosine hydroxylase-expressing neurons in the brainstem, as well as longevity in Mecp2 knockout mice (164).
Dietary choline supplementation improved motor coordination and locomotor activity in male Mecp2 mutant mice, and enhanced grip strength in female Mecp2 mutant mice (165). Increased NGF protein levels in the striatum (166) and of N-acetylaspartate content, as measured by NMR spectroscopy (167) suggests improved neuronal proliferation and survival after choline supplementation in Mecp2 mutant mice.
In several neurological disorders, environmental enrichment has beneficial effects on various behavioral and cellular phenotypes, including increased levels of BDNF expression (168, 169). Potentially related to the ability of either Bdnf overexpression (113) or increased BDNF levels after AMPAkine treatment (115) to improve RTT-like symptoms in Mecp2 deficient mice, rearing them in enriched environments also ameliorated some of their behavioral and synaptic phenotypes. For example, the cerebellar and hippocampal/amygdala-based learning deficits, as well as the reduced motor dexterity and decreased anxiety levels characteristic of heterozygous Mecp2tm1Tam females were prevented by their housing in larger-sized home cages with nesting material, a variety of objects with different textures, shapes and sizes, and running wheels starting at 4 weeks of age (84). Similarly, housing male mice of the Jaenisch Mecp2 mutant line in enriched environments starting at weaning (postnatal day 21) improved their locomotor activity, but not motor coordination or contextual or cued fear conditioning. Curiously, MRI revealed a reduction in ventricular volume after environmental enrichment in both Mecp2 mutant and wildtype littermates, without changes in total brain volume. Together with the known reduction in brain size in Mecp2 deficient mice, this observation suggests that environmental enrichment selectively increased grey and white matter (170).
Intriguingly, more robust effects were obtained when environmental enrichment began earlier in the development of the pups and included their dams, which displayed enhanced maternal care behaviors. Rearing Jaenisch Mecp2 mutant mice and their dams from postnatal day 10 in enriched environments led to improved motor coordination and motor learning compared to control Mecp2 mutant mice kept in standard housing conditions. In addition, environmental enrichment prevented the deficit of TBS-induced LTP in layer II/III of slices from primary somatosensory cortex typical of Mecp2 mutant mice kept in standard housing. Despite not being different between wildtype and Mecp2 mutant mice, the density of spine synapses in layer III of the primary sensory cortex, as well as of parallel fiber-Purkinje cell synapses in the molecular layer of the cerebellum were higher in mice reared in enriched environments than in standard housing, suggesting that Mecp2 deficient neurons are still capable of structural plasticity. Consistent with previous reports in rats and mice, this protocol of early environmental intervention increased the levels of BDNF mRNA and protein in the cerebral cortex of both wildtype and Mecp2 mutant mice (88).
Altogether, these successful therapeutic approaches that reversed many RTT-like symptoms in both Jaenisch Mecp2 mutant and Bird Mecp2 knockout mouse lines provide further support to the potential pharmacological reversal of neurodevelopmental disorders in adults (137).
6. SUMMARY
The wide range of clinical symptoms in RTT individuals and of phenotypes in MeCP2-based mouse models was initially thought to originate from unbalanced X chromosome inactivation. Ample evidence in mouse models now indicate that the role of MeCP2 in neuronal development and function is very different across various brain regions, suggesting that dysfunction in specific neuronal populations due to differential distribution of mutant MeCP2 also contributes to phenotypic variability. In addition, the recently uncovered role of Mecp2 dysfunction in glial cells leading to neuronal pathology cannot be overlooked, raising more questions regarding the primary deficits that initiate the cascade of events leading to clinical symptoms. Also, the realization that MeCP2 acts as both a repressor and activator of potentially thousands of genes has increased the complexity of this once thought simple monogenetic disorder. Finally, none of the experimental approaches tested in MeCP2-based mouse models fully reversed their RTT-like phenotypes, suggesting additional molecular and cellular deficits. In spite of these seemingly overwhelming limitations in our state of knowledge, we should remind ourselves that we have learned more in the last decade since the discovery that MeCP2 mutations cause RTT than in the preceding 30 years from the first description by Andreas Rett. Following this trajectory, it is likely that rational therapies grounded on basic scientific knowledge will be available for RTT individuals within the next decade.
ACKNOWLEDGEMENTS
We thank Dr. Carolyn Schanen for useful comments on the manuscript. Supported by NIH grants from NINDS (NS40593, NS057780, NS-065027) and NICHD (U54 grant HD061222 and IDDRC grant HD38985), IRSF, and the Civitan Foundation.
REFERENCES
- 1.Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood] Wien Med Wochenschr. 1966;116(37):723–726. [PubMed] [Google Scholar]
- 2.Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol. 1983;14(4):471–479. doi: 10.1002/ana.410140412. [DOI] [PubMed] [Google Scholar]
- 3.Laurvick CL, de Klerk N, Bower C, Christodoulou J, Ravine D, Ellaway C, Williamson S, Leonard H. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148(3):347–352. doi: 10.1016/j.jpeds.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 4.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56(3):422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 5.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 6.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69(6):905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 7.Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, Epplen JT. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42(2):e12. doi: 10.1136/jmg.2004.023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friez MJ, Jones JR, Clarkson K, Lubs H, Abuelo D, Bier JA, Pai S, Simensen R, Williams C, Giampietro PF, Schwartz CE, Stevenson RE. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics. 2006;118(6):e1687–e1695. doi: 10.1542/peds.2006-0395. [DOI] [PubMed] [Google Scholar]
- 9.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A(5):1079–1088. doi: 10.1002/ajmg.a.33184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Girard M, Couvert P, Carrie A, Tardieu M, Chelly J, Beldjord C, Bienvenu T. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet. 2001;9(3):231–236. doi: 10.1038/sj.ejhg.5200618. [DOI] [PubMed] [Google Scholar]
- 11.Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, Hanefeld F, Engel W. MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am J Hum Genet. 2001;68(5):1093–1101. doi: 10.1086/320109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10(10):981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol. 2005;20(9):747–753. doi: 10.1177/08830738050200090901. [DOI] [PubMed] [Google Scholar]
- 14.Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol (Berl) 1988;76(2):142–158. doi: 10.1007/BF00688098. [DOI] [PubMed] [Google Scholar]
- 15.Reiss AL, Faruque F, Naidu S, Abrams M, Beaty T, Bryan RN, Moser H. Neuroanatomy of Rett syndrome: a volumetric imaging study. Ann Neurol. 1993;34(2):227–234. doi: 10.1002/ana.410340220. [DOI] [PubMed] [Google Scholar]
- 16.Bauman ML, Kemper TL, Arin DM. Microscopic observations of the brain in Rett syndrome. Neuropediatrics. 1995;26(2):105–108. doi: 10.1055/s-2007-979737. [DOI] [PubMed] [Google Scholar]
- 17.Bauman ML, Kemper TL, Arin DM. Pervasive neuroanatomic abnormalities of the brain in three cases of Rett’s syndrome. Neurology. 1995;45(8):1581–1586. doi: 10.1212/wnl.45.8.1581. [DOI] [PubMed] [Google Scholar]
- 18.Ronnett GV, Leopold D, Cai X, Hoffbuhr KC, Moses L, Hoffman EP, Naidu S. Olfactory biopsies demonstrate a defect in neuronal development in Rett’s syndrome. Ann Neurol. 2003;54(2):206–218. doi: 10.1002/ana.10633. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong D, Dunn JK, Antalffy B, Trivedi R. Selective dendritic alterations in the cortex of Rett syndrome. J Neuropathol Exp Neurol. 1995;54(2):195–201. doi: 10.1097/00005072-199503000-00006. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong DD, Dunn K, Antalffy B. Decreased dendritic branching in frontal, motor and limbic cortex in Rett syndrome compared with trisomy 21. J Neuropathol Exp Neurol. 1998;57(11):1013–1017. doi: 10.1097/00005072-199811000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Kaufmann WE, Naidu S, Budden S. Abnormal expression of microtubule-associated protein 2 (MAP-2) in neocortex in Rett syndrome. Neuropediatrics. 1995;26(2):109–113. doi: 10.1055/s-2007-979738. [DOI] [PubMed] [Google Scholar]
- 22.Kaufmann WE, MacDonald SM, Altamura CR. Dendritic cytoskeletal protein expression in mental retardation: an immunohistochemical study of the neocortex in Rett syndrome. Cereb Cortex. 2000;10(10):992–1004. doi: 10.1093/cercor/10.10.992. [DOI] [PubMed] [Google Scholar]
- 23.Blue ME, Naidu S, Johnston MV. Altered development of glutamate and GABA receptors in the basal ganglia of girls with Rett syndrome. Exp Neurol. 1999;156(2):345–352. doi: 10.1006/exnr.1999.7030. [DOI] [PubMed] [Google Scholar]
- 24.Blue ME, Naidu S, Johnston MV. Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol. 1999;45(4):541–545. doi: 10.1002/1531-8249(199904)45:4<541::aid-ana21>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Pan JW, Lane JB, Hetherington H, Percy AK. Rett syndrome: 1H spectroscopic imaging at 4.1 Tesla. J Child Neurol. 1999;14(8):524–528. doi: 10.1177/088307389901400808. [DOI] [PubMed] [Google Scholar]
- 26.Belichenko PV, Oldfors A, Hagberg B, Dahlstrom A. Rett syndrome: 3-D confocal microscopy of cortical pyramidal dendrites and afferents. Neuroreport. 1994;5(12):1509–1513. [PubMed] [Google Scholar]
- 27.Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, Kudo S, Armstrong DL, Percy AK, Pozzo-Miller L. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2009;35(2):219–233. doi: 10.1016/j.nbd.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufmann WE, Worley PF, Taylor CV, Bremer M, Isakson PC. Cyclooxygenase-2 expression during rat neocortical development and in Rett syndrome. Brain Dev. 1997;19(1):25–234. doi: 10.1016/s0387-7604(96)00047-2. [DOI] [PubMed] [Google Scholar]
- 29.Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186(4169):1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- 30.Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39(1):29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- 31.Chapleau CA, Larimore JL, Theibert A, Pozzo-Miller L. Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J Neurodev Disord. 2009;1(3):185–196. doi: 10.1007/s11689-009-9027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18(11):6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 35.Nan X, Cross S, Bird A. Gene silencing by methyl-CpG-binding proteins. Novartis Found Symp. 1998:2146–2116. doi: 10.1002/9780470515501.ch2. discussion 16–21, 46–50. [DOI] [PubMed] [Google Scholar]
- 36.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102(49):17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35(2):243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 39.Reichwald K, Thiesen J, Wiehe T, Weitzel J, Poustka WA, Rosenthal A, Platzer M, Stratling WH, Kioschis P. Comparative sequence analysis of the MECP2-locus in human and mouse reveals new transcribed regions. Mamm Genome. 2000;11(3):182–190. doi: 10.1007/s003350010035. [DOI] [PubMed] [Google Scholar]
- 40.Kriaucionis S, Bird A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004;32(5):1818–1823. doi: 10.1093/nar/gkh349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mnatzakanian GN, Lohi H, Munteanu I, Alfred SE, Yamada T, MacLeod PJ, Jones JR, Scherer SW, Schanen NC, Friez MJ, Vincent JB, Minassian BA. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36(4):339–341. doi: 10.1038/ng1327. [DOI] [PubMed] [Google Scholar]
- 42.Jung BP, Jugloff DG, Zhang G, Logan R, Brown S, Eubanks JH. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol. 2003;55(1):86–96. doi: 10.1002/neu.10201. [DOI] [PubMed] [Google Scholar]
- 43.Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27(3):306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Dragich JM, Kim YH, Arnold AP, Schanen NC. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J Comp Neurol. 2007;501(4):526–542. doi: 10.1002/cne.21264. [DOI] [PubMed] [Google Scholar]
- 45.Bebbington A, Anderson A, Ravine D, Fyfe S, Pineda M, de Klerk N, Ben-Zeev B, Yatawara N, Percy A, Kaufmann WE, Leonard H. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70(11):868–875. doi: 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- 46.Neul JL, Fang P, Barrish J, Lane J, Caeg EB, Smith EO, Zoghbi H, Percy A, Glaze DG. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70(16):1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glaze DG, Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, Lane JB, Geerts SP, Annese F, Graham J, McNair L, Lee HS. Epilepsy and the natural history of Rett syndrome. Neurology. 2010;74(11):909–912. doi: 10.1212/WNL.0b013e3181d6b852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Percy AK, Lane JB, Childers J, Skinner S, Annese F, Barrish J, Caeg E, Glaze DG, MacLeod P. Rett syndrome: North American database. J Child Neurol. 2007;22(12):1338–1341. doi: 10.1177/0883073807308715. [DOI] [PubMed] [Google Scholar]
- 49.Archer HL, Whatley SD, Evans JC, Ravine D, Huppke P, Kerr A, Bunyan D, Kerr B, Sweeney E, Davies SJ, Reardon W, Horn J, MacDermot KD, Smith RA, Magee A, Donaldson A, Crow Y, Hermon G, Miedzybrodzka Z, Cooper DN, Lazarou L, Butler R, Sampson J, Pilz DT, Laccone F, Clarke AJ. Gross rearrangements of the MECP2 gene are found in both classical and atypical Rett syndrome patients. J Med Genet. 2006;43(5):451–456. doi: 10.1136/jmg.2005.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collins AL, Levenson JM, Vilaythong AP, Richman R, Armstrong DL, Noebels JL, David Sweatt J, Zoghbi HY. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 51.Kankirawatana P, Leonard H, Ellaway C, Scurlock J, Mansour A, Makris CM, Dure LSt, Friez M, Lane J, Kiraly-Borri C, Fabian V, Davis M, Jackson J, Christodoulou J, Kaufmann WE, Ravine D, Percy AK. Early progressive encephalopathy in boys and MECP2 mutations. Neurology. 2006;67(1):164–166. doi: 10.1212/01.wnl.0000223318.28938.45. [DOI] [PubMed] [Google Scholar]
- 52.del Gaudio D, Fang P, Scaglia F, Ward PA, Craigen WJ, Glaze DG, Neul JL, Patel A, Lee JA, Irons M, Berry SA, Pursley AA, Grebe TA, Freedenberg D, Martin RA, Hsich GE, Khera JR, Friedman NR, Zoghbi HY, Eng CM, Lupski JR, Beaud, Cheung SW, Roa BB, et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet Med. 2006;8(12):784–792. doi: 10.1097/01.gim.0000250502.28516.3c. [DOI] [PubMed] [Google Scholar]
- 53.Tate P, Skarnes W, Bird A. The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat Genet. 1996;12(2):205–208. doi: 10.1038/ng0296-205. [DOI] [PubMed] [Google Scholar]
- 54.Sauer B. Inducible gene targeting in mice using the Cre/lox system. Methods. 1998;14(4):381–392. doi: 10.1006/meth.1998.0593. [DOI] [PubMed] [Google Scholar]
- 55.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23(24):5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 57.Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13(12):1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 58.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23(1):99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 59.Samaco RC, Fryer JD, Ren J, Fyffe S, Chao HT, Sun Y, Greer JJ, Zoghbi HY, Neul JL. A partial loss of function allele of methyl-CpG-binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet. 2008;17(12):1718–1727. doi: 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, Tam PP. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129(Pt 4):887–898. doi: 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- 61.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 62.Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101(16):6033–6038. doi: 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Minichiello L, Korte M, Wolfer D, Kuhn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24(2):401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 64.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123(3):493–505. doi: 10.1016/j.cell.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 65.Fyffe SL, Neul JL, Samaco RC, Chao HT, Ben-Shachar S, Moretti P, McGill BE, Goulding EH, Sullivan E, Tecott LH, Zoghbi HY. Deletion of Mecp2 in Sim1-expressing neurons reveals a critical role for MeCP2 in feeding behavior, aggression, and the response to stress. Neuron. 2008;59(6):947–958. doi: 10.1016/j.neuron.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lindeberg J, Usoskin D, Bengtsson H, Gustafsson A, Kylberg A, Soderstrom S, Ebendal T. Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis. 2004;40(2):67–73. doi: 10.1002/gene.20065. [DOI] [PubMed] [Google Scholar]
- 67.Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, Greer JJ, Percy A, Glaze DG, Zoghbi HY, Neul JL. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A. 2009;106(51):21966–21971. doi: 10.1073/pnas.0912257106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott MM, Wylie CJ, Lerch JK, Murphy R, Lobur K, Herlitze S, Jiang W, Conlon RA, Strowbridge BW, Deneris ES. A genetic approach to access serotonin neurons for in vivo and in vitro studies. Proc Natl Acad Sci U S A. 2005;102(45):16472–16477. doi: 10.1073/pnas.0504510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adachi M, Autry AE, Covington HE, 3rd, Monteggia LM. MeCP2-mediated transcription repression in the basolateral amygdala may underlie heightened anxiety in a mouse model of Rett syndrome. J Neurosci. 2009;29(13):4218–4227. doi: 10.1523/JNEUROSCI.4225-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science. 1999;286(5441):950–952. doi: 10.1126/science.286.5441.950. [DOI] [PubMed] [Google Scholar]
- 71.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 72.Jin J, Bao X, Wang H, Pan H, Zhang Y, Wu X. RNAi-induced down-regulation of Mecp2 expression in the rat brain. Int J Dev Neurosci. 2008;26(5):457–465. doi: 10.1016/j.ijdevneu.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 73.Wood L, Gray NW, Zhou Z, Greenberg ME, Shepherd GM. Synaptic circuit abnormalities of motor-frontal layer 2/3 pyramidal neurons in an RNA interference model of methyl-CpG-binding protein 2 deficiency. J Neurosci. 2009;29(40):12440–12448. doi: 10.1523/JNEUROSCI.3321-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lawson-Yuen A, Liu D, Han L, Jiang ZI, Tsai GE, Basu AC, Picker J, Feng J, Coyle JT. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007:11801–11806. doi: 10.1016/j.brainres.2007.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jentarra GM, Olfers SL, Rice SG, Srivastava N, Homanics GE, Blue M, Naidu S, Narayanan V. Abnormalities of cell packing density and dendritic complexity in the MeCP2 A140V mouse model of Rett syndrome/X-linked mental retardation. BMC Neurosci. 2010;1119 doi: 10.1186/1471-2202-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hotta A, Cheung AY, Farra N, Vijayaragavan K, Seguin CA, Draper JS, Pasceri P, Maksakova IA, Mager DL, Rossant J, Bhatia M, Ellis J. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat Methods. 2009;6(5):370–376. doi: 10.1038/nmeth.1325. [DOI] [PubMed] [Google Scholar]
- 77.Ricceri L, De Filippis B, Laviola G. Mouse models of Rett syndrome: from behavioural phenotyping to preclinical evaluation of new therapeutic approaches. Behav Pharmacol. 2008;19(5–6):501–517. doi: 10.1097/FBP.0b013e32830c3645. [DOI] [PubMed] [Google Scholar]
- 78.Tao J, Wu H, Sun YE. Deciphering Rett syndrome with mouse genetics, epigenomics, and human neurons. Int Rev Neurobiol. 2009:89147–89160. doi: 10.1016/S0074-7742(09)89007-7. [DOI] [PubMed] [Google Scholar]
- 79.Young JI, Zoghbi HY. X-chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of rett syndrome. Am J Hum Genet. 2004;74(3):511–520. doi: 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Metcalf BM, Mullaney BC, Johnston MV, Blue ME. Temporal shift in methyl-CpG binding protein 2 expression in a mouse model of Rett syndrome. Neuroscience. 2006;139(4):1449–1460. doi: 10.1016/j.neuroscience.2006.01.060. [DOI] [PubMed] [Google Scholar]
- 81.Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146(3):907–921. doi: 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 82.Bissonnette JM, Knopp SJ. Effect of inspired oxygen on periodic breathing in methy-CpG-binding protein 2 (Mecp2) deficient mice. J Appl Physiol. 2008;104(1):198–204. doi: 10.1152/japplphysiol.00843.2007. [DOI] [PubMed] [Google Scholar]
- 83.Jugloff DG, Vandamme K, Logan R, Visanji NP, Brotchie JM, Eubanks JH. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum Mol Genet. 2008;17(10):1386–1396. doi: 10.1093/hmg/ddn026. [DOI] [PubMed] [Google Scholar]
- 84.Kondo M, Gray LJ, Pelka GJ, Christodoulou J, Tam PP, Hannan AJ. Environmental enrichment ameliorates a motor coordination deficit in a mouse model of Rett syndrome--Mecp2 gene dosage effects and BDNF expression. Eur J Neurosci. 2008;27(12):3342–3350. doi: 10.1111/j.1460-9568.2008.06305.x. [DOI] [PubMed] [Google Scholar]
- 85.Ward BC, Agarwal S, Wang K, Berger-Sweeney J, Kolodny NH. Longitudinal brain MRI study in a mouse model of Rett Syndrome and the effects of choline. Neurobiol Dis. 2008;31(1):110–119. doi: 10.1016/j.nbd.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 86.Belichenko NP, Belichenko PV, Mobley WC. Evidence for both neuronal cell autonomous and nonautonomous effects of methyl-CpG-binding protein 2 in the cerebral cortex of female mice with Mecp2 mutation. Neurobiol Dis. 2009;34(1):71–77. doi: 10.1016/j.nbd.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 87.D’Cruz JA, Wu C, Zahid T, El-Hayek Y, Zhang L, Eubanks JH. Alterations of cortical and hippocampal EEG activity in MeCP2-deficient mice. Neurobiol Dis. 2010;38(1):8–16. doi: 10.1016/j.nbd.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 88.Lonetti G, Angelucci A, Morando L, Boggio EM, Giustetto M, Pizzorusso T. Early environmental enrichment moderates the behavioral and synaptic phenotype of MeCP2 null mice. Biol Psychiatry. 2010;67(7):657–665. doi: 10.1016/j.biopsych.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 89.Roux JC, Panayotis N, Dura E, Villard L. Progressive noradrenergic deficits in the locus coeruleus of Mecp2 deficient mice. J Neurosci Res. 2010;88(7):1500–1509. doi: 10.1002/jnr.22312. [DOI] [PubMed] [Google Scholar]
- 90.Isoda K, Morimoto M, Matsui F, Hasegawa T, Tozawa T, Morioka S, Chiyonobu T, Nishimura A, Yoshimoto K, Hosoi H. Postnatal changes in serotonergic innervation to the hippocampus of methyl-CpG-binding protein 2-null mice. Neuroscience. 2010;165(4):1254–1260. doi: 10.1016/j.neuroscience.2009.11.036. [DOI] [PubMed] [Google Scholar]
- 91.Hagberg B, Goutieres F, Hanefeld F, Rett A, Wilson J. Rett syndrome: criteria for inclusion and exclusion. Brain Dev. 1985;7(3):372–373. doi: 10.1016/s0387-7604(85)80048-6. [DOI] [PubMed] [Google Scholar]
- 92.Moretti P, Levenson JM, Battaglia F, Atkinson R, Teague R, Antalffy B, Armstrong D, Arancio O, Sweatt JD, Zoghbi HY. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26(1):319–327. doi: 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59(5):468–476. doi: 10.1016/j.biopsych.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 94.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 95.Klauck SM, Lindsay S, Beyer KS, Splitt M, Burn J, Poustka A. A mutation hot spot for nonspecific X-linked mental retardation in the MECP2 gene causes the PPM-X syndrome. Am J Hum Genet. 2002;70(4):1034–1037. doi: 10.1086/339553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nan X, Hou J, Maclean A, Nasir J, Lafuente MJ, Shu X, Kriaucionis S, Bird A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A. 2007;104(8):2709–2714. doi: 10.1073/pnas.0608056104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12(3):311–317. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matarazzo V, Cohen D, Palmer AM, Simpson PJ, Khokhar B, Pan SJ, Ronnett GV. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol Cell Neurosci. 2004;27(1):44–58. doi: 10.1016/j.mcn.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 99.Fukuda T, Itoh M, Ichikawa T, Washiyama K, Goto Y. Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J Neuropathol Exp Neurol. 2005;64(6):537–544. doi: 10.1093/jnen/64.6.537. [DOI] [PubMed] [Google Scholar]
- 100.Smrt RD, Eaves-Egenes J, Barkho BZ, Santistevan NJ, Zhao C, Aimone JB, Gage FH, Zhao X. Mecp2 deficiency leads to delayed maturation and altered gene expression in hippocampal neurons. Neurobiol Dis. 2007;27(1):77–89. doi: 10.1016/j.nbd.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tropea D, Giacometti E, Wilson NR, Beard C, McCurry C, Fu DD, Flannery R, Jaenisch R, Sur M. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A. 2009;106(6):2029–2034. doi: 10.1073/pnas.0812394106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Belichenko PV, Wright EE, Belichenko NP, Masliah E, Li HH, Mobley WC, Francke U. Widespread changes in dendritic and axonal morphology in Mecp2-mutant mouse models of Rett syndrome: evidence for disruption of neuronal networks. J Comp Neurol. 2009;514(3):240–258. doi: 10.1002/cne.22009. [DOI] [PubMed] [Google Scholar]
- 103.Degano AL, Pasterkamp RJ, Ronnett GV. MeCP2 deficiency disrupts axonal guidance, fasciculation, and targeting by altering Semaphorin 3F function. Mol Cell Neurosci. 2009;42(3):243–254. doi: 10.1016/j.mcn.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56(1):58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol. 2010;103(5):2470–2481. doi: 10.1152/jn.00601.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102(35):12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng V, Matagne V, Banine F, Frerking M, Ohliger P, Budden S, Pevsner J, Dissen GA, Sherman LS, Ojeda SR. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16(6):640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- 108.Zhang L, He J, Jugloff DG, Eubanks JH. The MeCP2-null mouse hippocampus displays altered basal inhibitory rhythms and is prone to hyperexcitability. Hippocampus. 2008;18(3):294–309. doi: 10.1002/hipo.20389. [DOI] [PubMed] [Google Scholar]
- 109.Kline DD, Ogier M, Kunze DL, Katz DM. Exogenous brain-derived neurotrophic factor rescues synaptic dysfunction in Mecp2-null mice. J Neurosci. 2010;30(15):5303–5310. doi: 10.1523/JNEUROSCI.5503-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gambino F, Khelfaoui M, Poulain B, Bienvenu T, Chelly J, Humeau Y. Synaptic maturation at cortical projections to the lateral amygdala in a mouse model of Rett syndrome. PLoS One. 2010;5(7):e11399. doi: 10.1371/journal.pone.0011399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Taneja P, Ogier M, Brooks-Harris G, Schmid DA, Katz DM, Nelson SB. Pathophysiology of locus ceruleus neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29(39):12187–12195. doi: 10.1523/JNEUROSCI.3156-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dani VS, Nelson SB. Intact long-term potentiation but reduced connectivity between neocortical layer 5 pyramidal neurons in a mouse model of Rett syndrome. J Neurosci. 2009;29(36):11263–11270. doi: 10.1523/JNEUROSCI.1019-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49(3):341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 114.Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, Zhang W. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99(1):112–121. doi: 10.1152/jn.00826.2007. [DOI] [PubMed] [Google Scholar]
- 115.Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci. 2007;27(40):10912–10917. doi: 10.1523/JNEUROSCI.1869-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zucker RS. Short-term synaptic plasticity. Ann Rev Neurosci. 1989:1213–1231. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]
- 117.Asaka Y, Jugloff DG, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21(1):217–227. doi: 10.1016/j.nbd.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 118.Boggio EM, Lonetti G, Pizzorusso T, Giustetto M. Synaptic determinants of Rett syndrome. Frontiers in Synaptic Neuroscience. 2010 doi: 10.3389/fnsyn.2010.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44(1):5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 120.Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29(16):5051–5061. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Larimore JL, Chapleau CA, Kudo S, Theibert A, Percy AK, Pozzo-Miller L. Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol Dis. 2009;34(2):199–211. doi: 10.1016/j.nbd.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci U S A. 1991;88(17):7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jugloff DG, Jung BP, Purushotham D, Logan R, Eubanks JH. Increased dendritic complexity and axonal length in cultured mouse cortical neurons overexpressing methyl-CpG-binding protein MeCP2. Neurobiol Dis. 2005;19(1–2):18–27. doi: 10.1016/j.nbd.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 125.Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52(2):255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li H, Chen Y, Jones AF, Sanger RH, Collis LP, Flannery R, McNay EC, Yu T, Schwarzenbacher R, Bossy B, Bossy-Wetzel E, Bennett MV, Pypaert M, Hickman JA, Smith PJ, Hardwick JM, Jonas EA. Bcl-xL induces Drp1-dependent synapse formation in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105(6):2169–2174. doi: 10.1073/pnas.0711647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Dragich J, Schanen C, Tamura M. Functional analyses of MeCP2 mutations associated with Rett syndrome using transient expression systems. Brain Dev. 2001;23 Suppl 1:S165–S173. doi: 10.1016/s0387-7604(01)00345-x. [DOI] [PubMed] [Google Scholar]
- 128.Kudo S, Nomura Y, Segawa M, Fujita N, Nakao M, Schanen C, Tamura M. Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J Med Genet. 2003;40(7):487–493. doi: 10.1136/jmg.40.7.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, Woodcock CL. Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem. 2008;283(29):20523–20534. doi: 10.1074/jbc.M803021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29(4):525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 131.Nelson ED, Kavalali ET, Monteggia LM. MeCP2-dependent transcriptional repression regulates excitatory neurotransmission. Curr Biol. 2006;16(7):710–716. doi: 10.1016/j.cub.2006.02.062. [DOI] [PubMed] [Google Scholar]
- 132.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16(6):1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 133.Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366(6455):569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- 134.Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci. 2009;29(23):7395–7403. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A. 2007;104(6):1931–1936. doi: 10.1073/pnas.0610593104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30(15):5346–5356. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ehninger D, Li W, Fox K, Stryker MP, Silva AJ. Reversing neurodevelopmental disorders in adults. Neuron. 2008;60(6):950–960. doi: 10.1016/j.neuron.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 139.Rueda N, Florez J, Martinez-Cue C. Chronic pentylenetetrazole but not donepezil treatment rescues spatial cognition in Ts65Dn mice, a model for Down syndrome. Neurosci Lett. 2008;433(1):22–27. doi: 10.1016/j.neulet.2007.12.039. [DOI] [PubMed] [Google Scholar]
- 140.Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415(6871):526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- 141.Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15(21):1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 142.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28(21):5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63(4):444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100(18):10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42(6):947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 147.Dolen G, Osterweil E, Rao BS, Smith GB, Auerbach BD, Chattarji S, Bear MF. Correction of fragile X syndrome in mice. Neuron. 2007;56(6):955–962. doi: 10.1016/j.neuron.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ. Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci. 2007;10(3):280–282. doi: 10.1038/nn1845. [DOI] [PubMed] [Google Scholar]
- 149.Belichenko PV, Dahlstrom A. Studies on the 3-dimensional architecture of dendritic spines and varicosities in human cortex by confocal laser scanning microscopy and Lucifer yellow microinjections. J Neurosci Methods. 1995;57(1):55–61. doi: 10.1016/0165-0270(94)00125-z. [DOI] [PubMed] [Google Scholar]
- 150.Armstrong DD. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev. 2002;8(2):72–76. doi: 10.1002/mrdd.10027. [DOI] [PubMed] [Google Scholar]
- 151.Cobb S, Guy J, Bird A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans. 2010;38(2):498–506. doi: 10.1042/BST0380498. [DOI] [PubMed] [Google Scholar]
- 152.Alvarez-Saavedra M, Saez MA, Kang D, Zoghbi HY, Young JI. Cell-specific expression of wild-type MeCP2 in mouse models of Rett syndrome yields insight about pathogenesis. Hum Mol Genet. 2007;16(19):2315–2325. doi: 10.1093/hmg/ddm185. [DOI] [PubMed] [Google Scholar]
- 153.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302(5646):885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 154.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 155.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41(1):108–118. [PubMed] [Google Scholar]
- 156.Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2(1):24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- 157.Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD. From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem. 2002;9(5):224–237. doi: 10.1101/lm.51202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10(2):86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15(10):1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- 160.Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci. 2000;20(1):8–21. doi: 10.1523/JNEUROSCI.20-01-00008.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lynch G, Gall CM. Ampakines and the threefold path to cognitive enhancement. Trends Neurosci. 2006;29(10):554–562. doi: 10.1016/j.tins.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 162.Wang H, Chan SA, Ogier M, Hellard D, Wang Q, Smith C, Katz DM. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in Mecp2 null mice. J Neurosci. 2006;26(42):10911–10915. doi: 10.1523/JNEUROSCI.1810-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zheng WH, Quirion R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem. 2004;89(4):844–852. doi: 10.1111/j.1471-4159.2004.02350.x. [DOI] [PubMed] [Google Scholar]
- 164.Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci. 2007;25(7):1915–1922. doi: 10.1111/j.1460-9568.2007.05466.x. [DOI] [PubMed] [Google Scholar]
- 165.Nag N, Berger-Sweeney JE. Postnatal dietary choline supplementation alters behavior in a mouse model of Rett syndrome. Neurobiol Dis. 2007;26(2):473–480. doi: 10.1016/j.nbd.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 166.Nag N, Mellott TJ, Berger-Sweeney JE. Effects of postnatal dietary choline supplementation on motor regional brain volume and growth factor expression in a mouse model of Rett syndrome. Brain Res. 2008:1237101–1237109. doi: 10.1016/j.brainres.2008.08.042. [DOI] [PubMed] [Google Scholar]
- 167.Ward BC, Kolodny NH, Nag N, Berger-Sweeney JE. Neurochemical changes in a mouse model of Rett syndrome: changes over time and in response to perinatal choline nutritional supplementation. J Neurochem. 2009;108(2):361–371. doi: 10.1111/j.1471-4159.2008.05768.x. [DOI] [PubMed] [Google Scholar]
- 168.van Praag H, Kempermann G, Gage FH. Neural consequences of environmental enrichment. Nat Rev Neurosci. 2000;1(3):191–198. doi: 10.1038/35044558. [DOI] [PubMed] [Google Scholar]
- 169.Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7(9):697–709. doi: 10.1038/nrn1970. [DOI] [PubMed] [Google Scholar]
- 170.Nag N, Moriuchi JM, Peitzman CG, Ward BC, Kolodny NH, Berger-Sweeney JE. Environmental enrichment alters locomotor behaviour and ventricular volume in Mecp2 1lox mice. Behav Brain Res. 2009;196(1):44–48. doi: 10.1016/j.bbr.2008.07.008. [DOI] [PubMed] [Google Scholar]