

Abstract
Biofunctionalization of silicon substrates is important to the development of silicon-based biosensors and devices. Compared to conventional organosiloxane films on silicon oxide intermediate layers, organic monolayers directly bound to the non-oxidized silicon substrates via Si-C bonds enhance the sensitivity of detection and the stability against hydrolytic cleavage. Such monolayers presenting a high density of terminal alkynyl groups for bioconjugation via copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC, a “click” reaction) were reported. However, yields of the CuAAC reactions on these monolayer platforms were low. Also, the non-specific adsorption of proteins on the resultant surfaces remained a major obstacle for many potential biological applications. Herein, we report a new type of “clickable” monolayers grown by selective, photo-activated surface hydrosilylation of α,ω-alkenynes, where the alkynyl terminal is protected with a trimethylgermanyl (TMG) group, on hydrogen-terminated silicon substrates. The TMG groups on the film are readily removed in aqueous solutions in the presence of Cu(I). Significantly, the degermanylation and the subsequent CuAAC reaction with various azides could be combined into a single step in good yields. Thus, oligo(ethylene glycol) (OEG) with an azido-tag was attached to the TMG-alkyne surfaces, leading to OEG-terminated surfaces that reduced the non-specific adsorption of protein (fibrinogen) by >98%. The CuAAC reaction could be performed in microarray format to generate arrays of mannose and biotin with varied densities on the protein-resistant OEG background. We also demonstrated that the monolayer platform could be functionalized with mannose for highly specific capturing of living targets (Escherichia coli expressing fimbriae) onto the silicon substrates.
Introduction
Modification of silicon substrates with ultrathin organic films to allow for specific interactions with biological targets is important for the development of silicon-based bioelectrical sensors and devices,1,2 nanoparticle probes,3,4 nanowire sensors,5 photonic devices,6,7 cantilever sensors,8,9 microarrays,10,11 microfluidic12 and silicon-neuron interfaces.2,13 These silicon-based transducers interconvert specific biomolecular interactions with electrical, mechanical, or optical signals of the silicon devices. Ideal thin film platforms on silicon substrates should allow specific binding of biological targets. To block nonspecific binding, the silicon substrates are commonly modified with organosiloxane films presenting oligo- or poly(ethylene glycol) (OEG or PEG) on the oxide surface of the substrates.14 However, the protein-resistance and stability of these films are not satisfactory, probably due to the relatively low packing density of the films and the high density of defects resulted from the interaction of silanols with the hydrophilic OEG chains.14,15 Ourselves and others have developed monolayers presenting OEG, which are directly bound on non-oxidized silicon substrates via Si–C bonds.7,9,16–19 Formation of the Si–C bonds is via surface hydrosilylation on hydrogen-terminated silicon surfaces,20–22 using OEG25 terminated alkenes, such as 1 in Scheme 1.9,17–19,23 Our OEG-terminated monolayers are highly protein-resistant and stable in phosphate-buffered saline (PBS).19 Herein, we describe the development of a monolayer platform presenting OEG-alkyne on silicon substrates, which allow efficient bioconjugation using copper-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC, a “click” reaction).24–26
Scheme 1.

Preparation of the TMG-terminated Film (A) from the Alkenyne 2 and Its Deprotection to the Ethynyl-presenting Film B and Direct CuAAC Reactions with the Azides 3–7 Promoted by Cu+ and the Ligand 10 to Form Films Presenting CF3 (C), OEG (D), Mannose (E), Glucose (F) and Biotin (G)
Among a variety of reactions for biofunctionalization on surfaces,10,27 CuAAC reaction is specific and bioorthogonal, and can be performed under physiological conditions.25,26,28 It has been used on a wide variety of substrates.4,29–32 To use CuAAC reaction on alkylated silicon substrates, we need to incorporate either azido or alkynyl groups on the surface. Azido-presenting monolayers on silicon were prepared from H–Si surfaces through two steps: chlorination33 or hydrosilylation with Br-terminated alkenes34 followed by substitution with NaN3. Direct attachment of N3-alkenes onto H–Si surfaces by hydrosilylation has not been reported, and failed in our attempts, likely because the azido groups readily decompose during photo- or thermally-activated hydrosilylation via a highly reactive nitrenen intermediate.35
Alternative to azido-presenting monolayers, two types of alkynyl-presenting monolayers bound on silicon via Si–C bonds were reported.30,32,36,37 The first type was prepared by ethynylation of chlorinated Si(111) surfaces, providing a nearly complete coverage of ethynyl groups directly bound to the silicon substrate. The close packing of the rigid ethynyl groups on the atomically flat surface likely prevented the CuAAC reaction; the reaction could only occur at the step edges and defect sites, leading to a low overall yield (7%).32,36 The second type was prepared by Gooding and co-workers elegantly from a commercially available distal diyne (1,8-nonadiyne) by thermally activated hydrosilylation.30,37 The subsequent grafting of an OEG-azide to the monolayer via CuAAC reaction proceeded with a modest yield (42%–51%). Unfortunately, the protein resistance of the resultant OEG-terminated films was not satisfactory (adsorbing ~25% monolayer of bovine serum albumin), likely due to the low density of the OEG chains grafted on the hydrophobic alkynyl surface.30 The reasons for the lower yields of the CuAAC reactions on alkynyl vs azido surfaces were unclear, and could be due to steric hindrance, side reactions, and/or polymerization of the well-ordered alkynes.25 Gooding and co-workers showed that decreasing the density of the alkynyl chains by co-deposition with alkyl chains increased the yields of the subsequent CuAAC reaction up to 90%, attributed to the decrease of the above factors.37 However, this approach might not provide sufficient density of OEG chains for resisting non-specific adsorption of proteins.
Herein we report a versatile “clickable” monolayer platform that addresses the above issues. This platform is easily grown by photo-activated hydrosilylation of an enyne, where the alkynyl terminal is masked with a trimethylgermanyl (TMG) group, on hydrogen-terminated silicon surfaces. The removal of the TMG group and the subsequent CuAAC reaction can be performed in a single step in good yields. Significantly, non-specific adsorption of proteins on the resultant OEG surfaces was reduced by >98%, attributable to the unique alkynyl-OEG-alkyl platform and the efficient grafting of the OEG chains onto the platform. The reaction could be performed in microarray format to attach azido-labeled molecules (e.g. mannose and biotin) with varied densities on an OEG background to allow specific binding of targeted molecules. We also demonstrate that the monolayer platforms could be functionalized with mannose to specifically capture living targets (E. coli expressing mannose-binding fimbria) onto the silicon substrates.
Results and Discussion
Monolayer Preparation and Deprotection
Our approach to preparation of alkynyl-presenting (“clickable”) monolayers on silicon substrates is based on the selective hydrosilylation of α,ω-alkenynes on H–Si surfaces. In this approach, the terminal alkynyl group needs to be masked with a bulky protecting group since it is usually more reactive than the alkene group.38 The most common protecting groups for terminal alkynes are trialkylsilyl groups that are readily removed with F− in protic solvents. We initially tested several trialkylsilyl groups, including the fluorine-containing ones for monitoring their removal on the films by x-ray photoelectron spectroscopy (XPS). Unfortunately, the desilylation on these monolayers with F− in various solvents was sluggish, requiring a high concentration of F−, long reaction time and high temperatures (data not shown).
In search for a protecting group for terminal alkynes that could be removed under very mild, neutral conditions, we turned our attention to trimethylgermanyl (TMG) group.39 Cai, Ernst, and Vasella found that TMG group on terminal alkynes could be readily removed in protic solvents in the presence of catalytic amounts of Ag+ or Cu+.39 The reaction probably starts by the formation of Ag+ or Cu+ complexes with the protected alkyne leading to a β-vinyl cation that is more stabilized by Ge than by Si through hyperconjugation (β-effect).40 The subsequent degermylation followed by protonation of the metal acetylide intermediate provides the deprotected alkyne. In this work, the monolayers presenting C≡C-TMG groups (film A, Scheme 1) were prepared from the alkenyne 2 by photo-activated hydrosilylation on H-terminated silicon (111) surfaces.21 The films were characterized by ellipsometry, contact angle, atomic force microscopy (AFM), and XPS. The ellipsometric thickness of the monolayer was 56 ± 1 Å, close to the estimated molecular length (59 Å). Selected XPS spectra of the TMG-terminated films A (Scheme 1) are presented in Figure 1a–d. The survey scan of the film A (Figure 1a) shows the presence of C, O, Ge and Si. The narrow scan in the C1s region (Figure 1b) displays two deconvoluted signals at 286.7 eV and 285.0 eV, assigned to the etheric (C–O) and the rest of the carbon atoms, respectively. The ratio of areas derived from curve-fitting was 1.36, similar to the stoichiometric ratio (1.38). A narrow scan of the Ge3d region (Figure 1c, empty circle) showed a strong emission at 31.4 eV, assigned to the TMG group. The ratio of C/O/Ge was found to be 1:0.30:0.029, similar to the stoichiometric value of 1:0.29:0.026. A narrow scan of the Si2p region (Figure 1d, empty circle) showed the absence of any emission at 101–104 eV regions, thus no detectable oxide or suboxide silicon was present. The water contact angle of the film A was 62° ± 1°, and remained similar (59° ± 1°) upon removal of the TMG group to form film B. In comparison, a higher water contact angle of ~87° was reported for alkynyl-terminated monolayers.31 The lower water contact angles for both films A and B may be due to the amorphous or liquid-like state of the OEG chains, as illustrated in Scheme 1 for film A, which allows for a portion of the hydrophilic OEG chains to dynamically interact with water at the interface.
Figure 1.

Selected XPS data obtained on the films A before and after degermanylation and CuAAC reactions. XPS survey (a) and narrow scan for C1s (b, with deconvolution) of the films A, and narrow scans for Ge3d (c), Si2p (d), F1s (e) and N1s (f, with deconvolution) before (empty circle) and after (solid dot) CuAAC reaction with the CF3-terminated azide 3.
A typical atomic force microscopy (AFM) image of film A is shown in Figure 2a. Remarkably, the atomic steps of the silicon (111) substrate underneath the 56 Å thick film are clearly visible, indicating that the films were homogenous. The corresponding root mean square (rms) roughness was 0.34 nm.
Figure 2.
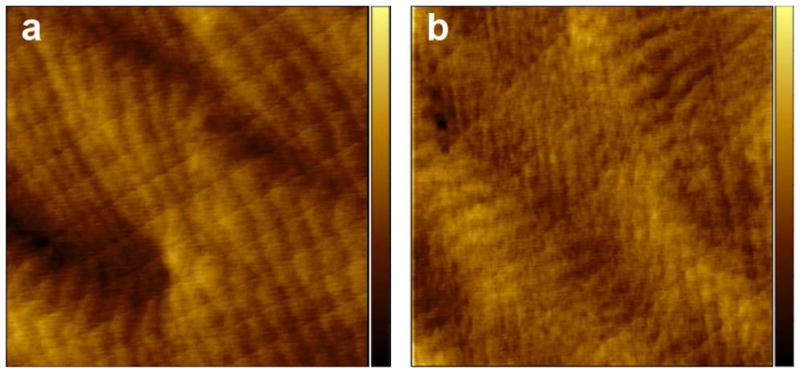
Tapping mode AFM images (3×3 μm2) of the TMG-alkynyl-terminated film A before (a) and after (b) CuAAC reaction with the OEG-azide 4. The z-scale (contrast) for both images is 3 nm.
The deprotection of the alkynyl groups was monitored ex situ by the decrease of the Ge3d signal intensity (Figure 1c, solid dots). Indeed, the degermanylation was greatly promoted by Cu+ in aqueous solution. At a copper concentration of 1.25 mM, it was completed within 30 min. Ascorbic acid served to reduce the air-oxidized copper to the catalytically active Cu+. We found that Ag+ was more efficient for the degermenylation, but did not facilitate the subsequent CuAAC reaction. R emarkably, Cu+ ligands that enhance the CuAAC reaction (see below) did not affect the degermanylation.
Direct CuAAC Reactions on the TMG-alkynyl-terminated Films
The main advantage of using TMG protecting group is that its removal proceeds faster than the CuAAC reaction, both being promoted by Cu+. Hence, they can be combined into one step for direct attachment of the azides 3–7 to the films A (Scheme 1). Indeed, XPS data (Figure 1) supported that the films A underwent CuAAC reaction with the CF3-terminated azide 3 (5 mM) in the presence of Cu(MeCN)4PF6 (1.25 mM) and ascorbic acid (25.0 mM). The F1s signal appeared at 690 eV (Figure 1e) and N1s at 401 eV (Figure 1f), accompanied by the reduction of the Ge3d signal intensity by 95% (Figure 1c). The N1s signal was deconvoluted and fitted to three peaks assigned to CONH (400.1 eV), N–N=N (400.8 eV), and N–N=N (401.7 eV), the ratio of the peak areas being about 1.2:2:1. The assignment of the N1s signals from the triazole ring is supported by the reported XPS data and density-functional theory calculation for some aromatic compounds containing sp2 N atoms bonded to two or three atoms, showing that the N1s signal from the former is ~1 eV lower than the latter.41 No signal was present at ~403 eV corresponding to the central, electron-deficient N-atom in the azido group, indicating no physisorption of 3 in the film.42 The atomic concentration ratio N/F was 1.3, consistent with the value of 1.33 from the molecular formula. Based on the C/F and C/N ratios, the reaction yield was estimated to be ~42% (Table 1). This value is similar to the yield (50%) derived from the increase of ellipsometric thickness (10 ± 2 Å) vs the calculated increase of molecular length (Table 1). Narrow scan of the Si2p region showed no detectable SiOx species in the 102–104 eV region (Figure 1d, solid dot).
Table 1.
XPS and Ellipsometry Derived Yields for the Grafting of the Azides 3–6 under CuAAC Reaction Conditionsa,b onto the TMG-alkynyl Films A to Form Films C–F
| Films | XPS |
Ellipsometry |
|||||
|---|---|---|---|---|---|---|---|
| C/F ratio | Yieldc (%) | C/N ratio | Yieldd,e (%) | Δdexpf (Å) | Δdcalcg (Å) | Yieldh (%) | |
| Ca | 32 ± 6 | 43 | 25 ± 5 | 41d | 10 ± 2 | 20 | 50 |
| Cb | 20 ± 4 | 76 | 17 ± 3 | 65d | 13 ± 2 | 20 | 65 |
| Db | 19 ± 4 | 75e | 10 ± 2 | 14 | 71 | ||
| Eb | 22 ± 4 | 72e | 13 ± 2 | 21 | 62 | ||
| Fb | 22 ± 4 | 72e | 12 ± 2 | 21 | 57 | ||
Azide 3 (5 mM), Cu(MeCN)4PF6 (1.25 mM) and ascorbic acid (25 mM) in EtOH/H2O 1:1, 25°C in air, 12 h.
Azide (5 mM, 3 for C, 4 for D, 5 for E, 6 for F), Cu(MeCN)4PF6 (1.25 mM), the ligand 10 (12.5 mM) and ascorbic acid (25 mM) in EtOH/H2O 1:1, 25°C in nitrogen, 4 h.
Yield (x%) is derived from C/F = (35 + 14x%)/3x%, where C/F is the atomic ratio measured by XPS with a random uncertainty of ~20%,43 3 is the number of F-atom in the azide 3, and 35 and 14 are the number of C-atom in the alkyne (after degermenylation of 2) and the azide 3, respectively.
Yield (x%) is derived from C/N = (35 + 14x%)/4x%, where C/N is the atomic ratio measured by XPS with a random uncertainty of ~20%,43 4 is the number of N-atom in the azide 3, and 35 and 14 are the number of C-atom in the degermenyled alkyne and the azide 3, respectively.
Yield (x%) is derived from C/N = (35 + NC x%)/3x%, where C/N is the atomic ratio measured by XPS with a random uncertainty of ~20%,43 3 is the number of N-atom in the azides 4–6, 35 is the number of C-atom in the degermenyled alkyne, NC is the number of C-atom in the corresponding azide (10 for 4 and 18 for 5 and 6).
Δdexp is the difference of the film thickness measured by ellipsometry before and after grafting of the corresponding azides 3–6 to the film A. The standard deviations of thickness measurement were within ±1 Å, leading to an uncertainty of ±2 Å for Δdexp.
Δ dcalc is the calculated increase of the molecular length of the TMG-alkyne 2 after CuAAC reaction with the corresponding azides. The molecular length increased from 59 Å of 2 to 79 Å after coupling with 3, 73 Å with 4, and 80 Å with 5 and 6.
Yield (%) = Δ dexp/Δ dcalc.
Minimizing Oxidative Degradation of the Films
Although it was convenient that the azides could be directly grafted to the TMG-alkynyl surfaces A, the reaction time (12 h) was long, and the yield (~42%) was unsatisfactory. Similarly low efficiency was reported for CuAAC reactions on other alkynyl-presenting surfaces,30,31,44 and was attributed to steric hindrance.30 During optimization of the reaction conditions, we found that O2 substantially decreased the yields, likely due to the facile oxidation of Cu+ to the catalytically inactive Cu2+ that may also promote the homocoupling of the adjacent alkynes.45 Furthermore, the redox cycle of Cu+/Cu2+ in the presence of O2 and sodium ascorbate generates oxy radicals26 that may degrade the OEG film.19 Hence, we performed the reactions in a N2 environment. Next, we tested a series of copper concentrations in the range of 0.3–10 mM. When the copper concentration was below 1 mM, the reaction was sluggish. Notably, the yields were not affected by the copper concentrations in the range of 2.5–10 mM. After the reaction, the harmful copper residue could be removed by washing with an EDTA solution, as confirmed by the absence of the Cu2p3/2 signal near 933 eV.
Improved Copper Catalyst
Recently, several series of Cu+ ligands have been investigated to accelerate CuAAC reactions.25,46,47 Among them, the commercially available tris-triazole 846 (TBTA, Scheme 1) has been most widely used. Unfortunately, we found that this ligand was ineffective for promoting surface CuAAC reactions in our systems, probably due to the steric hindrance of the ligand and the surface alkyne groups. We then tested several smaller ligands similar to the monotriazole 9 reported by Fokin and coworkers,46 and identified the most efficient ligand 10. The OEG chain in 10 renders the Cu+ complex water soluble, and the electron-donating NHCH3 group maintains a high catalytic activity of the complex. Indeed, grafting of the CF3-terminated azide 3 onto the TMG-ethynyl-terminated films A was greatly accelerated by the ligand 10, as shown by Figure 3 plotting the increase of the atomic concentration ratio F/C of the film over time in the presence and absence of the ligand 10. In the presence of 10, the reaction was completed in ~2 h as compared to 12 h without the ligand. The yield (65–76%) of the optimized reaction leading to film C was estimated by the F/C and N/C ratio, and the increase of the ellipsometric thickness (Table 1). The yield derived from the F/C ratio is higher, likely due to the ignorance of the attenuation factor leading to an overestimate of the F/C ratio, especially for the dense OEG top layer where the CF3 terminal groups are populated closer to the film surface.
Figure 3.
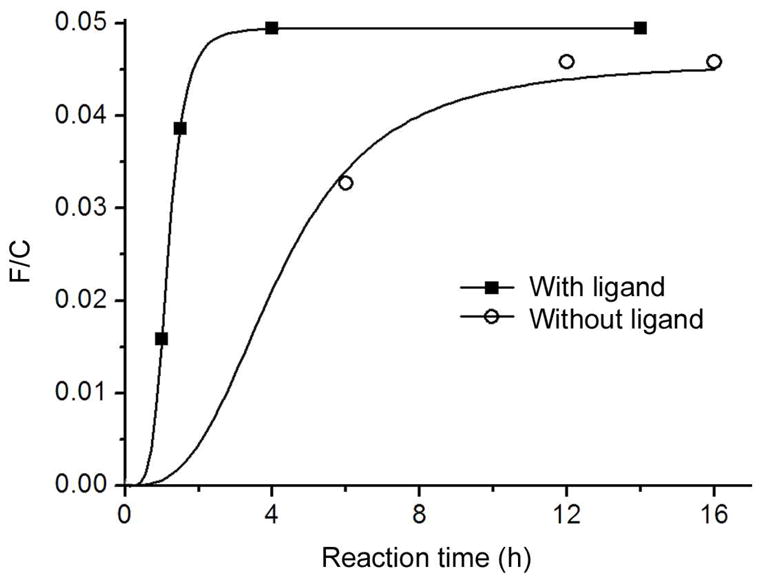
Progress of the CuAAC reaction on the TMG-alkynyl-terminated films A with the CF3-terminated azide 3, monitored ex situ by the F/C ratio of the film after various reaction time in the presence (square) and absence (circle) of the ligand 10 under otherwise identical conditions: Cu(MeCN)4PF6 (1.25 mM), ascorbic acid (25 mM), the azide 3 (5 mM) and the ligand 10 (12.5 mM) in EtOH/H2O 1:1 at 25°C. Each data point was obtained by reacting a film A in the reaction mixture for the given time, followed by cleaning and measuring of the F/C ratio of the film by XPS. The curve serves to guide the eyes.
Preparation of Films D–F
Using the above optimized conditions, the azides 4–6 were attached onto the TMG-terminated films A to provide the films presenting OEG (D), mannose (E) and glucose (F) (Scheme 1). The yields of the reactions were estimated from the XPS C/N ratio, and the increase of the ellipsometric thickness. As shown in Table 1, the yields derived by both methods were consistent within the ~20% random uncertainty43 for XPS measurement of the N1s signals and the ± 2 Å uncertainty for measurement of the increase of thickness. The yield (71–75%) for grafting the OEG-azide 4 was a substantial improvement over the reported ones on other systems.30 Most importantly, the resultant OEG-presented surfaces D were highly protein-resistant (see below). The slightly lower efficiency for grafting the sugars 5 and 6 is probably due to the steric hindrance of the sugar.
Films Presenting OEG (D)
Upon the CuAAC reaction leading to film D, the ellipsometric thickness was increased to 68 ± 1 Å, similar to the calculated molecular length of 73 Å, indicating a high density of the OEG chains grafted onto the film A, although determination of the exact OEG density on such thick films is beyond the scope of this work. The homogeneity of the films was maintained after the reaction, as indicated by the AFM image (Figure 2b) showing the atomic steps of the underlying silicon substrate, and by the small rms roughness of 0.35 nm. XPS data show the disappearance of the Ge3d signal (Figure 4a) and the appearance of the N1s signal (Figure 4b) that can be deconvoluted into two peaks at binding energies of 400.1 eV and 401.1 eV with an intensity ratio of 2:1, corresponding to the triazole moieties. The C1s signal is deconvoluted into two peaks at 286.3 eV for C–O and C–N and at 284.8 eV for the alkyl carbon atoms.
Figure 4.

Selected XPS narrow scans for Ge3d, N1s, and C1s of the films D and E prepared from A via CuAAC reaction with the azides 4 and 5 (Scheme 1), respectively. The data for films D include Ge3d (a, solid dots for film D vs empty circles for film A before the reaction), N1s (b, with deconvolution), N1s before (c, dots behind the squares) and after (c, squares) treatment with a 0.1% fibrinogen solution vs the N1s signal of a monolayer of fibrinogen (triangle) adsorbed on a H-Si (111) surface, and C1s (d, with deconvolution). The data for the films E include the deconvoluted C1s (e) and the N1s (f) signals.
Protein Resistance of Films D
The protein resistance of the above OEG-modified films D was evaluated by XPS measurement of the amount of adsorbed fibrinogen after incubation in a 0.1% fibrinogen solution in PBS for 1 h, followed by gentle washing with Millipore water for only ~30 seconds. The increase of the N1s signal intensity relative to that of a standard monolayer of fibrinogen17 indicated that only about (1.6 ± 0.8)% (n = 4) monolayer of the protein was adsorbed on the OEG-modified film D. Note that fibrinogen possesses ~4300 nitrogen atoms, leading to a detection limit of ~0.8% monolayer. Indeed, the N1s signals before and after treatment with the fibrinogen solution almost completely overlap to each other (Figure 4c, the squares and dots behind the squares). Factors influencing the protein-resistance of OEG monolayers are still not well understood.48 The high protein resistance of film D may be associated with the appropriate density of the OEG chains and their amorphous/liquid-like state, as illustrated in Scheme 1, which promotes tight binding of water.48
Films-presenting Mannose (E) and Glucose (F)
Upon grafting the mannose-azides 5 onto the TMG-terminated films A, the ellipsometric thickness of the film was increased to 71 ± 1 Å, not much behind the calculated molecular length of ~80 Å. The yield of the reaction was estimated to be 62–72% (Table 1). Considering the relative large size of the mannose moiety, the grafting is quite efficient. The water contact angle on the resultant film E decreased from 62° ± 1° to 33° ± 2°. XPS narrow scan for C1s of the films E show the increase of the etheric C1s signal at 286.3 eV. The N1s signal (Figure 4f) can be deconvoluted into two peaks at binding energies of 400.0 eV for N–N=N and 401.1 eV for N–N=N with an intensity ratio of 1:2. Unreacted mannose-azides were not present after the CuAAC reaction, as no apparent peak was observed near 403 eV. The thickness, contact angle and XPS data for the glucose-presenting films F were similar to those of the mannose-presenting films E.
Specific Adherence of Bacteria to Mannose-presenting Surfaces (E)
To demonstrate that the thin film platforms can be functionalized via CuAAC reaction to capture specific living biological targets, we used the mannose-modified film E to interact with E. coli 83972 strains with or without mannose-binding type-1 fimbrae (fim+ or fim−). Previously, self-assembled alkanethiol monolayers presenting mannose on gold substrate surfaces were used to capture E. coli expressing fimbrae,49 albeit without comparison with the bacteria that do not possess fimbrae. The mannose-presenting films E were incubated overnight in Luria Bertani media containing either fim+ or fim− E. coli 83972. As controls, films presenting either glucose (F) or ethynyl groups (upon degermanylation of A) were likewise exposed to these organisms under identical conditions. As shown by Figure 5a,f, the fim+ E. coli nearly fully covered the mannose-presenting surfaces, while the fim− strain did not adhere to the surfaces (Figure 5b,f). In addition, very few fim+ E. coli attached on the glucose-presenting surface (Figure 5c,f). Furthermore, no fim+ E. coli were seen on the OEG-alkynyl-terminated surface B (Figure 5d,f). Finally, pre-incubation of the fim+ E. coli strain with mannose substantially reduced their subsequent adherence to the mannose-presenting surface (Figure 5e,f). These results clearly demonstrate that the binding of fim+ E. coli is specifically between the mannose binding receptors on the bacterial fimbrae and the mannose presented on the surface E. The reason for the ability of OEG-alkynyl-presenting film B to repel fim+ E. coli is unclear. Understanding the factors influencing bacterial adhesion to surfaces is in its infancy.50 In general, modification of surfaces with OEG and PEG reduces bacterial adhesion to various extents depending on the bacterial species.51
Figure 5.

Fluorescent images (a–e) of various modified surfaces incubated with fim+ and fim− E. coli, and a plot (f) of the numbers of E. coli in all images with a standard deviation on these surfaces. The combinations depicted are: the mannose-presenting film E and fim+ E. coli (a, bacterial count: 11313 ± 1241 for f), film E and fim− E. coli (b, bacterial count: 1 ± 1 for f), the glucose-presenting films F and fim+ E. coli (c, bacterial count: 7 ± 1 for f), the ethynyl-presenting films B and fim+ E. coli (d, bacterial count: 0 ± 0 for f), and film E and the fim+ E. coli that had been pre-saturated with mannose in the media (e, bacterial count: 627 ± 352 for f). Each image is representative of up to 20 images obtained on random locations at the sample surface (for examples, see Figures S2–S6 in Supporting Information).
Attachment of Biotin and Mannose in Microarray Format
The versatility of the TMG-alkynyl-terminated monolayers A was demonstrated by performing the multi-component CuAAC reactions in microarray format (Scheme 2). Biotin and mannose with an azido handle (compounds 7 and 5) were mixed with the OEG azide 4 at ratios of 1:0, 1:1 and 1:9, together with other reagents at a ratio of azide/CuSO4/ligand 10/ascorbic acid 1:1.6:11:19, and were then spotted on the film. The spotting of the mixture of reagents and the subsequent reactions on the surface were performed in an anaerobic chamber under N2 atmosphere with a relative humidity of 60% for 4 h. The remaining surface was then back-filled with OEG via CuAAC reaction with the OEG-N3 4 to resist non-specific adsorption of proteins. The samples were then incubated in solutions of avidin and Concanavalin A (Con A), both labeled with fluorescein isothiocyanate (FITC). Selective binding of the proteins to the ligands immobilized via CuAAC reaction is shown in the fluorescent images (Scheme 2). The amount of bound proteins decreases with the ratio of the biotin azide 7 or the mannose azide 5 relative to the OEG azide 4. The control experiment with avidin-FITC saturated with biotin, or FITC-ConA saturated with mannose, showed no binding to the biotin- or mannose-presenting spots, thus establishing that the bindings were specific.
Scheme 2.

Attachment of the Biotin-N3 7 and Mannose-N3 5 with the OEG-N3 4 on the TMG-alkynylterminated Films A via CuAAC Reaction, Followed by Back-filling with the OEG-N3 4 and Binding with FITC-labeled Avidin and Con A.
Conclusion
We have developed a versatile monolayer platform presenting trimethylgermanyl (TMG)-protected alkynyl groups on silicon substrates that allows for direct tethering of biomolecules via CuAAC reaction in good yields. Significantly, the efficient grafting of OEG chains onto this platform provided an OEG-terminated surface that is highly resistant to non-specific adsorption of proteins, thus addressing the key issue of non-specific binding on the functionalized monolayers on non-oxidized silicon. Moreover, the CuAAC reaction mixtures can be spotted on the platform and the rest of the surface subsequently be passivated with OEG-azide to provide arrays/patterns of biomolecules with controlled composition on an inert background. We have also shown that upon attaching a mannose-azide to the monolayer platform, the resultant mannose-presenting surfaces can specifically capture E. coli expressing mannose-binding fimbriae. Furthermore, organogermanium has a low toxicity (it has been used in dietary supplements).56 We expect that this “clickable” platform can be applied for biofunctionalization of a wide range of silicon-based materials, including porous membrane, nanoparticles and nanowires.
Experimental Section
Materials
Sulfuric acid, 30% hydrogen peroxide solution, 40% ammonium fluoride solution, dichloromethane, N,N,N′,N′-tetramethylethane-1,2-diamine (EDTA), ascorbic acid, sodium ascorbate, Cu(MeCN)4PF6, CuSO4, fluoresceine isothiocyanate (FTIC)-avidin, fibrinogen and FTIC-Con A were purchased from Sigma-Aldrich, Silicon (111) wafers from Silicon Quest Int’l. Inc., and absolute ethanol from Alfa Aesar. The synthesis of compounds 2–7 are provided in Supporting Information.
Ellipsometry
Thickness measurements were performed on a Multiskop system (Optrel GmbH, Germany) or an Auto EL III ellipsometer (Rudolph Research) equipped with a 632.8 nm He-Ne laser source at an incident angle of 60° or 70°. The optical constants of the substrate were determined with a piece of freshly prepared H-Si(111) wafer (n = 3.839 and k = 0.016). The thicknesses of the monolayers were determined with assumed refractive indices of 1.45 for the organic monolayer. At least three measurements in random locations were taken for each sample, and the mean values were reproducible within ± 1 Å.
Estimation of Molecular Length
The molecular length was estimated by molecular mechanics modeling with MM2 in Chem3D Ultra 10.0 (CambridgeSoft).
Contact Angle Goniometry
Contact angles were measured on a Rame-Hart Model 100 goniometer under ambient conditions. Both edges of 3 drops of the contacting liquids (water) were measured on random locations of the surface for each sample.
X-ray Photoelectron Spectroscopy (XPS)
XPS was performed with a PHI 5700 X-ray photoelectron spectrometer equipped with a monochromatic Al Kα X-ray source (1486.7 eV) at a take-off angle (TOA) of 45° from the film surface. The spectrometer was operated both at high and low resolutions with window pass energies of 23.5 eV and 187.85 eV, respectively. Electron binding energies were calibrated with respect to the C1s line at 286.4 eV (C-C) or the Si2p line at 99.0 eV. The atomic concentrations were estimated by the PHI Multipak 5.0 software (Physical Electronics) using the standard procedure including the Shirley background subtraction, and corrections with the corresponding Scofield atomic sensitivity factors, assuming a homogenous distribution of the atoms to a depth of a few nanometers. The signal deconvolution was performed first by Shirley background subtraction, followed by non-linear fitting to mixed Gaussian-Lorentzian functions with 80% Gaussian and 20 % Lorentzian character.
Atomic Force Microscopy (AFM)
AFM imaging of the surfaces was performed using a MultiMode Nanoscope IIIa AFM (Digital Instruments Inc., Santa Barbara, CA). Images were acquired in tapping mode using a silicon nitride cantilever (MikroMasch, San Jose, CA) with a resonance frequency of 132.9 KHz and a nominal force constant of 1.75 N/m.
Fluorescence Microscopy
Fluorescent images were obtained with an Olympus BX 51 fluorescence microscope. Images were processed using QCapture software (QImaging Co.).
Preparation of H-Si(111) Substrates
Single side polished, p-type (boron-doped, 1–10 Ω-cm resistivity) silicon (111) wafers (Silicon Quest Int’l. Inc.) were cut into pieces of 2×2 cm2, cleaned with Piranha solution (concentrated H2SO4/30% H2O2 3:1 v/v) for 20–30 min at ~80 °C to remove organic contaminates. Caution: Piranha solutions react violently with organic materials and should be handled with extreme care. The freshly cleaned sample was immersed in an Argon-saturated, 40% NH4F solution for 20 min followed by rapid rinse with Argon-saturated Millipore water and dried with a stream of nitrogen.
Monolayers Terminated with TMG-C≡C Groups
The apparatus and procedure for surface hydrosilylation was described elsewhere.20 Briefly, a freshly prepared H-Si(111) substrate was placed on top of a z-translational manipulator inside a home-made vacuum chamber. After degassing for 10 min at 10−4 Torr, the sample was brought in contact with a droplet (ca. 2–3 mg) of the alkene 2 on a quartz window, forming a uniform layer of the alkene sandwiched by the quartz window and the silicon substrate. Hydrosilylation was performed under 254 nm UV illumination with a handhold illuminator (Spectroline Co.) for 2 h. The sample was washed thoroughly with dichloromethane and absolute ethanol followed by drying under a stream of Argon.
Removal of the TMG Protecting Groups
Under N2 environment, the above monolayers presenting TMG-alkynyl groups were immersed in a solution of CuSO4 (1.25 mM), sodium ascorbate (25 mM) and the ligand 10 (12.5 mM) in degassed water, or a solution of Cu(MeCN)4PF6 (1.25 mM) or AgNO3 (1.25 mM), ascorbic acid (25 mM) and the ligand 10 (12.5 mM) in degassed MeOH/EtOH/H2O (2:1:1) for 10–60 min, followed by washing with Millipore water and immersion in 25 mM EDTA solution, sonication in EtOH/MeOH (1:1) for 30 s and then in Millipore water for 30 s, and drying with a stream of nitrogen.
Surface CuAAC Reactions
Under nitrogen, a TMG-terminated substrate A was immersed in a solution of Cu(MeCN)4PF6 (1.25 mM), an azide (5 mM), the copper ligand 10 (12.5 mM) and ascorbic acid (25 mM) in degassed methanol/water (1:1 v/v). Alternatively, the reaction could be performed in a solution of CuSO4 (1.25 mM), an azide (5 mM), the copper ligand 10 (12.5 mM) and sodium ascorbate (25 mM) in degassed water. Both conditions gave similar results. After incubation for 4 h, the sample was taken out and immersed in 25 mM EDTA solution, sonicated for 10 s, and thoroughly washed with Millipore water and then ethanol, and dried under a stream of argon.
Protein Resistance
XPS N1s signal intensity on an OEG-terminated film D (Scheme 1) was first measured. Immediately after the measurement, this sample and a freshly prepared hydrogen-terminated silicon (111) substrate were individually incubated in a fibrinogen solution (1 mg/mL in 0.01 M PBS buffer (pH 7.4), prepared without excessive shaking to avoid formation of long-lasting bubbles and possible denaturing of the protein) for 1 h. The sample was taken out and immediately washed with Millipore water for ~ 30 s, and dried with a flow of argon. The ellipsometric thickness of the protein film on the H-Si(111) surface was 61 ± 2 Å, corresponding to a monolayer of the protein.17 Both dried films were immediately subjected to measurement of the N1s signal intensity. The protein resistance of the film D is calculated by the increase of the N1s signal intensity after protein adsorption divided by the N1s signal intensity of the protein monolayer, and the data were obtained from four experiments.
Surface CuAAC Reactions in Microarray Format
Solutions of a mixture of azides (the biotin azide 7 or the mannose azide 5 mixed with the OEG azide 4 at a molar ratio of 5/4 or 7/4 = 1:0, 1:1, and 1:9, and total concentration of 3.33 mM), CuSO4 (5.33 mM), the copper ligand 10 (37.0 mM), and sodium ascorbate (63.6 mM) in a 10:1 (v/v) mixed solution of PBS buffer and Micro Spotting Solution Plus 2x (TeleChem International, Inc., Sunnyvale, CA) were spotted on a TMG-terminated surface A using a Spotbot 2 Personal Microarray Robot (TeleChem International, Inc.) with a microarray spotting pin (946MP16). The spotter was placed in an anaerobic chamber filled with nitrogen, and the relative humidity in the chamber of the spotter was 60%. After spotting, the sample was allowed to incubate for 4 h in the chamber. The 10 nL droplets did not dry out during this period due to the presence of the above spotting solution that decreases evaporation. The samples were then rapidly washed with 10 mM EDTA solution (6 mL), PBS buffer (6 mL) and water, and immediately immersed in a solution of the OEG-N3 4 (3.33 mM), CuSO4 (5.33 mM), the copper ligand 10 (37.0 mM), and ascorbic acid (63.6 mM) in PBS buffer. The sample was allowed to incubate under N2 for 4 h, and then immersed in 10 mM EDTA for 10 min, followed by washing with water (6 mL) and drying in a stream of N2.
Binding of Targeted Molecules
The above microarray samples were immersed in a solution of FITC-avidin (0.5 mg/mL) or FITC-Con A (0.5 mg/mL) in PBS buffer for 30 min in a humidified chamber. The sample was washed with water and dried immediately with a stream of Argon. Fluorescent images of the microarrays were acquired using a GeneTAC UC-4 Array Scanner (Genomic Solutions) with a 488 nm excitation and 512 nm emission bandpass filter.
Specific Bacterial Adherence on Mannose-Modified Surfaces
Derivative strains of E. coli 8397252 expressing type 1 fimbriae (fim+) or without type 1 fimbriae (fim−) were used in this study. E. coli 83972 is a nonpathogenic strain52 that has been studied in vivo as a means to prevent catheter-associated urinary tract infection.53,54 To create fim+ E. coli 83972 that binds to mannose, we transformed the wildtype E. coli 83972 with pSH2 encoding type 1 fimbriae55 and pGreen encoding green fluorescent protein (GFP). We have previously confirmed over-expression of type 1 fimbriae by this strain.54 To create fim− E. coli 83972 that does not bind to mannose but has the same fluorescence and antibiotic resistance profile as fim+ E. coli 83972, we transformed wild-type E. coli 83972 with the empty pACYC vector and pGreen.
The ability of the derivative strains of E. coli 83972 to adhere to variously modified silicon substrates was assessed by the following assay. A silicon sample was placed in a 5 mL solution of 20 μg/mL chloramphenicol and 100 μg/mL ampicillin in Luria-Bertani (LB) broth (Difco Laboratories, Maryland). The broth was inoculated with a single colony of the given strain of bacteria from an agar plate and incubated with rocking at 37 °C overnight. The sample was rinsed 3 times in water prior to fluorescent imaging. Images of up to 20 randomly chosen visual fields were obtained for each sample. To confirm that the adherence of the bacteria was due to specific mannose-receptor binding, the fim+ E. coli 83972 was pre-incubated in the LB broth containing 50 mM mannose for 1 h (to saturate the mannose binding sites) prior to addition of a mannose-presenting substrate.
Supplementary Material
Acknowledgments
This work was supported by The Welch Foundation (E-1498), the NSF CAREER Award (CTS-0349228 to CC) and grant DMR-0706627, NIH R21 HD058985, NIH R21EY018303, Alliance for Nanohealth W81XWH-09-2-0139, and the Texas Center for Superconductivity at the University of Houston. The E. coli strain creation and adherence testing was supported by grants VA CDA-2 RR&D B4623 and NIH R21 DK077313.
Footnotes
Supporting Information Available. Synthesis of 2–7, AFM image of H-Si(111) sample surface, and fluorescence images of various modified surfaces incubated with fim+ and fim− E. coli. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hamers RJ. Annu Rev Anal Chem. 2008;1:707–736. doi: 10.1146/annurev.anchem.1.031207.112916. [DOI] [PubMed] [Google Scholar]; Ciampi S, Gooding JJ. Chem-Eur J. 2010;16:5961–5968. doi: 10.1002/chem.200903316. [DOI] [PubMed] [Google Scholar]; Yang WS, Butler JE, Russell JN, Hamers RJ. Analyst. 2007;132:296–306. doi: 10.1039/b612201a. [DOI] [PubMed] [Google Scholar]; Shalek AK, Robinson JT, Karp ES, Lee JS, Ahn DR, Yoon MH, Sutton A, Jorgolli M, Gertner RS, Gujral TS, MacBeath G, Yang EG, Park H. Proc Natl Acad Sci U S A. 2010;107:1870–1875. doi: 10.1073/pnas.0909350107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ainslie KM, Desai TA. Lab Chip. 2008;8:1864–1878. doi: 10.1039/b806446f. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vilan A, Yaffe O, Biller A, Salomon A, Kahn A, Cohen D. Adv Mater. 2010;22:140–159. doi: 10.1002/adma.200901834. [DOI] [PubMed] [Google Scholar]; Touahir L, Moraillon A, Allongue P, Chazalviel JN, de Villeneuve CH, Ozanam F, Solomon I, Gouget-Laemmel AC. Biosens Bioelectron. 2009;25:952–955. doi: 10.1016/j.bios.2009.08.046. [DOI] [PubMed] [Google Scholar]
- 2.Hochberg LR, Serruya MD, Friehs GM, Mukand JA, Saleh M, Caplan AH, Branner A, Chen D, Penn RD, Donoghue JP. Nature. 2006;442:164–171. doi: 10.1038/nature04970. [DOI] [PubMed] [Google Scholar]
- 3.He Y, Kang ZH, Li QS, Tsang CHA, Fan CH, Lee ST. Angew Chem-Int Edit. 2009;48:128–132. doi: 10.1002/anie.200802230. [DOI] [PubMed] [Google Scholar]; He Y, Su YY, Yang XB, Kang ZH, Xu TT, Zhang RQ, Fan CH, Lee ST. J Am Chem Soc. 2009;131:4434–4438. doi: 10.1021/ja808827g. [DOI] [PubMed] [Google Scholar]; Shiohara A, Hanada S, Prabakar S, Fujioka K, Lim TH, Yamamoto K, Northcote PT, Tilley RD. J Am Chem Soc. 2010;132:248–253. doi: 10.1021/ja906501v. [DOI] [PubMed] [Google Scholar]; Erogbogbo F, Yong KT, Roy I, Xu GX, Prasad PN, Swihart MT. ACS Nano. 2008;2:873–878. doi: 10.1021/nn700319z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Okamoto H, Kumai Y, Sugiyama Y, Mitsuoka T, Nakanishi K, Ohta T, Nozaki H, Yamaguchi S, Shirai S, Nakano H. J Am Chem Soc. 2010;132:2710–2718. doi: 10.1021/ja908827z. [DOI] [PubMed] [Google Scholar]; Orosco MM, Pacholski C, Miskelly GM, Sailor MJ. Adv Mater. 2006;18:1393–1396. [Google Scholar]; Tilley RD, Yamamoto K. Adv Mater. 2006;18:2053–2056. [Google Scholar]
- 4.Rosso-Vasic M, Spruijt E, Popovic Z, Overgaag K, van Lagen B, Grandidier B, Vanmaekelbergh D, Dominguez-Gutierrez D, De Cola L, Zuilhof H. J Mater Chem. 2009;19:5926–5933. [Google Scholar]
- 5.Gao XPA, Zheng GF, Lieber CM. Nano Lett. 2010;10:547–552. doi: 10.1021/nl9034219. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ben Ishai M, Patolsky F. J Am Chem Soc. 2009;131:3679–3689. doi: 10.1021/ja808483t. [DOI] [PubMed] [Google Scholar]; Bunimovich YL, Shin YS, Yeo WS, Amori M, Kwong G, Heath JR. J Am Chem Soc. 2006;128:16323–16331. doi: 10.1021/ja065923u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cohen-Karni T, Timko BP, Weiss LE, Lieber CM. Proc Natl Acad Sci U S A. 2009;106:7309–7313. doi: 10.1073/pnas.0902752106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Martinez JA, Misra N, Wang YM, Stroeve P, Grigoropoulos CP, Noy A. Nano Lett. 2009;9:1121–1126. doi: 10.1021/nl8036504. [DOI] [PubMed] [Google Scholar]; Patolsky F, Zheng G, Lieber CM. Nanomedicine. 2006;1:51–65. doi: 10.2217/17435889.1.1.51. [DOI] [PubMed] [Google Scholar]; Wang WU, Chen C, Lin KH, Fang Y, Lieber CM. Proc Natl Acad Sci U S A. 2005;102:3208–3212. doi: 10.1073/pnas.0406368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez SD, Derfus AM, Schwartz MP, Bhatia SN, Sailor MJ. Biomaterials. 2009;30:26–34. doi: 10.1016/j.biomaterials.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sailor MJ, Wu EC. Adv Funct Mater. 2009;19:3195–3208. [Google Scholar]; Qu YQ, Liao L, Li YJ, Zhang H, Huang Y, Duan XF. Nano Lett. 2009;9:4539–4543. doi: 10.1021/nl903030h. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ouyang H, Christophersen M, Viard R, Miller BL, Fauchet PM. Adv Funct Mater. 2005;15:1851–1859. [Google Scholar]
- 7.Kilian KA, Bocking T, Gaus K, Gal M, Gooding JJ. Biomaterials. 2007;28:3055–3062. doi: 10.1016/j.biomaterials.2007.03.014. [DOI] [PubMed] [Google Scholar]; Kilian KA, Bocking T, Ilyas S, Gaus K, Jessup W, Gal M, Gooding JJ. Adv Funct Mater. 2007;17:2884–2890. [Google Scholar]; Kilian KA, Boecking T, Gooding JJ. Chem Commun. 2009:630–640. doi: 10.1039/b815449j. [DOI] [PubMed] [Google Scholar]
- 8.Ilic B, Yang Y, Craighead HG. Appl Phys Lett. 2004;85:2604–2606. [Google Scholar]; Singamaneni S, LeMieux MC, Lang HP, Gerber C, Lam Y, Zauscher S, Datskos PG, Lavrik NV, Jiang H, Naik RR, Bunning TJ, Tsukruk VV. Adv Mater. 2008;20:653–680. [Google Scholar]
- 9.Yam CM, Xiao ZD, Gu JH, Boutet S, Cai CZ. J Am Chem Soc. 2003;125:7498–7499. doi: 10.1021/ja0350435. [DOI] [PubMed] [Google Scholar]; Gu JH, Xiao ZD, Yam CM, Qin GT, Deluge M, Boutet S, Cai CZ. Biophys J. 2005;89:L31–L33. doi: 10.1529/biophysj.105.069104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jonkheijm P, Weinrich D, Schroder H, Niemeyer CM, Waldmann H. Angew Chem-Int Edit. 2008;47:9618–9647. doi: 10.1002/anie.200801711. [DOI] [PubMed] [Google Scholar]
- 11.Cha T, Guo A, Jun Y, Pei DQ, Zhu XY. Proteomics. 2004;4:1965–1976. doi: 10.1002/pmic.200300747. [DOI] [PubMed] [Google Scholar]; Nijdam AJ, Cheng MMC, Geho DH, Fedele R, Herrmann P, Killian K, Espina V, Petricoin EF, Liotta LA, Ferrari M. Biomaterials. 2007;28:550–558. doi: 10.1016/j.biomaterials.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 12.Domachuk P, Tsioris K, Omenetto FG, Kaplan DL. Adv Mater. 2010;22:249–260. doi: 10.1002/adma.200900821. [DOI] [PubMed] [Google Scholar]; Stern E, Vacic A, Rajan NK, Criscione JM, Park J, Ilic BR, Mooney DJ, Reed MA, Fahmy TM. Nat Nanotechnol. 2010;5:138–142. doi: 10.1038/nnano.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fromherz P. Solid-State Electron. 2008;52:1364–1373. [Google Scholar]
- 14.Jo S, Park K. Biomaterials. 2000;21:605–616. doi: 10.1016/s0142-9612(99)00224-0. [DOI] [PubMed] [Google Scholar]; Lee SW, Laibinis PE. Biomaterials. 1998;19:1669–1675. doi: 10.1016/s0142-9612(98)00044-1. [DOI] [PubMed] [Google Scholar]
- 15.Dekeyser CM, Buron CC, Mc Evoy K, Dupont-Gillain CC, Marchand-Brynaert J, Jonas AM, Rouxhet PG. J Colloid Interface Sci. 2008;324:118–126. doi: 10.1016/j.jcis.2008.05.002. [DOI] [PubMed] [Google Scholar]; Harbers GM, Emoto K, Greef C, Metzger SW, Woodward HN, Mascali JJ, Grainger DW, Lochhead MJ. Chem Mat. 2007;19:4405–4414. doi: 10.1021/cm070509u. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hoffmann C, Tovar GEM. J Colloid Interface Sci. 2006;295:427–435. doi: 10.1016/j.jcis.2005.10.005. [DOI] [PubMed] [Google Scholar]; Tsukagoshi T, Kondo Y, Yoshino N. Colloid Surf B-Biointerfaces. 2007;54:82–87. doi: 10.1016/j.colsurfb.2006.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Lasseter TL, Cai W, Hamers RJ. Analyst. 2004;129:3–8. doi: 10.1039/b307591e. [DOI] [PubMed] [Google Scholar]; Wang C, Landis EC, Franking R, Hamers RJ. Acc Chem Res. 2010 doi: 10.1021/ar100011f. in press. [DOI] [PubMed] [Google Scholar]; Clare TL, Clare BH, Nichols BM, Abbott NL, Hamers RJ. Langmuir. 2005;21:6344–6355. doi: 10.1021/la050362q. [DOI] [PubMed] [Google Scholar]; Bocking T, Kilian KA, Hanley T, Ilyas S, Gaus K, Gal M, Gooding JJ. Langmuir. 2005;21:10522–10529. doi: 10.1021/la051191s. [DOI] [PubMed] [Google Scholar]; Bocking T, Killan KA, Gaus K, Gooding JJ. Langmuir. 2006;22:3494–3496. doi: 10.1021/la060331a. [DOI] [PubMed] [Google Scholar]; Yu QK, Qin GT, Darne C, Cai CZ, Wosik W, Pei SS. Sens Actuator A-Phys. 2006;126:369–374. [Google Scholar]
- 17.Yam CM, Lopez-Romero JM, Gu JH, Cai CZ. Chem Commun. 2004:2510–2511. doi: 10.1039/b401499e. [DOI] [PubMed] [Google Scholar]; Yam CM, Gu JH, Li S, Cai CZ. J Colloid Interface Sci. 2005;285:711–718. doi: 10.1016/j.jcis.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Gu JH, Yam CM, Li S, Cai CZ. J Am Chem Soc. 2004;126:8098–8099. doi: 10.1021/ja048405x. [DOI] [PubMed] [Google Scholar]
- 19.Qin GT, Cai CZ. Chem Commun. 2009:5112–5114. doi: 10.1039/b911155g. [DOI] [PubMed] [Google Scholar]
- 20.Linford MR, Fenter P, Eisenberger PM, Chidsey CED. J Am Chem Soc. 1995;117:3145–3155. [Google Scholar]; Buriak JM. Chem Rev. 2002;102:1271–1308. doi: 10.1021/cr000064s. [DOI] [PubMed] [Google Scholar]
- 21.Cicero RL, Linford MR, Chidsey CED. Langmuir. 2000;16:5688–5695. [Google Scholar]
- 22.Ciampi S, Harper JB, Gooding JJ. Chem Soc Rev. 2010;39:2158–2183. doi: 10.1039/b923890p. [DOI] [PubMed] [Google Scholar]
- 23.Qin GT, Zhang R, Makarenko B, Kumar A, Rabalais W, Romero JML, Rico R, Cai CZ. Chem Commun. 2010;46:3289–3291. doi: 10.1039/b925708j. [DOI] [PubMed] [Google Scholar]
- 24.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem-Int Edit. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Hein JE, Fokin VV. Chem Soc Rev. 2010;39:1302–1315. doi: 10.1039/b904091a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meldal M, Tornoe CW. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 26.Hong V, Presolski SI, Ma C, Finn MG. Angew Chem-Int Edit. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong LS, Khan F, Micklefield J. Chem Rev. 2009;109:4025–4053. doi: 10.1021/cr8004668. [DOI] [PubMed] [Google Scholar]
- 28.Hudalla GA, Murphy WL. Langmuir. 2009;25:5737–5746. doi: 10.1021/la804077t. [DOI] [PMC free article] [PubMed] [Google Scholar]; Le Droumaguet C, Wang C, Wang Q. Chem Soc Rev. 2010;39:1233–1239. doi: 10.1039/b901975h. [DOI] [PubMed] [Google Scholar]
- 29.Bryan MC, Fazio F, Lee HK, Huang CY, Chang A, Best MD, Calarese DA, Blixt C, Paulson JC, Burton D, Wilson IA, Wong CH. J Am Chem Soc. 2004;126:8640–8641. doi: 10.1021/ja048433f. [DOI] [PubMed] [Google Scholar]; Lutz JF. Angew Chem-Int Edit. 2007;46:1018–1025. doi: 10.1002/anie.200604050. [DOI] [PubMed] [Google Scholar]; Nandivada H, Jiang XW, Lahann J. Adv Mater. 2007;19:2197–2208. [Google Scholar]; Nebhani L, Barner-Kowollik C. Adv Mater. 2009;21:3442–3468. [Google Scholar]; Collman JP, Devaraj NK, Chidsey CED. Langmuir. 2004;20:1051–1053. doi: 10.1021/la0362977. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gallant ND, Lavery KA, Amis EJ, Becker ML. Adv Mater. 2007;19:965–969. [Google Scholar]; Fleming DA, Thode CJ, Williams ME. Chem Mat. 2006;18:2327–2334. [Google Scholar]; Li HM, Cheng FO, Duft AM, Adronov A. J Am Chem Soc. 2005;127:14518–14524. doi: 10.1021/ja054958b. [DOI] [PubMed] [Google Scholar]; O’Reilly RK, Joralemon MJ, Wooley KL, Hawker CJ. Chem Mat. 2005;17:5976–5988. [Google Scholar]; Prakash S, Long TM, Selby JC, Moore JS, Shannon MA. Anal Chem. 2007;79:1661–1667. doi: 10.1021/ac061824n. [DOI] [PubMed] [Google Scholar]; White MA, Johnson JA, Koberstein JT, Turro NJ. J Am Chem Soc. 2006;128:11356–11357. doi: 10.1021/ja064041s. [DOI] [PubMed] [Google Scholar]; Zhang Y, Luo SZ, Tang YJ, Yu L, Hou KY, Cheng JP, Zeng XQ, Wang PG. Anal Chem. 2006;78:2001–2008. doi: 10.1021/ac051919+. [DOI] [PubMed] [Google Scholar]; Devadoss A, Chidsey CED. J Am Chem Soc. 2007;129:5370–5371. doi: 10.1021/ja071291f. [DOI] [PubMed] [Google Scholar]; Duckworth BP, Xu JH, Taton TA, Guo A, Distefano MD. Bioconjugate Chem. 2006;17:967–974. doi: 10.1021/bc060125e. [DOI] [PubMed] [Google Scholar]; Sun XL, Stabler CL, Cazalis CS, Chaikof EL. Bioconjugate Chem. 2006;17:52–57. doi: 10.1021/bc0502311. [DOI] [PubMed] [Google Scholar]; Santos CM, Kumar A, Zhang W, Cai CZ. Chem Commun. 2009:2854–2856. doi: 10.1039/b821148e. [DOI] [PubMed] [Google Scholar]; Kumar A, Erasquin UJ, Qin G, Li K, Cai CZ. Chem Commun. 2010;46:5746–5748. doi: 10.1039/c0cc00784f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciampi S, Bocking T, Kilian KA, James M, Harper JB, Gooding JJ. Langmuir. 2007;23:9320–9329. doi: 10.1021/la701035g. [DOI] [PubMed] [Google Scholar]
- 31.Lin PC, Ueng SH, Tseng MC, Ko JL, Huang KT, Yu SC, Adak AK, Chen YJ, Lin CC. Angew Chem-Int Edit. 2006;45:4286–4290. doi: 10.1002/anie.200600756. [DOI] [PubMed] [Google Scholar]
- 32.Rohde RD, Agnew HD, Yeo WS, Bailey RC, Heath JR. J Am Chem Soc. 2006;128:9518–9525. doi: 10.1021/ja062012b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao PG, Xu K, Heath JR. J Am Chem Soc. 2008;130:14910–14911. doi: 10.1021/ja804448p. [DOI] [PubMed] [Google Scholar]
- 34.Haensch C, Hoeppener S, Schubert US. Nanotechnology. 2008:19. doi: 10.1088/0957-4484/19/03/035703. [DOI] [PubMed] [Google Scholar]; Giacomo MA, Fabrizio C, Franco D, Robertino Z, Maurizio C, Francesca IM. J Nanosci Nanotechnol. 2010;10:2901–2907. doi: 10.1166/jnn.2010.1409. [DOI] [PubMed] [Google Scholar]
- 35.Scriven EFV. Azides and nitrenes: reactivity and utility. Academic Press; Orlando, FL: 1984. [Google Scholar]
- 36.Hurley PT, Nemanick EJ, Brunschwig BS, Lewis NS. J Am Chem Soc. 2006;128:9990–9991. doi: 10.1021/ja061969b. [DOI] [PubMed] [Google Scholar]
- 37.Ciampi S, Eggers PK, Le Saux G, James M, Harper JB, Gooding JJ. Langmuir. 2009;25:2530–2539. doi: 10.1021/la803710d. [DOI] [PubMed] [Google Scholar]
- 38.Ng A, Ciampi S, James M, Harper JB, Gooding JJ. Langmuir. 2009;25:13934–13941. doi: 10.1021/la901526e. [DOI] [PubMed] [Google Scholar]; Scheres L, Arafat A, Zuilhof H. Langmuir. 2007;23:8343–8346. doi: 10.1021/la701359k. [DOI] [PubMed] [Google Scholar]
- 39.Cai CZ, Vasella A. Helvetica Chimica Acta. 1995;78:732–757. [Google Scholar]; Ernst A, Gobbi L, Vasella A. Tetrahedron Lett. 1996;37:7959–7962. [Google Scholar]
- 40.Dallaire C, Brook MA. Organometallics. 1990;9:2873–2874. [Google Scholar]; Eaborn C, Walton DRM. J Organomet Chem. 1964;2:95–97. [Google Scholar]
- 41.Ito E, Oji H, Araki T, Oichi K, Ishii H, Ouchi Y, Ohta T, Kosugi N, Maruyama Y, Naito T, Inabe T, Seki K. J Am Chem Soc. 1997;119:6336–6344. [Google Scholar]; Alfredsson Y, Brena B, Nilson K, Ahlund J, Kjeldgaard L, Nyberg M, Luo Y, Martensson N, Sandell A, Puglia C, Siegbahn H. J Chem Phys. 2005;122:6. doi: 10.1063/1.1924539. [DOI] [PubMed] [Google Scholar]
- 42.Collman JP, Devaraj NK, Eberspacher TPA, Chidsey CED. Langmuir. 2006;22:2457–2464. doi: 10.1021/la052947q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrovykh DY, Kimura-Suda H, Tarlov MJ, Whitman LJ. Langmuir. 2004;20:429–440. doi: 10.1021/la034944o. [DOI] [PubMed] [Google Scholar]
- 44.Lin PC, Ueng SH, Yu SC, Jan MD, Adak AK, Yu CC, Lin CC. Org Lett. 2007;9:2131–2134. doi: 10.1021/ol070588f. [DOI] [PubMed] [Google Scholar]
- 45.Eglinton G, McCrae W. Adv Org Chem. 1963;4:225. [Google Scholar]
- 46.Chan TR, Hilgraf R, Sharpless KB, Fokin VV. Org Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 47.Rodionov VO, Presolski SI, Gardinier S, Lim YH, Finn MG. J Am Chem Soc. 2007;129:12696–12704. doi: 10.1021/ja072678l. [DOI] [PubMed] [Google Scholar]; Rodionov VO, Presolski SI, Diaz DD, Fokin VV, Finn MG. J Am Chem Soc. 2007;129:12705–12712. doi: 10.1021/ja072679d. [DOI] [PubMed] [Google Scholar]
- 48.He Y, Chang Y, Hower JC, Zheng J, Chen SF, Jiang S. Phys Chem Chem Phys. 2008;10:5539–5544. doi: 10.1039/b807129b. [DOI] [PubMed] [Google Scholar]; Zolk M, Eisert F, Pipper J, Herrwerth S, Eck W, Buck M, Grunze M. Langmuir. 2000;16:5849–5852. [Google Scholar]
- 49.Qian XP, Metallo SJ, Choi IS, Wu HK, Liang MN, Whitesides GM. Anal Chem. 2002;74:1805–1810. doi: 10.1021/ac011042o. [DOI] [PubMed] [Google Scholar]; Barth KA, Coullerez G, Nilsson LM, Castelli R, Seeberger PH, Vogel V, Textor M. Adv Funct Mater. 2008;18:1459–1469. [Google Scholar]; Pieters RJ. Med Res Rev. 2007;27:796–816. doi: 10.1002/med.20089. [DOI] [PubMed] [Google Scholar]; Rozhok S, Shen CKF, Littler PLH, Fan ZF, Liu C, Mirkin CA, Holz RC. Small. 2005;1:445–451. doi: 10.1002/smll.200400072. [DOI] [PubMed] [Google Scholar]
- 50.Burton EA, Sirnon KA, Hou SY, Ren DC, Luk YY. Langmuir. 2009;25:1547–1553. doi: 10.1021/la803261b. [DOI] [PubMed] [Google Scholar]; Cheng G, Li GZ, Xue H, Chen SF, Bryers JD, Jiang SY. Biomaterials. 2009;30:5234–5240. doi: 10.1016/j.biomaterials.2009.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris LG, Tosatti S, Wieland M, Textor M, Richards RG. Biomaterials. 2004;25:4135–4148. doi: 10.1016/j.biomaterials.2003.11.033. [DOI] [PubMed] [Google Scholar]; Roosjen A, Busscher HJ, Nordel W, van der Mei HC. Microbiology-(UK) 2006;152:2673–2682. doi: 10.1099/mic.0.29005-0. [DOI] [PubMed] [Google Scholar]; Chapman RG, Ostuni E, Liang MN, Meluleni G, Kim E, Yan L, Pier G, Warren HS, Whitesides GM. Langmuir. 2001;17:1225–1233. [Google Scholar]; Kingshott P, Wei J, Bagge-Ravn D, Gadegaard N, Gram L. Langmuir. 2003;19:6912–6921. [Google Scholar]; Ostuni E, Chapman RG, Liang MN, Meluleni G, Pier G, Ingber DE, Whitesides GM. Langmuir. 2001;17:6336–6343. [Google Scholar]
- 52.Andersson P, Engberg I, Lidinjanson G, Lincoln K, Hull R, Hull S, Svanborg C. Infect Immun. 1991;59:2915–2921. doi: 10.1128/iai.59.9.2915-2921.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sunden F, Hakansson L, Ljunggren E, Wullt B. Int J Antimicrob Agents. 2006;28:S26–S29. doi: 10.1016/j.ijantimicag.2006.05.007. [DOI] [PubMed] [Google Scholar]; Trautner BW, Hull RA, Thornby JI, Darouiche RO. Infect Control Hosp Epidemiol. 2007;28:92–94. doi: 10.1086/510872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Trautner BW, Cevallos ME, Li HG, Riosa S, Hull RA, Hull SI, Tweardy DJ, Darouiche RO. J Infect Dis. 2008;198:899–906. doi: 10.1086/591093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hull RA, Gill RE, Hsu P, Minshew BH, Falkow S. Infect Immun. 1981;33:933–938. doi: 10.1128/iai.33.3.933-938.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tao SH, Bolger PM. Regul Toxicol Pharm. 1997;25:211–219. doi: 10.1006/rtph.1997.1098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.