

Abstract
This paper endeavors to clarify the current requirements and status of regulatory approval for chemoprevention (risk reduction) drugs and discusses possible improvements to the regulatory pathway for chemoprevention. Covering a wide range of topics in as much depth as space allows, this report is written in a style to facilitate the understanding of non-scientists and to serve as a framework for informing the directions of experts engaged more deeply with this issue. Key topics we cover here are as follows: a history of definitive cancer chemoprevention trials and their influence on the evolution of regulatory assessments; a brief review of the long-standing success of pharmacologic risk reduction of cardiovascular diseases and its relevance to approval for cancer risk reduction drugs; the use and limitations of biomarkers for developing and the approval of cancer risk reduction drugs; the identification of individuals at a high(er) risk for cancer and who are appropriate candidates for risk reduction drugs; business models that should incentivize pharmaceutical-industry investment in cancer risk reduction; a summary of scientific and institutional barriers to development of cancer risk reduction drugs; and a summary of major recommendations that should help facilitate the pathway to regulatory approval for pharmacologic cancer risk reduction drugs.
An ounce of prevention is worth a pound of cure.
Benjamin Franklin
Introduction
In June 2006, C-Change convened the Cancer Prevention Research Summit of oncologists, pharmaceutical researchers, and other scientists, government representatives, public policy experts, and patient advocates to discuss how best to unleash the potential of the promising field of cancer chemoprevention. Four barriers to research and development of chemoprevention drugs were identified as major impediments to progress in the field: uncertain reimbursement for new agents; limitations in current patent law and intellectual property protection; limitations in emerging prevention science, evolving designs of clinical trials, and processes of drug approval; and limited public participation in clinical trials. Proceedings from the summit were published (1).
Following this groundbreaking meeting, C-Change established the three following complementary subcommittees/task forces to report to the Chemoprevention Advisory Committee in developing solutions to these financial, legal, scientific, and regulatory barriers to the field: Chemoprevention Patent Law Advisory Subcommittee, Chemoprevention Reimbursement Subcommittee, and the Chemoprevention Clinical Trials and Biomarkers Subcommittee. C-Change believes that these task forces will be successful in this quest, thus helping to unleash the lifesaving potential of chemoprevention drugs through more-effective research, development, and delivery.
Dr. Steven H. Woolf has commented insightfully on the potential power of prevention (2), as follows: “A more direct strategy for confronting both spending and disease burden is to mitigate the problem at its source by preventing the early onset of disease.”
The Influence of Randomized Controlled Cancer Chemoprevention Trials on the Evolution of Regulatory Assessments
Over the past three decades, the following four major classes of drugs have produced positive results in definitive randomized controlled trials (RCTs) of chemopreventive agents: Retinoids, inhibitors of hormone action, cyclooxygenase-2 (COX-2)-specific and other nonsteroidal anti-inflammatory drugs (NSAIDs), and cancer-related–disease vaccines. The advancement of two molecular-targeted drugs (celecoxib and raloxifene) and of an immuno-modulatory agent (human papillomavirus [HPV] vaccine) to U.S. regulatory approval for cancer risk reduction are detailed in Supplementary appendices 1 (celecoxib, raloxifene) and 2 (HPV vaccine).
Although RCTs of retinoids in the settings of oral intraepithelial neoplasia (IEN) and cervical IEN (3, 4), and (at a high dose) in the prevention of second head and neck malignancies (5) were positive, these initial “proof of principle” successes did not lead to regulatory approval. Less-toxic retinoids or lower doses of the toxic, active ones in clinical trials with cancer and other end points in various settings have been negative (6-11), and so retinoids have not been adopted for cancer risk reduction or pursued for regulatory approval by the pharmaceutical industry.
The history of breast cancer chemoprevention is particularly informative in that it is highly effective, produced the first specific U.S. Food and Drug Administration (FDA) approval of cancer chemoprevention, and yet has been little adopted by at-risk women. Tamoxifen clearly reduced breast malignancies in women with increased risk as determined by the Gail nomogram in the large phase-III Breast Cancer Prevention Trial (12) and subsequently was approved by the U.S. Food and Drug Administration (FDA) for risk reduction. Nevertheless, women and doctors did not adopt tamoxifen for this indication because they were concerned about adverse effects, mainly increased endometrial cancer and thrombotic events. Later results of the randomized clinical Study of Tamoxifen and Raloxifene (STAR; ref. 13) showed that raloxifene was equivalent to tamoxifen in reducing breast cancer in postmenopausal women at the same Gail risk, with lesser toxicity. Raloxifene also has been FDA-approved for breast cancer risk reduction but has encountered resistance to acceptance by women and doctors for reasons that are less clear than those involving tamoxifen. Very recent long-term follow-up of STAR has strengthened the risk-benefit profiles of both raloxifene and tamoxifen for breast cancer prevention (14, 15), possibly reopening the public dialog on the merits of these two important cancer chemoprevention agents.
The 2003 results of the Prostate Cancer Prevention Trial (PCPT) showed that finasteride reduced prostate cancer overall by 25% but apparently also increased high-grade tumors, which obviated its acceptance in this setting (16). Subsequent careful analysis showed that the excess of high-grade tumors was probably due to biopsy artifacts (17-19). Although the initial concerns and patent limitations precluded the seeking of regulatory approval of finasteride for risk reduction, these clarifying studies regarding biopsy artifacts and recently reported RCT results showing that dutasteride reduced prostate cancer by 23% with no apparent increase in high-grade cancer (20) increase the likelihood of an approval of a secondary regulatory indication for finasteride or dutasteride for cancer risk reduction.
Recent trials of COX-2 inhibitors highlight the complexity of chemoprevention and the need for close collaboration among all stakeholders in developing and educating the public about acceptable risk profiles for given chemopreventive agents in specific cancer risk settings. Celecoxib was approved by the FDA in 1988 for reducing polyp burden in patients with the high-risk genetic condition familial adenomatous polyposis (21) and was effective in a lower-risk group as well (22, 23). An excess of serious cardiovascular events, however, temporarily halted all clinical trials of COX-2-selective compounds for prevention (the National Cancer Institute [NCI] allowed testing for cancer prevention to resume after deeming it safe to do so). A subsequent detailed meta-analysis of COX-2-selective trials clearly demonstrated that the patients who had a drug-related cardiovascular event (especially on more-frequent and higher doses) had an increased baseline risk for cardiovascular disease (characterization using simple clinical criteria; ref. 24). The same cardiovascular risk also might track with less-selective NSAIDs, but the data on this issue are incomplete and inconclusive.
Another example of the complexity of chemopreventive risk-benefit is provided by a randomized placebo-controlled trial of combined low doses of difluoromethylornithine (DFMO), a polyamine synthesis inhibitor, and sulindac (an NSAID). The combination produced dramatic reductions in all adenomas (70%), advanced adenomas (92%), and multiple adenomas (95%) in patients with prior adenomas (25), along with generally minimal toxicity but a non-statistically significant excess of cardiovascular events. As in the case of celecoxib, subsequent analysis showed that the excess of cardiovascular events with this combination was limited to individuals who had a high baseline cardiovascular risk based on previously described simple standard clinical criteria (26). Larger trials will be needed to confirm the relative risk-benefit profile of the combination since the absolute numbers of cases (recurrent adenomas, especially advanced adenomas) was low.
Is the Long-standing Regulatory-approval Success of Cardiovascular Risk-reduction Drugs Relevant to Cancer Risk-reduction Drugs?
The short answer is yes and no. Over six decades ago, the therapeutic paradigm for the treatment of cardiovascular disease began to include a chemopreventive risk reduction approach (27, 28). The first task, of course, was to identify modifiable factors that would influence the outcome of cardiovascular disease. Blood pressure and cholesterol levels were widely accepted as risk factors only after results of the Framingham study were published in 1961 (29), although basic science supporting them as such had been accumulating for over 30 years (reviewed in ref. 30). Despite Framingham, however, modulation of these risk factors as surrogates for cardiovascular disease outcome was not generally accepted until many years later (reviewed in refs. 31, 32), following the results of large clinical trials in the 1970s for blood pressure (refs. 33, 34; reviewed in ref. 35) and in the 1980s for cholesterol (reviewed in ref. 36). These studies have been refined up to the present (37, 38), resulting in a steady and marked reduction in mortality from cardiovascular diseases (Fig. 1). Current estimates attribute about 50% of this decline to chemoprevention, i.e., early pharmacologic intervention to interrupt the atherogenic process. This success in lowering mortality from cardiovascular disease and the ageing of the population ironically have made cancer-related deaths the number one U.S. health hazard of the twenty-first century.
Fig. 1.
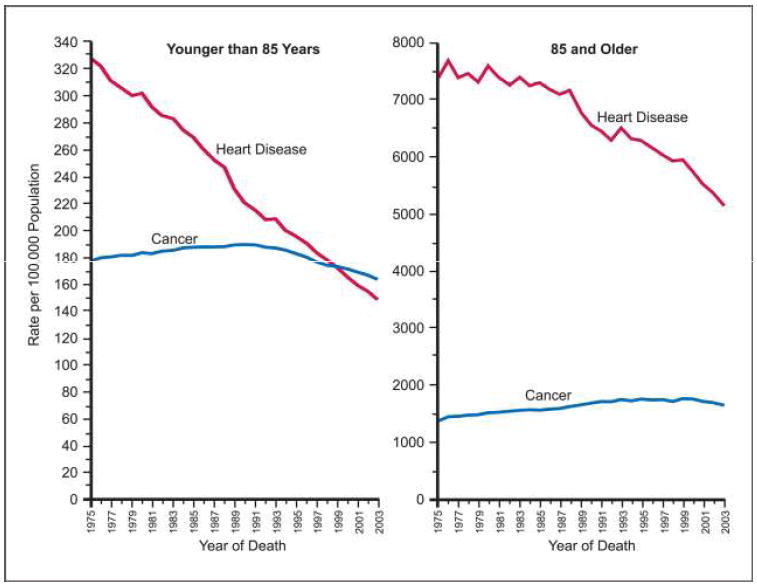
Death rates from cancer and heart disease for ages younger than 85 and 85 and older. Rates are age-adjusted to the 2000 U.S. standard population (U.S. Mortality Public Use Data Tapes 1060 to 2001, National Center for Health Statistics, Center for Disease Control and Prevention, 2004).
Can a similar approach improve the outcomes of patients at a higher risk for cancer?
In the last few years, cancer-related death rates began falling in the U.S. Much of this improvement is believed to be due to improved screening for and early detection of common tumors, such as cervix, colorectal, and breast cancers. Modest gains also have occurred in overall survival from treatment of metastatic cancer, but at a high cost. For example, the five-year survival of metastatic colorectal cancer patients is still only about 5%, but each patient’s treatment costs are approaching $150,000 or more. We can do better, and using the great leaps of the past two decades in our understanding of the pathogenesis of cancer to better identify and prevent cancer in at-risk individuals will help us to do so.
Screening modalities are an important tool for identifying pre-cancers or cancers at the earliest possible stage so as to interrupt or ablate the pathogenic process. Screening approaches have facilitated the earlier detection of several cancers, including cervix, breast, colon, and possibly prostate cancers, which not only is directly beneficial but also should enhance the opportunities to identify high-risk individuals and groups most likely to benefit from chemoprevention.
Why has the early management of oncologic conditions using pharmacologic intervention lagged behind cardiovascular prevention?
Simply put, knowledge of the pathogenesis of cardiovascular disease and biomarkers (cholesterol, blood pressure) that predict cardiovascular disease and allow effective prevention preceded advances in the same understanding of carcinogenesis by 20–25 years, as has the development of effective cardiovascular pharmacologic interventions (Table 1). Although we now have efficacious chemopreventive drugs, we do not yet have reliable, validated surrogate biomarkers for cancer. The lack of reliable biomarkers is a major hurdle and has slowed the development of the field enormously since the true end point (cancer) takes a long time to reach. It is time to bring our knowledge of carcinogenesis and clinical trials to bear on this problem and begin to replicate the success of our cardiovascular colleagues.
Table 1.
Development of antihypertensive and anticancer risk reduction drugs over time1
| ANTIHYPERTENSIVE DRUGS FROM THE 1930S ONWARD | CANCER CHEMOPREVENTION DRUGS FROM 1980s ONWARD | |
|---|---|---|
| 1930s | Veratrum alkaloids | |
| 1940s | Thiocyanates | 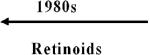 |
| Ganglion blocking agents | ||
| Catecholamine depletors (Rauwolfia derivatives) | ||
| 1950s | Vasodilators (Hydralazine) | 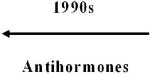 |
| Peripheral sympathetic inhibitors (guanethidine) | ||
| Monoamine oxidase inhibitors | ||
| Diuretics | ||
| 1960s | Central a2-agonists (sympathetic nervous system inhibitors) | |
| 1970 | ß-Adrenergic inhibitors | 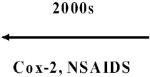 |
| a-Adrenergic inhibitors | ||
| a-ß-Blockers | ||
| Converting enzyme inhibitors | ||
| 1980s | Calcium channel blockers | |
| 1990s | Angiotensin II (AT1) receptor antagonists | |
Abbreviation: Cox-2, cyclooxygenase 2.
Adapted from Moser M, “Evolution of the Treatment of Hypertension from the 1940s to JNC V,” Am J Hypertens 1997;10:2S-8S, and reproduced with permission of Marvin Moser.
The second major hurdle discussed here for the development of cancer risk reduction drugs emanates from the initial conceptual basis of oncologic treatment and the philosophy that evolved from this early basis, an impediment that is not generally appreciated. The era of modern oncology was founded on observations of leucopenia and bone marrow toxicity in soldiers exposed to mustard gas in World Wars I and II. These observations were translated into medical benefits, for example, dramatic therapeutic successes with high doses of combined cytotoxic drugs against childhood leukemias, Hodgkin’s disease, and non-Hodgkin’s lymphomas, and testicular cancer. Such work provided a strategic and conceptual framework for cancer management that has persisted to this day. The advent of targeted agents, starting with hormonal manipulators, has modified this approach, but combinations of toxic drugs remain the prevailing paradigm for cancer management. Given the great threat that advanced cancer poses to the patient’s welfare, the acceptance of toxicity by both patients and practitioners has been high.
The understandable focus on treating disease that is life threatening in the near term had an unanticipated consequence on the types of drugs initially used for cancer chemoprevention, a term coined in 1976 by Sporn (39). The initial forays into cancer chemoprevention did not sufficiently account for the impact of toxicity on the acceptance of a strategy which did not adequately define the drugs’ risk/benefit ratio. The level and nature of toxicities acceptable for treatment of advanced disease are not acceptable in the prevention setting, a concept that seems obvious in retrospect but was largely ignored in the development and choice of first- and second-generation chemoprevention agents. A survey of the timeline of the historical development of anti-hypertensive agents and a comparison of this timeline with that of cancer chemoprevention (which started 25 years later) suggest that cancer chemoprevention may not be doing so badly after all (Table 1).
The third major hurdle to be discussed here is the very demanding path that is required for regulatory approval, although this process is just as rigorous for cardiovascular disease prevention as for cancer prevention. Assessing the balance of risk-benefit stands at the core of regulatory review. In therapy-drug development, we define benefit as the quantitative assessment of improvement in quality of life, duration of life, or both. The benefit is balanced with the risk of a given intervention. We define risk as adverse consequences in quality or duration of life that result from a therapeutic intervention. The regulatory review process also takes into consideration the risk of not intervening and the risk-risk concept of the potential effect on other organs.
For cancer chemoprevention (in contrast to therapy), however, there are the additional issues of difficulty in being able to accurately select patients at risk for developing cancer and in monitoring the effect a drug on that risk. These issues require us to develop clinically meaningful biomarkers. Without reliable biomarkers of a preventative effect (such as cholesterol and blood pressure for the risk of heart disease), patients are blinded to benefit and thus have little incentive to take a drug. Indeed, many patients who may benefit from tamoxifen for preventing breast cancer have declined to do so. Tamoxifen has some serious, albeit rare, side effects, and many patients avoided its use in the absence of any marker of personal benefit during chronic treatment. Increasingly, too, insurance companies and Centers for Medicare & Medicaid Services are likely to demand proof that the right patient is receiving the right drug, whether for cancer treatment or cancer risk reduction. An acceptable trade-off of risk and benefit needs to be defined by the regulatory agencies in collaboration with the academic, patient, and pharmaceutical communities if we are to achieve more rapid progress in the arena of cancer chemoprevention. A word of caution, however: Even cardiovascular surrogates, for example, high-density lipoprotein (HDL) cholesterol and arrhythmias, have not always been predictive (33). Large-scale clinical trials with cardiovascular surrogate end points were needed because the event rate was low in the very large at-risk population, and the assessment of risk-benefit was essential for regulatory evaluation and approval.
Defining cancer risk, drug safety and risk, and clinical endpoints are hurdles that must be cleared before drug approval is feasible. Showing that cancer risk from no intervention outweighs any risk from cancer risk reduction, and further that cancer risk reduction is “better” than an alternative intervention because it is less invasive, having less risk of toxicity and/or providing better quality of life, are imperative.
Changes in the presence or number of cancers and an acceptable risk-benefit ratio have been the “gold standard” for regulatory approval. Changes in the presence or number of IENs, or “precancers,” however, may, with an acceptable risk/benefit ratio, also lead to approval in well-defined situations (Table 2) and has been the subject of much discussion (40-42). The challenge to achieving regulatory approval in IEN settings is high and risky: regulatory precedents are few (see Table 2) and IEN is very heterogeneous. Therefore, this area is unattractive for pharmaceutical development, particularly because the reimbursement status for such treatments is uncertain. To change this situation, it will be necessary to develop credible validated biomarkers that are as easy to measure as blood pressure or cholesterol and that span the continuum of carcinogenesis from intrinsic constitutive genetic changes to histologically identifiable changes.
Table 2.
FDA-approved chemoprevention of human cancers
| 1978 | Bladder CIS | BCG |
| 1990s | Actinic keratoses | Diclofenac |
| FAP – polyps | Celecoxib | |
| Barrett’s esophagus | Photofrin | |
| 2000s | Breast cancer | Tamoxifen |
| Raloxifene | ||
| Cervix cancer | Vaccines |
Abbreviations: FDA, Food and Drug Administration; CIS, carcinoma in situ; BCG, Bacillus Calmette-Guérin; FAP, familial adenomatous polyposis.
The Use and Limitations of Biomarkers in the Development of Cancer Risk Reduction Drugs
A major challenge to the continued development of agents for cancer risk reduction is to define clinical trial endpoints that correlate with, or provide, clinical benefit to at-risk populations and that demonstrate efficacy. In some well-defined high-risk cohorts such as patients who have had a previous malignancy, cancer incidence is a feasible end point. In other settings, cancer incidence endpoints are not practical or ethical. As a result, the development of cancer risk reduction drugs has relied heavily on a variety of biomarkers to demonstrate presumptive efficacy in early-phase clinical trials; this developmental process is detailed in Supplementary Appendix 3.
The term biomarker is often used rather cavalierly in the field, invested frequently with variable and casual, and too often with wrong, meanings (43). The nosology of biomarkers can be represented in a number of ways, one of which is presented in Fig. 2. The number of biomarkers is immense if not infinite. The question is whether a particular biomarker (or set of biomarkers) reliably estimates the endpoint of interest (i.e., a cancer or, in some cases, a precancer). The pathways of carcinogenesis are complex, and therefore a particular intermediate biomarker may not be a true surrogate for cancer at the end of a molecular and/or histological pathway along which it develops. However, the utility of a biomarker for drug development is related to its accuracy in predicting a particular cancer (or potentially IEN).
Fig. 2.
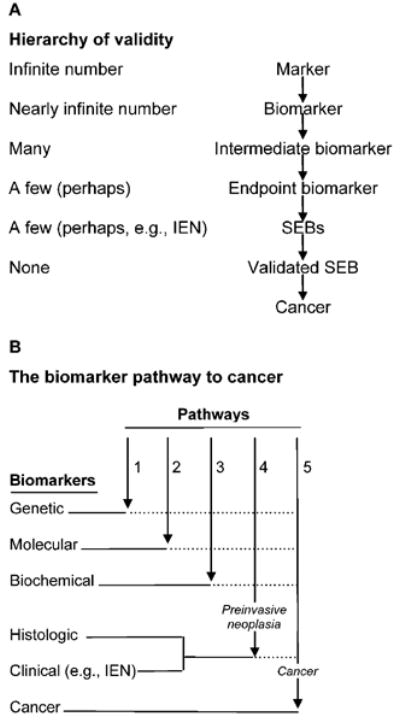
A nosology of biomarkers. A, the hierarchy of validity highlights the critical lack of validated surrogate endpoint biomarkers (SEBs) available for chemoprevention research. B, determining when a biomarker is on the path of and becomes a true surrogate for the cancer endpoint is a challenging task. As indicated by the dotted lines following them, only genetic, molecular, biochemical, and histologic and clinical biomarkers that ultimately transverse pathways 4 and 5 would be SEBs. Cancer biomarkers may not be present at earlier stages of the pathway to cancer and thus may not serve as SEBs. Modified from ref. 43 and reprinted with permission of Springer.
An important, frequently ignored distinction in the development of biomarkers is between their function in prognosis and that in predicting drug effectiveness (for which they are called “predictive” biomarkers), as has been discussed in detail elsewhere (43, 44). Modulation of a prognostic biomarker may not predict the usefulness of a candidate drug; likewise, a biomarker that is a specific drug target may not be prognostic for the endpoint or even on the pathway down which cancer develops. The issue of biomarkers and disease management has been taken up by many groups resulting, for example, in a detailed “prototypical” process for creating evidentiary standards for biomarkers and diagnostics for disease processes (including cancer; Supplementary Appendix 4). Whether this general schema is relevant to any particular organ site remains to be tested.
Biomarkers are used primarily in the following two ways in the field of cancer risk reduction: (1) As a modulatable targets for candidate risk reduction drugs that may be related directly (e.g., an enzyme) to the action of the drug or indirectly to a relevant general effect (e.g., proliferation, apoptosis) and (2) as surrogates for the final endpoint of histologic precancer or cancer.
Reductions in IENs already have demonstrated the potential efficacy of cancer risk reduction drugs. However, IENs have different rates of progression to cancer, and cannot be globally lumped together—some IENS may be appropriate endpoints for the development of a particular drug, others may be less informative. IEN regression is most likely to be useful as an endpoint if its associated rate of progression to cancer is high. Histologic phenotypes alone, however, are not sufficiently accurate to characterize the malignant potential of premalignancies. Therefore, other data such as genotype and gene-expression profiles may be needed to identify the truly high-risk lesions within the premalignant cohort (45-47). In later phases of drug development, trials are randomized and controlled, with endpoints measured at a time when results in treated subjects are expected to be significantly different from controls; for example, prevention of sporadic colorectal adenomas is determined after three years of treatment (44, 45).
To date, no biochemical or molecular biomarker has been demonstrated to be a true surrogate for IEN or cancer incidence. Therefore, the current use of a tissue or serum biomarker in early-phase trials cannot be viewed as a surrogate but only as a tool to demonstrate that a drug produces an effect that is related to the mechanistic action of the compound. This information is critical, however, prior to embarking on phase-III, or definitive, clinical trials (discussed further in Supplementary Appendix 3). The failure to demonstrate that an agent can modulate its putative molecular or biochemical target in the relevant organ generally indicates either that further development of this agent for cancer chemoprevention in this organ is unwarranted or that the studied biomarker is not related to the agent’s action.
Identifying a High Risk of Cancer and Thus Appropriate Candidates for Risk Reduction Drugs
Measuring factors for predicting the development of cancer was largely qualitative until the first attempts to quantify this process were made in the late 1980s (48-50). The lessons learned from 20 years of trying to develop biomarkers for predicting drug efficacy against cancer development and the results from chemoprevention trials to date produce a sobering conclusion: “The development of validated biomarkers is a long and difficult process fraught with blind alleys and wrong turns” (51). Although rapid progress in developing and testing multiple genomic and proteomic biomarkers may allow more “personalized” preventive medicine (52), this hoped-for result has yet to be realized in the clinic (a contrary view is discussed in ref. 53). The field of cardiovascular medicine faced a similar critical point vis à vis the identification of biomarkers over 50 years ago, and this impasse was not overcome in cardiology until the results of the Framingham study clearly identified cholesterol and hypertension as markers of a high risk of cardiovascular disease (29). Not only did these factors mark risk, they could be modulated in reducing risk, which allowed the development of effective cardiovascular-disease risk-reducing drugs. In step with this example, progress in the further development of cancer risk reduction drugs should accelerate with efforts to emphasize identifying markers that indicate a high risk for particular types of cancer and can be modulated in reducing cancer risk.
The topic of at-risk populations has been reviewed in detail elsewhere (50, 51, 54). From a practical and regulatory point of view, several levels of cancer risk are identifiable (Fig. 3). In a decreasing order, these general risk categories are as follows:
A strong hereditable genetic risk for malignancy, such as retinoblastoma, xeroderma pigmentosa, or familial adenomatous polyposis.
A prior common cancer for which the risk of a second cancer is high (including in the breast, colon, lung).
Pre-existing clinical evidence of a precancer confirmed by cytology or histopathology from biopsies (reviewed in ref. 55). This cohort can be further enriched by testing for relevant molecular markers.
A substantially increased risk for a particular cancer based on logistic regression risk models (relative risk [RR] > 3.0). The Gail Model is the archetypal such model and has been widely used in assessing breast-cancer risk (56). Models for lung (57, 58), prostate (59), and melanoma (60) cancer risk also have been developed but not widely utilized except in the research setting.
An RR of 1.5–3.0 for a particular cancer. Classical epidemiologic studies have been used in general to identify individuals at such risks from population-based studies.
A low, but elevated risk (RR = 1.1–1.5) for a particular cancer in a large number of individuals. For example, differences in heritable single nucleotide polymorphisms (SNPs) have identified a large number of individuals at a low risk for cancer and many other conditions (61).
Fig. 3.
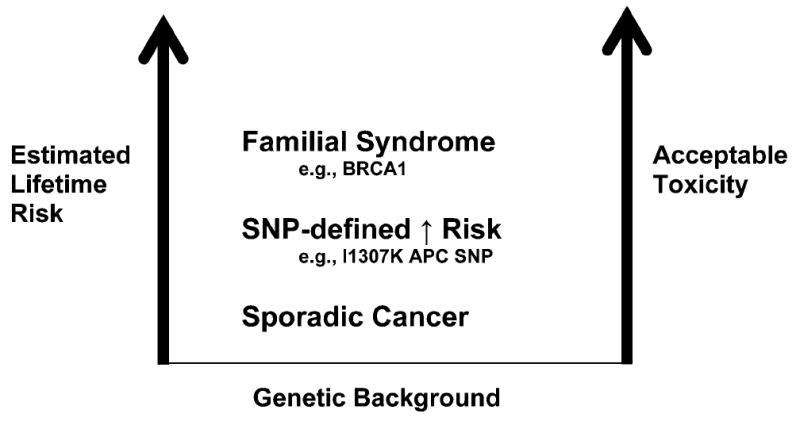
Qualitative ranking of cancer risk reduction interventions based on drug toxicity. The level of toxicity that would be acceptable to the patient increases with conditions that lead to a higher risk of cancer.
A strategic approach to cancer risk reduction and the therapeutic index needs to be developed. In the case of breast cancer risk, a continuous variable risk-assessment tool, the Gail Scale, provides quantitative risk estimates that are useful in assessing risk-benefit for cancer risk reduction. The development of quantitative continuous variable risk-assessment algorithms should improve the selection of cancer risk reduction interventions. Pharmacogenetic variables such as those regulated by SNPs or due to polymorphic metabolism of drugs also should improve these selections. For example, differential metabolism of drugs promises to improve the selection (and enrich the population) of individuals most likely to benefit from targeted risk reduction therapies (61, 62). This approach might provide a basis for future regulatory approval of cancer chemoprevention agents in clearly defined subsets of at-risk individuals and may lead to identifying individuals with a favorable therapeutic index for cancer risk reduction.
The number of participants available for, and the generalizability of, definitive studies increases as the RR decreases, but the tolerance for toxicity decreases markedly. We can either perform trials in a small number of high-risk patients, who are frequently hard to identify but whose tolerance of toxicity is higher, or we can conduct trials in large, low-risk groups who are willing to accept only minimal toxicity. After identifying high-risk individuals, determining the RR of toxicity of the candidate agent should be of paramount importance, a painful lesson that recently was relearned with COX-2 selective inhibitors for colorectal adenoma prevention (63). The recent development of toxicity self-reporting may provide in the future individualized meaning to the risk side of the risk-benefit issue (64). Patient self-assessment of mild-to-moderate toxicities allows a better understanding of what is important to individual patients, unfiltered through the prism of health-care providers and thus “personalized toxicity.”
General limitations of categorizing cancer-chemoprevention approaches by RR should be acknowledged. Prevalence can be low if the cancer has a high RR but is uncommon and may be considerable if the cancer has a low RR but is common. Cancer is many different diseases; a single pharmaceutical compound is almost certainly unlikely to be useful for preventing multiple cancer types, a situation quite different from prevention in atherogenesis and cardiovascular disease risk. Many types of cancer are uncommon. An RR of 3 may sound high, but if the cancer is rare, even a 10-fold increased RR leads to few cases. The limitations of RR also apply to adverse effects, as, for example, in the case of endometrial cancer risk and tamoxifen—the increase was greater than 3-fold, although the number of cases was very small compared with the number of breast cancers prevented. Great variation in prevalences of different tumor types makes it difficult to formulate guidelines on what RR warrants pharmaceutical intervention. This difficulty is compounded in cases of IEN, where associations of IEN with cancer development frequently are not clearly defined. Nevertheless, the FDA has approved IEN as an endpoint of cancer risk reduction trials on a case by case basis, or when an agent demonstrates substantial effectiveness and an acceptable risk-benefit profile (Table 2).
The following 1981 recommendation of the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee for cardiovascular-disease preventive agents is a good starting point for re-examining in 2010 the paradigm for cancer risk reduction (65):
This committee previously recommended, and the Food and Drug Administration concurred, that approval of lipid altering agents should be based on a drug’s biochemical efficacy in decreasing serum lipids. Attempts to establish clinical efficacy in the prevention of coronary artery disease or other manifestations of atherosclerosis would require prolonged observations and hamper research and development of this class of drugs.
It cannot be emphasized strongly enough that lowering blood pressure or cholesterol levels was accepted as efficacy biomarkers only after extensive evidence from a series of randomized clinical trials demonstrated benefit in reducing the number of cardiovascular events; some experts still do not trust cholesterol levels as surrogates for serious cardiovascular disease (66).
Business Models for Incentivizing Pharmaceutical Investment in Cancer Risk Reduction
The pharmaceutical industry views drug development through several prisms, including that of enabling patients to live longer and healthier lives, and in so doing, improve public health. This model was successfully promoted for the adoption of cardiovascular risk reduction drugs. There also are the scientific and medical challenges of unmet medical needs. It is also necessary, however, to show investors and stockholders a return on considerable investment at a time when developing a new drug has surpassed the billion-dollar threshold.
Business investment in cancer prevention involves many disincentives. It is difficult to predict a return on investment in the current cancer-prevention environment, where definitive trials generally are large and the acceptable side-effects profile of a drug is problematical because prevention involves healthy people. Furthermore, prevention trials can require years to decades to complete, during which time the intellectual property clock continues to tick. Even given the most efficient milestones, a successful cancer prevention drug will be protected with limited (or no) exclusivity. Nevertheless, many experts in the field of public health argue convincingly that prevention is necessary if we are to have a significant impact on cancer mortality. Stroke and cardiac disease have emphatically demonstrated the impact of prevention.
What incentives, then, can motivate the pharmaceutical industry to invest in cancer risk reduction? As discussed in detail by Grabowski and Moe (67), the business disincentives currently greatly outweigh any potential for return on investment. Still, there are models where government and society have changed the rules of engagement to provide incentives for investment by the pharmaceutical industry in specific areas.
For example, the Best Pharmaceutical Act for Children grants six months of intellectual property protection in exchange for early testing of investigational agents in pediatric populations. Another innovative approach would be to reset the patent clock to begin with “first-in-human” trials. The national Vaccine Injury Compensation Program mitigates liability for companies working in the area of vaccines. Programs such as these could also be implemented to spur development of new agents for cancer risk reduction.
The Orphan Drug Act includes tax credits for the cost of clinical research and allows seven years of marketing exclusivity for drugs developed for rare diseases. This Act provides one of the most promising tools to facilitate interest in successful development of chemoprevention drugs for important labeled indications. These regulations were written in 1983 (and subsequently amended), when it was recognized that adequate drugs for many rare diseases and conditions were not being developed. The basis for the Orphan-Drug incentives includes the following premises: 1) Relatively small sales in comparison with drug development costs because few patients are affected by a rare disease or condition; 2) some promising Orphan Drugs would not be developed without changes in applicable federal laws to provide financial incentives; and 3) incentives to develop Orphan Drugs is in the public interest. The Act defined rare diseases or conditions as “any disease or condition which affects less than 200,000 persons in the US or affects more than 200,000 persons in the US and for which there is no reasonable expectation that the cost of developing and making available a drug for such disease or condition will be recovered from sales in the US of such drug.”
The Orphan Drug Act has been used to develop cancer risk reduction drugs in familial malignancies with fewer than 200,000 cases, such as familial adenomatous polyposis. Orphan-drug–like incentives are a mechanism that should be considered for stimulating further investment in cancer risk-reducing agent development.
In a dialogue between C-Change and the FDA about overcoming the disincentives to cancer-prevention drug development, the FDA informally has offered constructive ideas. For example, it suggested extending the patent life for cancer chemopreventive agents. After approval, a drug could be prescribed without advertising and other promotional efforts for a year. Postponing these marketing efforts would limit the number of “real-world” patients exposed to the drug and potentially would allow the detection of early safety signals in a broader population than that of the clinical trial that led to the FDA approval. If the safety profile in this environment conforms with that demonstrated in the clinical trial or suggests an amended FDA approval to exclude certain people (e.g., with poor renal function), the drug could then be marketed with new exclusivity for a finite period of, perhaps, ten years.
The U.S. Congress has considered a model to incentivize industry investment in effective treatment for bio-terrorism attacks. One would hope that such treatments never will be needed or demanded by the public, and it is difficult to envision a reasonable return on investment in them. The potential solution to this disincentive would be to give the manufacturer of an approved anti-bioterrorism drug six months of added exclusivity for any other single drug in its portfolio, a so-called “wild card” patent. In exchange, the company would release the technology for the new treatment into the public domain. This approach might be particularly applicable to some chemopreventives, where the cost of chronic treatment would need to be low. Indeed, either approach, extended patent life or a patent “wild card,” might be offered to a company sponsoring a new cancer chemoprevention agent for FDA approval.
In summary, incentives for chemoprevention of cancer should be considered to make the case for business investment feasible by strengthening intellectual, data, and/or patent protections, extending the period of time for investment recovery, reducing liability risks, and/or creating tax advantages.
Summary of Scientific and Institutional Barriers to Developing Cancer Risk Reduction Drugs
Assessments of the risk-benefit of cancer risk-reduction drugs pose special challenges and barriers that are not encountered with most cancer therapeutic agents. These challenges include high therapeutic index requirements, the long latency period to cancer endpoints, patient adherence barriers, complex risk assessments, inadequate patent protection, and uncertain insurance reimbursement.
High therapeutic index
Pre-invasive carcinogenesis is a chronic process which generally requires prolonged exposure to an intervention. The potential toxicity of an intervention aimed at delaying or reversing transformation must be acceptable to individuals who are asymptomatic yet may benefit from an extended (years-long) intervention. Models of long-term treatment with disease-risk reductions, such as hypertension control, suggest that mild, tolerable toxicity associated with a long-term intervention is acceptable. Hypertension, a validated surrogate endpoint for efficacy in cardiovascular-preventive interventions, is accepted by the clinical, regulatory, and third-party payer groups. Based on this professional consensus, the public now accepts mildly to moderately toxic interventions and their associated costs for cardiovascular-disease prevention. Patient consensus forums conducted by advocacy-group participants in clinical trials might help address some of the issues surrounding risk-benefit considerations.
Long latency to cancer
Assessing the effectiveness of an agent for reducing cancer incidence requires years-long trials involving thousands of participants in most cases. It is not economically feasible to test a large number of potentially effective drugs based on this strategy, given the large number of available promising preventive agents. Biomarker endpoints as guides during the early phases of drug development are necessary to enhance throughput and reduce the cost and time of clinical efficacy testing. A critical issue of this effort will be efficient biomarker validation. One way to facilitate this validation is to include biomarkers in most phase III trials in order to gather data on their potential as surrogate markers for regulatory decision making, and to fund these biomarker-related trial costs in a rigorous way. Long-term follow-up of participants in carefully conducted randomized cancer chemoprevention trials also should be routine so that the long-term benefit of the intervention can be assessed after its discontinuation and consequently decreased toxicity. For example, reductions of second malignancies and IENs have persisted long after drug discontinuation in randomized clinical trials in settings of the colon, head and neck, and breast (68-70).
Adherence
Cancer risk-reducing agents should be designed for ease of use and engaging the willingness of participants to follow a treatment plan. Interventions with sufficiently prolonged half-lives may minimize the biological impact of a dropped dose on the physiological target. Minimal toxicity and strong personal commitment to a preventive goal also enhances adherence. Future studies should actively incorporate baseline measures of attitude, desire for health behavior change, and motivational approaches to improve adherence; incorporating potentially healthful behavior changes also would support the risk-reduction effort. In sum, adherence is critical to the success of a cancer risk reduction intervention.
Complex cancer-risk assessment
Highly penetrant but infrequent, inherited genetic mutations are major risks for certain breast and colon cancers. Genetic testing for some such risks (e.g., the risk of breast-ovarian cancer syndromes or of cancer related to familial adenomatous polyposis) has identified high-risk subjects who appear to benefit from chemoprevention interventions, although not without psychological and social ramifications such as depression, anxiety, low self-esteem, and stigmatization. Much more commonly, risk assessment is based on data sets that include epidemiologic associations with cancer, such as personal and family history of cancers, environmental exposures, and lifestyle variables such as diet, exercise, and smoking. These variables are amenable to health behavior interventions via counseling and education, which can decrease risk (or increase early detection for people with a family history). Pharmacologic risk reduction should be viewed as an adjunct to these essential primary prevention efforts.
Inadequate patent protection
The many years of research and development required to establish cancer-chemoprevention safety and efficacy often consume most or all of the limited period of patent protection and data exclusivity. Under these circumstances, drug companies are reluctant to assume the risk of funding (alone or in collaboration with federal and other agencies) this research because of the difficulty in recouping their investment. For example, there is only a five-year period of exclusivity for an orphan claim on behalf of a new molecular entity whose patent life is exhausted but has new uses in prevention. In the case of preventing a rare disease, two years would be added to the five years of extra protection, but even seven years would likely be insufficient to recoup a return on investment in the rare-disease setting. Patents may also be extended for up to five years based on development and FDA review time. Importantly a drug with known biological effects already approved for other uses would receive only 3 years of added exclusivity. This significant barrier has been discussed elsewhere by Grabowski and Moe (67). Extended patent protection would allow market forces to set prices, leaving it up to patients and providers to decide if the expenses are worth the benefits. Given likely high costs of using a chemopreventive agent, its broad use likely would occur only after the agent becomes generic, further disincentivizing investment. Clearly, a new intellectual property model is needed to encourage capital investment in drugs that lower cancer risk.
Uncertain insurance reimbursement
Once a risk reduction drug has been developed, approved, and made available to the market, there is no assurance it will be prescribed by physicians or covered by insurance. Two recent examples are tamoxifen for breast cancer prevention and finasteride for prostate cancer prevention. Both were proven to be effective but, because of concerns about side effects or other issues, have not been prescribed or used for cancer prevention to any great extent. Reimbursement issues and evidence-based medicine have been discussed in detail elsewhere by Pyenson et al. (71).
Summary of Major Recommendations for Facilitating the Pathway to Regulatory Approval for Cancer Risk Reduction Drugs
The following recommendations are intended to facilitate regulatory approval of cancer chemoprevention drugs:
The framework for the development of pharmacological cancer risk reduction should emulate the long-standing and accepted model for cardiovascular disease.
Refining risk models (e.g., the Gail model) for identifying high-risk individuals should be encouraged to allow better evidence-based drug approvals vis à vis the risk of intervention versus the risk of no intervention.
The goal of early-phase chemopreventive-drug trials should be modulation of a biologically relevant or molecular or biochemical endpoint by a drug with minimal toxicity.
Reduction of the risk, or regression, of an IEN, along with acceptable toxicity, in a randomized phase IIb trial, where the endpoint was determined with regulatory agency input, may provide the basis for accelerated approval in specific cases, where the progression rate of the IEN to cancer is well established and sufficiently high. Accelerated approvals coupled with clearer policies for confirmatory trials and post-approval surveillance to obtain long-term safety and efficacy should be the goal.
With no currently validated surrogate biomarkers for chemopreventive-drug development, randomized phase-IIb and -III trials need to include biomarker measurements for the purpose of gathering data on their potential as surrogate markers in regulatory decision making (and funding the costs for these biomarker studies in a rigorous way).
As has been done in cardiovascular chemoprevention trials, long-term follow-up of participants in carefully conducted randomized phase-III cancer chemoprevention trials needs to be done, as has occurred recently.
The major challenge in 2010 for the development of chemopreventive agents is parsing the trade-off between drug risks/adverse effects and potential benefits. Risk-benefit considerations are intimately intertwined with identifying cohorts at a high-enough risk to tolerate some toxicity with pharmacologic or other risk-reducing interventions. Genetic parameters (often referred to as pharmacogenetics or pharmacogenomics), classical epidemiologic assessment, clinical factors, and risk modeling should help identify higher-risk individuals and facilitate objective considerations of the risk-benefit equation. Whereas significant toxicity may be acceptable in the setting of advanced disease, it is not acceptable in the setting of high-risk, otherwise healthy individuals. Risk profiles that apply more to individuals (“personalized risk”) than to a population will further improve risk-benefit considerations. Absent effective risk identification across a substantial range of cancers, large, long, and expensive randomized clinical trials will be necessary to establish chemopreventive efficacy and tolerability.
Increased support is needed for post-approval drug safety surveillance by the FDA. The ability to identify post-approval adverse events would help the FDA in approving many useful cancer-chemoprevention drugs that might otherwise never be available.
Incentives such as those included in the Orphan Drug Act should be extended to the development of cancer risk-reduction drugs. Consideration also should be given to a period of market-exclusivity protection balanced against the interests of competition and new research.
A balanced message approved by major stakeholders should be the basis for campaigns for preventing breast and colon cancer with a focus on medical approaches (14, 15). This approach would emulate what has been done for cardiovascular disease prevention and would provide a fair presentation of this complex topic.
We need to learn from our cardiovascular colleagues how to educate the public about risk (72). Extensive education campaigns led by the National Heart, Blood and Lung Institute and American Heart Association have led to widespread professional and pubic awareness of cardiovascular risk factors. As do people with high blood pressure and lipid levels, people at risk for developing cancer might accept beneficial preventive drugs with acceptable levels of toxicity if properly educated about their risks. Although the toxicity of cancer chemoprevention may be minimized in the future by early interventions and lower doses or by new agents with lower and more acceptable toxicity profiles, physicians and the lay public need to understand that minimal-to-no toxicity is a long-term goal that will not be achieved readily or in time for many people who could benefit in the nearer term from interventions with either mild, or rare serious, toxicities. For example, modern social media such as “Twitter groups” are being used to enhance educational efforts and adherence (D. Hershman, personal communication).
Geyman (73) has dramatically described the perfect storm that baby boomers face as the “cancer generation.” We need to address this storm in an intelligent and cost-effective manner. As recently adopted in a policy statement by ASCO, “Drugs that reduce risk should be a key approach along with primary prevention, screening, and early detection in the management of human cancer” (74).
Supplementary Material
Acknowledgments
We thank Janis DeJohn for administrative assistance and cardiovascular epidemiologist Nathan Wong, Ph.D. (UC Irvine), for assistance in understanding the history of cardiovascular prevention.
Grant Support Supported in part by P30CA62203 (to Frank Meyskens).
Appendix
The full membership of the C-Change Chemoprevention Clinical Trials and Biomarkers Subcommittee is as follows: Chao Family Comprehensive Cancer Center: Frank L. Meyskens, Jr., M.D. (Co-Chair); AstraZeneca Oncology: Gregory A. Curt, M.D. (Co-Chair), Joseph D. Purvis, M.D.; University of Michigan Health System: Dean E. Brenner, M.D.; University of Texas M. D. Anderson Cancer Center: Powell H. Brown, M.D., Ph.D., Ernest Hawk, M.D., M.P.H., Scott M. Lippman, M.D.; National Cancer Institute: James Crowell, PhD, Gary J. Kelloff, M.D., Samir N. Khleif, M.D., Eva Szabo, M.D.; Cancer Research UK: Jack Cusick, M.D.; Arizona Cancer Center: Robert T. Dorr, Ph.D.; Pfizer, Inc.: Craig Eagle, M.D.; University of Washington: Scott S. Emerson, M.D., Ph.D.; University of Pittsburgh School of Medicine: Olivera Finn, Ph.D.; OSI Pharmaceuticals, Inc.: Neil Gibson, Ph.D.; Abbott Laboratories, Inc.: Gary Gordon, M.D., Ph.D.; Intrexon Corporation: Ronald B. Herberman, M.D.; University of California, Irvine: Claude L’Enfant, M.D., Christine E. McLaren, Ph.D.; American Association of Cancer Research: Mark Mendenhall, J.D.; Eli Lilly and Company: Colleen Mockbee, R.Ph.; GlaxoSmithKline: Clet Niyikiza, Ph.D; Nodality, Inc.: David R. Parkinson, M.D.; CCS Associates: Caroline C. Sigman, Ph.D.; Lombardi Comprehensive Cancer Center, Georgetown University: Louis M. Weiner, M.D.; Geisinger Medical Clinic: Victor Vogel, M.D.; RCY Medicine: Robert C. Young, M.D; Subcommittee Staff Support: Alison P. Smith, B.A., B.S.N., R.N.
Footnotes
Disclosure of Potential Conflicts of Interest F. Meyskens is a co-founder of Cancer Chemoprevention Pharmaceuticals. G. Gordon has ownership interest in Abbott Pharmaceuticals and Pfizer Pharmaceuticals and has an employee relationship (other than primary affiliation) with Abbott Pharmaceuticals. R. Herberman has an employee relationship (other than primary affiliation) with Interon Corporation. No other potential conflicts of interest were disclosed.
References
- 1.Herberman RB, Pearce HL, Lippman SM, Pyenson BS, Alberts DS. Cancer chemoprevention and cancer preventive vaccines--a call to action: leaders of diverse stakeholder groups present strategies for overcoming multiple barriers to meet an urgent need. Cancer Res. 2006;66:11540–9. doi: 10.1158/0008-5472.CAN-06-4122. [DOI] [PubMed] [Google Scholar]
- 2.Woolf SH. The power of prevention and what it requires. JAMA. 2008;299:2437–9. doi: 10.1001/jama.299.20.2437. [DOI] [PubMed] [Google Scholar]
- 3.Hong WK, Endicott J, Itri LM, et al. 13-cis-retinoic acid in the treatment of oral leukoplakia. N Engl J Med. 1986;315:1501–5. doi: 10.1056/NEJM198612113152401. [DOI] [PubMed] [Google Scholar]
- 4.Meyskens FL, Jr, Surwit E, Moon TE, et al. Enhancement of regression of cervical intraepithelial neoplasia II (moderate dysplasia) with topically applied all-trans-retinoic acid: a randomized trial. J Natl Cancer Inst. 1994;86:539–43. doi: 10.1093/jnci/86.7.539. [DOI] [PubMed] [Google Scholar]
- 5.Hong WK, Lippman SM, Itri LM, et al. Prevention of second primary tumors with isotretinoin in squamous-cell carcinoma of the head and neck. N Engl J Med. 1990;323:795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 6.Decensi A, Torrisi R, Bruno S, et al. Randomized trial of fenretinide in superficial bladder cancer using DNA flow cytometry as an intermediate end point. Cancer Epidemiol Biomarkers Prev. 2000;9:1071–8. [PubMed] [Google Scholar]
- 7.Khuri FR, Lee JJ, Lippman SM, et al. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. J Natl Cancer Inst. 2006;98:441–50. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- 8.Lippman SM, Lee JJ, Martin JW, et al. Fenretinide activity in retinoid-resistant oral leukoplakia. Clin Cancer Res. 2006;12:3109–14. doi: 10.1158/1078-0432.CCR-05-2636. [DOI] [PubMed] [Google Scholar]
- 9.Brewster AM, Lee JJ, Clayman GL, et al. Randomized trial of adjuvant 13-cis-retinoic acid and interferon alfa for patients with aggressive skin squamous cell carcinoma. J Clin Oncol. 2007;25:1974–8. doi: 10.1200/JCO.2006.05.9873. [DOI] [PubMed] [Google Scholar]
- 10.Sabichi AL, Lerner SP, Atkinson EN, et al. Phase III prevention trial of fenretinide in patients with resected non-muscle-invasive bladder cancer. Clin Cancer Res. 2008;14:224–9. doi: 10.1158/1078-0432.CCR-07-0733. [DOI] [PubMed] [Google Scholar]
- 11.Lippman SM, Lee JJ, Karp DD, et al. Randomized phase III intergroup trial of isotretinoin to prevent second primary tumors in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2001;93:605–18. doi: 10.1093/jnci/93.8.605. [DOI] [PubMed] [Google Scholar]
- 12.Fisher B, Costantino JP, Wickerham DL, et al. Tamoxifen for the prevention of breast cancer: current status of the National Surgical Adjuvant Breast and Bowel Project P-1 study. J Natl Cancer Inst. 2005;97:1652–62. doi: 10.1093/jnci/dji372. [DOI] [PubMed] [Google Scholar]
- 13.Vogel VG, Costantino JP, Wickerham DL, et al. National Surgical Adjuvant Breast and Bowel Project (NSABP). Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other outcomes: the NSABP Study of Tamoxifen and Raloxifene (STAR) P-2 trial. JAMA. 2006;295:2727–41. doi: 10.1001/jama.295.23.joc60074. [DOI] [PubMed] [Google Scholar]
- 14.Vogel VG, Costantino JP, Wickerham DL, et al. National Surgical Adjuvant Breast and Bowel Project. Update of the National Surgical Adjuvant Breast and Bowel Project Study of Tamoxifen and Raloxifene (STAR) P-2 Trial: preventing breast cancer. Cancer Prev Res. 2010;3:696–706. doi: 10.1158/1940-6207.CAPR-10-0076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hortobagyi GN, Brown PH. Two good choices to prevent breast cancer: great taste, less filling. Cancer Prev Res. 2010;3:681–5. doi: 10.1158/1940-6207.CAPR-10-0101. [DOI] [PubMed] [Google Scholar]
- 16.Thompson IM, Goodman PJ, Tangen CM, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 17.Redman MW, Tangen CM, Goodman PJ, Lucia MS, Coltman CA, Thompson IM. Finasteride does not increase the risk of high-grade prostate cancer: a bias-adjusted modeling approach. Cancer Prev Res. 2008;1:174–81. doi: 10.1158/1940-6207.CAPR-08-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Logothetis CJ, Schellhammer PF. High-grade prostate cancer and the prostate cancer prevention trial. Cancer Prev Res. 2008;1:151–2. doi: 10.1158/1940-6207.CAPR-08-0085. [DOI] [PubMed] [Google Scholar]
- 19.Lucia MS, Epstein JI, Goodman PJ, et al. Finasteride and high-grade prostate cancer in the Prostate Cancer Prevention Trial. J Natl Cancer Inst. 2007;99:1375–83. doi: 10.1093/jnci/djm117. [DOI] [PubMed] [Google Scholar]
- 20.Andriole GL, Bostwick DG, Brawley OW, et al. Effect of dutasteride on the risk of prostate cancer. N Eng J Med. 2010;362:1192–1202. doi: 10.1056/NEJMoa0908127. [DOI] [PubMed] [Google Scholar]
- 21.Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–52. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 22.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–84. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 23.Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885–95. doi: 10.1056/NEJMoa061652. [DOI] [PubMed] [Google Scholar]
- 24.Solomon SD, Wittes J, Finn PV, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation. 2008;117:2104–13. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyskens FL, McLaren CE, Pelot D, et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: A randomized placebo-controlled, double-blind trial. Cancer Prev Res. 2008;1:9–11. doi: 10.1158/1940-6207.CAPR-08-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zell JA, Pelot D, Chen WP, McLaren CE, Gerner EW, Meyskens FL. Risk of cardiovascular events in a randomized placebo-controlled, double-blind trial of difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas. Cancer Prev Res Mar. 2010:209–12. doi: 10.1158/1940-6207.CAPR-08-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hay J. The significance of a raised blood pressure. The Brit Med J. 1931:43–7. doi: 10.1136/bmj.2.3679.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Page IH. Low cholesterol—low fat diets in prevention and treatment of atherosclerosis. Med Clin North Am. 1952;36:1995–9. [PubMed] [Google Scholar]
- 29.Kannel WB, Dawber TR, Kagan A, Revotskie N, Stokes J. Factors of risk in the development of coronary heart disease – six-year follow-up experience. The Framingham Study. Ann of Int Med. 1961;55:33–50. doi: 10.7326/0003-4819-55-1-33. [DOI] [PubMed] [Google Scholar]
- 30.Dock W. Atherosclerosis; inevitable or controllable? Can Med Assoc J. 1953;69:355–63. [PMC free article] [PubMed] [Google Scholar]
- 31.Wong ND, Cupples LA, Ostfeld AM, Levy D, Kannel WB. Risk factors for long-term coronary prognosis after initial myocardial infarction: the Framingham Study. Am J of Epid. 1989;130:469–80. doi: 10.1093/oxfordjournals.aje.a115360. [DOI] [PubMed] [Google Scholar]
- 32.Wong ND, Black HR, Gardin JM. Preventive Cardiology. McGraw-Hill; 2005. [Google Scholar]
- 33.Veterans Administration Cooperative Study Group on antihypertensive Agents. Effects of treatment on morbidity and hypertension: Results in patients with diastolic pressures averaging 115 through 129 millimeters of mercury. JAMA. 1967;202:1028–34. [PubMed] [Google Scholar]
- 34.Veterans Administration Cooperative Study Group on Antihypertensive Agents. Effects of treatment on morbidity and hypertension II: Results in patients with diastolic blood pressure averaging 90 through 114 millimeters of mercury. JAMA. 1970;213:1143–52. [PubMed] [Google Scholar]
- 35.Chobanian AV, Bakris GL, Black HR, et al. The seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; The JNC 7 Report. JAMA. 2003;289:2560–72. doi: 10.1001/jama.289.19.2560. [DOI] [PubMed] [Google Scholar]
- 36.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 37.Colhoun HM, Betteridge DJ, Furrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomized placebo-controlled trial. Lancet. 2004;364:685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 38.Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomized placebo-controlled trial. Lancet. 2004;364:685–96. doi: 10.1016/S0140-6736(04)16895-5. [DOI] [PubMed] [Google Scholar]
- 39.Sporn MB. Approaches to prevention of epithelial cancer during the preneoplastic period. Cancer Res. 1976;36:2699–702. [PubMed] [Google Scholar]
- 40.Kelloff GJ, Johnson JR, Crowell JA, et al. Approaches to the development and marketing approval of drugs that prevent cancer. Cancer Epidemiol Biomarkers Prev. 1995;4:1–10. [PubMed] [Google Scholar]
- 41.O’Shaughnessy JA, Kelloff GJ, Gordon GB, et al. Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clin Cancer Res. 2002;8:314–46. [PubMed] [Google Scholar]
- 42.Kelloff GJ, Sigman CC. Assessing intraepithelial neoplasia and drug safety in cancer-preventive drug development. Nat Rev Cancer. 2007;7:508–18. doi: 10.1038/nrc2154. [DOI] [PubMed] [Google Scholar]
- 43.Meyskens FL, Szabo E. How should we move the field of chemopreventive agent development forward in a productive manner? Recent Results Cancer Res. 2005;166:113–24. doi: 10.1007/3-540-26980-0_9. [DOI] [PubMed] [Google Scholar]
- 44.Kelloff GJ, Sigman CC, Johnson KM, et al. Perspectives on surrogate end points in the development of drugs that reduce the risk of cancer. Cancer Epidemiol Biomarkers Prev. 2000;9:127–37. [PubMed] [Google Scholar]
- 45.Kelloff GJ, Schilsky RL, Alberts DS, et al. Colorectal adenomas: a prototype for the use of surrogate end points in the development of cancer prevention drugs. Clin Cancer Res. 2004;10:3908–18. doi: 10.1158/1078-0432.CCR-03-0789. [DOI] [PubMed] [Google Scholar]
- 46.Califano J, van der Riet P, Westra W, et al. Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res. 1996;56:2488–92. [PubMed] [Google Scholar]
- 47.Mao L, El-Naggar AK, Papadimitrakopoulou V, Shin DM, et al. Phenotype and genotype of advanced premalignant head and neck lesions after chemopreventive therapy. J Natl Cancer Inst. 1998;90:1545–51. doi: 10.1093/jnci/90.20.1545. [DOI] [PubMed] [Google Scholar]
- 48.Lippman SM, Bassford TL, Meyskens FL., Jr A quantitatively scored cancer-risk assessment tool: its development and use. J Cancer Edu. 1992;7:15–36. doi: 10.1080/08858199209528139. [DOI] [PubMed] [Google Scholar]
- 49.Lippman SM, Bassford, Meyskens FL. Quantitative assessment of cancer risk. Texas Med. 1988;8:48–53. [PubMed] [Google Scholar]
- 50.Meyskens FL., Jr Strategies for prevention of cancer in humans. Oncology. 1992;6:15–24. [PubMed] [Google Scholar]
- 51.Lippman SM, Lee JJ. Reducing the “risk” of chemoprevention: defining and targeting high risk--2005 AACR Cancer Research and Prevention Foundation Award Lecture. Cancer Res. 2006;66:2893–903. doi: 10.1158/0008-5472.CAN-05-4573. [DOI] [PubMed] [Google Scholar]
- 52.Blackburn EH, Tisty TD, Lippman SM. Unprecedented opportunities and promise for cancer prevention research. Cancer Prev Res. 2010;3:394–402. doi: 10.1158/1940-6207.CAPR-10-0051. [DOI] [PubMed] [Google Scholar]
- 53.Le Fanu J. Disappointments of the double helix: a master theory. J R Soc Med. 2010;103:43–5. doi: 10.1258/jrsm.2009.09k077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meyskens FL., Jr American Society of Preventive Oncology Distinguished Career Achievement Lecture 2006--Enjoy the journey: the long and winding road of chemoprevention agent development. Cancer Epidemiol Biomarkers Prev. 2006;15:2038–41. doi: 10.1158/1055-9965.EPI-06-0609. [DOI] [PubMed] [Google Scholar]
- 55.Kelloff GJ, Lippman SM, Dannenberg AJ, et al. Progress in chemoprevention drug development: the promise of molecular biomarkers for prevention of intraepithelial neoplasia and cancer--a plan to move forward. Clin Cancer Res. 2006;12:3661–97. doi: 10.1158/1078-0432.CCR-06-1104. [DOI] [PubMed] [Google Scholar]
- 56.Gail MH, Brinton LA, Byar DP, et al. Projecting individualized probabilities of developing breast cancer for white females who are being examined annually. J Natl Cancer Inst. 1989;81:1879–86. doi: 10.1093/jnci/81.24.1879. [DOI] [PubMed] [Google Scholar]
- 57.Spitz MR, Hong WK, Amos CI, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst. 2007;99:715–26. doi: 10.1093/jnci/djk153. [DOI] [PubMed] [Google Scholar]
- 58.Cassidy A, Myles JP, van Tongenen M, et al. The LLP risk prediction model for lung cancer. Brit J Cancer. 2008;98:270–6. doi: 10.1038/sj.bjc.6604158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snow DC, Klein EA. Use of nomograms for early detection in prostate cancer. J Natl Comp Cancer Netw J Natl Cancer Inst. 2010;8:271–6. doi: 10.6004/jnccn.2010.0018. [DOI] [PubMed] [Google Scholar]
- 60.Fears TR, Guerry DT, Pfeiffer RM, et al. Identifying individuals at high risk of melanoma: a practical predictor of absolute risk. J Clin Oncol. 2006;24:3590–6. doi: 10.1200/JCO.2005.04.1277. [DOI] [PubMed] [Google Scholar]
- 61.Overdevest JB, Theodorescu D, Lee JK. Utilizing the molecular gateway: the path to personalized cancer management. Clin Chem. 2009;55:684–97. doi: 10.1373/clinchem.2008.118554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hemminki K, Forsti A, Lorenzo Bermejo J. Single nucleotide polymorphisms (SNPs) are inherited from parents and they measure heritable events. J Carcinog. 2005;4:2. doi: 10.1186/1477-3163-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006;6:130–40. doi: 10.1038/nrc1801. [DOI] [PubMed] [Google Scholar]
- 64.Goodwin PJ, Black JT, Bordelean LJ, Gan PA. Health-related quality of life measurements in randomized clinical trials in breast cancer – taking stock. J Natl Cancer Inst. 2003;95:263–281. doi: 10.1093/jnci/95.4.263. [DOI] [PubMed] [Google Scholar]
- 65.Minutes of the Endocrinologic and Metabolic Drug Advisory Committee. Rockville, MD: Oct 15, 1981. US FDA Document No. F82-25307:16. [Google Scholar]
- 66.Steinberg D. The pathogenesis of atherosclerosis: An interpretive history of the cholesterol controversy. J Lipid Res. 2006;47:1–14. doi: 10.1194/jlr.R500014-JLR200. [DOI] [PubMed] [Google Scholar]
- 67.Grabowski HG, Moe JL. Impact of economic, regulatory and patent policies on innovation in cancer chemoprevention. Cancer Prev Res. 2008;1:84–90. doi: 10.1158/1940-6207.CAPR-08-0048. [DOI] [PubMed] [Google Scholar]
- 68.Bertagnolli MM, Eagle CJ, Zauber AG, et al. Adenoma Prevention with Celecoxib Study Investigators. Five-year efficacy and safety analysis of the aAdenoma Prevention with Celecoxib Trial. Cancer Prev Res. 2009;2:310–21. doi: 10.1158/1940-6207.CAPR-08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brenner SE, Pajak TF, Lippman SM, Earley C, Hong WK. Prevention of second primary tumors with isotretinoin in patients with squamous cell carcinoma of the head and neck: long-term follow-up. J Natl Cancer Inst. 1994;86:140–1. doi: 10.1093/jnci/86.2.140. [DOI] [PubMed] [Google Scholar]
- 70.Cuzick J. Long-term follow-up in cancer prevention tirals (It ain’t over “til it’s over) Cancer Prev Res. 2010;3:689–91. doi: 10.1158/1940-6207.CAPR-10-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pyenson B, Campbell KP, Ramsey S, Slotnik J, Zon R, Smith AP, Tyus S. Considering reimbursement for cancer preventive agents. [11/18/2008];C-Change. 2008 at http://www.milliman.com/expertise/healthcare/publications/published/pdfs/considering-reimbursement-for-cancerPA-06–18–08.pdf.
- 72.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel of Detection, Evaluation and Treatment of High blood cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 73.Geyman J. The Cancer Generation: Baby Boomers forming a perfect storm. Common Courage Press; 2009. [Google Scholar]
- 74.Zon RT, Goss E, Vogel V, et al. American Society of Clinical Oncology Policy Statement: the role of the oncologist in cancer prevention and risk assessment. J Clin Oncol. 2009;27:986–93. doi: 10.1200/JCO.2008.16.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.