

Abstract
Parathyroid cancer is an uncommon malignancy and rare cause of primary hyperparathyroidism with a high morbidity and patient death in advanced cases usually resulting from intractable hypercalcemia. Inactivation of the HRPT2/CDC73 gene, encoding the putative tumor suppressor protein parafibromin and discovered in the context of the hyperparathyroidism-jaw tumor syndrome (HPT-JT), is a common, somatic genetic event in most parathyroid cancers. Some 25% of patients with apparently sporadic parathyroid cancer carry germline HRPT2/CDC73 mutation. Germline DNA analysis for HRPT2/CDC73 mutation is recommended in all patients with parathyroid cancer because of the potential benefit for offspring and other first-degree relatives. The histopathologic diagnosis of parathyroid cancer is non-specific unless vascular, lymphatic, capsular or soft tissue invasion are seen, or metastases are clinically evident. Immunohistochemical analysis of parathyroid tumors for loss of parafibromin expression offers promise as a diagnostic tool. En bloc tumor resection offers the highest chance of cure in patients with suspected parathyroid carcinoma. No adjuvant chemotherapy regimen has yet proven effective, and the role of local adjuvant radiotherapy is being evaluated. Metastatic disease can be palliated with surgical debulking. Medical therapy with the calcimimetic cinacalcet and bisphosphonates can ameliorate hypercalcemia in patients with inoperable disease.
Introduction
Parathyroid carcinoma is a rare malignancy and an uncommon cause of primary hyperparathyroidism (HPT). Although most parathyroid carcinomas secrete parathyroid hormone and cause hypercalcemia, a small fraction are non-functional. The malignant character of these tumors can be difficult to diagnose preoperatively, and is sometimes only recognized months to years later when the disease recurs. Parathyroid carcinoma is associated with higher serum calcium and PTH levels than primary HPT due to benign adenoma, and is more likely to be symptomatic at the time of presentation. Most of the histological features of parathyroid carcinoma are not specific, and the diagnosis may depend on demonstration of either local invasion, or metastases to regional lymph nodes or distant sites. The prevalence of parathyroid carcinoma is higher in kindreds with the hyperparathyroidism-jaw tumor (HPT-JT) syndrome. Germline mutation of the HRPT2/CDC73 gene can be demonstrated in most patients with HPT-JT as well as in about a quarter of patients with apparently sporadic parathyroid cancer. Somatic loss or mutation of HRPT2/CDC73 is present in the majority of sporadic parathyroid cancers as well.
The best opportunity for cure is complete surgical resection at the initial operation. Although findings of a fibrous capsule and tissue invasion may offer clues to the diagnosis, often the affected gland is grossly indistinguishable from a benign atypical adenoma. Adequate surgical approach is therefore dependent on pre-operative suspicion and the experience of the surgeon. Parathyroid carcinoma usually follows an indolent but progressive course, manifested by local recurrence initially and distant metastases later. Metastatic disease most commonly affects the lung or bone. The major morbidity of parathyroid carcinoma is due to complications of hypercalcemia, such as neuropsychiatric symptoms, cardiac arrhythmias, renal failure and pathologic fractures. Surgical resection of local recurrences and distant metastases is not curative, but may relieve symptoms and reduce serum calcium levels with effects lasting months to years depending on the extent of disease. Chemotherapy and radiation are generally not effective, but newer modalities such as transcatheter arterial embolization and radiofrequency ablation are promising palliative therapies in selected patients. Medical therapy with the calcimimetic cinacalcet and intravenous bisphosphonates are useful adjuncts for control of hypercalcemia.
Epidemiology and Demographics
Parathyroid carcinoma is a rare cause of primary HPT. Early single institution case series suggested that it was responsible for 2–5% of all cases,1–3 but these figures are likely overestimates resulting from referral and publication biases, and/or the use of variable diagnostic criteria. Data from the largest single-institution study, a comprehensive literature review and recent tumor registries indicate that it accounts for less than 1% of all cases in most of the world.4–6 The incidence of parathyroid carcinoma was 5.1% of cases of primary HPT in a national survey in Japan in 1981, indicating that there may be significant regional variation.7
It is an extraordinarily uncommon malignancy, with an annual incidence of approximately 3.5–5.7 per 10 million population.8 It accounted for 0.005% of all malignancies in the National Cancer Database from 1985–1995.9 Males account for approximately 50% of all cases.2,3,8,9 The mean age at diagnosis compiled from published reports is 44–48 years1–3 with a range of 12–90. Classically the disease is described as occurring about 10 years earlier than benign primary HPT, but this was not confirmed by the findings of the two largest registry studies to date, in which the mean age at diagnosis was between 54 and 56 years.8,9
Etiology and pathophysiology
The etiology of parathyroid cancer, like that of other malignancies, likely involves the interaction of multiple environmental and genetic factors. Exposure to radiation, especially during childhood, increases the risk of benign parathyroid disease10–12 as well as concurrent thyroid and parathyroid neoplasia,13–15 but whether such exposure plays an etiologic factor in parathyroid carcinoma remains unclear. Case reports and retrospective identification of several cases of parathyroid carcinoma in patients exposed to radiation have appeared in the literature in the last three decades but, because of the paucity of cases involved and the variable diagnostic criteria employed, a clear inference of causality is not possible.3,16–18
Recent understanding of the genetics of parathyroid cancer has resulted mainly from the clinical and molecular genetic characterization of HPT-JT, a rare autosomal dominant familial cancer syndrome in which affected individuals may develop primary HPT due to benign or malignant parathyroid tumors, cemento-ossifying fibrous tumors of the maxilla and/or mandible, and less commonly renal cysts or tumors and/or uterine tumors.19–22 Parathyroid carcinoma is present in approximately 15% of those with primary HPT due to HPT-JT. The trait has variable and incomplete penetrance since some 10% of gene carriers have no clinical manifestations (Fig. 1).
Figure 1.

The variable penetrance of germline HRPT2/CDC73 mutation results in a spectrum of different clinical phenotypes. Some 25% of patients with apparently sporadic parathyroid cancer have germline mutation of HRPT2/CDC73 (2), while 10% of known or obligate mutation carriers may be asymptomatic (1). Other manifestations of germline HRPT2/CDC73 mutation include familial isolated hyperparathyroidism, with or without individuals in the kindred with parathyroid cancer (3), and the hyperparathyroidism-jaw tumor syndrome with primary hyperparathyroidism due to benign adenomas (85%) or parathyroid cancer (15%), cemento-ossifying tumors of the maxilla or mandible, renal cysts or tumors, and uterine tumors (4).
In a majority of kindreds with classic HPT-JT manifestations an inactivating mutation of the HRPT2/CDC73 gene, located at 1q31, can be demonstrated.23 The HRPT2/CDC73 gene contains 17 exons that encode the protein parafibromin, a putative tumor suppressor protein consisting of 531 amino acids with weak homology to the yeast transcriptional regulatory protein CDC73p.24 Mutations are scattered throughout the coding region, and most are predicted to cause inactivation or premature truncation of the protein product.25 A subset of kindreds with familial isolated primary HPT (FIHP) also harbor germline HRPT2/CDC73 mutation indicating this condition may be a forme fruste of HPT-JT26–34 (Fig. 1).
Because of the high incidence of parathyroid carcinoma in HPT-JT and in germline HRPT2/CDC73 mutation-positive FIHP kindreds,26,31–33 mutation in HRPT2/CDC73 was investigated in sporadic cases of parathyroid cancer. Mutational analyses of tumors from patients with no family history and histologically confirmed parathyroid carcinoma demonstrates HRPT2/CDC73 somatic mutations in 60–100% of cases.35–37 In many parathyroid cancers analyzed, two distinct HRPT2/CDC73 mutations or a single mutation in combination with 1q31 LOH can be demonstrated, consistent with a tumor suppressor mechanism and providing evidence that HRPT2/CDC73 mutation is an early event in malignant transformation.35,37 Molecular genetic analysis of parathyroid carcinomas reveals mutations in HRPT2/CDC73 that are also present in the germline DNA in as many as 20–30% of cases, suggesting that a significant percentage of apparently sporadic carcinomas may, like a subset of FIHP, represent incomplete expressions of HPT-JT (Fig. 1).28,37,38 Because of the potential benefit for offspring and other first-degree relatives, germline DNA analysis for HRPT2/CDC73 mutation is recommended in all patients with parathyroid cancer.28,37,38
Biallelic mutational inactivation of HRPT2/CDC73 cannot be demonstrated in all parathyroid cancers, even when those tumors lack parafibromin expression, making epigenetic gene silencing by promoter hypermethylation or histone acetylation a potentially relevant mechanism: hypermethylation is not frequently observed however.39,40 Recent studies suggest that dysregulation of several microRNAs may contribute to the pathogenesis of parathyroid cancers harboring HRPT2/CDC73 mutation.41
Mutational inactivation of HRPT2/CDC73 appears to be a specific marker of parathyroid malignancy. Although reported, HRPT2/CDC73 mutation or 1q31 LOH in parathyroid adenomas is quite uncommon.23,35,42 Gene profiling studies43 and analysis of chromosomal imbalances in parathyroid tumors44 also suggest that the molecular changes leading to malignancy are distinct from those leading to benign parathyroid neoplasia.
Parafibromin, the product of the HRPT2/CDC73 gene, is a ubiquitously expressed protein whose function as a tumor suppressor is not well understood. It is the human homolog of the yeast Cdc73 protein which is a component of a transcriptional-regulatory Paf1 complex.24 Mammalian parafibromin is part of a homologous PAF1 complex and appears to have roles in RNA polymerase II-mediated gene transcription and histone methylation in the promoter and coding regions of specific genes.45 A posttranscriptional role for parafibromin in the maturation and processing of mRNA 3’-ends has also been proposed.46,47 A role for parafibromin in the repression of cyclin D1 expression has been explored48–50, but studies of parathyroid tumors found no correlation between HRPT2/CDC73 mutation or loss of parafibromin expression with cyclin D1 upregulation.51,52
Parathyroid carcinoma has been only rarely linked to the genes implicated in the familial HPT syndromes, apart from those syndromes associated with HRPT2/CDC73 mutation. Several cases of parathyroid carcinoma have been reported in multiple endocrine neoplasia type 1 (MEN1) patients both with53,54 and without55,56 documented germline MEN1 mutation. In addition somatic MEN1 mutation has been reported in a small subset of parathyroid carcinomas.38 Metastatic parathyroid carcinoma has been reported in a patient with multiple endocrine neoplasia type 2A (MEN2A) and a germline RET mutation.57
Alterations of other genes besides HRPT2/CDC73 have been studied for possible roles in the malignant transformation of parathyroid tissue. Loss of function of the retinoblastoma (RB1) tumor suppressor has been considered in the etiology of parathyroid cancer since a study that found LOH at the RB1 locus on chromosome 13q and/or abnormal RB1 expression in a majority of parathyroid cancers.58 In a follow-up study however, direct RB1 gene sequencing in parathyroid carcinomas failed to identify any somatic mutations suggesting RB1 loss was not a frequent event in the progression to parathyroid cancer.59 In other studies RB1 protein expression alone was not found to help distinguish benign from malignant parathyroid tumors,60 although it may be useful in conjunction with other markers.61,62 Somatic mutations in the exons representing the conserved regions of the P53 tumor suppressor gene were not found in parathyroid carcinomas,63,64 even though allelic loss at the P53 locus and/or abnormal P53 protein expression were found in a minority of parathyroid cancers.64
The loss or gain of specific regions of chromosomal DNA detected by techniques such as comparative genomic hybridization (CGH) suggests the existence of currently unidentified parathyroid tumor suppressors and oncogenes relevant to parathyroid cancer. Investigators have found loss of DNA on chromosomes 1p, 9p, 13q and/or 17 in malignant parathyroid tumors indicating the potential presence of novel parathyroid tumor suppressor genes at these loci.44,65 The presence of currently unknown oncogenes on chromosome 5 or at loci 1q, 19p, and Xq is suggested by the demonstration of specific chromosomal gain in parathyroid cancer.44,65
There have been several case reports of parathyroid carcinoma developing in the context of secondary or tertiary HPT due to chronic kidney disease (CKD) or other conditions. Several cases of parathyroid carcinoma in the context of CKD have had distant metastases documented by surgical pathologic analysis.66–68 Interestingly, the expression of HRPT2/CDC73 was preserved in the majority of primary and metastatic tumors from 5 patients with CKD-associated parathyroid carcinoma, suggesting a pathway to malignancy in this setting that does not involve biallelic HRPT2/CDC73 inactivation 69.
Clinical presentation of parathyroid cancer
The clinical presentation of parathyroid carcinoma is usually related to symptoms caused by the effects of markedly elevated serum PTH and hypercalcemia, rather than mass effects due to local infiltration or distant metastases. Systemic symptoms of hypercalcemia include fatigue, weakness, weight loss, anorexia, nausea, vomiting, polyuria, polydipsia and depression. Manifestations of hyperparathyroid bone disease include osteitis fibrosa cystica, subperiostial bone resorption, “salt and pepper” skull, absence of the lamina dura, diffuse osteopenia, osteoporosis, bone pain, or pathological fractures,70 and is present in in 22–91% of patients with parathyroid carcinoma at the time of diagnosis.1–4,6,71,72 Formerly a common manifestation of all types of primary HPT, bone involvement is now rare in benign disease, which is more commonly asymptomatic at presentation, and detected incidentally from routine laboratory studies.73 Renal complications such as nephrolithiasis, nephrocalcinosis, reduced glomerular filtration rate, or renal colic are diagnosed in 32–60% of parathyroid carcinoma patients.1–4,6,71,72 Renal involvement alone is not specific for malignancy. Concomitant bone and kidney disease is seen in 40-50% of patients at presentation,2,4,6 and is very unusual in benign primary HPT.70 Other associated clinical signs of HPT may also be seen in malignant disease, including anemia and peptic ulcer disease.70,74 Pancreatitis is reported in 4–6%.2,3,6,71 Carcinoma must be considered in the differential diagnosis of any patient presenting with acute primary HPT or parathyroid crisis.70 Almost unheard of prior to the advent of the multichannel chemistry autoanalyzer in 1974, asymptomatic disease at presentation has been reported in about 2% of patients with parathyroid cancer in the larger subsequent literature reviews,2,3,6 and may actually be increasing in frequency. Two more recent single-institution series reported asymptomatic presentation of parathyroid cancer in 7 and 30% of their patients.4,71 Few signs on physical examination are specific to differentiate parathyroid carcinoma from benign adenoma or multigland disease. A palpable neck mass, which is reported in 15–50% of patients with parathyroid carcinoma is extremely rare in benign conditions.1–3,6,71,72 Hoarseness can be a sign of recurrent laryngeal nerve palsy due to local invasion. It is very uncommon in benign primary HPT, and highly suggestive of malignancy.70
Laboratory testing and imaging in parathyroid cancer
No single laboratory finding is diagnostic of parathyroid carcinoma, but certain findings may offer clues that it ought to be included in the differential diagnosis of a patient presenting with primary HPT. The mean serum calcium in parathyroid cancer patients reported in the last three decades is 14.6–15.0 mg/dL3,4,72 and about 60–65% of patients present with a calcium level greater than 14 mg/dL.3,4,72 A subset of functional parathyroid cancers demonstrate normal serum calcium despite elevations of PTH at the time of diagnosis.75 Intact PTH levels in parathyroid cancer are on average 5–10 times the upper limit of the normal range,2–4,72,76 but no threshold level for malignancy has been defined. Alkaline phosphatase is usually elevated, and serum phosphorus is often low normal, but can be below the normal reference range. Severe primary HPT associated with benign disease is uncommon, but it can be difficult to distinguish on clinical grounds from parathyroid carcinoma.
Identification of new laboratory markers that may help differentiate malignant from benign primary HPT is an ongoing area of research. Elevated plasma human chorionic gonadotropin (HCG) has been documented in parathyroid carcinoma,77 but also can be seen in benign primary HPT. Urinary HCG, specifically the hyperglycosylated HCG fraction, is elevated in some patients with parathyroid carcinoma, but normal in benign disease.78 The recently identified N-terminal PTH immunoreactive species, less hydrophobic than intact PTH 1-84 on reverse-phase liquid chromatography, is overproduced in some parathyroid cancers.79
Preoperative imaging is frequently helpful for tumor localization but in the evaluation of primary parathyroid tumors cannot reliably distinguish benign from malignant disease. Ultrasonographic features of parathyroid tumors including inhomogeneity, hypoechogenicity, and irregularity of borders have been reported to correlate with malignancy (Fig. 2),80 but such characteristics are not always present in parathyroid cancers.81 Fine needle aspiration of cervical masses should be avoided in patients with suspected parathyroid cancer because of the documented risk of cutaneous or subcutaneous seeding along the needle track.82,83
Figure 2.
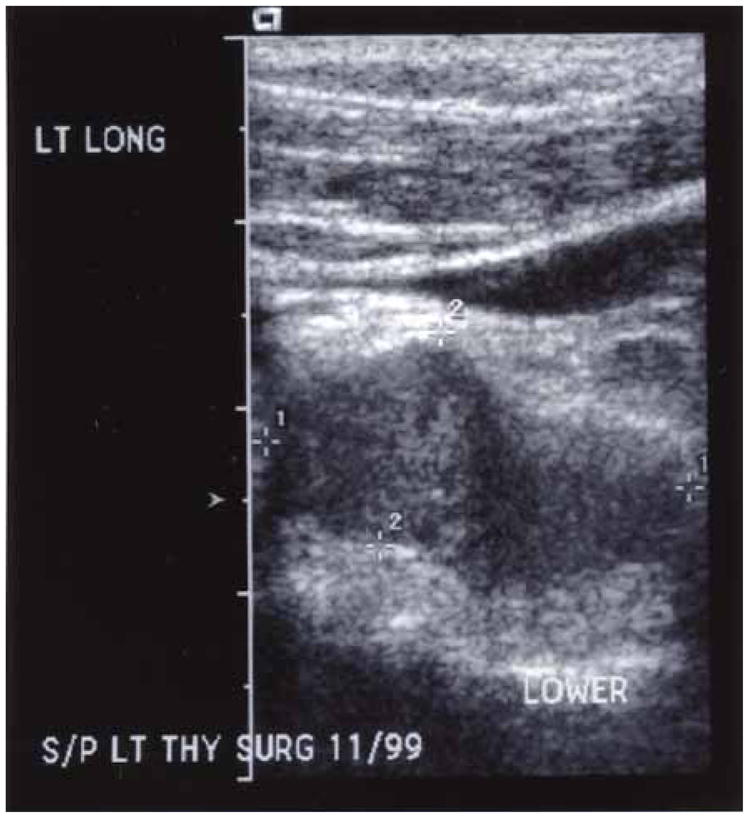
Neck ultrasound in a patient with recurrent parathyroid carcinoma. The lesion shows typical features associated with parathyroid carcinoma (inhomogeneity, hypoechogenicity, and irregularity of borders).
Surgical findings
The diagnosis of parathyroid carcinoma is often suspected due to characteristic findings at the time of initial surgery. The tumor usually has a firm or hard consistency, is sometimes lobulated, and is usually surrounded by a dense fibrous capsule that gives it a white, grey-white or grey-brown hue. Parathyroid carcinoma often adheres to and infiltrates adjacent structures.1,84,85 The most common sites of local invasion are the ipsilateral thyroid gland, strap muscles, ipsilateral recurrent laryngeal nerve, esophagus and trachea.6 Lymph node metastases is present at the initial operation in 3–19%,3,6,8,9,72 and distant metastases are present in 3–4%.6,8,71
Frozen section is not helpful to distinguish benign from malignant disease, and excisional biopsy is not recommended due to the risk of intraoperative seeding of tissue leading to parathyromatosis.3 In some cases, carcinoma may be difficult to differentiate from adenoma,86 which is usually of soft consistency, round or oval and reddish brown in color.70 Surgeons do not recognize the presence of cancer in some 25% of cases.84 Although exceedingly rare, carcinoma of multiple glands has been documented in 3 cases in the literature, emphasizing the importance of four-gland exploration at the initial operation to avoid a reoperation for missed disease.87–89
Diagnosis
In the absence of obvious metastatic disease the distinction between parathyroid carcinoma and adenoma is difficult utilizing microscopic histopathologic criteria. The principal histologic features that distinguish parathyroid carcinoma from adenoma, identified by Schantz and Castleman based on their examination of 70 parathyroid cancers, were the presence of parenchymal mitoses, trabeculated parenchyma including often thick fibrous bands, and capsular or vascular invasion.90 Because any of these individual features can also be found in atypical parathyroid adenomas, capsular or lymphovascular invasion remains the most specific histopathologic feature of primary parathyroid cancer (Fig. 3).
Figure 3.
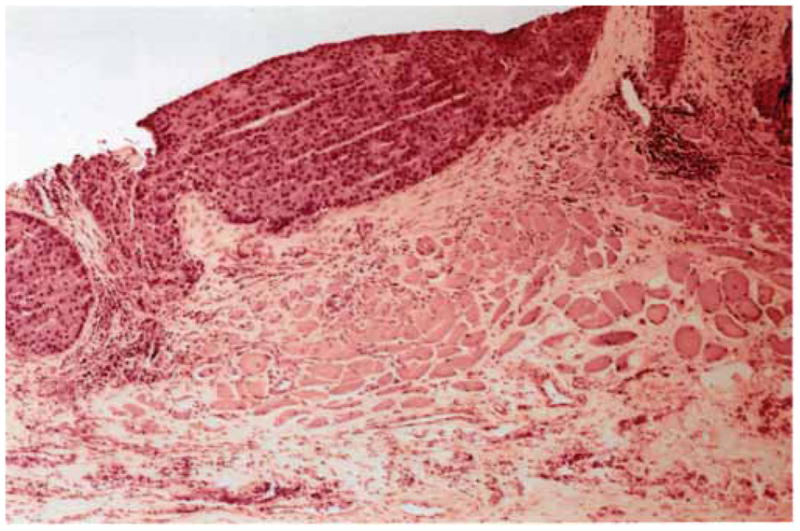
Hematoxylin and eosin histologic section of parathyroid carcinoma invading adjacent soft tissue and muscle (4X magnification).
Because of the difficulty of distinguishing benign from malignant parathyroid tumor based on morphological criteria alone, considerable effort has been directed at identifying immunohistochemical tumor markers useful to this end. Given the strong evidence linking HRPT2/CDC73 inactivation with parathyroid malignancy, the most promising marker to date has been parafibromin, either used alone or in conjunction with other markers. The original report found that loss of nuclear expression of parafibromin could identify definite parathyroid malignancies with 96% sensitivity and 99% specificity and noted that absent nuclear staining also characterized HPT-JT associated adenomas.91 Subsequent investigators have also found loss of parafibromin expression to be a highly useful marker of parathyroid malignancy, although with generally lower specificity and sensitivity.52,92,93 Clark and co-workers found that the combination of markers that includes loss of parafibromin and RB1 expression and overexpression of galectin-3 was most specific for parathyroid cancers.62 Strong staining for protein gene product 9.5 was found to be at least as sensitive and specific for parathyroid malignancy as complete loss of parafibromin nuclear expression and was additionally positive in an HPT-JT-associated parathyroid tumor expressing a missense mutant of parafibromin.94
The cell proliferative marker Ki-67 (target of the MIB-1 monoclonal antibody), often in conjunction with other antigens, has proven useful in some laboratories to identify a subset of parathyroid cancers. Several investigators noted a uniquely high proliferative rate and Ki-67 labeling index in parathyroid malignancies and proposed a cutoff point that helps to distinguish parathyroid cancers,61,95 although others challenge the diagnostic utility of the Ki-67 labeling index as a sole criterion because of significant overlap with adenomas.60 The combination of other markers with the Ki-67 labeling index, including underexpression of p27Kip196 or CaSR,97 or galectin-3 overexpression98 may improve its predictive value. The Ki-67 index may also be useful as a prognostic factor for parathyroid cancer recurrence.99
Recently loss of the Wnt pathway components adenomatous polyposis coli (APC) and glycogen synthase kinase 3-β were found to be specific markers of parathyroid malignancy, with lost APC immunoreactivity reported to be 100% specific and 75% sensitive for the detection of parathyroid cancer.100
Non-functional parathyroid carcinoma
Non-functional parathyroid carcinoma is extremely rare. Fewer than 20 cases have been reported in the literature since 1929,101 including a case in the context of FIHP with germline mutation of HRPT2/CDC73.31 Patients are normocalcemic at diagnosis and, in all those in whom it was measured, PTH is within the normal range. About 80% present with a neck mass, and other common findings at presentation include dysphagia, hoarseness or vocal cord paralysis, and dyspnea.101,102 Grossly, tumors are usually described as similar in appearance to functional parathyroid carcinoma, and they also appear similar histologically.102 Tumor immunohistochemistry demonstrating the presence of PTH, and absence of thyroglobulin, thyroid transcription factor 1 and calcitonin may help ascertain the diagnosis.74,103,104
Treatment of parathyroid cancer: surgical
Most authors recommend en bloc resection at the initial surgery to offer the best chance for cure. This approach requires recognition of the possibility of malignancy during exploration. Adequate surgical excision requires removal of the ipsilateral thyroid lobe and isthmus, skeletonization of the trachea, and excision of any skeletal muscle intimately related to the tumor. The surgeon must be careful not to rupture the capsule of the tumor to avoid seeding of the surgical field.1 If the recurrent laryngeal nerve is involved and not functioning, it should be resected. Central neck and ipsilateral modified functional radical neck resection are usually performed only if enlarged or abnormal appearing lymph nodes are found to be involved on frozen section analysis.3,84
Because of the rarity of the disease and the infrequency of pre-operative diagnosis, no prospective data exists regarding the initial surgical approach. Although en bloc resection is recommended, in practice it is the first surgical procedure performed in only about 12% of cases.8 While some retrospective case series and reviews support the contention that en bloc surgical resection at the initial operation decreases recurrence,3,4,6,7,76,105 this finding has not been corroborated by others.8,71 Up to 86% of patients may have inadequate surgical resection at the initial operation.9
With persistent and recurrent disease, it is important to exclude metastases to the lung or bone before proceeding to repeat neck exploration. Prior to neck exploration, at least two non-invasive localizing studies should be performed. If non-invasive studies are negative or equivocal, selective venous catheterization with PTH sampling should be pursued to confirm the finding of a non-invasive study.84 Patients should also have a direct laryngoscopy to evaluate their vocal cord function before reoperation for persistent and recurrent disease.
Following preoperative localization, treatment of locally recurrent disease, although not expected to be curative, is successful in improving symptoms and lowering serum calcium levels in 68–86%.4,76,99 Subsequent re-operations generally have lower success rates, but may result in successful palliation of selected patients. Because of the rarity of the disease and its indolent nature, survival benefit has not been definitively demonstrated with re-operation. Complications of both initial and repeat neck operations include recurrent laryngeal nerve injury, transient and permanent hypoparathyroidism, esophageal or tracheal injury and neck hematoma.84
Multiple authors have reported palliative effects of resection of distant metastases, particularly in the lung1,106 and bone.107 If metastatic disease is localized, resection reduces tumor burden, improves symptoms and may lower or even normalize serum calcium and PTH.108,109 With widely metastatic parathyroid carcinoma, reoperation is not likely to improve symptoms or reduce the serum calcium.84
Treatment of parathyroid cancer: radiotherapy
Parathyroid carcinoma is generally not believed to be radiosensitive, and radiation treatment as primary therapy has not been demonstrated to have a significant effect in either the neck or at sites of distant metastases. Several case reports, however, have suggested that adjuvant radiation treatment after surgical treatment may reduce the chance of local recurrence.4,71 It is difficult to know, however, the efficacy of adjuvant radiotherapy as some patients with parathyroid cancer may have long disease-free intervals after parathyroidectomy alone.
Treatment of parathyroid cancer: other modalities
Unresectable disease due to diffuse metastases in the lung has been treated with multiple sessions of radiofrequency ablation (RFA) in a patient with parathyroid carcinoma.110 A combination of RFA and transcatheter arterial embolization has been used similarly to treat multiple metastatic lesions in the liver in a single patient.111 Improvement in both serum calcium and PTH levels were demonstrated following treatment in both reports. Recurrent disease in the neck has been treated with ultrasound-guided percutaneous alcohol injection and short-term improvements in calcium and PTH levels were achieved.112
In general, chemotherapy has not been demonstrated to be beneficial in parathyroid carcinoma. Short-term responses have been demonstrated with single-agent dacarbazine113 and combination chemotherapy regimens of fluorouracil, cyclophosphamide, and dacarbazine114 in patients with metastatic disease, but a survival benefit was not demonstrated.
Because the major morbidity and ultimate cause of death in most patients with parathyroid carcinoma is severe hypercalcemia, medical management has focused on controlling calcium levels in patients with persistent disease. Acute hypercalcemia is treated with standard therapies such as intravenous hydration, furosemide, calcitonin, glucocorticoids, mithramycin, and hemodialysis, but these treatments are ineffective for long-term management, with effects lasting days to several months at best. Short-term decreases in serum calcium have been demonstrated with the intravenous bisphosphonates pamidronate115 and zoledronate.116 Initial treatment appears to be clinically significant in some patients, but the effect generally diminishes with subsequent infusions. Oral bisphosphonates have not been reported to be effective in the management of hypercalcemia related to parathyroid carcinoma.
Calcimimetics have emerged as a more effective solution to mitigate the hypercalcemia of parathyroid cancer. After promising results with a first-generation calcimimetic,117 a second-generation calcimimetic, cinacalcet, was shown to decrease serum calcium by 1 mg/dL in 62% of patients enrolled in an open-label, single-arm trial. Patients tolerated total daily doses up to 360 mg. In responders, the magnitude of decrease in calcium levels was greatest in those with the highest baseline calcium levels. Interestingly, decreases in serum calcium were achieved despite no significant decrease in serum PTH.118
Immunization with synthetic human and bovine PTH peptides resulted in production of anti-PTH antibodies and decrease in the serum calcium of more than 1 mmol/L in a patient with parathyroid cancer and unresectable metastases.119 Tumor shrinkage, in addition to an improvement in serum calcium and PTH, has been demonstrated in another patient.120
It is likely that denosumab, a monoclonal antibody targeting the receptor activator of nuclear factor kappa-b ligand with potent anti-resorptive actions in bone, will be tested for efficacy in controlling the hypercalcemia of parathyroid cancer.121
Natural history of parathyroid cancer
Following initial surgery, anywhere from 25–80% of patients with parathyroid cancer develop local recurrence. Recurrence is detected on average 2–4 years after the initial operation, and these patients have a median survival on the order of 5–6 years after the initial diagnosis.2–4,6,71,76,122 Approximately 25% of patients develop distant metastases at some point during the disease. Rarely, they are present at the time of diagnosis. At the other extreme, metastases have been documented as long as 20 years after the original diagnosis following a period of quiescence.123 Five year survival rates from case series and registry data of the last 20 years are fairly consistent at 76–85%, and 10 year survival rates range anywhere from 49–77%.8,9,71,76 The most compelling data regarding treatment comes from reports of complete surgical (en bloc) resection at the initial operation. Anywhere from 28–50% of these patients remain alive with no recurrent disease at follow-up.3,4,6,105 Although modalities such as surgery, adjuvant radiation, RFA, and calcimimetics have demonstrated responses in clinical parameters, insufficient information is available to determine the effects of these therapies on survival. The prognosis appears to be worse in patients with non-functional parathyroid carcinoma since local invasion and distant metastases appear to be more likely at the time of diagnosis.101,102
Conclusions
Parathyroid carcinoma is a rare endocrine malignancy whose recognition requires a high index of suspicion based on clinical features, such as a palpable neck mass and severe primary HPT. Histologic features can be non-specific. Diagnosis is definitive when local invasion, lymph node or distant metastases are demonstrated. Somatic mutation of HRPT2/CDC73 is a common finding in sporadic parathyroid carcinomas, and because some 25% of apparently sporadic cases harbor germline HRPT2/CDC73 mutation, DNA analysis should be offered to all patients. Pre-operative suspicion is crucial since parathyroid carcinoma may be difficult to distinguish from benign adenoma intraoperatively and en bloc resection is potentially curative. Adjuvant radiation therapy following surgical resection is an area of ongoing investigation. Local recurrence is common with possible late occurrence of distant metastases, especially to lung and bone. Surgical resection, arterial embolization, or radiofrequency ablation of persistent or recurrent disease is rarely curative, but may reduce serum calcium. Cinacalcet and intravenous bisphosphonates are the most effective medical treatments currently available for the hypercalcemia of recurrent or persistent parathyroid cancer.
Acknowledgments
The authors thank Dr. Stephen J. Marx for his critical reading of the manuscript, and our colleagues Drs. Lee S. Weinstein, Monica C. Skarulis and Sunita K. Agarwal of the National Institute of Diabetes and Digestive and Kidney Diseases for their ongoing support and encouragement. The Intramural Research Program of the National Cancer Institute and the National Institute of Diabetes and Digestive and Kidney Diseases supported this research.
Financial Support: The Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases and the National Cancer Institute supported this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holmes EC, Morton DL, Ketcham AS. Parathyroid carcinoma: a collective review. Ann Surg. 1969;169:631–40. doi: 10.1097/00000658-196904000-00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shane E, Bilezikian JP. Parathyroid carcinoma: a review of 62 patients. Endocr Rev. 1982;3:218–26. doi: 10.1210/edrv-3-2-218. [DOI] [PubMed] [Google Scholar]
- 3.Obara T, Fujimoto Y. Diagnosis and treatment of patients with parathyroid carcinoma: an update and review. World J Surg. 1991;15:738–44. doi: 10.1007/BF01665308. [DOI] [PubMed] [Google Scholar]
- 4.Wynne AG, van Heerden J, Carney JA, Fitzpatrick LA. Parathyroid carcinoma: clinical and pathologic features in 43 patients. Medicine (Baltimore) 1992;71:197–205. [PubMed] [Google Scholar]
- 5.Ruda JM, Hollenbeak CS, Stack BC., Jr A systematic review of the diagnosis and treatment of primary hyperparathyroidism from 1995 to 2003. Otolaryngol Head Neck Surg. 2005;132:359–72. doi: 10.1016/j.otohns.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Koea JB, Shaw JH. Parathyroid cancer: biology and management. Surg Oncol. 1999;8:155–65. doi: 10.1016/s0960-7404(99)00037-7. [DOI] [PubMed] [Google Scholar]
- 7.Fujimoto Y, Obara T, Ito Y, Kanazawa K, Aiyoshi Y, Nobori M. Surgical treatment of ten cases of parathyroid carcinoma: importance of an initial en bloc tumor resection. World J Surg. 1984;8:392–400. doi: 10.1007/BF01655086. [DOI] [PubMed] [Google Scholar]
- 8.Lee PK, Jarosek SL, Virnig BA, Evasovich M, Tuttle TM. Trends in the incidence and treatment of parathyroid cancer in the United States. Cancer. 2007;109:1736–41. doi: 10.1002/cncr.22599. [DOI] [PubMed] [Google Scholar]
- 9.Hundahl SA, Fleming ID, Fremgen AM, Menck HR. Two hundred eighty-six cases of parathyroid carcinoma treated in the U.S. between 1985–1995: a National Cancer Data Base Report. The American College of Surgeons Commission on Cancer and the American Cancer Society. Cancer. 1999;86:538–44. doi: 10.1002/(sici)1097-0142(19990801)86:3<538::aid-cncr25>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 10.Schneider AB, Gierlowski TC, Shore-Freedman E, Stovall M, Ron E, Lubin J. Dose-response relationships for radiation-induced hyperparathyroidism. The Journal of clinical endocrinology and metabolism. 1995;80:254–7. doi: 10.1210/jcem.80.1.7829622. [DOI] [PubMed] [Google Scholar]
- 11.Holmberg E, Wallgren A, Holm LE, Lundell M, Karlsson P. Dose-response relationship for parathyroid adenoma after exposure to ionizing radiation in infancy. Radiat Res. 2002;158:418–23. doi: 10.1667/0033-7587(2002)158[0418:drrfpa]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 12.Rasmuson T, Damber L, Johansson L, Johansson R, Larsson LG. Increased incidence of parathyroid adenomas following X-ray treatment of benign diseases in the cervical spine in adult patients. Clinical endocrinology. 2002;57:731–4. doi: 10.1046/j.1365-2265.2002.01616.x. [DOI] [PubMed] [Google Scholar]
- 13.Tezelman S, Rodriguez JM, Shen W, Siperstein AE, Duh QY, Clark OH. Primary hyperparathyroidism in patients who have received radiation therapy and in patients who have not received radiation therapy. J Am Coll Surg. 1995;180:81–7. [PubMed] [Google Scholar]
- 14.McMullen T, Bodie G, Gill A, Ihre-Lundgren C, Shun A, Bergin M, et al. Hyperparathyroidism after irradiation for childhood malignancy. Int J Radiat Oncol Biol Phys. 2009;73:1164–8. doi: 10.1016/j.ijrobp.2008.06.1487. [DOI] [PubMed] [Google Scholar]
- 15.Katz A, Braunstein GD. Clinical, biochemical, and pathologic features of radiation-associated hyperparathyroidism. Arch Intern Med. 1983;143:79–82. [PubMed] [Google Scholar]
- 16.Christmas TJ, Chapple CR, Noble JG, Milroy EJ, Cowie AG. Hyperparathyroidism after neck irradiation. Br J Surg. 1988;75:873–4. doi: 10.1002/bjs.1800750914. [DOI] [PubMed] [Google Scholar]
- 17.Ireland JP, Fleming SJ, Levison DA, Cattell WR, Baker LR. Parathyroid carcinoma associated with chronic renal failure and previous radiotherapy to the neck. J Clin Pathol. 1985;38:1114–8. doi: 10.1136/jcp.38.10.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mashburn MA, Chonkich GD, Chase DR, Petti GH., Jr Parathyroid carcinoma: two new cases--diagnosis, therapy, and treatment. Laryngoscope. 1987;97:215–8. doi: 10.1288/00005537-198702000-00015. [DOI] [PubMed] [Google Scholar]
- 19.Jackson CE, Norum RA, Boyd SB, Talpos GB, Wilson SD, Taggart RT, et al. Hereditary hyperparathyroidism and multiple ossifying jaw fibromas: a clinically and genetically distinct syndrome. Surgery. 1990;108:1006–12. [PubMed] [Google Scholar]
- 20.Bradley KJ, Hobbs MR, Buley ID, Carpten JD, Cavaco BM, Fares JE, et al. Uterine tumours are a phenotypic manifestation of the hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2005;257:18–26. doi: 10.1111/j.1365-2796.2004.01421.x. [DOI] [PubMed] [Google Scholar]
- 21.Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2003;253:634–42. doi: 10.1046/j.1365-2796.2003.01168.x. [DOI] [PubMed] [Google Scholar]
- 22.Mallette LE, Malini S, Rappaport MP, Kirkland JL. Familial cystic parathyroid adenomatosis. Ann Intern Med. 1987;107:54–60. doi: 10.7326/0003-4819-107-1-54. [DOI] [PubMed] [Google Scholar]
- 23.Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nature genetics. 2002;32:676–80. doi: 10.1038/ng1048. [DOI] [PubMed] [Google Scholar]
- 24.Shi X, Chang M, Wolf AJ, Chang CH, Frazer-Abel AA, Wade PA, et al. Cdc73p and Paf1p are found in a novel RNA polymerase II-containing complex distinct from the Srbp-containing holoenzyme. Mol Cell Biol. 1997;17:1160–9. doi: 10.1128/mcb.17.3.1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newey PJ, Bowl MR, Thakker RV. Parafibromin--functional insights. J Intern Med. 2009;266:84–98. doi: 10.1111/j.1365-2796.2009.02107.x. [DOI] [PubMed] [Google Scholar]
- 26.Simonds WF, Robbins CM, Agarwal SK, Hendy GN, Carpten JD, Marx SJ. Familial isolated hyperparathyroidism is rarely caused by germline mutation in HRPT2, the gene for the hyperparathyroidism-jaw tumor syndrome. J Clin Endocrinol Metab. 2004;89:96–102. doi: 10.1210/jc.2003-030675. [DOI] [PubMed] [Google Scholar]
- 27.Villablanca A, Calender A, Forsberg L, Hoog A, Cheng JD, Petillo D, et al. Germline and de novo mutations in the HRPT2 tumour suppressor gene in familial isolated hyperparathyroidism (FIHP) J Med Genet. 2004;41:e32. doi: 10.1136/jmg.2003.012369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab. 2004;89:5583–91. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 29.Bradley KJ, Cavaco BM, Bowl MR, Harding B, Young A, Thakker RV. Utilisation of a cryptic non-canonical donor splice site of the gene encoding PARAFIBROMIN is associated with familial isolated primary hyperparathyroidism. J Med Genet. 2005;42:e51. doi: 10.1136/jmg.2005.032201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradley KJ, Cavaco BM, Bowl MR, Harding B, Cranston T, Fratter C, et al. Parafibromin mutations in hereditary hyperparathyroidism syndromes and parathyroid tumours. Clinical endocrinology. 2006;64:299–306. doi: 10.1111/j.1365-2265.2006.02460.x. [DOI] [PubMed] [Google Scholar]
- 31.Guarnieri V, Scillitani A, Muscarella LA, Battista C, Bonfitto N, Bisceglia M, et al. Diagnosis of parathyroid tumors in familial isolated hyperparathyroidism with HRPT2 mutation: implications for cancer surveillance. J Clin Endocrinol Metab. 2006;91:2827–32. doi: 10.1210/jc.2005-1239. [DOI] [PubMed] [Google Scholar]
- 32.Mizusawa N, Uchino S, Iwata T, Tsuyuguchi M, Suzuki Y, Mizukoshi T, et al. Genetic analyses in patients with familial isolated hyperparathyroidism and hyperparathyroidism-jaw tumour syndrome. Clinical endocrinology. 2006;65:9–16. doi: 10.1111/j.1365-2265.2006.02534.x. [DOI] [PubMed] [Google Scholar]
- 33.Kelly TG, Shattuck TM, Reyes-Mugica M, Stewart AF, Simonds WF, Udelsman R, et al. Surveillance for early detection of aggressive parathyroid disease: carcinoma and atypical adenoma in familial isolated hyperparathyroidism associated with a germline HRPT2 mutation. J Bone Miner Res. 2006;21:1666–71. doi: 10.1359/jbmr.060702. [DOI] [PubMed] [Google Scholar]
- 34.Masi G, Barzon L, Iacobone M, Viel G, Porzionato A, Macchi V, et al. Clinical, genetic, and histopathologic investigation of CDC73-related familial hyperparathyroidism. Endocr Relat Cancer. 2008;15:1115–26. doi: 10.1677/ERC-08-0066. [DOI] [PubMed] [Google Scholar]
- 35.Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, Chen J, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. Journal of medical genetics. 2003;40:657–63. doi: 10.1136/jmg.40.9.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, Cianferotti L, et al. Genetic analyses of the HRPT2 gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. The Journal of clinical endocrinology and metabolism. 2004;89:5583–91. doi: 10.1210/jc.2004-0294. [DOI] [PubMed] [Google Scholar]
- 37.Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–9. doi: 10.1056/NEJMoa031237. [DOI] [PubMed] [Google Scholar]
- 38.Haven CJ, van Puijenbroek M, Tan MH, Teh BT, Fleuren GJ, van Wezel T, et al. Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clinical endocrinology. 2007;67:370–6. doi: 10.1111/j.1365-2265.2007.02894.x. [DOI] [PubMed] [Google Scholar]
- 39.Hahn MA, Howell V, Gill A, Clarkson A, Weaire-Buchanan G, Robinson B, et al. CDC73/HRPT2 CpG island hypermethylation and mutation of 5'-untranslated sequence are uncommon mechanisms of silencing parafibromin in parathyroid tumors. Endocr Relat Cancer. 2009 doi: 10.1677/ERC-09-0291. [DOI] [PubMed] [Google Scholar]
- 40.Hewitt KM, Sharma PK, Samowitz W, Hobbs M. Aberrant methylation of the HRPT2 gene in parathyroid carcinoma. Ann Otol Rhinol Laryngol. 2007;116:928–33. doi: 10.1177/000348940711601210. [DOI] [PubMed] [Google Scholar]
- 41.Corbetta S, Vaira V, Guarnieri V, Scillitani A, Eller-Vainicher C, Ferrero S, et al. Differential expression of microRNAs in human parathyroid carcinomas compared with normal parathyroid tissue. Endocr Relat Cancer. 2009 doi: 10.1677/ERC-09-0134. [DOI] [PubMed] [Google Scholar]
- 42.Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. The Journal of clinical endocrinology and metabolism. 2005;90:5015–7. doi: 10.1210/jc.2005-0717. [DOI] [PubMed] [Google Scholar]
- 43.Haven CJ, Howell VM, Eilers PH, Dunne R, Takahashi M, van Puijenbroek M, et al. Gene expression of parathyroid tumors: molecular subclassification and identification of the potential malignant phenotype. Cancer Res. 2004;64:7405–11. doi: 10.1158/0008-5472.CAN-04-2063. [DOI] [PubMed] [Google Scholar]
- 44.Kytölä S, Farnebo F, Obara T, Isola J, Grimelius L, Farnebo LO, et al. Patterns of chromosomal imbalances in parathyroid carcinomas. Am J Pathol. 2000;157:579–86. doi: 10.1016/S0002-9440(10)64568-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, et al. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Molecular and cellular biology. 2005;25:612–20. doi: 10.1128/MCB.25.2.612-620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farber LJ, Kort EJ, Wang P, Chen J, Teh BT. The tumor suppressor parafibromin is required for posttranscriptional processing of histone mRNA. Mol Carcinog. 2009 doi: 10.1002/mc.20591. [DOI] [PubMed] [Google Scholar]
- 47.Rozenblatt-Rosen O, Nagaike T, Francis JM, Kaneko S, Glatt KA, Hughes CM, et al. The tumor suppressor Cdc73 functionally associates with CPSF and CstF 3' mRNA processing factors. Proc Natl Acad Sci U S A. 2009;106:755–60. doi: 10.1073/pnas.0812023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodard GE, Lin L, Zhang JH, Agarwal SK, Marx SJ, Simonds WF. Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene. 2005;24:1272–6. doi: 10.1038/sj.onc.1208274. [DOI] [PubMed] [Google Scholar]
- 49.Yang YJ, Han JW, Youn HD, Cho EJ. The tumor suppressor, parafibromin, mediates histone H3 K9 methylation for cyclin D1 repression. Nucleic acids research. 2010;38:382–90. doi: 10.1093/nar/gkp991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao J, Yart A, Frigerio S, Perren A, Schraml P, Weisstanner C, et al. Sporadic human renal tumors display frequent allelic imbalances and novel mutations of the HRPT2 gene. Oncogene. 2007;26:3440–9. doi: 10.1038/sj.onc.1210131. [DOI] [PubMed] [Google Scholar]
- 51.Juhlin C, Larsson C, Yakoleva T, Leibiger I, Leibiger B, Alimov A, et al. Loss of parafibromin expression in a subset of parathyroid adenomas. Endocr Relat Cancer. 2006;13:509–23. doi: 10.1677/erc.1.01058. [DOI] [PubMed] [Google Scholar]
- 52.Cetani F, Ambrogini E, Viacava P, Pardi E, Fanelli G, Naccarato AG, et al. Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol. 2007;156:547–54. doi: 10.1530/EJE-06-0720. [DOI] [PubMed] [Google Scholar]
- 53.Shih RY, Fackler S, Maturo S, True MW, Brennan J, Wells D. Parathyroid carcinoma in multiple endocrine neoplasia type 1 with a classic germline mutation. Endocr Pract. 2009;15:567–72. doi: 10.4158/EP09045.CRR1. [DOI] [PubMed] [Google Scholar]
- 54.Sato M, Miyauchi A, Namihira H, Bhuiyan MM, Imachi H, Murao K, et al. A newly recognized germline mutation of MEN1 gene identified in a patient with parathyroid adenoma and carcinoma. Endocrine. 2000;12:223–6. doi: 10.1385/ENDO:12:3:223. [DOI] [PubMed] [Google Scholar]
- 55.Agha A, Carpenter R, Bhattacharya S, Edmonson SJ, Carlsen E, Monson JP. Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: two case reports of an unrecognised entity. J Endocrinol Invest. 2007;30:145–9. doi: 10.1007/BF03347413. [DOI] [PubMed] [Google Scholar]
- 56.Dionisi S, Minisola S, Pepe J, De Geronimo S, Paglia F, Memeo L, et al. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc. 2002;77:866–9. doi: 10.4065/77.8.866. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins PJ, Satta MA, Simmgen M, Drake WM, Williamson C, Lowe DG, et al. Metastatic parathyroid carcinoma in the MEN2A syndrome. Clinical endocrinology. 1997;47:747–51. doi: 10.1046/j.1365-2265.1997.3421147.x. [DOI] [PubMed] [Google Scholar]
- 58.Cryns VL, Thor A, Xu HJ, Hu SX, Wierman ME, Vickery AL, Jr, et al. Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med. 1994;330:757–61. doi: 10.1056/NEJM199403173301105. [DOI] [PubMed] [Google Scholar]
- 59.Shattuck TM, Kim TS, Costa J, Yandell DW, Imanishi Y, Palanisamy N, et al. Mutational analyses of RB and BRCA2 as candidate tumour suppressor genes in parathyroid carcinoma. Clinical endocrinology. 2003;59:180–9. doi: 10.1046/j.1365-2265.2003.01814.x. [DOI] [PubMed] [Google Scholar]
- 60.Farnebo F, Auer G, Farnebo LO, Teh BT, Twigg S, Aspenblad U, et al. Evaluation of retinoblastoma and Ki-67 immunostaining as diagnostic markers of benign and malignant parathyroid disease. World J Surg. 1999;23:68–74. doi: 10.1007/s002689900567. [DOI] [PubMed] [Google Scholar]
- 61.Vargas MP, Vargas HI, Kleiner DE, Merino MJ. The role of prognostic markers (MiB-1, RB, and bcl-2) in the diagnosis of parathyroid tumors. Mod Pathol. 1997;10:12–7. [PubMed] [Google Scholar]
- 62.Fernandez-Ranvier GG, Khanafshar E, Tacha D, Wong M, Kebebew E, Duh QY, et al. Defining a molecular phenotype for benign and malignant parathyroid tumors. Cancer. 2009;115:334–44. doi: 10.1002/cncr.24037. [DOI] [PubMed] [Google Scholar]
- 63.Hakim JP, Levine MA. Absence of p53 point mutations in parathyroid adenoma and carcinoma. The Journal of clinical endocrinology and metabolism. 1994;78:103–6. doi: 10.1210/jcem.78.1.8288693. [DOI] [PubMed] [Google Scholar]
- 64.Cryns VL, Rubio MP, Thor AD, Louis DN, Arnold A. p53 abnormalities in human parathyroid carcinoma. The Journal of clinical endocrinology and metabolism. 1994;78:1320–4. doi: 10.1210/jcem.78.6.8200932. [DOI] [PubMed] [Google Scholar]
- 65.Agarwal SK, Schrock E, Kester MB, Burns AL, Heffess CS, Ried T, et al. Comparative genomic hybridization analysis of human parathyroid tumors. Cancer Genet Cytogenet. 1998;106:30–6. doi: 10.1016/s0165-4608(98)00049-1. [DOI] [PubMed] [Google Scholar]
- 66.Tominaga Y, Numano M, Uchida K, Sato K, Asano H, Haba T, et al. Lung metastasis from parathyroid carcinoma causing recurrent renal hyperparathyroidism in a hemodialysis patient: report of a case. Surg Today. 1995;25:984–6. doi: 10.1007/BF00312388. [DOI] [PubMed] [Google Scholar]
- 67.Djema AI, Mahmoud MD, Collin P, Heymann MF. Tertiary hyperparathyroidism: parathyroid cancer with liver metastases in a hemodialyzed patient. Nephrologie. 1998;19:121–3. [PubMed] [Google Scholar]
- 68.Khan MW, Worcester EM, Straus FH, 2nd, Khan S, Staszak V, Kaplan EL. Parathyroid carcinoma in secondary and tertiary hyperparathyroidism. J Am Coll Surg. 2004;199:312–9. doi: 10.1016/j.jamcollsurg.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 69.Tominaga Y, Tsuzuki T, Matsuoka S, Uno N, Sato T, Shimabukuro S, et al. Expression of parafibromin in distant metastatic parathyroid tumors in patients with advanced secondary hyperparathyroidism due to chronic kidney disease. World J Surg. 2008;32:815–21. doi: 10.1007/s00268-007-9458-8. [DOI] [PubMed] [Google Scholar]
- 70.Shane E. Clinical review 122: Parathyroid carcinoma. J Clin Endocrinol Metab. 2001;86:485–93. doi: 10.1210/jcem.86.2.7207. [DOI] [PubMed] [Google Scholar]
- 71.Busaidy NL, Jimenez C, Habra MA, Schultz PN, El-Naggar AK, Clayman GL, et al. Parathyroid carcinoma: a 22-year experience. Head Neck. 2004;26:716–26. doi: 10.1002/hed.20049. [DOI] [PubMed] [Google Scholar]
- 72.Fernandez-Ranvier GG, Khanafshar E, Jensen K, Zarnegar R, Lee J, Kebebew E, et al. Parathyroid carcinoma, atypical parathyroid adenoma, or parathyromatosis? Cancer. 2007;110:255–64. doi: 10.1002/cncr.22790. [DOI] [PubMed] [Google Scholar]
- 73.Silverberg SJ, Bilezikian JP. The diagnosis and management of asymptomatic primary hyperparathyroidism. Nat Clin Pract Endocrinol Metab. 2006;2:494–503. doi: 10.1038/ncpendmet0265. [DOI] [PubMed] [Google Scholar]
- 74.Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res. 2008;23:1869–80. doi: 10.1359/jbmr.081018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Messerer CL, Bugis SP, Baliski C, Wiseman SM. Normocalcemic parathyroid carcinoma: an unusual clinical presentation. World J Surg Oncol. 2006;4:10. doi: 10.1186/1477-7819-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kebebew E, Arici C, Duh QY, Clark OH. Localization and reoperation results for persistent and recurrent parathyroid carcinoma. Arch Surg. 2001;136:878–85. doi: 10.1001/archsurg.136.8.878. [DOI] [PubMed] [Google Scholar]
- 77.Stock JL, Weintraub BD, Rosen SW, Aurbach GD, Spiegel AM, Marx SJ. Human chorionic gonadotropin subunit measurement in primary hyperparathyroidism. J Clin Endocrinol Metab. 1982;54:57–63. doi: 10.1210/jcem-54-1-57. [DOI] [PubMed] [Google Scholar]
- 78.Rubin MR, Bilezikian JP, Birken S, Silverberg SJ. Human chorionic gonadotropin measurements in parathyroid carcinoma. Eur J Endocrinol. 2008;159:469–74. doi: 10.1530/EJE-08-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rubin MR, Silverberg SJ, D'Amour P, Brossard JH, Rousseau L, Sliney J, Jr, et al. An N-terminal molecular form of parathyroid hormone (PTH) distinct from hPTH(1 84) is overproduced in parathyroid carcinoma. Clin Chem. 2007;53:1470–6. doi: 10.1373/clinchem.2007.085506. [DOI] [PubMed] [Google Scholar]
- 80.Hara H, Igarashi A, Yano Y, Yashiro T, Ueno E, Aiyoshi Y, et al. Ultrasonographic features of parathyroid carcinoma. Endocr J. 2001;48:213–7. doi: 10.1507/endocrj.48.213. [DOI] [PubMed] [Google Scholar]
- 81.Tamler R, Lewis MS, LiVolsi VA, Genden EM. Parathyroid carcinoma: ultrasonographic and histologic features. Thyroid. 2005;15:744–5. doi: 10.1089/thy.2005.15.744. [DOI] [PubMed] [Google Scholar]
- 82.Spinelli C, Bonadio AG, Berti P, Materazzi G, Miccoli P. Cutaneous spreading of parathyroid carcinoma after fine needle aspiration cytology. J Endocrinol Invest. 2000;23:255–7. doi: 10.1007/BF03343718. [DOI] [PubMed] [Google Scholar]
- 83.Agarwal G, Dhingra S, Mishra SK, Krishnani N. Implantation of parathyroid carcinoma along fine needle aspiration track. Langenbecks Arch Surg. 2006;391:623–6. doi: 10.1007/s00423-006-0095-8. [DOI] [PubMed] [Google Scholar]
- 84.Kebebew E. Parathyroid carcinoma. Curr Treat Options Oncol. 2001;2:347–54. doi: 10.1007/s11864-001-0028-2. [DOI] [PubMed] [Google Scholar]
- 85.Wang CA, Gaz RD. Natural history of parathyroid carcinoma. Diagnosis, treatment, and results. Am J Surg. 1985;149:522–7. doi: 10.1016/s0002-9610(85)80050-7. [DOI] [PubMed] [Google Scholar]
- 86.DeLellis RA. Parathyroid carcinoma: an overview. Adv Anat Pathol. 2005;12:53–61. doi: 10.1097/01.pap.0000151319.42376.d4. [DOI] [PubMed] [Google Scholar]
- 87.Obara T, Fujimoto Y, Hirayama A, Kanaji Y, Ito Y, Kodama T, et al. Flow cytometric DNA analysis of parathyroid tumors with special reference to its diagnostic and prognostic value in parathyroid carcinoma. Cancer. 1990;65:1789–93. doi: 10.1002/1097-0142(19900415)65:8<1789::aid-cncr2820650820>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 88.Kameyama K, Takami H. Double parathyroid carcinoma. Endocr J. 2003;50:477–9. doi: 10.1507/endocrj.50.477. [DOI] [PubMed] [Google Scholar]
- 89.Sahasranam P, Tran MT, Mohamed H, Friedman TC. Multiglandular parathyroid carcinoma: a case report and brief review. South Med J. 2007;100:841–4. doi: 10.1097/SMJ.0b013e318073ca37. [DOI] [PubMed] [Google Scholar]
- 90.Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer. 1973;31:600–5. doi: 10.1002/1097-0142(197303)31:3<600::aid-cncr2820310316>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 91.Tan MH, Morrison C, Wang P, Yang X, Haven CJ, Zhang C, et al. Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin Cancer Res. 2004;10:6629–37. doi: 10.1158/1078-0432.CCR-04-0493. [DOI] [PubMed] [Google Scholar]
- 92.Gill AJ, Clarkson A, Gimm O, Keil J, Dralle H, Howell VM, et al. Loss of nuclear expression of parafibromin distinguishes parathyroid carcinomas and hyperparathyroidism-jaw tumor (HPT-JT) syndrome-related adenomas from sporadic parathyroid adenomas and hyperplasias. Am J Surg Pathol. 2006;30:1140–9. doi: 10.1097/01.pas.0000209827.39477.4f. [DOI] [PubMed] [Google Scholar]
- 93.Juhlin CC, Villablanca A, Sandelin K, Haglund F, Nordenstrom J, Forsberg L, et al. Parafibromin immunoreactivity: its use as an additional diagnostic marker for parathyroid tumor classification. Endocr Relat Cancer. 2007;14:501–12. doi: 10.1677/ERC-07-0021. [DOI] [PubMed] [Google Scholar]
- 94.Howell VM, Gill A, Clarkson A, Nelson AE, Dunne R, Delbridge LW, et al. Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. The Journal of clinical endocrinology and metabolism. 2009;94:434–41. doi: 10.1210/jc.2008-1740. [DOI] [PubMed] [Google Scholar]
- 95.Abbona GC, Papotti M, Gasparri G, Bussolati G. Proliferative activity in parathyroid tumors as detected by Ki-67 immunostaining. Hum Pathol. 1995;26:135–8. doi: 10.1016/0046-8177(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 96.Erickson LA, Jin L, Wollan P, Thompson GB, van Heerden JA, Lloyd RV. Parathyroid hyperplasia, adenomas, and carcinomas: differential expression of p27Kip1 protein. Am J Surg Pathol. 1999;23:288–95. doi: 10.1097/00000478-199903000-00007. [DOI] [PubMed] [Google Scholar]
- 97.Haven CJ, van Puijenbroek M, Karperien M, Fleuren GJ, Morreau H. Differential expression of the calcium sensing receptor and combined loss of chromosomes 1q and 11q in parathyroid carcinoma. J Pathol. 2004;202:86–94. doi: 10.1002/path.1489. [DOI] [PubMed] [Google Scholar]
- 98.Bergero N, De Pompa R, Sacerdote C, Gasparri G, Volante M, Bussolati G, et al. Galectin-3 expression in parathyroid carcinoma: immunohistochemical study of 26 cases. Hum Pathol. 2005;36:908–14. doi: 10.1016/j.humpath.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 99.Iihara M, Okamoto T, Suzuki R, Kawamata A, Nishikawa T, Kobayashi M, et al. Functional parathyroid carcinoma: Long-term treatment outcome and risk factor analysis. Surgery. 2007;142:936–43. doi: 10.1016/j.surg.2007.09.014. discussion 43 e1. [DOI] [PubMed] [Google Scholar]
- 100.Juhlin CC, Haglund F, Villablanca A, Forsberg L, Sandelin K, Branstrom R, et al. Loss of expression for the Wnt pathway components adenomatous polyposis coli and glycogen synthase kinase 3-beta in parathyroid carcinomas. International journal of oncology. 2009;34:481–92. [PubMed] [Google Scholar]
- 101.Wilkins BJ, Lewis JS. Non-Functional Parathyroid Carcinoma: A Review of the Literature and Report of a Case Requiring Extensive Surgery. Head Neck Pathol. 2009;3:140–9. doi: 10.1007/s12105-009-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fernandez-Ranvier GG, Jensen K, Khanafshar E, Quivey JM, Glastonbury C, Kebebew E, et al. Nonfunctioning parathyroid carcinoma: case report and review of literature. Endocr Pract. 2007;13:750–7. doi: 10.4158/EP.13.7.750. [DOI] [PubMed] [Google Scholar]
- 103.Ordonez NG, Ibanez ML, Samaan NA, Hickey RC. Immunoperoxidase study of uncommon parathyroid tumors. Report of two cases of nonfunctioning parathyroid carcinoma and one intrathyroid parathyroid tumor-producing amyloid. Am J Surg Pathol. 1983;7:535–42. doi: 10.1097/00000478-198309000-00004. [DOI] [PubMed] [Google Scholar]
- 104.Yamashita H, Noguchi S, Murakami N, Toda M, Adachi M, Daa T. Immunohistological study of nonfunctional parathyroid carcinoma. Report of a case. Acta Pathol Jpn. 1992;42:279–85. doi: 10.1111/j.1440-1827.1992.tb02542.x. [DOI] [PubMed] [Google Scholar]
- 105.Wang CA, Gaz RD. Natural history of parathyroid carcinoma. Diagnosis, treatment, and results. Am J Surg. 1985;149:522–7. doi: 10.1016/s0002-9610(85)80050-7. [DOI] [PubMed] [Google Scholar]
- 106.Obara T, Okamoto T, Ito Y, Yamashita T, Kawano M, Nishi T, et al. Surgical and medical management of patients with pulmonary metastasis from parathyroid carcinoma. Surgery. 1993;114:1040–8. discussion 8–9. [PubMed] [Google Scholar]
- 107.Koyano H, Shishiba Y, Shimizu T, Suzuki N, Nakazawa H, Tachibana S, et al. Successful treatment by surgical removal of bone metastasis producing PTH: new approach to the management of metastatic parathyroid carcinoma. Intern Med. 1994;33:697–702. doi: 10.2169/internalmedicine.33.697. [DOI] [PubMed] [Google Scholar]
- 108.Flye MW, Brennan MF. Surgical resection of metastatic parathyroid carcinoma. Ann Surg. 1981;193:425–35. doi: 10.1097/00000658-198104000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fujimoto Y, Obara T, Ito Y, Kodama T, Nobori M, Ebihara S. Localization and surgical resection of metastatic parathyroid carcinoma. World J Surg. 1986;10:539–47. doi: 10.1007/BF01655520. [DOI] [PubMed] [Google Scholar]
- 110.Tochio M, Takaki H, Yamakado K, Uraki J, Kashima M, Nakatsuka A, et al. A Case Report of 20 Lung Radiofrequency Ablation Sessions for 50 Lung Metastases from Parathyroid Carcinoma Causing Hyperparathyroidism. Cardiovasc Intervent Radiol. 2009 doi: 10.1007/s00270-009-9730-4. [DOI] [PubMed] [Google Scholar]
- 111.Artinyan A, Guzman E, Maghami E, Al-Sayed M, D'Apuzzo M, Wagman L, et al. Metastatic parathyroid carcinoma to the liver treated with radiofrequency ablation and transcatheter arterial embolization. J Clin Oncol. 2008;26:4039–41. doi: 10.1200/JCO.2007.15.9038. [DOI] [PubMed] [Google Scholar]
- 112.Montenegro FL, Chammas MC, Juliano AG, Cernea CR, Cordeiro AC. Ethanol injection under ultrasound guidance to palliate unresectable parathyroid carcinoma. Arq Bras Endocrinol Metabol. 2008;52:707–11. doi: 10.1590/s0004-27302008000400019. [DOI] [PubMed] [Google Scholar]
- 113.Calandra DB, Chejfec G, Foy BK, Lawrence AM, Paloyan E. Parathyroid carcinoma: biochemical and pathologic response to DTIC. Surgery. 1984;96:1132–7. [PubMed] [Google Scholar]
- 114.Bukowski RM, Sheeler L, Cunningham J, Esselstyn C. Successful combination chemotherapy for metastatic parathyroid carcinoma. Arch Intern Med. 1984;144:399–400. [PubMed] [Google Scholar]
- 115.Vega EM, Mautalen CA. Intravenous pamidronic acid in hypercalcemia due to parathyroid carcinoma. Medicina (B Aires) 1991;51:106–10. [PubMed] [Google Scholar]
- 116.Szmuilowicz ED, Utiger RD. A case of parathyroid carcinoma with hypercalcemia responsive to cinacalcet therapy. Nat Clin Pract Endocrinol Metab. 2006;2:291–6. doi: 10.1038/ncpendmet0189. quiz 7. [DOI] [PubMed] [Google Scholar]
- 117.Collins MT, Skarulis MC, Bilezikian JP, Silverberg SJ, Spiegel AM, Marx SJ. Treatment of hypercalcemia secondary to parathyroid carcinoma with a novel calcimimetic agent. J Clin Endocrinol Metab. 1998;83:1083–8. doi: 10.1210/jcem.83.4.4726. [DOI] [PubMed] [Google Scholar]
- 118.Silverberg SJ, Rubin MR, Faiman C, Peacock M, Shoback DM, Smallridge RC, et al. Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3803–8. doi: 10.1210/jc.2007-0585. [DOI] [PubMed] [Google Scholar]
- 119.Bradwell AR, Harvey TC. Control of hypercalcaemia of parathyroid carcinoma by immunisation. Lancet. 1999;353:370–3. doi: 10.1016/S0140-6736(98)06469-1. [DOI] [PubMed] [Google Scholar]
- 120.Betea D, Bradwell AR, Harvey TC, Mead GP, Schmidt-Gayk H, Ghaye B, et al. Hormonal and biochemical normalization and tumor shrinkage induced by anti-parathyroid hormone immunotherapy in a patient with metastatic parathyroid carcinoma. J Clin Endocrinol Metab. 2004;89:3413–20. doi: 10.1210/jc.2003-031911. [DOI] [PubMed] [Google Scholar]
- 121.Lumachi F, Brunello A, Roma A, Basso U. Cancer-induced hypercalcemia. Anticancer Res. 2009;29:1551–5. [PubMed] [Google Scholar]
- 122.Sandelin K, Auer G, Bondeson L, Grimelius L, Farnebo LO. Prognostic factors in parathyroid cancer: a review of 95 cases. World J Surg. 1992;16:724–31. doi: 10.1007/BF02067369. [DOI] [PubMed] [Google Scholar]
- 123.Hundley JC, Albertson DA, Bradley RF, Levine EA. Resection of pulmonary metastasis from parathyroid carcinoma. Am Surg. 2003;69:779–83. [PubMed] [Google Scholar]