

Abstract
In addition to supporting rapid nerve conduction, myelination nurtures and stabilizes axons and protects them from acute toxic insults. One myelin molecule that protects and sustains axons is myelin-associated glycoprotein (MAG). MAG is expressed on the innermost wrap of myelin, apposed to the axon surface, where it interacts with axonal receptors that reside in lateral membrane domains including gangliosides, the GPI-anchored Nogo receptors, and β1-integrin. We report here that MAG protection extends beyond the axon to the neurons from which those axons emanate, protecting them from excitotoxicity. Compared to wild type mice, Mag-null mice displayed markedly increased seizure activity in response to intraperitoneal injection of kainic acid, an excitotoxic glutamate receptor agonist. Mag-null mice also had larger lesion volumes in response to intrastriatal injection of the excitotoxin N-methyl-D-aspartate (NMDA). Prior injection of a soluble form of MAG partially protected Mag-null mice from NMDA-induced lesions. Hippocampal neurons plated on proteins extracted from wild-type rat or mouse myelin were resistant to kainic acid-induced excitotoxicity, whereas neurons plated on proteins from Mag-null myelin were not. Protection was reversed by anti-MAG antibody and replicated by addition of soluble MAG. MAG-mediated protection from excitotoxicity was dependent on Nogo receptors and β1-integrin. We conclude that MAG engages membrane-domain resident neuronal receptors to protect neurons from excitotoxicity, and that soluble MAG mitigates excitotoxic damage in vivo.
Keywords: Excitotoxicity, kainic acid, N-methyl-D-aspartate, myelin, seizure, neuroprotection
Axon function is regulated by support cells, including oligodendrocytes (in the CNS) and Schwann cells (in the PNS) that wrap axons with myelin, an insulating structure of multilayered membrane (Lemke 1992; Edgar and Garbern 2004). Myelin supports rapid nerve conduction by providing segmental insulation, controlling the structure and distribution of ion channels at nodes of Ranvier, and regulating the axon cytoskeleton (Poliak and Peles 2003; Hsieh et al. 1994). Myelin also nurtures the axons it enwraps; demyelination results in axon degeneration (Nave and Trapp 2008). In addition to these supportive roles, residual myelin debris inhibits axon regeneration at sites of injury, especially in the CNS (Sandvig et al. 2004; Yiu and He 2006). The various effects of myelin on axons involve a rich network of communications among molecules expressed on the periaxonal surface of myelin and complementary receptors located on the axon surface.
One molecule that regulates myelin-axon interactions is myelin-associated glycoprotein (MAG), a minor constituent of CNS and PNS myelin (Quarles 2007). MAG is expressed selectively on periaxonal myelin membranes (Trapp et al. 1989), leading to speculation that it might be required for myelination. Although Mag-null mice were subsequently found to elaborate abundant functional myelin (Li et al. 1994; Montag et al. 1994), they proved revealing in that they display late onset progressive axonal atrophy and increased Wallerian degeneration in both CNS and PNS. This led to the proposal that MAG stabilizes myelinated axons (Fruttiger et al. 1995; Pan et al. 2005; Nguyen et al. 2009). Mag-null mice also display reduced neurofilament phosphorylation and spacing, reduced axon caliber, increased unmyelinated axons in the CNS and progressive disruption of nodes of Ranvier in the PNS (Yin et al. 1998; Montag et al. 1994; Susuki et al. 2007).
Recently, MAG was found to protect axons from acute toxicity induced by a variety of known axonopathic agents, including the industrial neurotoxin acrylamide, the cancer chemotherapeutic agent vincristine, and inflammatory mediators (Nguyen et al. 2009). These data identify MAG as one of the molecules on myelin responsible for its stabilizing and protective effects on axons. In addition to, or perhaps related to MAG’s stabilizing effects, MAG also inhibits axon regeneration after CNS injury, impeding functional recovery (Sandvig et al. 2004; Yiu and He 2006; Quarles 2009; Lee et al. 2010).
MAG, on myelin, exerts its axon stabilizing and axon inhibitory actions by binding to one or more receptors on the axon. Functional MAG receptors on axons include the major brain gangliosides GD1a and GT1b, the glycosylphosphatidylinositol (GPI)-anchored Nogo receptors (NgR1, NgR2), β1-integrin, and the paired immunoglobulin-like receptor B (PirB) (Yiu and He 2006; Schnaar and Lopez 2009; Goh et al. 2008; Atwal et al. 2008). Most MAG receptors (gangliosides, NgRs and β1-integrin) partition into lateral membrane domains, and “lipid rafts” have been implicated as required components of MAG signaling (Yu et al. 2004; Vinson et al. 2003; Venkatesh et al. 2005; Fujitani et al. 2005). Although the roles of each MAG receptor are not fully resolved, MAG appears to engage its different receptors in a cell-type dependent manner to accomplish diverse tasks (Venkatesh et al. 2007; Mehta et al. 2007; Mehta et al. 2010).
In this report we describe a new biological role of MAG: neuroprotection against excitotoxicity. Pharmacological characterization of the receptors responsible for MAG protection of cultured hippocampal neurons from excitotoxicity indicates that this neuroprotective role is mediated by Nogo receptors and β1-integrin. The data expand our understanding of the contribution of myelination to neuronal health, extend the protective effects of MAG from axons to the neurons from which those axons emanate, and provide evidence that soluble MAG can be a neuroprotective agent.
Materials and methods
Materials
Phosphatidylinositol-specific phospholipase C (PI-PLC, B. Cereus) and Y-27632 (Rho kinase inhibitor) were from Sigma-Aldrich Corp., St. Louis, MO. NEP1–40 (NgR blocking peptide) (GrandPre et al. 2002), and TAT-Pep5 (p75NTR inhibitor, (Yamashita and Tohyama 2003) were from EMD Biosciences, La Jolla, CA. Sialidase (V. cholerae) was produced as described (Moustafa et al. 2004). Anti-MAG monoclonal antibody (mAb 513) was generated from the hybridoma as described (Poltorak et al. 1987). Kainic acid (KA) was from A.G. Scientific, Inc., San Diego, CA. β1-integrin specific function-blocking antibody (Ha2/5) was from BD Biosciences, San Jose, California. MAG-human Fc chimera (MAG-Fc) was purchased from R&D Systems, Minneapolis, MN or was overexpressed in mammalian cells using a vector (Collins et al. 2000), stably transfected into Flp-InTM_CHO cells (Invitrogen, Carlsbad, CA), and then purified from culture supernatant by Protein-G chromatography and dialyzed against Dulbecco’s phosphate-buffered saline (PBS).
Animals
Mag-null founder mice, kindly provided by Dr. Bruce Trapp, The Cleveland Clinic Foundation, Cleveland, OH, were constructed by disruption of exon 5 of the Mag gene as previously reported (Li et al. 1994). Mutant mice were repeatedly back-crossed onto a C57BL/6 background to obtain 99.5% strain purity (Pan et al. 2005). Comparisons were made between Mag-null mice and age-matched C57BL/6 wild type (WT) mice.
Kainate-induced seizures
All procedures were approved by the Johns Hopkins Animal Care and Use Committee consistent with federal law and NIH regulations. Johns Hopkins Medical Institutions are accredited by the American Association for Accreditation of Laboratory Animal Care. Seizures were induced by intraperitoneal injection of 14–16 week old C57BL/6 WT and Mag-null mice with 25 mg/kg KA in a final volume of 300 μl PBS. Control mice received PBS only. Seizure activity was recorded for 2 min every 12 min in the first 2 h, and every 20 min thereafter until seizures ceased. Seizures were quantitatively scored as previously described (Liu et al. 1999): 0, normal behavior; 1, ceases exploring, grooming and sniffing (becomes motionless); 2 forelimb and/or tail extension, rigid posture; 3, myoclonic jerks of the head and neck with brief twitching or repetitive movements, head bobbing or “wet-dog shakes”; 4, forelimb clonus and partial rearing and falling; 5, forelimb clonus, continuous rearing and falling; 6 tonic-clonic movements, loss of posture. Animals that died from seizure activity were assigned the highest quantitative ranking for the remainder of the observation period.
NMDA excitotoxicity
WT and Mag-null mice (3 to 5 months old) were injected stereotaxically in the right striatum with 15 nmol of NMDA in 0.3 μl PBS or vehicle (Ahmad et al. 2006). The needle was then retracted, the hole plugged with bone wax, and the wound sutured. Throughout the surgical procedures and recovery mice were maintained at 37°C. For MAG-Fc treatment, Mag-null mice were first injected in the striatum with 1 μl of MAG-Fc (15 mg/ml in PBS) or vehicle. The needle was left in place for 5 min, and then was retracted. One hour later, excitotoxicity was induced by injection of NMDA at the same site. Forty-eight hours after NMDA injection, mice were subjected to magnetic resonance imaging (MRI, see below), and then were transcardially perfused with phosphate-buffered saline followed by 4% paraformaldehyde. Brains were removed, post-fixed in paraformaldehyde overnight, cryoprotected in sucrose (30%) for 3 days, and frozen in dry ice-cooled 2-methylbutane. Cryostat sections were collected on slides and stained with cresyl violet to estimate lesion volumes. Images were collected and analyzed using SigmaScan Pro 5.0 and SigmaStat 2.0 (Systat Software Inc., Richmond, CA) (Ahmad et al. 2006).
MRI
Anesthetized mice were imaged using a 9.4 Tesla NMR spectrometer (Bruker Biospin, Billerica, MA, USA). Mice were placed in a holder with bite bar and head holder and maintained at 30 °C. T2-weighted magnetic resonance images of brain lesions were acquired with a fast spin echo sequence: echo time = 40 ms, repetition time ≅ 5000 ms with respiratory gating, echo train length = 4, number of signal average = 6, total 50 slices. Images were segmented semiautomatically with Amira software (Mercury Computer Systems, Chelmsford, MA). Brain lesion areas were manually segmented and volumes were obtained by multiplying the number of pixels from selected hypointense areas in the T2-weighted images with the size of each individual pixel.
Hippocampal neurons
Isolation and culture of primary adult rat hippocampal neurons was modified from a prior report (Brewer 1997). Media were from Invitrogen or BrainBits (Springfield, IL). Postnatal day 4–5 Sprague–Dawley rats were decapitated and hippocampi were rapidly dissected into 2 ml of ice-cold isolation medium (Hibernate A:B27 (98:2)). Meninges and white matter were removed, and then the tissue was chopped with a razor and transferred to a 15-ml conical tube. The tissue pieces were allowed to settle, the supernatant removed, and 5 ml of a 37°C solution containing 2 mg/ml of papain (Worthington, Lakewood, NJ) in isolation medium were added. The tissue was incubated for 30 min at 37°C with gentle agitation. After allowing the tissue to settle, the papain solution was removed and 2 ml of isolation medium were added. After 5 min, hippocampi fragments were disaggregated by trituration with a fire-polished Pasteur pipette. After 2 min to allow any remaining tissue pieces to settle, the supernatant was collected and replaced with fresh isolation medium. The trituration procedure was repeated twice, and the supernatants combined and applied atop a step gradient of 15% and 35% NycoprepTM Universal (Axis-Shield, Oslo, Norway) in isolation medium. After centrifugation (800 × g, 15 min, ambient temperature), cells at the 15%–35% Nycodenz interface were collected and diluted into 5 ml Hibernate A medium. Cells were collected by centrifugation (300 × g, 8 min), resuspended in 3 ml of culture medium (Neurobasal A:B27 (98:2) containing 0.5 mM glutamine, 100 U/ml penicillin, 0.05 mg/ml streptomycin and 5 ng/ml of recombinant human FGF2 (Chemicon International, Temecula, California)), and passed through a 40 μm cell strainer (BD Biosciences). Cells were plated at 24,000 cells/well (96-well plates, Nunc, Roskilde, Denmark) onto control surfaces (coated for 1 h with 125 μg/ml of poly-D-lysine (MW >300,000, Sigma)) or the same surfaces that were further adsorbed with mild detergent extract of brain myelin exactly as described (Mehta et al. 2007).
Kainic acid excitotoxicity in vitro
The medium of 48-h hippocampal cultures was replaced with medium containing 130 μM KA. Half of the medium was removed 24-h later and replaced with medium containing 10 μg/ml propidium iodide to label dead cells (Brana et al. 2002). Thirty min later, the cells were washed twice with Neurobasal A medium and fixed with 2% paraformaldehyde in PBS for 12–16 h. After fixation cells were permeabilized using 0.1% Triton X-100 in PBS containing 2% horse serum (blocking buffer). Neurons were immunostained with anti-β-tubulin antibody (TUJ1, 1:2000, Covance, Berkeley, CA) and fluorescein-conjugated anti-mouse IgG (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA). Four random microscopic fields equidistant from the edges of the well were captured for image analysis using a Nikon epifluorescence microscope and a CoolSNAP HQ2 camera (Photometrix, Tucson, AZ). Live neurons (TUJ1-positive, propidium iodide-negative) were quantified. Antibodies, enzymes and pharmacological agents were added 1 h after cell plating and again upon addition of KA-containing medium. MAG-Fc was added only upon addition of KA. None of the treatments reduced cell numbers in control cultures. PI-PLC efficacy was confirmed by reduction in immunostaining of the GPI anchored protein Thy-1. Sialidase efficacy was confirmed by elimination of GD1a immunostaining (Lunn et al. 2000). TAT-Pep5 efficacy was confirmed by its ability to block proNGF-mediated apoptosis in hippocampal neurons (Lee et al. 2001).
Results
Mag-null mice display increased susceptibility to excitotoxicity
Two independent measures were used to quantify excitotoxicity in vivo: seizure activity in response to systemically administered kainate, and brain lesion volume induced by stereotaxic striatal injection of NMDA. In the seizure model, KA was administered intraperitoneally and seizure activity was quantified. Injection of 25 mg/kg of KA resulted in greater seizure activity in Mag-null than in WT mice (Fig. 1). More Mag-null mice reached higher seizure scores, and their seizure duration was increased. Treatment with KA resulted in lethal seizures in 38% of the Mag-null mice, but none of the WT mice. Total seizure activity (combined seizure length and severity) was significantly greater in Mag-null mice than in WT. To test MAG neuroprotection in a more direct model of excitotoxic neuronal death in vivo, the susceptibility of WT and Mag-null mice to lesions induced by intrastriatal injection of NMDA was quantified (Kemp and McKernan 2002). Stereotaxic injection of NMDA into the striatum produced a brain lesion (Fig. 2) whose volume reached a plateau within 48 h. Mag-null mice displayed increased average brain lesion volume (6.7 mm3) compared to WT mice (5.6 mm3), a significant increase of 20% (p < 0.05).
Fig. 1.

Mag-null mice display increased susceptibility to kainic acid-induced seizures. (Left) Seizures were induced in WT (n = 7) and Mag-null mice (n = 8) by intraperitoneal injection of KA (25 mg/kg). Seizure activity was video recorded and quantified as follows: 0, normal behavior; 1, ceases exploring, grooming and sniffing (becomes motionless); 2 forelimb and/or tail extension, rigid posture; 3, myoclonic jerks of the head and neck with brief twitching or repetitive movements, head bobbing or “wet-dog shakes”; 4, forelimb clonus and partial rearing and falling; 5, forelimb clonus, continuous rearing and falling; 6 tonic-clonic movements, loss of posture. Mice that died due to seizure were scored at the highest level for the remainder of the observation period. Each trace represents an individual mouse. The traces represent seizure activity recorded every 12–20 min over a 4-h observation period. (Upper Right) Pie charts indicate the percent of mice reaching the indicated maximum seizure score for each geneotype. (Lower Right) Total seizure activity (mean ± SEM) is presented as the sum of the seizure scores at each time analyzed. Mag-null mice displayed significantly greater seizure activity (*, p <0.02).
Fig. 2.

Mag-null mice display increased susceptibility to excitotoxicity induced by intrastriatal injection of NMDA. WT (n = 6) and Mag-null (n = 8) mice were injected in the striatum with 0.3 μl PBS containing 15 nmol NMDA to induce a brain lesion. After 48 h, animals were sacrificed, perfused, fixed, then brains were removed, sectioned, and stained with cresyl violet and lesion volumes quantified. (Left) Representative images of brain sections stained with cresyl violet. The lesion area is outlined. (Right) Quantification of the lesion volumes (mean ± SEM) induced by intrastriatal injection of NMDA was determined by an observer blinded to the genotype. Mag-null mice displayed significantly greater lesion volume (*, p < 0.025).
MAG treatment improves the response of Mag-null mice to NMDA-induced lesions
Mag-null mice were pre-treated with MAG-Fc, a soluble chimeric protein bearing the extracellular domain of MAG fused to human Fc. One hour after intrastriatal injection of MAG-Fc or vehicle (control), mice received NMDA at the same site. Lesion volumes were determined 48 h later via MRI. The same mice were then perfusion fixed, and cryostat brain sections were stained with cresyl violet. Lesion volumes, independently determined by MRI and microscopic image analyses, revealed significant protection from excitotoxicity by injection of MAG-Fc (Fig. 3).
Fig. 3.

Soluble chimeric MAG (MAG-Fc) reduces the susceptibility of Mag-null mice to NMDA-induced excitotoxicity. Mag-null mice were injected in the striatum with 1 μl of MAG-Fc chimera (15 mg/ml, n = 6) or vehicle (saline, n = 8) as indicated. One hour later, 0.3 μl containing 15 nmole of NMDA was injected into the same site to induce a brain lesion. After an additional 48 h, animals were scanned by MRI. Mice were then perfusion-fixed and their brains were removed, sectioned, and stained with cresyl violet to determine lesion volumes. (Left) Representative MRI images of similar anatomical brain regions showing NMDA-induced brain lesions in Mag-null mice pre-injected with saline (control) or MAG-Fc. (Right) Quantification of brain lesion volumes obtained by cresyl violet staining and by MRI (of the same set of mice) was determined by observers blinded to the genotype. MAG-Fc-treated mice displayed significantly smaller lesion volumes (*, p < 0.05).
MAG protects cultured hippocampal neurons from kainate excitotoxicity
Hippocampal neurons were cultured 48 h on control or myelin-adsorbed surfaces. Kainate (130 μM) was then added for 24 h, resulting in 35% loss of hippocampal neurons on control surfaces, the remaining cells displaying shortened neurites (Fig. 4). Kainate-induced neuronal loss on myelin-adsorbed surfaces was markedly and significantly reduced, and a function blocking anti-MAG monoclonal antibody (mAb 513) reversed myelin protection, implicating MAG as the agent responsible for myelin-mediated protection. This was confirmed using surfaces adsorbed with extracts from WT and Mag-null mice; WT mouse myelin was neuroprotective whereas Mag-null myelin was not (Fig. 5). Consistent with its efficacy in vivo, MAG-Fc was also neuroprotective in vitro (Fig. 6).
Fig. 4.

MAG protects hippocampal neurons from kainic acid-induced excitotoxicity in vitro. (Left) Hippocampal neurons were plated onto control surfaces or the same surfaces adsorbed with detergent-extracted proteins from rat brain myelin. As indicated, 1 h after plating, some cultures were treated with 10 μg/ml anti-MAG mAb. After 48 h, the medium was replaced with fresh control medium or medium containing 130 μM kainic acid (KA) to induce excitotoxicity. After an additional 24 h, the cultures were incubated for 30 min with medium containing propidium iodide, and then were fixed and stained with anti-tubulin mAb (TUJ1). Representative fluorescent micrographs are presented as reverse gray scale images to enhance clarity. (Right) Cell survival (mean ± SEM) was quantified (as described in the text) and normalized with respect to control surfaces. Live cell counts from 4 microscopic fields from each of 9 microwells from three independent experiments were averaged. Growth on myelin-adsorbed surfaces provided significant neuroprotection from KA (*, p < 0.001) compared to cells grown on control surfaces. Addition of anti-MAG antibody reversed this protection, resulting in cell loss that was not significantly different from KA-treated cells grown on control surfaces (†, p > 0.7).
Fig. 5.
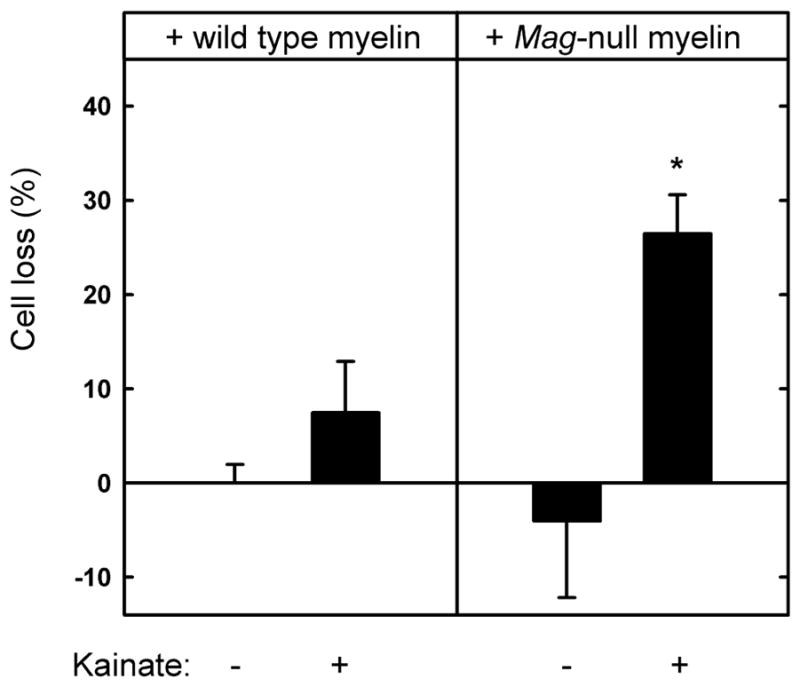
Myelin extract from Mag-null mice has diminished capacity to protect hippocampal neurons from kainic acid-induced excitotoxicity. Hippocampal neurons were plated on surfaces adsorbed with detergent-extracted proteins from wild type or Mag-null mouse brain myelin. After 48 h, the medium was replaced with fresh medium containing 130 μM KA. After an additional 24 h, the cultures were incubated for 30 min with medium containing propidium iodide and then fixed and stained with TUJ1 mAb. Cell survival (mean ± SEM) was quantified (as described in the text) and normalized with respect to control surfaces. Live cell counts from 4 microscopic fields from each of 9 microwells from three independent experiments were averaged. Statistically significant KA toxicity was only observed when cells were plated on Mag-null myelin (*, p < 0.02 compared to matched culture in the absence of KA). The average number of live cells on Mag-null wells in the absence of KA exceeded those on control cells, resulting in negative values, although the difference from zero (equal to control wells) was not statistically significant.
Fig. 6.
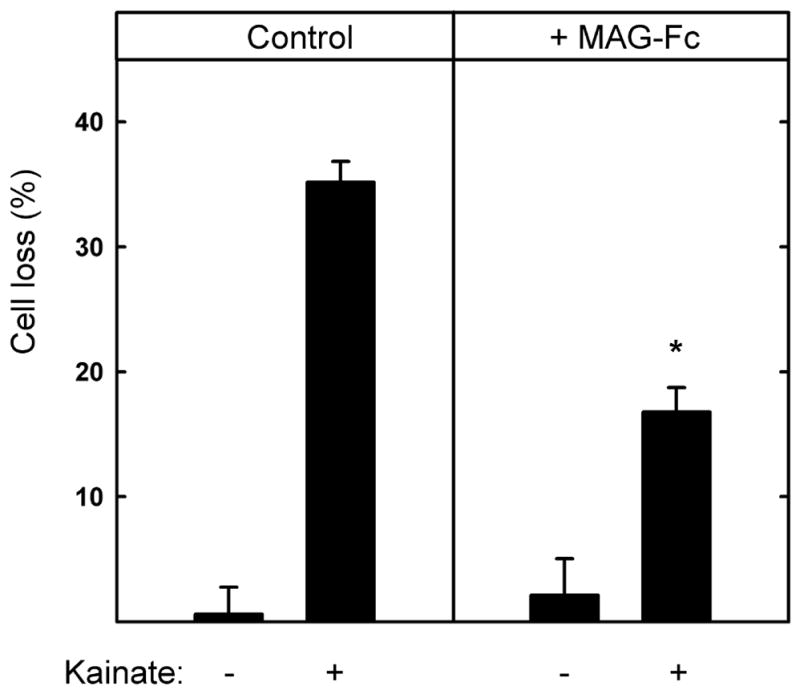
Soluble chimeric MAG (MAG-Fc) partially protects hippocampal neurons from kainic acid-induced excitotoxicity. Hippocampal neurons were grown for 48 h on control surfaces, then (as indicated) the medium was replaced with fresh medium containing 130 μM KA with or without MAG-Fc chimera (25 μg/ml). After an additional 24 h, the cultures were incubated for 30 min with medium containing propidium iodide and then fixed and stained with TUJ1 mAb. Cell survival (mean ± SEM) was quantified and normalized with respect to the control. The presence of MAG-Fc resulted in a statistically significant reduction in KA-induced toxicity as compared to cells treated with KA in the absence of MAG-Fc (*, p < 0.001).
Receptors and pathways mediating MAG protection from excitotoxicity
Different receptors and signaling pathways mediate different MAG functions (Yiu and He 2006; Sandvig et al. 2004; Schnaar and Lopez 2009; Goh et al. 2008; Atwal et al. 2008; Venkatesh et al. 2007; Mehta et al. 2007; Mehta et al. 2010). To investigate which MAG receptors are responsible for MAG protection from kainate excitotoxicity, hippocampal neurons cultured on myelin surfaces were pretreated with pharmacological agents or enzymes that interfere with MAG receptors and signaling molecules: Sialidase disrupts gangliosides; PI-PLC cleaves NgRs; NEP1–40 peptide blocks MAG-NgR binding; mAb Ha2/5 disrupts β1-integrin; TAT-Pep5 disrupts p75NTR; and Y-27632 inhibits the RhoA effector ROCK.
Sialidase did not significantly reverse MAG protection, whereas PI-PLC, NEP1–40, and the function-blocking anti-β1-integrin mAb robustly reversed MAG protection (Fig. 7). The p75NTR inhibitor TAT-Pep5 was without effect, but the ROCK inhibitor Y-27632 significantly reversed MAG-mediated neuroprotection. These data imply that MAG protects hippocampal neurons from KA-induced excitotoxicity primarily via NgR and β1-integrin receptors that transmit intracellular signals via transducers (but not p75NTR) that link MAG binding to RhoA activation.
Fig. 7.

MAG-mediated protection from kainic acid-induced excitotoxicity is predominantly via NgR and β1-integrin. Hippocampal neurons were plated on control surfaces or the same surfaces adsorbed with detergent-extracted rat myelin. As indicated, 1 h after plating the following agents were added to the cultures: 10 mU/ml sialidase, 0.5 U/ml PI-PLC, 1 μM NEP1–40, 2.5 μg/ml function-blocking anti-β1-integrin receptor mAb, 100 nM TAT-Pep5, or 5 μM Y-27632. After 48 h, the media were replaced with fresh media containing the same agents plus 130 μM KA. After an additional 24 h, the cultures were incubated for 30 min with medium containing propidium iodide and then fixed and stained with TUJ1 mAb. Cell survival (mean ± SEM) was quantified and normalized with respect to the control. Neurons cultured on myelin-adsorbed surfaces were significantly protected from KA-induced toxicity (see Fig. 4). Significant reversal of myelin-mediated protection was observed upon addition of PI-PLC, NEP1–40, anti-β1-integrin mAb, and the ROCK inhibitor Y-27632 (*, p < 0.001 compared to KA-treated cultures on myelin-adsorbed surfaces without added agents). Statistically significant reversal of protection was not observed upon treatment with sialidase or TAT-Pep5.
Discussion
This study reveals a newfound role for myelin in protecting neurons from excitotoxicity. Whereas prior studies established that myelin protects the axons it ensheathes (Nave and Trapp 2008; Nguyen et al. 2009; Mehta et al. 2010), the current study extends that protection to the neuronal cell bodies from which those axons emanate. In particular we demonstrate that myelin-associated glycoprotein, which is expressed selectively on the innermost wrap of myelin apposing the axon (Quarles 2007), protects neurons from excitotoxicity as evidenced by: (i) the differential susceptibility of wild type and Mag-null neurons to two in vivo excitotoxicity models; (ii) the ability of locally injected MAG-Fc to diminish excitotoxic damage; (iii) the ability of a function-blocking anti-MAG antibody to reverse myelin protection in vitro; (iv) the ability of MAG-Fc to protect neurons in vitro; and (v) the lack of protection in vitro using extracts of Mag-null myelin.
In addition to defining a new neuroprotective role for MAG, studies were performed to investigate which neuronal receptors are involved. Knowledge of neuronal receptors for MAG derives primarily from studies characterizing MAG inhibition of axon regeneration (Cao et al. 2009; Yiu and He 2006). The two best characterized inhibitory axonal receptors for MAG are gangliosides (GD1a and GT1b) and NgRs (NgR1 and NgR2), although PirB and integrins have also been implicated (Atwal et al. 2008; Goh et al. 2008). Mice lacking complex gangliosides (including GD1a and GT1b), displayed a similar pattern of axonal degeneration to Mag-null mice (Sheikh et al. 1999, Pan et al. 2005; Nguyen et al. 2009), and gangliosides were found to mediate the axonal protective effects of MAG against vincristine, acrylamide, and inflammatory toxicity (Nguyen et al. 2009; Mehta et al. 2010). However, these gangliosides on cultured HNs do not appear to be required for MAG protection against excitotoxicity. Because different MAG receptors function on different cell types (Mehta et al. 2007; Venkatesh et al. 2007), a role for gangliosides in mediating neuroprotective effects against excitotoxicity on other neuronal cell types cannot be excluded. We previously described that gangliosides (along with NgRs) are functional receptors for MAG inhibition of neurite outgrowth from the same HNs in vitro (Mehta et al. 2007). Together with the current data, these studies confirm the previous observation that MAG can exert different biological effects on the same neurons by engaging different axonal receptors (Mehta et al. 2010).
The current study supports a role for NgRs in protecting neurons against excitotoxicity. It has been reported that MAG interacts with two members of the NgR family, NgR1 and NgR2, in a signaling complex with Lingo-1 and p75NTR or alternatively Taj/Troy (Schnaar and Lopez 2009; Park et al. 2005). Although the protective effect of MAG through NgRs was confirmed using PI-PLC and NEP 1–40, additional transducer molecule(s) were not identified since treatment of HNs with TAT-Pep5, a specific p75NTR signaling inhibitor, did not reverse MAG protection despite the prior observation that TAT-Pep5 partially reverses MAG inhibition of neurite outgrowth from HNs (Mehta et al. 2007). One possibility is that NgRs engage Taj/Troy (rather than p75NTR) as the transducer molecule for the protective effect of MAG, or that NgRs recruit an as yet unidentified molecule. Whichever subsequent signaling pathways are involved, the data reported here support a newfound role for NgRs in neuroprotection.
Consistent with the current findings, β1-integrin activation by laminin was previously found to promote resistance of hippocampal neurons to glutamate-induced excitotoxicity (Gary and Mattson 2001). In those studies, PI3-kinase was implicated in protective signaling, whereas focal adhesion kinase phosphorylation was not. Preliminary results (data not shown) using Wortmanin, a specific inhibitor of PI3K, suggest that MAG protection against excitotoxicity engages the PI3K pathway in HNs in vitro. Together, these data suggest that neuronal integrins may promote survival downstream of cell interactions with the extracellular matrix or with myelin using similar signaling pathways. Additional studies will be required to more fully establish the signaling pathways involved, as well as where the signaling molecules reside (cell body and/or axon).
In the studies reported here, treatment with an anti-MAG antibody reversed myelin protection of KA-induced excitotoxicity in vitro. In contrast, intracerebroventricular administration of anti-MAG antibody provided neuroprotection in models of stroke and traumatic brain injury (Irving et al. 2005; Thompson et al. 2006). It was noted that MAG signaling is bidirectional, and antibody-induced crosslinking of cell surface MAG protected oligodendrocytes from glutamate-induced cytotoxicity (Irving et al. 2005). It remains to be determined whether the neuroprotective effect of anti-MAG antibody is secondary to its direct effects on oligodendroglia, or due to inhibition of axon-myelin interactions. The current studies reveal that delivery of MAG-Fc reduced tissue excitotoxic damage in mice lacking MAG. The finding that MAG protects neurons from acute excitotoxicity reveals a mechanism by which oligodendroglia nurture not only axons but also the cell bodies from which those axons emanate. In addition to highlighting the importance of intact axon-myelin interactions to neuronal and axonal health, the results imply that delivery of molecularly expressed MAG might be beneficial in disorders where excitotoxicity plays a role in disease pathophysiology.
Acknowledgments
The authors thank Zhipeng Hou for expert assistance with MRI image acquisition and analysis and Andres Hurtado for critical reading of the manuscript.
Funding
This work was supported by the following grants from the National Institutes of Health: NS037096 (RLS), NS046400 (SD), AG022971 (SD), and NS059529 (JZ).
Abbreviations used
- MAG
myelin-associated glycoprotein
- MRI
magnetic resonance imaging
- NMDA
N-methyl-D-aspartate
- PI-PLC
phosphatidylinositol-specific phospholipase C
- WT
wild type
References
- Ahmad AS, Zhuang H, Dore S. Heme oxygenase-1 protects brain from acute excitotoxicity. Neuroscience. 2006;141:1703–1708. doi: 10.1016/j.neuroscience.2006.05.035. [DOI] [PubMed] [Google Scholar]
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 2008;322:967–970. doi: 10.1126/science.1161151. [DOI] [PubMed] [Google Scholar]
- Brana C, Benham C, Sundstrom L. A method for characterising cell death in vitro by combining propidium iodide staining with immunohistochemistry. Brain Res Brain Res Protoc. 2002;10:109–114. doi: 10.1016/s1385-299x(02)00201-5. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Cao Z, Gao Y, Deng K, Williams G, Doherty P, Walsh FS. Receptors for myelin inhibitors: Structures and therapeutic opportunities. Mol Cell Neurosci. 2009;43:1–14. doi: 10.1016/j.mcn.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Collins BE, Fralich TJ, Itonori S, Ichikawa Y, Schnaar RL. Conversion of cellular sialic acid expression from N-acetyl- to N-glycolylneuraminic acid using a synthetic precursor, N-glycolylmannosamine pentaacetate: inhibition of myelin-associated glycoprotein binding to neural cells. Glycobiology. 2000;10:11–20. doi: 10.1093/glycob/10.1.11. [DOI] [PubMed] [Google Scholar]
- Edgar JM, Garbern J. The myelinated axon is dependent on the myelinating cell for support and maintenance: molecules involved. J Neurosci Res. 2004;76:593–598. doi: 10.1002/jnr.20063. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Montag D, Schachner M, Martini R. Crucial role for the myelin-associated glycoprotein in the maintenance of axon-myelin integrity. Eur J Neurosci. 1995;7:511–515. doi: 10.1111/j.1460-9568.1995.tb00347.x. [DOI] [PubMed] [Google Scholar]
- Fujitani M, Kawai H, Proia RL, Kashiwagi A, Yasuda H, Yamashita T. Binding of soluble myelin-associated glycoprotein to specific gangliosides induces the association of p75NTR to lipid rafts and signal transduction. J Neurochem. 2005;94:15–21. doi: 10.1111/j.1471-4159.2005.03121.x. [DOI] [PubMed] [Google Scholar]
- Gary DS, Mattson MP. Integrin signaling via the PI3-kinase-Akt pathway increases neuronal resistance to glutamate-induced apoptosis. J Neurochem. 2001;76:1485–1496. doi: 10.1046/j.1471-4159.2001.00173.x. [DOI] [PubMed] [Google Scholar]
- Goh EL, Young JK, Kuwako K, Tessier-Lavigne M, He Z, Griffin JW, Ming GL. [beta]-1-Integrin mediates myelin-associated glycoprotein signaling in neuronal growth cones. Mol Brain. 2008;1:10. doi: 10.1186/1756-6606-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPre T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Hsieh ST, Kidd GJ, Crawford TO, Xu Z, Lin WM, Trapp BD, Cleveland DW, Griffin JW. Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci. 1994;14:6392–6401. doi: 10.1523/JNEUROSCI.14-11-06392.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving EA, Vinson M, Rosin C, Roberts JC, Chapman DM, Facci L, Virley DJ, Skaper SD, Burbidge SA, Walsh FS, Hunter AJ, Parsons AA. Identification of neuroprotective properties of anti-MAG antibody: a novel approach for the treatment of stroke? J Cereb Blood Flow Metab. 2005;25:98–107. doi: 10.1038/sj.jcbfm.9600011. [DOI] [PubMed] [Google Scholar]
- Kemp JA, McKernan RM. NMDA receptor pathways as drug targets. Nat Neurosci. 2002;5(Suppl):1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- Lee JK, Geoffroy CG, Chan AF, Tolentino KE, Crawford MJ, Leal MA, Kang B, Zheng B. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 2010;66:663–670. doi: 10.1016/j.neuron.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lemke G. Myelin and myelination. In: Hall Z, editor. An Introduction to Molecular Neurobiology. Sinauer; Sunderland, MA: 1992. pp. 281–309. [Google Scholar]
- Li C, Tropak MB, Gerial R, Clapoff S, Abramow-Newerly W, Trapp B, Peterson A, Roder J. Myelination in the absence of myelin-associated glycoprotein. Nature. 1994;369:747–750. doi: 10.1038/369747a0. [DOI] [PubMed] [Google Scholar]
- Liu H, Cao Y, Basbaum AI, Mazarati AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci U S A. 1999;96:12096–12101. doi: 10.1073/pnas.96.21.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn MP, Johnson LA, Fromholt SE, Itonori S, Huang J, Vyas AA, Hildreth JE, Griffin JW, Schnaar RL, Sheikh KA. High-affinity anti-ganglioside IgG antibodies raised in complex ganglioside knockout mice: reexamination of GD1a immunolocalization. J Neurochem. 2000;75:404–412. doi: 10.1046/j.1471-4159.2000.0750404.x. [DOI] [PubMed] [Google Scholar]
- Mehta NR, Lopez PH, Vyas AA, Schnaar RL. Gangliosides and Nogo receptors independently mediate myelin-associated glycoprotein inhibition of neurite outgrowth in different nerve cells. J Biol Chem. 2007;282:27875–27886. doi: 10.1074/jbc.M704055200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta NR, Nguyen T, Bullen JW, Griffin JW, Schnaar RL. Myelin-associated glycoprotein (MAG) protects neurons from acute toxicity using a ganglioside-dependent mechanism. ACS Chem Neurosci. 2010;1:215–222. doi: 10.1021/cn900029p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montag D, Giese KP, Bartsch U, Martini R, Lang Y, Bluthmann H, Karthingasan J, Kirschner DA, Wintergerst ES, Nave K-A, Zielasek J, Toyka KV, Lipp HP, Schachner M. Mice deficient for the myelin-associated glycoprotein show subtle abnormalities in myelin. Neuron. 1994;13:229–246. doi: 10.1016/0896-6273(94)90472-3. [DOI] [PubMed] [Google Scholar]
- Moustafa I, Connaris H, Taylor M, Zaitsev V, Wilson JC, Kiefel MJ, von Itzstein M, Taylor G. Sialic acid recognition by Vibrio cholerae neuraminidase. J Biol Chem. 2004;279:40819–40826. doi: 10.1074/jbc.M404965200. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Mehta NR, Conant K, Kim K, Jones M, Calabresi PA, Melli G, Hoke A, Schnaar RL, Ming GL, Song H, Keswani SC, Griffin JW. Axonal protective effects of the myelin associated glycoprotein. J Neurosci. 2009;29:630–637. doi: 10.1523/JNEUROSCI.5204-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Fromholt SE, Hess EJ, Crawford TO, Griffin JW, Sheikh KA, Schnaar RL. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Exp Neurol. 2005;195:208–217. doi: 10.1016/j.expneurol.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang JF, He ZG. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Poliak S, Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- Poltorak M, Sadoul R, Keilhauer G, Landa C, Fahrig T, Schachner M. Myelin-associated glycoprotein, a member of the L2/HNK-1 family of neural cell adhesion molecules, is involved in neuron-oligodendrocyte and oligodendrocyte-oligodendrocyte interaction. J Cell Biol. 1987;105:1893–1899. doi: 10.1083/jcb.105.4.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarles RH. Myelin-associated glycoprotein (MAG): past, present and beyond. J Neurochem. 2007;100:1431–1448. doi: 10.1111/j.1471-4159.2006.04319.x. [DOI] [PubMed] [Google Scholar]
- Quarles RH. A Hypothesis About the Relationship of Myelin-Associated Glycoprotein’s Function in Myelinated Axons to its Capacity to Inhibit Neurite Outgrowth. Neurochem Res. 2009;34:79–86. doi: 10.1007/s11064-008-9668-y. [DOI] [PubMed] [Google Scholar]
- Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: Expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Lopez PH. Myelin-associated glycoprotein and its axonal receptors. J Neurosci Res. 2009;87:3267–3276. doi: 10.1002/jnr.21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susuki K, Baba H, Tohyama K, Kanai K, Kuwabara S, Hirata K, Furukawa K, Furukawa K, Rasband MN, Yuki N. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia. 2007;55:746–757. doi: 10.1002/glia.20503. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Marklund N, LeBold DG, Morales DM, Keck CA, Vinson M, Royo NC, Grundy R, McIntosh TK. Tissue sparing and functional recovery following experimental traumatic brain injury is provided by treatment with an anti-myelin-associated glycoprotein antibody. Eur J Neurosci. 2006;24:3063–3072. doi: 10.1111/j.1460-9568.2006.05197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Andrews SB, Cootauco C, Quarles R. The myelin-associated glycoprotein is enriched in multivesicular bodies and periaxonal membranes of actively myelinating oligodendrocytes. J Cell Biol. 1989;109:2417–2426. doi: 10.1083/jcb.109.5.2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Sheu SS, Giger RJ. Molecular dissection of the myelin-associated glycoprotein receptor complex reveals cell type-specific mechanisms for neurite outgrowth inhibition. J Cell Biol. 2007;177:393–399. doi: 10.1083/jcb.200702102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson M, Rausch O, Maycox PR, Prinjha RK, Chapman D, Morrow R, Harper AJ, Dingwall C, Walsh FS, Burbidge SA, Riddell DR. Lipid rafts mediate the interaction between myelin-associated glycoprotein (MAG) on myelin and MAG-receptors on neurons. Mol Cell Neurosci. 2003;22:344–352. doi: 10.1016/s1044-7431(02)00031-3. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci. 1998;18:1953–1962. doi: 10.1523/JNEUROSCI.18-06-01953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Guo W, Feng L. Segregation of Nogo66 receptors into lipid rafts in rat brain and inhibition of Nogo66 signaling by cholesterol depletion. FEBS Lett. 2004;577:87–92. doi: 10.1016/j.febslet.2004.09.068. [DOI] [PubMed] [Google Scholar]