

Abstract
MYC is a potential target for many cancers but is not amenable to existing pharmacological approaches. Inhibition of HMG-CoA reductase by statins has shown potential efficacy against a number of cancers. Here, we demonstrate that inhibition of HMG-CoA reductase by atorvastatin blocks both MYC phosphorylation and activation, suppressing tumor initiation and growth in vivo in a transgenic model of MYC-induced HCC as well as in human HCC-derived cell lines. To confirm specificity, we show that the anti-tumor effects of atorvastatin are blocked by co-treatment with the HMG-CoA reductase product, Mevalonate. Moreover, by using a novel molecular imaging sensor, we confirm that inhibition of HMG-CoA reductase blocks MYC phosphorylation in vivo. Importantly, the introduction of phosphorylation mutants of MYC at Ser62 or Thr58 into tumors blocks their sensitivity to inhibition of HMG-CoA reductase. Finally, we demonstrate that inhibition of HMG-CoA reductase suppresses MYC phosphorylation through Rac GTPase. Therefore, HMG-CoA reductase is a critical regulator of MYC phosphorylation, activation, and tumorigenic properties. The inhibition of HMG-CoA reductase may be a useful target for the treatment of MYC-associated HCC as well as other tumors.
Keywords: MYC, HMG-CoA Reductase, Hepatocellular Carcinoma
Introduction
Hepatocellular carcinoma (HCC) is one of the most common and generally incurable malignancies with an estimated 9% 5-year survival rate (1). Hepatocellular carcinogenesis is strongly associated with hepatitis B and C virus (HBV and HCV) infection, as well as other pathological conditions resulting in liver regeneration (2), which in turn facilitates the activation of specific oncogenes, most notably MYC (3). Thus, targeted inactivation of MYC may be an effective therapy for HCC (4–6). Indeed, we have reported recently that the conditional inactivation of MYC can be sufficient to induce sustained regression of HCC (7). However, there is no existing therapy for patients that targets MYC for the treatment of any cancer.
The MYC proto-oncogene family is comprised of c-MYC, N-MYC, and L-MYC and has been shown to impact almost every aspect of tumorigenesis, including promoting unrestricted cell proliferation, inhibiting cell differentiation, reducing cell adhesion, and enhancing metastasis, genomic instability and angiogenesis (8, 9). MYC functions as an oncogene upon over-expression, either due to increased expression of the myc gene or by increased stability of the MYC protein. MYC protein stability is regulated through the ubiquitin/26S proteasome pathway and the sequential phosphorylation of MYC at Serine 62 (S62) and Threonine 58 (T58) (10). The phosphorylation of S62 is mediated by the MAPK/ERK pathway and contributes to the stabilization of MYC. Subsequent phosphorylation of T58, mediated by GSK3β, promotes ubiquitin-dependent MYC degradation once S62 is dephosphorylated (11–13). Mutations in these phosphorylation sites that stabilize MYC protein have been identified in human cancers, thereby highlighting the importance of S62 and T58 phosphorylation as regulators of MYC in tumorigenesis (14, 15). Hence, targeting MYC phosphorylation could be useful as an anti-cancer therapy.
The enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) controls the rate-limiting step in the mevalonate pathway that is essential for cholesterol biosynthesis (16). Statins were initially utilized to inhibit HMG-CoA reductase as a means to reduce the serum cholesterol level (16). However, many studies have shown that inhibition of HMG-CoA reductase also has anti-tumor efficacy both in vitro and in vivo in multiple tumor types (17–20), including colorectal cancer (21, 22), breast cancer (23), melanoma (24), and lymphoma (25). Statins have been suggested to block tumor cell growth through the inhibition of proliferation and angiogenesis, induction of apoptosis, and repression of tumor metastasis (26).
Statins may mediate their anti-cancer properties through inhibition of the synthesis of lipid isoprenoid intermediates that are produced downstream of the mevalonate pathway, including farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) (19). Generally, FPP activates the Ras GTPase family while GGPP activates the Rho/Rac family by prenylating and anchoring them on the cell membrane (27). Both Ras (12, 27) and Rho/Rac family members (28) are required to phosphorylate MYC. Hence, we speculated, that by inhibiting these pathways, statins might therefore block MYC activation.
Here we show that the inhibition of HMG-CoA reductase by atorvastatin (AT) inhibits MYC phosphorylation and activation and thereby blocks MYC-induced HCC onset and tumor maintenance. Moreover, by using a novel molecular imaging sensor that noninvasively detects c-Myc phosphorylation, we found, both in vitro in human HCC cells and in vivo in mice, that AT inhibits MYC phosphorylation. Furthermore, the introduction of mutant MYC alleles that cannot be phosphorylated on S62 or T58 prevented AT from inhibiting the in vivo tumor growth of HCC. Finally, we provide evidence suggesting that AT may mediate these effects on MYC phosphorylation and activation by inhibiting Rac GTPase. Therefore, HMG-CoA reductase appears to be a critical regulator of MYC activation and may have potent activity against MYC-induced cancers.
Materials and Methods
Antibodies
The antibody to Ki67 was obtained from BD Biosciences (San Jose, CA); c-MYC, Cdk4, E2F1, and Rac1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); phospho-c-MYC was obtained from Cell Signaling Technology (Danvers, MA); antibodies to Tubulin and HA were from Sigma-Aldrich (St Louis, MO). Horseradishperoxidase-conjugated sheep anti-mouse IgG and sheep anti-rabbit IgG were obtained from GE Healthcare (Little Chalfond Buckinghamshire, UK) and Biotinylated anti-mouse IgG was from BD Biosciences.
Cell Lines
The Huh7 and HepG2 cell lines were obtained from the ATCC originally characterized by DNA profile and cytogenetic analysis and were passaged for less than 6 months in vitro.
Transgenic mice
The Tet-system was used previously to generate transgenic mice that conditionally express human c-MYC cDNA in hepatocytes, as described (7). MYC expression was induced by removing doxycycline (Dox, 100μg/ml) from the drinking water of mice. All animals were maintained and treated in accordance with the policies of Stanford University.
Atorvastatin treatment
Atorvastatin (AT, prescription formulation; Pfizer Inc, New York, NY) was resuspended in PBS. AT was administered orally in 100mg/kg doses with or without 20mg/kg mevalonate (MV) 3 times a week using 20-mm feeding needles (Popper and Sons, New Hyde Park, NY). PBS was administered as a negative control.
Purified AT (Sequoia Research Products, Pangbourne, United Kingdom) resuspended in 100% DMSO was used for in vitro studies.
Histology and immunohistochemistry
Tissues were fixed in 10% buffered formalin and embedded in paraffin. Sections of 5 μm were stained with hematoxylin-eosin (H&E), or analyzed by immunohistochemistry using the antibody to Ki67. DAB (Vector Laboratories, Redwood City, CA) was used to achieve color development.
Proliferation assay
Cells were seeded in 24-well plates (5000 cells per well) and incubated overnight. Next, cells were treated with PBS, AT (0.5, 1.0, 2.5, 5.0, 10, or 25 μM), 10 or 25μM AT and 100μM MV, 10 or 25μM AT and 10μM FPP, or 10 or 25μM AT and 10μM GGPP for 96 hours. Cell proliferation was evaluated using the3-(4, 5- dimethylthiazo-2-yl)-2, 5-diphenyltetrazolium bromide(MTT) assay. Data were from six replicated experiments.
Quantitative real-time polymerase chain reaction (qPCR)
HCC cells were treated with PBS, 20 ng/mL Dox, 10μM AT, or 10μM AT and 100μM MV for 24 hours. Total mRNA from HCC cells was extracted and purified using the RNeasy Mini Kit from Qiagen (Austin, TX) and quantified by spectrophotometer (Beckman Coulter, Fullerton, CA). cDNA was reverse-transcribed from 2μg of total mRNA using oligo-d(T) primers. Real-time PCR analysis was performed in an ABI PRIZM analyzer (Applied Biosystems, Foster City, CA).
Cell membrane fractionation and protein isolation
Cells (2x108) were washed 3 times with PBS and extracted in lysis buffer (50 mM Tris, 50 mM NaCl, 2 mM EDTA, 1 mM MgCl2, 10 mM NaF, 1 mM DTT, pH 7.4). Lysates were centrifuged at 36,000 rpm for 40 minutes using Beckman L8-70M ultracentrifuge. The membrane pellet was solubilized in immunoprecipitation buffer (0.15 M NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 10 mM Tris-HCl, pH 7.4) and incubated at 4°C for 30 minutes. The solution was centrifuged at 15,000 rpm for 10 minutes, and the supernatant was collected as the membrane protein fraction.
Viral infection
Ad-c-MYCWT, Ad-c-MYCS62A, and Ad-c-MYCT58A viruses were kindly provided as a gift from Dr Rosalie C. Sears (Oregon Health & Science University, Portland, OR). Briefly, Ad-c-MYCWT, Ad-c-MYCS62A, and Ad-c-MYCT58A adenovirus were cloned by inserting c-MYCWT (Ad-c-MYCWT), c-MYCS62A (Ad-c-MYCS62A) and c-MYCT58A (Ad-c-MYCT58A) cDNA into the pADEasy-1 backbone (Stratagene, La Jolla, CA). Transplanted tumor cells were infected as previously described (29). Briefly, SCID mice were injected with hepatocellular carcinoma cells isolated from LAP-tTA/TRE-MYC transgenic mice. Tumor masses were injected in three sites with Ad-c-MYCWT or Ad-c-MYCT58A once every week. Successful infection was confirmed by GFP co-expression in tumors (Supplementary information, Fig. S9). Mice were treated with 100μg/mL Dox to inactivate transgenic MYC expression. Tumor growth was measured using calipers three times a week for three weeks after viral injection.
Immunoprecipitation and immunoblotting
Cells were lysed in immunoprecipitation buffer (0.15 M NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 10 mM Tris-HCl, pH 7.4) and cleared lysates were immunoprecipitated with 2μg HA antibody. The precipitated proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose, and blotted with the antibodies indicated in the figures. Total MYC and phospho-MYC level were detected by immunoblotting, and their optical density (OD) was measured and normalized to actin band OD. The MYC phosphorylation level was determined by the ratio between phospho-MYC and total MYC.
Molecular imaging of c-Myc phosphorylation
A bioluminescent sensor system that can noninvasively detect c-Myc phosphorylation was utilized to detect the AT inhibitory effect in intact cells and living mice. The sensor system utilizes the fact that S62 phosphorylation of c-Myc is required for its interaction with GSK3β and detects the protein interaction between GSK3β and c-Myc to indirectly report the c-Myc phosphorylation using a split Firefly luciferase (FL) complementation system (30). Specific GSK3β and c-Myc fragments are fused with the inactive C-terminal and N-terminal fragment of the split FL respectively (GSK35-433-CFL/NFL-c-Myc). Phosphorylation-induced interaction between GSK3β and c-Myc brings the two split fragments into close proximity and recovery of the luciferase activity.
The sensor system has been validated both in intact cells and mouse xenograft model, which is described in an independent manuscript (31). For in vitro imaging, the sensor plasmids were transiently transfected into HuH7 and HepG2 cells using Superfect (Qiagen) and Lipofectamine2000 (Invitrogen) reagent respectively. Renilla luciferase gene was co-transfected for the control of the transfection efficiency. 24hrs after transfection, cells were treated with AT with different concentration as indicated for 18hrs. Bioluminescent imaging (BLI) was performed in IVIS 50 (Caliper life science, Hopkinton, MA) after adding 45μg/ml D-Luciferin (Promega) to the cells. Cells were lysed for RL assay (Promega) and western blot analysis after imaging. For in vivo liver tumor imaging, we used a hydrodynamic injection method as previously described (32). Briefly, 2ml of saline solution containing 25μg of the MYC sensor plasmid with CMV promoter was injected into the tail vein within 8 seconds. Mice were imaged in IVIS 200 22hrs after injection based on time course of the sensor expression as determined in control experiments (Supplementary information, Fig. S6 and S7).
Results
Inhibition of HMG-CoA reductase by atorvastatin suppresses MYC-induced HCC
The effects of inhibition of HMG-CoA reductase were examined on HCC growth in vitro and in vivo by administering atorvastatin (AT) to multiple HCC tumor cell lines derived from the LAP-tTA/TRE-MYC transgenic mice, a previously described conditional transgenic model of MYC-induced HCC using the Tet-system (7), in addition to human HCC cell lines. The murine cell lines were all generated from HCC isolated from dual transgenic animals. These cell lines are dependent upon high levels of human MYC expression, which can be inactivated upon treatment with doxycycline (Dox). Hence, in these transgenic tumor-derived cell lines, as a positive control for MYC inactivation, we used Dox to induce the suppression of transgenic MYC. Doses of AT used for our experiments were comparable to previously published studies (33, 34). Moreover, as control for nonspecific effects of statins, we confirmed that effects were reversed with co-treatment with the enzyme product of HMG-CoA reductase, mevalonate (MV). Note that because of marked differences in the pharmacokinetics between mouse and human, AT doses in mice are approximately 50-fold higher than the pharmacologically equivalent dose in humans (35, 36).
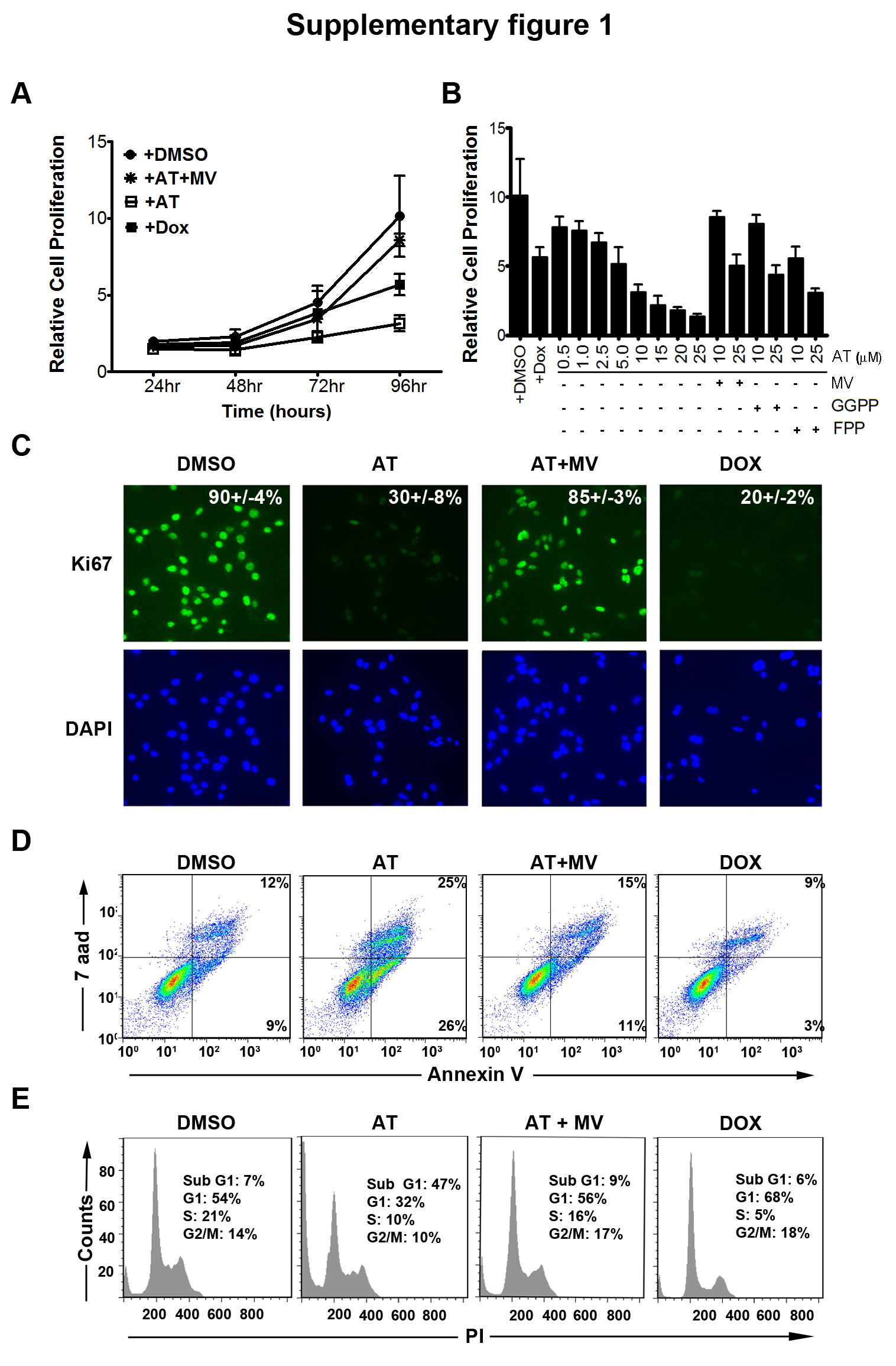
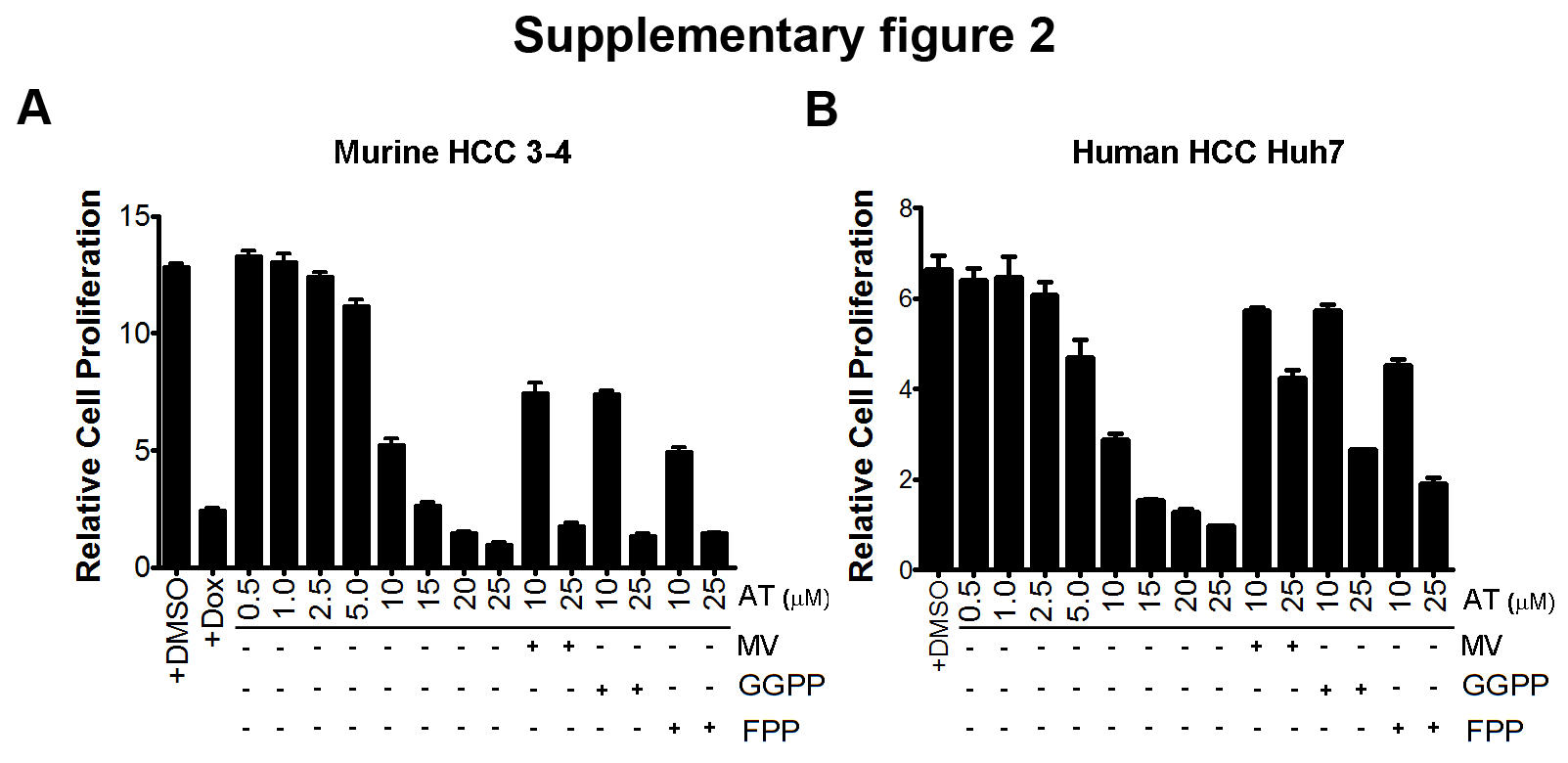
First, AT inhibited the in vitro proliferation of the MYC-induced HCC murine cell line 3-4 as measured by the MTT assay (Fig. 1a and Supplementary information, Fig. S2a; 75% decrease at 10μM and 92% decrease at 25μM AT at 96 hours of AT treatment, p<0.0001 each). Also, AT decreased the number of cells in S-phase and G2/M from 38% to 26% as assessed by propidium iodide staining (Fig. 1b, p=0.01), and reduced Ki67 immunofluorescence from 81.4% to 31.2% following 48 hours of 10μM AT treatment (Fig. 1c, p<0.0001). Importantly, the effects of AT on proliferation and cell cycle arrest were rescued by co-treatment with MV, the immediate downstream target of HMG-CoA reductase (Fig. 1a–c), confirming these effects are specific to inhibition of the cholesterol biosynthesis pathway. Moreover, AT was found to similarly inhibit proliferation and induce cell cycle arrest and apoptosis in two independently derived murine HCC cell lines, EC4 and HCC 4–4, in a dose-dependent manner (Supplementary information, Fig. S1–S3). Thus, AT inhibits proliferation and induces apoptosis of murine HCC tumor cells in vitro.
Figure 1. Inhibition of HMG-CoA reductase by atorvastatin (AT) suppresses growth of MYC-induced hepatocellular carcinoma (HCC) in vitro and in vivo.

a) AT inhibits the proliferation of a MYC-induced HCC tumor derived cell line, 3–4. MTT assay was performed every 24 hours for 4 days on HCC cells treated with 10μM AT, AT plus 100μM MV, or DMSO as a vehicle control. Cells were also treated with doxycycline (Dox) to inactivate transgenic MYC as a positive control. All experiments were repeated 3 times (p<0.0001). Error bars represent standard deviation. b) AT induces cell cycle arrest in murine HCC as assessed by FACS analysis of PI stained cells (p=0.01). Cells were treated with 10μM AT, AT plus MV, DMSO, or Dox for 48 hr. c) Immunofluorescence for Ki67 on HCC cells treated with 10μM AT for 48hr demonstrates that statin treatment inhibits HCC proliferation (p<0.0001). d) AT inhibits growth of MYC-induced HCC cells in vivo. Murine HCC cells were subcutaneously transplanted into FVB/N mice treated with PBS (n=5) or AT (n=5). p=0.0003. Error bars represent standard deviation. e) Representative images of mice treated with PBS (left), AT (middle), or AT plus MV (right) shows that AT suppresses growth of MYC-induced HCC in vivo.
Second, AT inhibited the growth of the murine HCC 3–4 cell line transplanted into syngeneic mice by up to 80% compared with treatment by PBS or AT and MV (Fig. 1d, e, p=0.0003). Note, that FVB/N mice treated with 100mg/kg AT did not exhibit any general toxicity and, in particular, had normal liver histology and serum bilirubin levels, demonstrating the clinical effects are not secondary nonspecific hepatotoxicity (Supplementary information, Fig. S4). Therefore, inhibition of HMG-CoA reductase by AT has potent in vivo anti-tumor activity against murine MYC-induced HCC.
Third, we interrogated the ability of AT to suppress growth in Huh7 cells, a human HCC cell line. AT blocked the in vitro growth of Huh7 cells over a 96-hour time course (Fig. 2a and Supplementary information, Fig. S2b; 69% decrease at 10μM and 86% decrease at 25μM AT, p<0.0001 each) while inhibiting cell cycle progression (Fig. 2b; 75% reduction in S-phase, p=0.003) and reducing Ki67 positivity (Fig. 2c; 58% reduction, p<0.0001) following 48 hours of 10μM AT treatment. Moreover, AT suppressed the in vivo growth of Huh7 cells (Fig. 2d; PBS versus AT, p=0.03; AT versus AT+MV, p=0.04; PBS versus AT+MV, p=0.8). Co-treatment with MV blocked the effects of statin treatment, confirming that the inhibition of human HCC by AT is specific to the suppression of HMG-CoA reductase (Fig. 2a–d).
Figure 2. Blocking HMG-CoA reductase via AT inhibits growth of human HCC cells in vitro and in vivo.

a) MTT analysis of the human HCC cell line, Huh7, treated with 10μM AT demonstrates significant inhibition of growth in vitro (p<0.0001). Error bars represent standard deviation. b) FACS analysis of PI-stained Huh7 cells reveals that AT suppresses cell cycle progression (p=0.0003). Cells were treated with 10μM AT for 48hr. c) Huh7 cells treated with 10μM AT for 48hr were examined by immunofluorescence for Ki67, demonstrating statin-induced reduction in proliferative cells (p<0.0001). d) Huh7 cells were transplanted intraperitoneally into SCID mice, and host animals were treated with PBS, AT, or AT with MV. A Kaplan-Meier curve demonstrates a significant increase in survival of animals treated with AT (PBS versus AT, p=0.03; AT versus AT+MV, p=0.04; PBS versus AT+MV, p=0.8).
Finally, we evaluated the ability of HMG-CoA reductase inhibition to block the initiation of MYC-induced HCC growth in vivo in the LAP-tTA/TRE-MYC transgenic mice treated with PBS, AT, or AT with MV (Fig. 3a, left). Treatment with 100mg/kg AT versus PBS significantly delayed tumor onset and increased survival (Fig. 3a; median survival increased from 80 days to 147 days, p<0.005). Importantly, MV treatment prevented AT from inhibiting tumorigenesis (Fig. 3a). Gross pathology revealed that AT markedly reduced the size and frequency of tumor nodules (Fig. 3b, Left). H&E staining revealed histologically normal liver tissue in AT-treated mice, suggesting a robust inhibition of disease onset (Fig. 3b, Middle). AT-treat animals also exhibited evidence of inhibited cell proliferation, as indicated by reduced Ki67 staining compared to PBS-treated mice (Fig. 3b, Right; 8 ± 3 positive cells versus 422 ± 23 positive cells per field, p<0.02). Hence, inhibition of HMG-CoA reductase by AT is potent at inhibiting MYC-induced liver tumorigenesis.
Figure 3. AT inhibition of HMG-CoA reductase suppresses MYC-induced hepatocellular tumorigenesis.
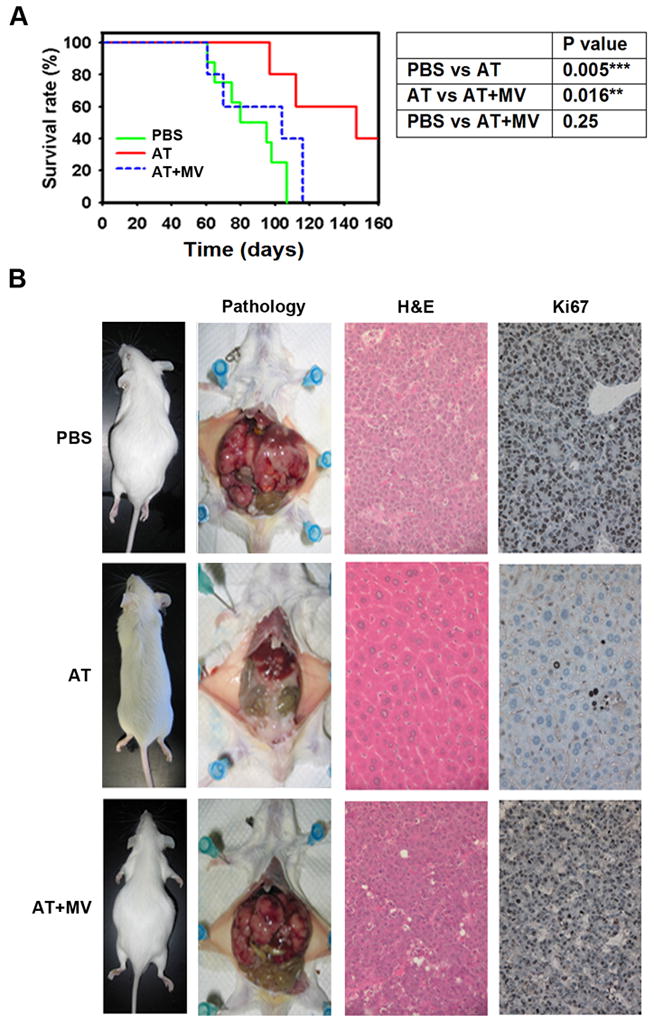
a) Kaplan-Meier survival curves of adult LAP-tTA/TRE-MYC mice treated with PBS (n=8), 100mg/kg AT (n=5), or AT with MV (n=5) three times per week demonstrates a significant increase in survival of animals treated with AT (PBS versus AT, p=0.005; AT versus AT/MV, p=0.016; PBS versus AT/MV, p=0.25). b) Representative pictures from each treatment group are shown. Gross anatomy reveals inhibition of tumor onset due to AT treatment. H&E staining shows that normal hepatic structure is maintained by AT treatment (middle row), which was reversed by MV treatment (bottom row). Immunohistochemistry for Ki67 shows a significant inhibition of proliferation due to AT (right column, 8 ± 3 positive cells versus 422 ± 23 positive cells per high power field; p<0.02).
Inhibition of HMG-CoA reductase suppresses MYC phosphorylation, stability, and transactivation
MYC activation has been shown to be regulated by phosphorylation (12). Thus, we considered that AT might exert its anti-neoplastic effects by inhibiting MYC phosphorylation. Indeed, AT, but not AT with MV, was found to induce a dose-dependent down-regulation of MYC phosphorylation in vitro upon 24 hours treatment (Fig. 4a; 29% reduction at 0.5μM, 34% at 1.0μM, 79% at 2.5μM, 83% at 5.0μM, 94% at 10 μM, and 97% at 25μM AT; p=0.004) as well as in vivo (Fig. 4b; 93% reduction). Moreover, AT blocked MYC phosphorylation in MYC-induced murine lymphoma, osteosarcoma and lung cancer, as well as in human breast cancer cell lines (data not shown). In turn, the de-phosphorylation was associated with a reduction in MYC protein levels in vitro (Fig. 4a; 17% reduction at 0.5μM, 40% at 1.0μM, 9% at 2.5μM, 41% at 5.0μM 69% at 10μM and 91% at 25μM AT, p=0.002) and in vivo (Fig. 4b; 57% reduction, p=0.04). Importantly, the inhibition of MYC phosphorylation by AT not only reduced MYC levels but appeared to dramatically inhibit MYC transcriptional activation as illustrated by the reduced mRNA expression of canonical target genes ODC and nucleolin, both in murine (Fig. 4c; 72% reduction for ODC, p=0.003; 76% reduction for nucleolin, p=0.03) and in human HCC upon 24 hours of 10μM AT treatment (Fig. 4d; 64% reduction for ODC, p=0.016; 59% reduction for nucleolin, p=0.008; Supplementary information, Fig. S8b). Thus, the inhibition of HMG-CoA reductase blocks MYC phosphorylation, reduces MYC protein levels, and inhibits MYC transactivation.
Figure 4. Inhibition of HMG-CoA reductase blocks MYC activity by reducing its phosphorylation.

a) Murine HCC cells were treated with indicated concentrations of AT with or without MV for 24 hours. MYC phosphorylation and expression are suppressed by AT treatment in a dose-dependent manner (p=0.002). Values are normalized to the DMSO control. Representative immunoblots are shown. b) Primary liver tissue of transgenic animals shows an AT-dependent suppression of MYC phosphorylation (p=0.04). Representative immunoblots are shown. Error bars represent standard deviation. c) qPCR analysis of MYC and MYC target gene mRNA expression in murine HCC cells. Treatment of cells with 10μM AT for 24 hours results in reduced MYC transcriptional activity as shown by 72% and 76% reduction in expression of ODC and nucleolin, respectively (p=0.003, p=0.03). Expression is normalized to ubiquitin and values are relative to DMSO control. Error bars represent standard deviation. d) qPCR analysis of MYC and MYC target gene mRNA expression in human Huh7 cells. Cells were treated with 10μM AT for 24hr and demonstrate suppression of MYC transcriptional activity as assessed by reductions of 64% for ODC and 59% for nucleolin expression (p=0.016, p=0.008). Expression is normalized to ubiquitin and values are relative to DMSO control. Error bars represent standard deviation.
Noninvasive molecular imaging to measure in vivo MYC phosphorylation
To evaluate the effects of inhibition of HMG-CoA reductase on MYC phosphorylation in situ in a living host, we utilized a novel molecular imaging sensor system (31). The sensor system consists of two parts: 1) a peptide corresponding to the phospho-regulated domain of MYC fused to the N-terminal domain of Firefly luciferase (FL) and 2) the C-terminal domain of FL fused to a peptide fragment of GSK3β that recognizes phospho-MYC (Fig. 5a). When co-expressed in an intact cell, MYC phosphorylation can be detected via interaction between the MYC and GSK3β peptides, thereby localizing the N- and C-termini of FL in close proximity, conferring luciferase activity. Renilla luciferase (RL) is co-transfected as a control for transfection efficiency. The full-length Firefly luciferase (FL) was also transfected independently into these cells as a control for the direct effect of AT on luciferase activity. We confirmed that this imaging method could detect the dose dependent reduction of c-Myc phosphorylation in human Huh7 and HepG2 cells upon 18hrs of AT treatment (Fig. 6b,c and Supplementary information, Fig. S5, p<0.0001).
Figure 5. A novel bioluminescence c-Myc phospho-sensor noninvasively demonstrates AT-dependent inhibition of MYC phosphorylation.

a) The N- and C-termini of split firefly luciferase (FL) were fused to the phospho-regulated domain of c- Myc and the corresponding domain of GSK3β, respectively. Phosphorylation of the c- Myc peptide results in FL enzymatic activity. b) Huh7 cells were transfected with the c-Myc phosphorylation sensor and treated with AT. Bioluminescent imaging (BLI) shows a dose-dependent inhibition of phospho-MYC (p<0.0001). Firefly luciferase (FL) activity was normalized to Renilla luciferase (RL) activity and plotted against AT concentration. Error bars represent standard deviation. c) Western blot analysis confirms AT-dependent suppression of phospho-c-Myc in transfected Huh7 cells. d) LAP-tTA/TRE-MYC transgenic mice were treated with AT or PBS (n=3), and hydrodynamic injection of the phospho-sensor followed by BLI shows AT-dependent inhibition of c-Myc phosphorylation in vivo (AT-treated mice day 0 versus day 15, p=0.038; PBS-treated mice day 0 versus day 15, p=0.638). e) FL activity was normalized to RL activity and plotted against days of AT treatment.
Figure 6. HCC transduced with S62A or T58A MYC phospho-mutants demonstrate reduced sensitivity to HMG-CoA reductase inhibition.

a) Murine HCC cells were infected with Ad-MYCWT (WT), Ad-MYCS62A (S62A), or Ad-MYCT58A (T58A) adenovirus, and HA-tagged MYC was immunoprecipitated using an antibody to the HA tag. Immunoblot analysis suggests that AT-dependent phospho-regulation of MYC is via S62. However, the inhibition of protein stability requires both S62 and T58 phospho- regulation. Cells were treated with 10μM AT for 24 hr. b) S62A partially and T58A completely abrogated AT inhibition of cell proliferation (S62A: PBS versus AT, p<0.0001; T58A: PBS versus AT p=0.8). Error bars represent standard deviation. c) HCC cells isolated from transgenic animals were transplanted into SCID mice, injected with Ad-MYCWT, Ad-MYCS62A, or Ad-MYCT58A once every week, and orally treated with PBS (n=6), AT (n=5), or AT with MV (n=5) together with Dox. Tumor growth was measured three times per week. d) In vivo growth kinetics of HCC infected with Ad-MYCWT show that AT inhibits tumor growth in vivo (Left, PBS versus AT, p=0.01; AT versus AT+MV, p=0.007). Error bars represent standard deviation. Infection with Ad-MYCS62A partially rescues growth inhibition due to AT (Middle, PBS versus AT, p=0.02; AT versus AT+MV, p=0.03). Error bars represent standard deviation. Ad-MYCT58A completely rescues AT-mediated growth inhibition of HCC (Right, PBS versus AT, p=0.56; AT versus AT+MV, p=0.03). Error bars represent standard deviation.
Next, this imaging sensor was used to monitor MYC phosphorylation in vivo. The sensor system was introduced in liver cells of LAP-tTA/TRE-MYC mice by hydrodynamic injection. Two groups of transgenic mice (n=3 each) had MYC activated at the same time and were treated with either AT or PBS. The MYC sensor was imaged at day 0 and day 15 post-treatment. The PBS-treated group showed no significant change of the sensor signal, while the AT-treated group showed 72% reduction of the sensor signal at day 15 of treatment (Fig. 5d,e; AT-treated mice day 0 versus day 15, p=0.038; PBS-treated day 0 versus day 15, p=0.638). Notably, AT-mediated inhibition of MYC phosphorylation in vivo was associated with a 44% and 56% down-regulation in the expression of downstream target genes, E2F1 and Cdk4 (Supplementary information, Fig. S8a), further demonstrating the inhibition of MYC activity. Hence, a novel imaging sensor was used to show that the inhibition of HMG-CoA reductase by AT inhibits MYC phosphorylation in vivo.
MYC phospho-mutants confer resistance to the inhibition of HMG-CoA Reductase
To examine if inhibition of MYC phosphorylation mediates the anti-cancer effect of AT, we introduced phospho-mutants of MYC into HCC tumor cell lines. First, recombinant adenovirus was used to express MYC that is mutated with either an Alanine substitution at either S62 or T58 (Ad-MYCS62A and Ad-MYCT58A) and hence cannot be regulated by phosphorylation at these sites (37). AT treatment at 10μM for 24 hours dramatically suppressed WT MYC but failed to significantly inhibit protein levels of either S62A MYC or T58A MYC (Fig. 6a; MYCS62A versus MYCS62A+AT, p=0.09; MYCT58A versus MYCT58A+AT, p=0.06). Thus, we concluded that phospho-regulation through either S62 or T58 is necessary for AT to suppress MYC expression.
Using an antibody specific for phospho-S62 and -T58, we show that AT inhibits phosphorylation in T58A MYC similar to wild-type MYC (Fig. 6a, 62% reduction for MYCWT, p=0.01; 68% reduction for MYCT58A, p=0.008). These data show that the reduction of phospho-MYC by AT appears to occur by preventing S62 phosphorylation. However, the reduction of total MYC protein cannot occur without T58 phosphorylation. In addition, HCC cells expressing the S62A MYC mutant were less sensitive whereas cells expressing T58A MYC were completely insensitive to the inhibition of proliferation upon AT treatment (Fig. 6b, 26% reduction for WT, p<0.001; 17% reduction for S62, p<0.0001; no reduction for T58, p=0.8). Thus, inhibition of HMG Co-A reductase suppresses MYC activation in a phosphorylation-dependent manner.
Next, we investigated whether the MYC phospho-mutants could suppress the ability of the inhibition of HMG Co-A reductase to block HCC tumor growth in vivo. Syngeneic hosts were transplanted with MYC-induced HCC cells that were then injected with Ad-MYCWT, Ad-MYCS62A, or Ad-MYCT58A. Tumor growth was monitored in response to AT, PBS, or AT with MV treatment (Fig. 6c). The adenoviral delivery of the MYC phospho-mutants was confirmed by co-expression of GFP (Supplementary information, Fig. S9). To suppress the conditional transgenic MYC expression, mice were treated with doxycycline, thereby resulting in the effective knock-in of the Ad-MYCWT, Ad-MYCS62A, or Ad-MYCT58A constructs into the HCC cells. HCC growth upon injection of Ad-MYCWT showed 66% inhibition by 100mg/kg AT but not by PBS or AT and MV treatment (Fig. 6d, Left, PBS versus AT, p=0.01; AT versus AT+MV, p=0.007). However, HCC tumors that were injected with Ad-MYCS62A exhibited only 44% inhibition of tumor growth upon treatment with AT (Fig. 6d, Middle, PBS versus AT, p=0.02; AT versus AT+MV, p=0.03). Tumors that were injected with Ad-MYCT58A showed complete rescue from sensitivity to AT treatment (Fig. 6d, Right, PBS versus AT, p=0.56; AT versus AT+MV, p=0.03). Therefore, MYC phosphorylation is necessary for AT to inhibit MYC-induced HCC tumor growth.
Inhibition of HMG-CoA reductase may block MYC activity through Rac GTPase
We examined if the inhibition of HMG-CoA reductase blocks MYC activation through Rac GTPases. Statins block production of the isoprenoids farnesyl pyrophosphate and geranylgeranyl pyrophosphate (23, 38, 39). FPP prenylates the Ras, Rheb, and PTP4A3 family while GGPP prenylates the Rac, Rho, and Cdc42 family of small GTPases (16). Previous studies suggest that Ras and Rac/Rho families of GTPases may contribute to the regulation of MYC phosphorylation (25). Thus, the inhibition of HMG-CoA reductase is likely to prevent MYC phosphorylation through these GTPases
To explore the role of GTPases in mediating inhibition of MYC phosphorylation, we performed several experiments. First, to control GTPase activity, we supplemented growth media with either FPP or GGPP before 96hrs AT treatment of MYC-induced murine HCC cells in vitro. Both MV and GGPP restored HCC cell proliferation to levels similar to DMSO controls (Fig. 7a; DMSO versus AT+MV, p=0.07; DMSO versus GGPP, p=0.053), whereas FPP demonstrated significantly less reduction in AT-mediated growth inhibition (DMSO versus FPP, p<0.002). Similarly, in Huh7 cells, GGPP was more potent than FPP in abrogating the effect of 96hrs of 10μM AT on proliferation (Fig. 7b; 16% reduction for DMSO versus AT+MV, p <0.01; 16% reduction for DMSO versus GGPP, p<0.01; 32% reduction for DMSO versus FPP, p<0.001). In addition, GGPP was more efficient in rescuing the transcription of multiple MYC target genes than FPP, including Cdk4 and E2F1, following 24 hours of 10μM AT treatment (Supplementary information, Fig. S10, 40–45% reduction for FPP, p<0.001; 25–60% increase for GGPP, p<0.03). MYC phosphorylation was restored to levels similar to PBS-treated controls when media containing 10μM AT was supplemented with GGPP but not with FPP (Fig. 7c; 76% reduction for FPP, p=0.001; 21% reduction for GGPP, p=0.02). Therefore, inhibition of HMG-CoA reductase appears to inactivate MYC through the inhibition of the Rho/Rac pathway.
Figure 7. HMG-CoA reductase influences MYC phosphorylation through a Rac GTPase-dependent mechanism.

a) Suppression of murine HCC growth in vitro upon 10μM AT treatment for 96 hours is rescued by GGPP treatment, as assessed by MTT (DMSO versus AT+MV, p=0.07; DMSO versus GGPP, p=0.053, DMSO versus FPP, p=0.002). Error bars represent standard deviation. b) GGPP treatment rescues the AT- dependent suppression in human HCC cell growth upon 10μM AT treatment (DMSO versus AT+MV, p=0.01; DMSO versus GGPP, p=0.01, DMSO versus FPP, p<0.001). Error bars represent standard deviation. c) GGPP treatment rescues AT-dependent inhibition of MYC phosphorylation. Representative immunoblots are shown. Error bars represent standard deviation. d) GGPP treatment prevents the decrease in the membrane accumulation of Rac induced by 24 hours of 10μM AT. e) AT treatment inhibits Rac activity. Rac activity was reduced by 77% as measured by pull-down assay. f) Our results suggest a model in which inhibition of HMG-CoA reductase by AT blocks prenylation and activation of small GTPases, specifically including Rac. AT-mediated inhibition of Rac likely results in reduction of phospho-S62 MYC. Dephosphorylation at S62 in the context of phospho-T58 thereby results in the ubiquitin-mediated degradation of MYC. As such, AT treatment ultimately results in the inhibition of MYC oncogenic activity and suppressed hepatocellular carcinogenesis.
Finally, we examined the potential role of the Rac/Rho/Cdc42 pathway as a mechanism by which AT suppresses MYC activation. First, we investigated if AT was influencing the membrane localization of Rac1. Indeed, 24-hour treatment with 10μM AT resulted in the delocalization of Rac1 from the plasma membrane (Fig. 7d; 83% reduction for AT, p<0.001). As a control, we showed that treatment with MV blocked these effects, as did treatment with GGPP (Fig. 7d; no change for PBS versus AT+MV, p=0.08; 37% increase for PBS versus GGPP, p<0.03). Notably, AT had little effect on Ras localization (data not shown). Second, using a Rac pull-down assay, 24 hours of 10 μM AT treatment was shown to reduce Rac1 activity by 77% (Fig. 7e). Thus, the inhibition of HMG-CoA reductase by AT appears to suppress activation of the Rac pathway, suggesting that AT in blocks MYC phosphorylation and activation via inhibition of Rac.
Discussion
Here, we demonstrate that MYC phosphorylation, activation, and thereby tumorigenic potential are regulated by HMG-CoA reductase. In particular, the inhibition of HMG-CoA reductase by statins suppresses MYC phosphorylation and activation. The consequences of these effects on MYC include preventing HCC initiation as well as inhibiting the in vivo growth of established murine and human HCC tumors. Moreover, statins, by blocking HMG-CoA reductase, inhibit GTPase activity, thereby resulting in MYC dephosphorylation and inactivation, which is essential for their anti-cancer therapeutic effect (Fig. 7f). Hence, the inhibition of HMG-CoA reductase by AT may be an effective strategy for the inhibition of MYC in the treatment and prevention of HCC.
Importantly, we confirmed the anti-tumor effects of statins that we observed both in vitro and in vivo were specific to HMG CoA reductase because they could be readily reversed by co-treatment with MV. We note that the AT doses we used in mice appear to be higher than generally used in humans. However, it is well known that, because of differences in the pharmacokinetics, murine doses have to be 50-fold higher than in humans (35, 36). Moreover, we confirmed that the anti-tumor dose of AT did not exhibit in vivo toxicity (Fig. S4) and hence is likely to be achievable in humans.
The inhibition of HMG-CoA reductase was found to block MYC phosphorylation. First, AT treatment blocks MYC phosphorylation in murine and human HCC cells both in vitro and in vivo. Concomitant treatment with MV abrogates the ability of AT to block MYC phosphorylation and activation and mediate its suppressive effect on HCC growth, indicating that the anti-neoplastic effect of AT is via the inhibition of HMG-CoA reductase (Fig. 4). Second, a novel phosphorylation sensor was used to demonstrate in situ in human HCC cells and in vivo in living mice (Fig. 5). This phospho-sensor may be a useful approach to develop therapies that target MYC phosphorylation. Third, mutations in two MYC phosphorylation sites, specifically S62 and T58, blocks the ability of AT to inhibit tumor growth in vivo. Therefore, HMG-CoA reductase activity is important to the regulation of MYC phosphorylation.
We infer that MYC phosphorylation is an essential component of the mechanism by which statins mediate their anti-neoplastic properties. The introduction of mutant MYC alleles in HCC tumor cells reduced their sensitivity to statins in vitro and in vivo. Specifically, the differential ability of S62A and T58A MYC to block the effect of AT in vitro and in vivo (Fig. 6) suggests that AT-mediated reduction in phospho-S62 would therefore result in MYC phosphorylated at only T58, which is rapidly degraded in a ubiquitin-dependent manner (12, 13). Our results are consistent with a role of S62 and T58 phosphorylation in MYC stability and transcriptional activity and, most importantly, their role in tumorigenesis (12). However, we note that S62/T58 phosphorylation has not always been found to regulate MYC stability (40). We conclude that the inhibition of MYC phosphorylation may be important to the mechanism by which the inhibition of HMG-CoA reductase by statins exerts its anti-neoplastic properties.
Many reports suggest that statins have anti-neoplastic properties (25, 41–44). Many mechanisms have been proposed including: the inhibition of the ErbB2 pathway (45), the blocking of the interaction between the lymphocyte function-associated antigen (LFA) and intercellular cell adhesion molecule-1 (ICAM-1) (46), the suppression of geranylgeranylation of the Rho family proteins (47), and the prevention of the prenylation of RhoA and downstream activation of focal adhesion kinase, AKT, and beta-catenin (23). Although we cannot preclude any of these possibilities, our results are consistent with the notion that the inhibition of HMG-CoA reductase by AT in HCC cells blocks MYC phosphorylation likely through the inhibition of small GTPases in the Rac pathway (Fig. 7).
Our results are the first to suggest that HMG-CoA reductase regulates MYC activation via Rac. We are currently investigating the signaling intermediates that may function between Rac and MYC to mediate the anti-neoplastic effect of AT. Previously, it has been suggested that Rac regulates MYC (28). Rac1 can inhibit protein phosphatase PP2A (48), which has been shown to dephosphorylate MYC at S62 (13). One possible mechanism suggested by our work is that AT inhibition of Rac can result in activation of PP2A, which subsequently dephosphorylates MYC at S62 and induces the ubiquitin-dependent degradation of T58-phosphorylated MYC (Fig. 7f).
Our results illustrate that the inhibition of HMG-CoA reductase by statins may be useful in the treatment and prevention of human HCC. HCC is increasing in incidence, has a generally dismal prognosis, and there are few treatment options (49). Statins were developed to inhibit HMG-CoA reductase in the liver to reduce cholesterol. Hence, they may be aptly suited for treating cancers of the liver. Moreover, statins are well tolerated in humans and may be useful in the prevention of HCC in patients at high risk (49). Notably, some clinical studies have suggested that that the statin, pravastatin, may have clinical activity in patients with HCC (50, 51), while other studies have not found clinical benefit (52). A possible explanation for this possible discrepancy in the benefit from statins is that clinical activity could depend upon the activation status of MYC. Also, AT may be a more effective statin for the treatment of HCC.
We conclude that MYC phosphorylation is a critical mechanism by which the inhibition of HMG-CoA Reductase by statins mediates their anti-neoplastic effects. We have shown that a novel molecular imaging sensor may be useful for the identification through high-throughput methods of new therapeutic agents that inhibit MYC phosphorylation and activation. Importantly, statins may be effective agents to inhibit MYC function as a treatment for HCC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We would like to dedicate this paper in memory of Julie Do. We would like to thank Dr Rosalie C. Sears (Oregon Health & Science University, Portland, OR) for kindly providing us with mutant Myc constructs. We also value the helpful comments provided by Dr. Stacey Adam and the members of the Felsher, Sylvester, and Gambhir laboratories.
Financial Support: This work was supported by National Institutes of Health grants CA89305–01A1, CA89305–0351, CA105102, and CA112973; Department of Defense grant PR080163, the Burroughs Wellcome Fund; the Leukemia and Lymphoma Society; the Damon Runyon Foundation (D.W.F.); the in vivo cancer molecular image center (ICMIC P50) at Stanford grant CA114747 (S.S.G, D.W.F.). Dr. Zhongwei Cao was supported by an American Liver Foundation (A.L.F.) fellowship. Dr. Hua-Fan Minogue is supported by a NIH R25T training grant. Dr. David I. Bellovin is supported by the NIH under NRSA fellowship F32-CA132312.
Footnotes
Conflicts of Interest: None
References
- 1.Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 2.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 3.Riehle KJ, Campbell JS, McMahan RS, et al. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J Exp Med. 2008;205:91–103. doi: 10.1084/jem.20070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coleman WB. Mechanisms of human hepatocarcinogenesis. Curr Mol Med. 2003;3:573–88. doi: 10.2174/1566524033479546. [DOI] [PubMed] [Google Scholar]
- 5.Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–11. doi: 10.1038/ng1481. [DOI] [PubMed] [Google Scholar]
- 6.Thorgeirsson SS, Factor VM, Snyderwine EG. Transgenic mouse models in carcinogenesis research and testing. Toxicology letters. 2000;112–113:553–5. doi: 10.1016/s0378-4274(99)00224-6. [DOI] [PubMed] [Google Scholar]
- 7.Shachaf CM, Kopelman AM, Arvanitis C, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–7. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 8.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 9.Kaposi-Novak P, Libbrecht L, Woo HG, et al. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009;69:2775–82. doi: 10.1158/0008-5472.CAN-08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salghetti SE, Kim SY, Tansey WP. Destruction of Myc by ubiquitin-mediated proteolysis: cancer-associated and transforming mutations stabilize Myc. EMBO J. 1999;18:717–26. doi: 10.1093/emboj/18.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 12.Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14. doi: 10.1101/gad.836800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh E, Cunningham M, Arnold H, et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat Cell Biol. 2004;6:308–18. doi: 10.1038/ncb1110. [DOI] [PubMed] [Google Scholar]
- 14.Bhatia K, Huppi K, Spangler G, Siwarski D, Iyer R, Magrath I. Point mutations in the c-Myc transactivation domain are common in Burkitt's lymphoma and mouse plasmacytomas. Nat Genet. 1993;5:56–61. doi: 10.1038/ng0993-56. [DOI] [PubMed] [Google Scholar]
- 15.Smith-Sorensen B, Hijmans EM, Beijersbergen RL, Bernards R. Functional analysis of Burkitt's lymphoma mutant c-Myc proteins. J Biol Chem. 1996;271:5513–8. doi: 10.1074/jbc.271.10.5513. [DOI] [PubMed] [Google Scholar]
- 16.Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM. Statins and cancer prevention. Nat Rev Cancer. 2005;5:930–42. doi: 10.1038/nrc1751. [DOI] [PubMed] [Google Scholar]
- 17.Shibata MA, Ito Y, Morimoto J, Otsuki Y. Lovastatin inhibits tumor growth and lung metastasis in mouse mammary carcinoma model: a p53-independent mitochondrial-mediated apoptotic mechanism. Carcinogenesis. 2004;25:1887–98. doi: 10.1093/carcin/bgh201. [DOI] [PubMed] [Google Scholar]
- 18.Feleszko W, Mlynarczuk I, Balkowiec-Iskra EZ, et al. Lovastatin potentiates antitumor activity and attenuates cardiotoxicity of doxorubicin in three tumor models in mice. Clin Cancer Res. 2000;6:2044–52. [PubMed] [Google Scholar]
- 19.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 20.Wong WW, Dimitroulakos J, Minden MD, Penn LZ. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis. Leukemia. 2002;16:508–19. doi: 10.1038/sj.leu.2402476. [DOI] [PubMed] [Google Scholar]
- 21.Narisawa T, Morotomi M, Fukaura Y, Hasebe M, Ito M, Aizawa R. Chemoprevention by pravastatin, a 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, of N-methyl-N-nitrosourea-induced colon carcinogenesis in F344 rats. Jpn J Cancer Res. 1996;87:798–804. doi: 10.1111/j.1349-7006.1996.tb02103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agarwal B, Rao CV, Bhendwal S, et al. Lovastatin augments sulindac-induced apoptosis in colon cancer cells and potentiates chemopreventive effects of sulindac. Gastroenterology. 1999;117:838–47. doi: 10.1016/s0016-5085(99)70342-2. [DOI] [PubMed] [Google Scholar]
- 23.Denoyelle C, Albanese P, Uzan G, et al. Molecular mechanism of the anti-cancer activity of cerivastatin, an inhibitor of HMG-CoA reductase, on aggressive human breast cancer cells. Cell Signal. 2003;15:327–38. doi: 10.1016/s0898-6568(02)00124-9. [DOI] [PubMed] [Google Scholar]
- 24.He L, Mo H, Hadisusilo S, Qureshi AA, Elson CE. Isoprenoids suppress the growth of murine B16 melanomas in vitro and in vivo. J Nutr. 1997;127:668–74. doi: 10.1093/jn/127.5.668. [DOI] [PubMed] [Google Scholar]
- 25.Shachaf CM, Perez OD, Youssef S, et al. Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis. Blood. 2007;110:2674–84. doi: 10.1182/blood-2006-09-048033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hindler K, Cleeland CS, Rivera E, Collard CD. The role of statins in cancer therapy. Oncologist. 2006;11:306–15. doi: 10.1634/theoncologist.11-3-306. [DOI] [PubMed] [Google Scholar]
- 27.Jackson SM, Ericsson J, Edwards PA. Signaling molecules derived from the cholesterol biosynthetic pathway. Subcell Biochem. 1997;28:1–21. doi: 10.1007/978-1-4615-5901-6_1. [DOI] [PubMed] [Google Scholar]
- 28.Chiariello M, Marinissen MJ, Gutkind JS. Regulation of c-myc expression by PDGF through Rho GTPases. Nat Cell Biol. 2001;3:580–6. doi: 10.1038/35078555. [DOI] [PubMed] [Google Scholar]
- 29.Mesri M, Wall NR, Li J, Kim RW, Altieri DC. Cancer gene therapy using a survivin mutant adenovirus. J Clin Invest. 2001;108:981–90. doi: 10.1172/JCI12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulmurugan R, Gambhir SS. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal Chem. 2003;75:1584–9. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan-Minogue H, Cao Z, Paulmurugan R, et al. Noninvasive molecular imaging of c-Myc activation in living mice. Proc Natl Acad Sci U S A. 107:15892–7. doi: 10.1073/pnas.1007443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 33.Black AE, Sinz MW, Hayes RN, Woolf TF. Metabolism and excretion studies in mouse after single and multiple oral doses of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor atorvastatin. Drug Metab Dispos. 1998;26:755–63. [PubMed] [Google Scholar]
- 34.Lubet RA, Boring D, Steele VE, Ruppert JM, Juliana MM, Grubbs CJ. Lack of efficacy of the statins atorvastatin and lovastatin in rodent mammary carcinogenesis. Cancer Prev Res (Phila) 2009;2:161–7. doi: 10.1158/1940-6207.CAPR-08-0134. [DOI] [PubMed] [Google Scholar]
- 35.Pinkel D. The use of body surface area as a criterion of drug dosage in cancer chemotherapy. Cancer Res. 1958;18:853–6. [PubMed] [Google Scholar]
- 36.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 37.Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol. 1994;14:5510–22. doi: 10.1128/mcb.14.8.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denoyelle C, Vasse M, Korner M, et al. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast cancer cell lines: an in vitro study. Carcinogenesis. 2001;22:1139–48. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- 39.Jakobisiak M, Bruno S, Skierski JS, Darzynkiewicz Z. Cell cycle-specific effects of lovastatin. Proc Natl Acad Sci U S A. 1991;88:3628–32. doi: 10.1073/pnas.88.9.3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henriksson M, Bakardjiev A, Klein G, Luscher B. Phosphorylation sites mapping in the N-terminal domain of c-myc modulate its transforming potential. Oncogene. 1993;8:3199–209. [PubMed] [Google Scholar]
- 41.Gniadecki R. Depletion of membrane cholesterol causes ligand-independent activation of Fas and apoptosis. Biochem Biophys Res Commun. 2004;320:165–9. doi: 10.1016/j.bbrc.2004.05.145. [DOI] [PubMed] [Google Scholar]
- 42.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105:739–45. doi: 10.1161/hc0602.103393. [DOI] [PubMed] [Google Scholar]
- 44.Tilkin-Mariame AF, Cormary C, Ferro N, et al. Geranylgeranyl transferase inhibition stimulates anti-melanoma immune response through MHC Class I and costimulatory molecule expression. FASEB J. 2005;19:1513–5. doi: 10.1096/fj.04-3482fje. [DOI] [PubMed] [Google Scholar]
- 45.Mueck AO, Seeger H, Wallwiener D. Effect of statins combined with estradiol on the proliferation of human receptor-positive and receptor-negative breast cancer cells. Menopause. 2003;10:332–6. doi: 10.1097/01.GME.0000055485.06076.00. [DOI] [PubMed] [Google Scholar]
- 46.Chan KK, Oza AM, Siu LL. The statins as anticancer agents. Clin Cancer Res. 2003;9:10–9. [PubMed] [Google Scholar]
- 47.Agarwal B, Halmos B, Feoktistov AS, et al. Mechanism of lovastatin-induced apoptosis in intestinal epithelial cells. Carcinogenesis. 2002;23:521–8. doi: 10.1093/carcin/23.3.521. [DOI] [PubMed] [Google Scholar]
- 48.ten Klooster JP, Leeuwen I, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J. 2007;26:336–45. doi: 10.1038/sj.emboj.7601518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular Carcinoma Incidence, Mortality, and Survival Trends in the United States From 1975 to 2005. J Clin Oncol. 2009 doi: 10.1200/JCO.2008.20.7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Graf H, Jungst C, Straub G, et al. Chemoembolization combined with pravastatin improves survival in patients with hepatocellular carcinoma. Digestion. 2008;78:34–8. doi: 10.1159/000156702. [DOI] [PubMed] [Google Scholar]
- 51.Kawata S, Yamasaki E, Nagase T, et al. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial. Br J Cancer. 2001;84:886–91. doi: 10.1054/bjoc.2000.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lersch C, Schmelz R, Erdmann J, et al. Treatment of HCC with pravastatin, octreotide, or gemcitabine--a critical evaluation. Hepatogastroenterology. 2004;51:1099–103. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.