

Abstract
Gaucher disease, a prevalent lysosomal storage disease, is caused by insufficient activity of acid β-glucosidase (GCase) and resultant glucosylceramide accumulation. Recently in Parkinson disease (PD) patients, heterozygous mutations in GCase have been associated with earlier onset and more progressive PD. To understand the pathogenic relationships between GCase variants and Parkinsonism, α-synuclein and ubiquitin distributions and levels in the brains of several mouse models containing GCase variants were evaluated by immunohistochemistry. Progressive α-synuclein and ubiquitin aggregate accumulations were observed in the cortex, hippocampus, basal ganglia, brainstem, and some cerebellar regions between 4-24 wks in mice that were homozygous for GCase [D409H (9H) or V394L (4L)] variants and also had a prosaposin hypomorphic (PS-NA) transgene. In 4L/PS-NA and 9H/PS-NA mice, this was coincident with progressive neurological manifestations and brain glucosylceramide accumulation. Ultrastructural studies showed electron dense inclusion bodies in neurons and axons of 9H/PS-NA brains. α-Synuclein aggregates were also observed in ventricular, brainstem, and cerebellar regions of older mice (>42-wk) with the GCase variant (D409H/D409H) without overt neurological disease. In a chemically induced GCase deficiency, α-synuclein aggregates and glucosylceramide accumulation also occurred. These studies demonstrate a relationship between glucosylceramide accumulation and α-synuclein aggregates, and implicate glucosylceramide accumulation as risk factor for the α-synucleinopathies.
Keywords: Glucocerebrosidase (GCase), lysosomal storage diseases, Parkinsonism, α-synuclein, ubiquitin
Introduction
Gaucher disease, an autosomal recessive disorder, is a common lysosomal storage disease [1, 2] that results from insufficient activity of acid β-glucosidase (GCase, encoded by the GBA1 gene) and resultant accumulation of its substrates glucosylceramide and glucosylsphingosine. Accumulations of glucosylceramide and glucosylsphingosine produce the visceral and CNS manifestations by as yet ill-defined mechanisms. Classically, the three clinical phenotypes include the “non-neuronopathic” (type 1), and “neuronopathic” (type 2 and 3) variants [1, 3, 4]. In all variants, glucosylceramide engorged visceral macrophages or “Gaucher cells” are a hallmark of the disease. In the “neuronopathic “variants, the CNS pathology includes neuronal cell death.
The distinction between the “nonneuronopathic” and “neuronopathic” variants has become somewhat blurred. Recent studies showed that Parkinson disease patients have a 3- to 7-fold increased risk of being heterozygous for GBA1 variants [5-13]. Such associations with Parkinsonism are not related to specific GBA1 mutations [14]. Parkinsonism also occurs in Gaucher disease type 1 or 3 patients and in heterozygotes for GBA1 mutations [6-8, 11], but the risk of GBA1 heterozygotes of developing Parkinsonism is unknown. Parkinson signs and symptoms include memory loss, resting tremor, uncontrolled movements, kinetic rigidity syndrome, asymmetric onset, horizontal myoclonus, supranuclear gaze palsy, typical progression rigidity, difficulty ambulating, and bradykinesia [5-13, 15]. This spectrum of manifestations is similar in persons with or without GBA1 mutations, but can be more severe in their presence. These observations indicate that mutant GBA1 even in heterozygotes, is a significant risk factor for potentiating the effects of Parkinsonism. Neither the basis for these effects or the general pathology and their relationships to glucosylceramide accumulation are known.
These findings contrast with the neuropathology of Gaucher Disease types 2 and 3 in which neuronal loss and degeneration are the most consistent findings, particularly in the basal ganglia, nuclei of the midbrain, pons and medulla, cerebellum, dentate nucleus and hypothalamus [16-19]. Cerebral cortical laminar necrosis [16, 19] and neuronal loss with astrogliosis [20, 21] also have been reported, but only in some type 2 patients. Importantly, α-synuclein inclusion-associated neurodegenerative lesions (α-synucleinopathies) were reported in similar brain regions from some PD patients who also had Gaucher disease type 1 [7, 15, 22].
α-Synuclein is a small presynaptic cytosolic protein that is abundant in nerve terminals of dopaminergic system. Its normal function is incompletely defined, but it has been implicated in dopamine metabolism and synaptic vesicle homeostasis [23-25]. Mutations in the gene for α-synuclein (e.g., Ala30Pro or Ala53Thr) have been implicated directly in the pathogenesis of Parkinson disease [26, 27], as has the over-expression of a human wild-type α-synuclein [28-30]. The presence of α-synuclein insoluble intracellular aggregates (Lewy bodies) is a feature of Parkinson disease and other neurodegenerative disorders [31, 32]. Although α-synuclein aggregates have been observed in several lysosomal diseases and their animal analogues, only GBA1 mutations show a clear and, potentially direct risk association with α-synucleinopathies and Parkinson disease for this reason these shared clinical and neuropathologic findings suggested that GBA1 mutations or glucosylceramide excess act as contributory risk factors that interfere with the clearance of or promote the aggregation of α-synuclein in some patients.
Here, Gba1 point-mutated mice bearing a prosaposin hypomorph (4L/PS-NA and 9H/PS-NA) [33] or having CBE induced-GCase deficiency [34] were used as models of Gaucher disease. Prosaposin is a precursor of the four saposins A, B, C, and D that are essential proteins for the optimal activity of selected glycosphingolipid hydrolyases [35]. Saposin C optimizes GCase hydrolysis of glucosylceramide and other substrates, as well as protecting GCase from proteolytic digestion [35, 36]. Saposin C's protective function accounts for the decreased GCase protein and activity with excess glucosylceramide in the CNS and visceral organs of 4L/PS-NA and 9H/PS-NA mice [33]. Extensive histological and immunohistological analyses of such mice showed a particular pattern of α-synuclein accumulation that implicates mutant GCase and/or excess glucosylceramide in the development of α-synuclein accumulation.
Materials and Methods
Materials
The following were from commercial sources: Conduritol B epoxide (CBE, Calbiochem, San Diego, La Jolla, CA). 4-methyl-umbelliferyl-β-D-glucopyranoside (4MU-Glc; Biosynth AG, Switzerland). Sodium taurocholate and Protease Inhibitor Cocktail (Calbiochem, La Jolla, CA). Triton X-100 (Sigma, St. Louis, MO). Antibody sources are as follows: mouse monoclonal anti-α-synuclein, rabbit polyclonal anti-mouse α-synuclein, rabbit polyclonal anti-ubiquitin and tyrosine hydroxylase (Abcam, Inc. Cambridge, MA). Rat monoclonal to CD68 (Serotec, Raleigh, NC). Mouse monoclonal anti-GFAP (Sigma, St. Louis, MO). Mouse monoclonal anti-β-actin antibody (Sigma, St. Louis, MO). Goat anti-rabbit/rat (Alexa-488, FITC), or Goat anti-mouse-biotinylated antibody with streptavidin-Alexa-610 (Molecular Probes, Irvine, CA). Hybond ECL Nitrocellulose Membrane (Amersham, Piscataway, NJ). 10% Bis-Tris Gel (Invitrogen, Carlsbad, CA). BCA Protein Assay Kit, Peroxidase-Conjugated Goat anti-mouse IgG, M-Per Mammalian Protein Extraction Reagent and ECL Kit (PIERCE, Rockford, IL).
Gaucher disease and other lysosomal disease mouse models
Several GCase, prosaposin, and other lysosomal disease mouse models were analyzed (Table 1). The homozygous point-mutated Gba1 mice, V394L (4L), D409H (9H), and D409V (9V), have normal life spans and no overt CNS disease [37]. Hypomorphic prosaposin mice (PS-NA) and prosaposin knockout mice (PS-/-) had glucosylceramide accumulation in the CNS with CNS degenerative disease [33, 38, 39]. PS-NA mice that were also homozygous for V394L or D409H (4L/PS-NA or 9H/PS-NA, respectively), exhibit greater excesses of glucosylceramide accumulation in the CNS than PS-NA mice [33] at least in part due to additive deficiencies of GCase activity. These mice were used here as models for chronic effects of GCase deficiency and glucosylceramide accumulation since no other viable models for the CNS variants of GCase deficiency available. Other mice with lysosomal storage diseases were used for comparative purposes and included those with individual saposin deficiencies (saposin A, B, C or D), Niemann-Pick type C1 (NPC1), mucopolysaccharidosis type I (MPS I), and lysosomal acid lipase (LAL) deficiency. NPC1 is characterized by intracellular accumulations of free cholesterol and glycosphingolipids, including glucosylceramide, in the endosomal/lysosomal compartment [40]. MPS I, α-L-iduronidase deficiency, leads to defective catabolism of the glycosaminoglycans heparan and dermatan, sulphate, and have secondary accumulations of GM2 in neurons [41]. Lysosomal acid lipase is essential for the hydrolysis of the triglycerides and cholesteryl esters in lysosomes and deficient mice do not exhibit overt CNS disease during their 8-10 month lifespan [42]. Age-matched wild-type mice in the FVB background were from Jackson Laboratory (FVB/NJ, Stock No 001800). The mice were maintained in microisolators in accordance with institutional guidelines under IACUC approval at the animal facility of Cincinnati Children's Hospital Research Foundation. Brains were collected from each genotype at the designated times for histological and biochemical analyses.
Table 1. Gaucher and LSD mouse models.
| Genotypes | Life span (wks) | GCase activity1(%WT) | Brain GC level2 | CNS phenotype | References |
|---|---|---|---|---|---|
| Variant Gaucher disease mice | |||||
| 9H/PS-NA | 22 | 34.2 | 2-fold 3 | Wobble/ataxia | [33] |
| 4L/PS-NA | 22 | 11.0 | 4-fold 3 | Wobble/ataxia | |
| GCase point mutant | |||||
| V394L/V394L | >100 | 27.4 | - | Normal | [37] |
| D409H/D409H | >100 | 25.6 | - | Normal | [34] |
| D409V/D409V | >100 | 22.5 | - | Normal | |
| CBE induced Gaucher disease mice | >16 | 12.9 | + | Wobble/ataxia/seizure | [34] |
| V394L/V394L | >16 | 13.1 | + | Wobble/ataxia/seizure | |
| D409H/D409H | >16 | 7.2 | + | Wobble/ataxia/seizure | |
| D409V/D409V | >16 | 10.2 | + | Wobble/ataxia/seizure | |
| WT | |||||
| LSD mouse models | |||||
| PS-NA | 28 | 88 | + | Wobble/ataxia | [39] |
| PS-/- | 4 | 80 | + | Ataxia/seizure | [36, 38] |
| LAL | 20 | N/A4 | N/A4 | Normal | [42] |
| NPC1 | 13 | N/A4 | + | Ataxia/seizure | [72] |
| MPS1 | 52 | N/A4 | N/A4 | Degeneration | [41, 73] |
GCase activity in the brain;
Glucosylceramide levels in the brain determined by TLC: (-) undetectable, (+) low level, (++) moderate level;
Glucosylceramide level compared with that in WT by LC/MS;
N/A, data not available.
Histological studies
For histological studies, 3 to 4 mice from each genotype at the indicated ages, a minimum of 2 sections from each tissue were examined. Brains were dissected after perfusion with 1 × PBS, pH 7.4 and 4% paraformaldehyde in 1 × PBS, pH 7.4. The brains were post fixed overnight in 10% formalin or 4% paraformaldehyde, and then processed to paraffin or frozen block, respectively. Karnovsky's fixative was used for ultrastructural studies. Serial coronal and sagittal sections of 8 μm thickness were processed for H&E, silver staining, and immunohistochemistry analyses. For immunofluorescence, paraffin sections were deparaffinized in xylene and frozen sections were fixed in 100% acetone at -20°C for 10 min. The sections were blocked with 10% goat serum/PBS plus 0.4% Triton X-100, and then incubated with primary antibody against α-synuclein (1:200), ubiquitin (1:500), glial fibrillary acidic protein (GFAP, 1:500), CD68 (1:300), or tyrosine hydroxylase (1:300) followed by successive incubations with the appropriate biotin-conjugated anti-rabbit or anti-mouse antibodies. The signals were detected by Alexa-488, FITC, or streptavidin-Alexa-610. Antibodies used for double staining were tested individually and showed similar signal intensities. Images were captured with a Zeiss Apotome microscope (AxioV200) at excitation of 506 nm (for Alexa-488, FITC) or 590 nm (for Alexa-610, Rhodamine). In general, α-synuclein signals (Alexa-610, red) were captured with a green background (with FITC filter) to distinguish the specific signals from autofluorescence, except in dual antibody studies. Cell nuclei were stained with DAPI (blue). For negative controls, respective sections were processed for immunoreactivity without primary antibody or with pre-immune (mouse or rabbit) serum as the primary antibodies. The signal locations in brain sections were according to The Mouse Brain in Stereotaxic Coordinates [43]. For immunohistochemistry study, brain sections were incubated with rat anti-mouse CD68 monoclonal antibody and detected with ABC Vectastain and DAB peroxidase substrate [33].
Immunoblots
The cerebral cortex tissues from 4L/PS-NA, 9H/PS-NA and WT brains were microdissected and homogenized (1:5, wt/vol) in Mammalian Protein Extraction Reagent (M-Per) with a douncer (10-12 strokes). Cortex lysate protein (100 μg) was subjected to 10% SDS-PAGE. Immunoblots were pre-treated with 3% BSA in PBS-0.05% Tween-20 for 1 h and then incubated with the primary antibody, mouse monoclonal anti-α-synuclein, (1:1,000) in PBS. The blot was then incubated with peroxidase-conjugated goat anti-mouse IgG, in 1% BSA-PBS (1:1,500) and developed using ECL kit. Pure α-synuclein protein (∼ 14 KD) was used as molecular weight reference. The developed immunoblots were stripped, and probed with anti-mouse β-actin as a loading control.
CBE-treated mice
Gba1 point mutated 4L, 9H and 9V mice or WT mice were intraperitoneally injected with 100 mg/kg/day of CBE [34, 44]. In short-term experiments, daily injections were initiated at postnatal day 5 and continued for 6 daily doses. The mice were sacrificed on day 12 or 2 months after last injection. In long-term experiments, 4L mice were injected daily beginning at postnatal day 15 for 24 or 36 daily doses and sacrificed the day after last injection. Mice were perfused with PBS and organs were harvested for enzyme activity, lipid and histological analyses.
GCase enzyme activity
For GCase activities, tissues (∼50 mg) were homogenized [1:10 (mg: μl) in 1% sodium taurocholate/Triton-X100 (TC/TX), sonicated (4°C, 30s × 3), and clarified (10,000×g; 5 min, 4°C) [45]. The assay mixtures were pre-incubated in the presence or absence of CBE (1 mM; 30 min at room temperature), and then substrate (4MU-Glc) was added and incubated (1 h, 37°C). Protein concentrations were determined using the BCA kit according to manufacturer's instructions.
Tissue lipid analyses
Anesthetized mice were perfused with 3 blood volumes of normal saline. The brains were immediately microdissected to isolate the cerebral cortices, cerebella, hippocampi, and brainstems. Brain tissues were homogenized in 1% TC/TX and protein concentrations were determined using the BCA kit. Glucosylceramide and glucosylsphingosine in the tissue extracts were analyzed by LC/MS (Lipidomic Core, University of South Carolina). The glucosylceramide and glucosylsphingosine amounts were normalized to the extract protein in each sample and expressed as pmol/mg protein. Three mice for each genotype were included in the analyses.
Results
Neurological and pathological phenotypes of Gaucher mouse models
The homozygotes point-mutated Gba1 (4L, 9H, and 9V) have 22-27% of WT GCase activity in the CNS as assayed with the 4-MU-Glc substrate, and no CNS glucosylceramide accumulation by TLC, or gross CNS phenotypes (Table 1) [37]. Crossing the 9H and 4L mice into hypomorphic prosaposin (PS-NA) mice led to severe neurological phenotypes and foreshortened life spans (∼22 wks). The resultant 9H/PS-NA and 4L/PS-NA mice appeared phenotypically normal until ∼10 wks, and then developed progressive ataxia, generalized tremor, and gross shaking to the point of falling over. Lack of rear limb control progressed to nearly complete paralysis by ∼20 wks. The lack of body control eventually resulted in the inability to eat and drink, with consequent malnutrition, dehydration, and death (∼22 wks). Histological studies of 9H/PS-NA brains showed degenerating neurons in the most regions of the brain including cerebral cortex (Fig. 1), which was highly similar to the changes in the PS-NA and 4L/PS-NA mice [33]. Ultrastructural analyses revealed electron dense amorphous inclusions in the cytoplasm of neurons and in axons of 9H/PS-NA cortex (Fig. 1D and F, arrows). Brain glucosylceramide and glucosylsphingosine levels in 4L/PS-NA and 9H/PS-NA mice were higher than those in age-matched WT or PS-NA mice (Fig. 2A). In cerebellum, glucosylceramide levels were ∼4-, 10-, or 6-fold higher in PS-NA, 4L/PS-NA, or 9H/PS-NA mice, respectively, than those in WT mice (Fig. 2A). In the hippocampus and brainstem, glucosylceramide levels were ∼2 (9H/PS-NA mice) to 4-fold (4L/PS-NA mice) higher than those in WT mice; PS-NA mice showed almost WT levels (Fig. 2A). Glucosylsphingosine levels were increased from 6 to 28- fold in 9H/PS-NA brains and ∼20 to 117-fold in 4L/PS-NA brains (Fig. 2B), whereas levels in PS-NA mice were comparable to WT levels. These biochemical and neurological findings in the 9H/PS-NA and 4L/PS-NA mice provided data for correlation of pathologic findings and potential insights of relationships between Gaucher disease and Parkinsonism.
Figure 1. Pathology in 9H/PS-NA brain.

Brain sections were from 20-wk 9H/PS-NA mice. Upper panels: H&E staining sections showed (A) Normal neuron cells in WT and (B) axonal spheroids (arrows) suggesting degenerative neurons in cerebral cortex of 9H/PS-NA mice. Lower panels: Electron micrographs of neurons in 9H/PS-NA and WT deep cortical layer. (C) Neuron in heterozygote littermate. (D) Degenerating neurons in 9H/PS-NA mice showed irregular nuclear membranes with condensed chromatin (n), multiple mitochondria (m) and filament structures, and electronic dense particles (arrows) in the cytoplasm. (E) The axons in 9H/PS-NA brain showed accumulations of complex electron dense inclusion bodies (arrows) that were amorphous. The myelin sheaths of axons appeared normal in 9H/PS-NA mice. The scale bars are shown in each image. n=nuclei.
Figure 2. Glucosylceramide and glucosylsphingosine in the brain of disease mice.

Cortex, cerebellum, hippocampus, and brainstem were dissected from 4L/PS-NA, 9H/PS-NA, PS-NA and WT mice for GSL analyses by LC/MS. (A) Glucosylceramide was 2 to 4-fold increased in cortex, cerebellum, hippocampus and brainstem of 9H/PS-NA and 4L/PS-NA mice, but only slightly increased in PS-NA mice. (B) Brain glucosylsphingosine was increased >10-20 fold in 4L/PS-NA, 3-5 fold in 4H/PS-NA, and unchanged in PS-NA.
Distribution of α-synuclein in brain sagittal section
Serial sagittal brain sections from 10 to 12-wk 9H/PS-NA, 4L/PS-NA, and PS-NA mice were examined by immunofluorescence microscopy to provide an overview of brain α-synuclein distribution. Such brain sections from 9H/PS-NA mice showed α-synuclein signals (Alexa-610, red) concentrated in two areas: 1) Deep cerebral cortical layer: the area overlaying the corpus callosum (cc) from dorsal tenia tecta (DTT) and ventral orbital cortex (VO) through dorsal peduncular cortex (DP), prelimbic cortex (PrL), cingulate cortex area 1 (Cg1), and in the secondary motor cortex (M2) to the retrosplenial agranular cortex (RSA) and retrosplenial granular cortex (RSGa) (Fig. 3 and supplemental Fig. 1). α-Synuclein signals were in ∼5-20 μm aggregates within these regions in 9H/PS-NA (Fig. 3A), A FITC filter was used to make the background green allowing visualization of the brain fields, and to distinguish the authentic signals (red) from auto-fluorescence (yellow or green) caused by lipofuscin. In the hippocampal region small (2-5 μm) dense α-synuclein particles were observed in CA3 region of 9H/PS-NA brains (Fig. 3B), in contrast to larger α-synuclein particulates in the cortex. Only weak and very small α-synuclein particles (<2 μm) were seen in these regions from age matched WT mice (Fig. 3C and D).
Figure 3. Multiple α-synuclein aggregates in cortex and hippocampus of 9H/PS-NA brains.

(A) Multiple α-synuclein particles (10-20 μm) in cortex. (B) Small dense α-synuclein particles (2-5 μm) within hippocampal CA3. (C and D) Corresponding brain sections from age-matched WT mice showing fine and low level α-synuclein signals. The size of α-synuclein aggregates were measured using an Apotome AxioV 200 microscope. Neuronal cell nuclei were stained with DAPI (blue). The scale bars are 20 μm.
In comparison to 9H/PS-NA mice, large α-synuclein particles (≥5 μm) were observed in cerebral cortex of 10-12 wk PS-NA and 4L/PS-NA mice (Fig. 4, arrows), but the signals were much weaker in PS-NA brains. Similar results were observed in the 18-wk age group (data not shown), No α-synuclein aggregates were observed in the same regions of age-matched WT mice (Fig. 4). Immunoblots for α-synuclein showed the presence of monomers in the cerebral cortex of 18-wk 4L/PS-NA, 9H/PS-NA and WT mice. However, two forms of α-synuclein oligomers were present in the cortices of 4L/PS-NA and 9H/PS-NA mice (Fig. 5).
Figure 4. α-Synuclein aggregates in brain.

Brain sections from 12-wk WT, PS-NA, 4L/PS-NA, and 9H/PS-NA mice were processed for immunofluorescence analyses with anti α-synuclein antibody conjugated with Alexa-610 (red). Images were taken from cerebrum (Cr) and hippocampus (Hp) using the same exposure condition. Fine 1-2 μm α-synuclein particles were present in each genotype of mouse brain. α-Synuclein aggregates (≥ 5 μm) were only observed in the cerebrum of 12-wk 4L/PS-NA and 9H/PS-NA mice (arrows) or the hippocampus of 12-wk 9H/PS-NA mice (arrows). Such aggregates were not seen in WT brains. Neuronal cell nuclei were stained by DAPI (blue). The scale bars are 20 μm.
Figure 5. α-Synuclein oligomers in 4L/PS-NA and 9H/PS-NA brains.

Immunoblots of WT, 4L/PS-NA, and 9H/PS-NA cerebral cortex (100 μg protein) from 18 wk mice (n=3) were developed with mouse-specific anti-α-synuclein monoclonal antibody (Top panel). α-Synuclein monomers were present in cortex from WT (lanes 1-2), 4L/PS-NA (lanes 3-5) and 9H/PS-NA (lanes 6-8), but two forms of α-synuclein oligomers were only in cortex from 4L/PS-NA (lane 3-5) and 9H/PS-NA (lane 6-8). Protein molecular weight markers (KD) are in the far right lane. The stripped and re-probed blot shows β-actin antibody as the loading control (bottom panel).
Distribution of α-synuclein in brain coronal section
Serial coronal sections of 9H/PS-NA and 4L/PS-NA brains were examined by immunofluorescence staining from Interaural 1.98 mm/Bregma -1.82 mm to Interaural 0.40 mm/Bregma -3.40 mm [43]. This analysis included the basal ganglia region and brainstem where the neuronal lesions had been found in Gaucher disease type 2 and Parkinson disease [46, 47]. In the coronal sections, the regions containing α-synuclein-positive aggregates are indicated with text-abbreviations in Figure 6A (Interaural 1.98 mm/Bregma -1.82 mm) or 6B (Interaural 0.40 mm/Bregma -3.40 mm) that were constructed from 18 to 20 serial black-white images taken prior to processing for immunofluorescence. In the cortex, many α-synuclein aggregates were observed near the external capsule (ec) and near the temporal association cortex (TeA), Piriform cortex (Pir) (Fig. 6A), and retrosplenial agranular cortex (RSA), retrosplenial granular cortex (RSG), secondary visual cortex mediolateral (V2ML), primary visual cortex (V1), secondary visual cortex lateral (V2L), primary auditory cortex (Au1), cingulum (cg), perirhinal cortex (PRh), lateral entorhinal cortex (LEnt) (Fig. 6B). The α-synuclein-positive particles were also observed in the amydaloid nuclear complex, i.e., the lateral amydaloid nucleus, dorsolateral (LaDL) and lateral amydaloid nucleus, ventromedial (LaVM), basomedial amydaloid nucleus posterior (BMP), medial amydaloid nucleus posterodorsal (MepD), and medial amydaloid nucleus posteroventral (MepV) (Fig. 6A). In brainstem, α-synuclein-positive aggregates were seen at the area of cerebral peduncle basal part (cp), subthalamic nucleus (STH), ventral posteromedial thalamic nucleus (VPM), posterior thalamic nuclear group (po), paraventricular thalamic nucleus (PV), central medial thalamic nucleus (CM) (Fig. 6A) and substantia nigra (SN), medial geniculate nucleus ventral part (MGV), interstitial nucleus of Cajal (InC), red nucleus parvicellular part (RPC) interpeduncular nucleus rostral subnucleus (IPR), interpeduncular nucleus caudal subnucleus (IPc) (Fig. 6B). In the hippocampal region the signals were localized to lateral habenular nucleus medial part (LHbM), fasciculus retroflexus (fr) (Fig. 6A), lacunosum molecular layer (LMo), stratum radiatum (Rad), molecular layer of dentate gyrus (MoL), pyramidal cell layer (Py) and subiculum (Fig. 6B).
Figure 6. Coronal distribution of α-synuclein in the brain of 9H/PS-NA mice.

Serial coronal brain sections of 10-wk 9H/PS-NA mice were photographed with a 4× objective under bright field before immunofluorescence staining processed. The brain sections were prepared from (A) Interaural 1.98 mm/Bregma -1.82 mm to (B) Interaural 0.40 mm/Bregma -3.40 mm including basal ganglia region and brainstem. Each figure is a hemisphere section composed by ∼20 images. After black/white photography, the sections were processed for immunofluorescence study using anti α-synuclein antibody. The regions containing α-synuclein positive aggregates were text-marked and the abbreviations are described in RESULTS.
The nature of the inclusion bodies in neurons
The α-synuclein aggregates in the brain were evaluated by silver staining since this has been used to identify pathological deposits in studies of Parkinson disease [48, 49]. In general, brain sections from 9H/PS-NA and 4L/PS-NA mice stained darker than those from control mice. The silver stained neurons were observed at multiple regions in 9H/PS-NA brains, but were less intense in the brains of 4L/PS-NA mice. Dense dark particles were found in the cytoplasm around the nuclei of neurons (Fig. 7A), suggesting aggregated proteins in the diseased mouse brain. To further characterize the silver stained aggregates, consecutive sections were probed with anti-α-synuclein and anti-ubiquitin antibodies. Some of these silver stained neurons showed α-synuclein (Fig. 7B) or ubiquitin (Fig. 7C) immunoreactivity positivity indicating intracellular aggregation of these proteins.
Figure 7. Aggregated cytoplasmic proteins in the brain neurons from 9H/PS-NA mice.
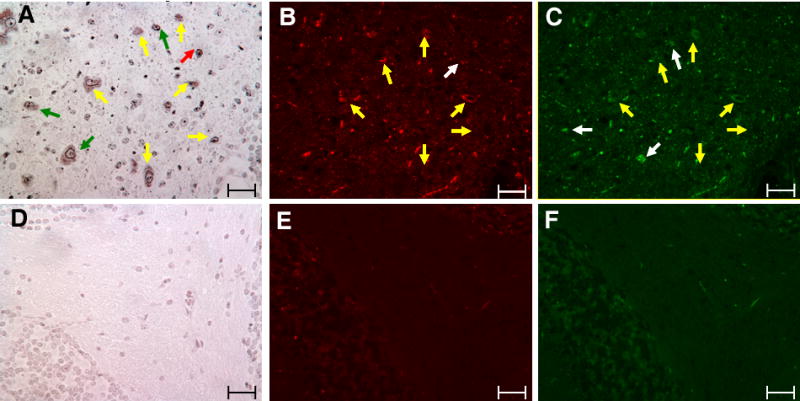
Serial brain sections from 23-wk 9H/PS-NA mice were processed for silver and α-synuclein and ubquitin immunofluorescence staining. Silver staining images show the presence of dense dark particles in cerebellar white matter of 9H/PS-NA mouse (A), but not in heterozygote littermate controls (D). The adjacent sections were processed for immunofluorescence staining using anti α-synuclein (Alexa-610, red) and anti ubiquitin (Alexa-488, green) antibodies. Arrows indicate the α-synuclein positive aggregates (B) or ubiquitin signals (C) in 9H/PS-NA sections. Yellow arrows indicate appositionally matched silver aggregates with both α-synuclein (B) and ubiquitin (C) Red arrows indicate the silver signals (A) that matched only with α-synuclein (B). The green arrow indicates that the silver signals in (A) matched only with ubiquitin signals (C). Brain tissues from heterozygote littermates were processed in parallel and no α-synuclein (E) or ubiquitin particles (F) were observed. The scale bars are 50 μm.
Age-associated α-synuclein accumulation
To characterize the developmental stages of α-synuclein accumulation, brain sections from 9H/PS-NA, 4L/PS-NA, PS-NA and WT mice at 4, 10-12, and 18-20 wks were examined. In general, fine α-synuclein signals (∼1 μm) were seen in brain of WT and diseased mice at all age groups. α-Synuclein positive particles were observed in 4-wk 9H/PS-NA brain, [e.g., 1-2 μm in OB, Cb and dentate gyrus; 3-5 μm in CPu (data not shown)]; and a few larger (6-7 μm) α-synuclein particles were found in cortex at that age (not shown). The aggregated α-synuclein particles became more widely distributed with increasing age, especially in 9H/PS-NA mice (Fig. 8). Larger amounts of α-synuclein particles (5 - 30 μm) were seen in 9H/PS-NA brains at 10-or 20-wks (Fig. 8), and the particle sizes increased with age. In PS-NA or 4L/PS-NA mice, a few of α-synuclein positive particles (2-4 μm or 4-6 μm) were seen in the cortex at 4 to 20 wks (data not shown).
Figure 8. Age associated α-synuclein accumulation in brain of 9H/PS-NA mice.

Brain sections from 10-, or 20-wk 9H/PS-NA mice were examined with anti-α-synuclein antibody. Various sized (5 to 32 μm) α-synuclein particles (Alex-610, red) were observed in olfactory bulb (OB), cerebellum (Cb), dentate gyrus (DG) and caudate putamen (CPu) regions as indicated. The size of some α-synuclein particles is increased from 10 to 20 wks as indicated in the figure. The images were taken using FITC filter as background (green). Neuron cell nuclei were stained by DAPI (blue). The scale bars are 20 μm.
α-Synuclein and ubiquitin accumulation
The relationships between α-synuclein and ubiquitin were studied in 9H/PS-NA (Fig. 9), 4L/PS-NA, PS-NA and WT mice. α-Synuclein aggregates appeared in brains at 4-wks, but the ubiquitin aggregates were detected at ∼10-12 wks. In the brain of 10-wk 9H/PS-NA, 4L/PS-NA or PS-NA mice, sporadic ubiquitin signals were observed in cortex, brainstem and midbrain region (data not shown). Larger sized (≤18 μm) ubiquitin signals were present in the brainstem, medulla, midbrain and cortex of 20-wk 9H/PS-NA mice (Fig 9D); the overlap of ubiquitin and α-synuclein signals was partial (Fig. 9F, arrows). A few small ubiquitin signals were observed in PS-NA and 4L/PS-NA brains (data not shown).
Figure 9. Expression of α-synuclein and ubiquitin in the brain.

Brain sections from 20-wk WT and 9H/PS-NA mice were examined with anti-ubiquitin (A and D, Alexa-488, green) and anti-α-synuclein (B and E, Alexa-610, red) antibodies as indicated. The panels show the midbrain region. Various sizes (5 – 17 μm) of ubiquitin and α-synuclein signals were seen only in 9H/PS-NA (D and E), and not in WT (A and B) brain. The ubiquitin signals were only partially co-localized with α-synuclein signals (arrow) in 9H/PS-NA (F) and not in WT (C). The scale bars are 20 μm.
Strong and dense GFAP staining, indicating astrogliosis, was in the cortex and ventricular area of 10-wk 9H/PS-NA mice (Fig. 10 top panels), but without colocalization with α-synuclein. α-Synuclein signals did not co-localize with CD68 signals (Fig. 10 lower panels), but some were embedded at the center of CD68 positive microglial cells, which suggested the phagocytosis of α-synuclein by microglial cells. These immunofluorescence studies indicated that α-synuclein was not present in astrocytes or microglial cells. Tyrosine hydroxylase-positive signals were mostly seen in basal ganglia and brainstem regions, but no significant differences were found between diseased and WT brains (not shown). In the 9H/PS-NA, 4L/PS-NA and PS-NA mice, no obvious relationships of tyrosine hydroxylase and α-synuclein signals were observed.
Figure 10. The association of α-synuclein signals with astrocytes and microglial cells.

Brain sections from 20-wk 9H/PS-NA mice were examined by dual antibody staining. (Upper panels) Cortical regions stained with monoclonal anti-mouse GFAP for astrocyte (Alexa-488, green)/polyclonal anti-mouse α-synuclein (Alexa-610, red). (Lower panels) Hippocampal CA2 regions stained with anti-CD68 for microglial cell (Alexa-488, green)/α-synuclein (Alexa-610, red). The GFAP signals did not colocalized with α-synuclein signals. α-Synuclein signals were not merged with CD68 signals, but some were within microglial cells (arrows). The scale bars are 20 μm. No significant α-synuclein, GFAP and CD68 signals were present in WT brain sections (not shown).
α-Synuclein accumulation in Gba1 point-mutated and CBE induced GCase deficiency
α-Synuclein aggregation was examined in selected Gba1 point-mutated mice (4L, 9H and 9V). Aggregated α-synuclein signals were only found in cerebellar and brainstem regions of 42-46 wk 9H mice, but not in age-matched 4L, 9V, or WT brains (Fig. 11). A GCase irreversible inhibitor, CBE, was used to generate mice with neuronopathic disease [34] in an effort to distinguish the potential for gain (malfolding) or loss (activity absence) of GCase. Short-term (6 day) CBE treatment of 4L, 9H, 9V and WT mice (100 mg/kg/day, intraperitoneal injection) from postnatal day 5 to 11 did not induce α-synuclein aggregates, either when harvested one day or 60 days after the last CBE injection. These mice did develop varying degrees of neuronopathic disease, e.g., shaking and paralysis after 6 injections [34]. Long-term daily CBE treatment of 4L mice (100 mg/kg/day of CBE from postnatal day 15 for 24 or 36 doses) led to hind limb paralysis and small amounts of α-synuclein accumulation in the olfactory bulb, brainstem, and PVP (paraventricular th nu, posterior) near D3V (dorsal 3rd ventricle) (Fig. 12). In these mice, glucosylceramide accumulation in brain can be appreciated by TLC analyses as compared to untreated 4L or WT mice (data not shown). Interestingly, by LC/MS brain glucosylsphingosine levels in these mice were 20- to 30-fold higher than those in untreated 4L mice (not shown).
Figure 11. α-Synuclein staining in the brains of older mice.

Serial brain sections from 42-wk WT (top) and 46-wk 9H (bottom) mice were examined with anti-α-synuclein antibody. Aggregated α-synuclein signals (Alexa-610, red) were detected in the brainstem (BS) regions of 9H mice (arrows). The corresponded sections from WT mice were processed in parallel (top). Some sporadic α-synuclein signals were observed in Cb, but not in BS of WT mice (data not shown). The scale bars were 20 μm (40×).
Figure 12. α-Synuclein in the brains of CBE-treated mice.

Brain sections from 8-wk 4L mice that had 24 or 36 daily injections with 100 mg CBE/kg/day were examined with anti-α-synuclein antibody. Aggregated α-synuclein signals (Alexa-610, red) were detected in ventricle (VC, lower left panel), brainstem (BS, upper right panel) and olfactory bulb (OB, lower right panel) in the injected mice (arrows), but not in untreated (no-CBE) 4L mice (upper left panel). The scale bars were 20 μm (40×).
α-Synuclein and ubiquitin in the brain of other mice with lysosomal storage diseases
Several lysosomal storage disease models were used as controls for these studies as summarized in Table 1 and below. Ubiquitin was found in the cortex and midbrain of 4-wk PS-/- mice, but α-synuclein aggregates were not observed. Sporadic 4-8 μm α-synuclein aggregates were present in the cortical, cerebellar, and brainstem areas of NPC1. Occasional larger (≤ 15 μm) α-synuclein and ubiquitin aggregates were present in the CPu area of NPC1 mice (data not shown). In MPS1 mice, ubiquitin aggregates >10 μm were in the cortex, SNC, OB, and brainstem areas; most α-synuclein aggregates were in SNC (≤10 μm), and cerebellum and brainstem (∼5 μm) (data not shown). In brains from LAL knockout or saposin (A, B, C, or D) deficient mice, α-synuclein and ubiquitin aggregates were not found (data not shown).
Discussion
Here, α-synuclein expression was examined in various Gba1 mutated mice with or without CNS phenotypes including: Gba1 point mutated mice, and hypomorphic prosaposin mice (PS-NA), with or without 9H or 4L, and in CBE - treated Gba1 mutated mice. The 9H/PS-NA mice had the most significant α-synuclein accumulation, particularly in the deeper layers of the cortex. Brain lipid analyses showed moderately increased levels of glucosylceramide in the cortex and other regions compared with PS-NA and point mutated Gba1 models; this suggested a correlation of glucosylceramide accumulation with significant α-synuclein aggregation. In comparison with 9H/PS-NA mice, similar glucosylceramide levels and neuronopathology, but less significant α-synucleinopathy were observed in 4L/PS-NA mice. This result indicates that glucosylceramide levels are one factor in the development of α-synucleinopathy and that additional pathophysiologic mechanisms are needed, e.g., the in vivo properties of mutant GCases, (4L or 9H). Interestingly, glucosylsphingosine accumulation in 4L/PS-NA is much higher (10 times) than that in 9H/PS-NA, but with greater α-synuclein accumulations in the latter. Thus, glucosylsphingosine is not likely to play a major role in the α-synucleinopathy, but rather it contributes to neuronal and cell death. The rapid neuronal degeneration and death induced by glucosylsphingosine may not support the time needed for α-synuclein aggregate deposition. A complex and chronic mechanism is implied for α-synucleinopathy development.
The chronicity of the α-synuclein accumulation is supported by its age-dependency in the 9H/PS-NA, 4L/PS-NA, and Gba1 point-mutated mice. Low levels of α-synuclein accumulation were detected as early as 4-wks in 9H/PS-NA mice with progression to substantial aggregates by 12-20 wks. Immunoblot analysis confirmed the formation of α-synuclein oligomers in 9H/PS-NA and 4L/PS-NA cortices that contribute to the aggregates. The short-lived kn-9H mice (14 days) [34] that were GCase null in the CNS and had glucosylceramide accumulation in the brain, did not have α-synuclein aggregates (data not shown). In CBE induced GCase deficiency in adult mice, α-synuclein aggregates only developed after long-term treatment. These observations indicate that α-synuclein aggregation is of a chronic progressive nature, consistent with the time-dependent formation of α-synuclein aggregates following an in vitro initial nucleation event [50, 51]. α-Synuclein aggregates could be formed under a number of conditions including simple solutions, incubation with lipids, in cell cultures, or in animal models of disease [52]. However, in vivo systems are complicated by interactions among multiple proteins/biomolecules involved in the formation of inclusions [52-54]. Here, ultrastructural analyses showed numerous inclusion bodies in neurons and the structure of these inclusions suggested aggregated proteins.
Various patterns of α-synuclein aggregation were observed in brains from the Gba1 mouse variants in these studies, particularly in 9H/PS-NA. Numerous large α-synuclein aggregates were present in the deep layers of cortex overlaying corpus callosum, whereas fine α-synuclein particles were in the hippocampal CA3 and dentate gyrus. The patterns of α-synuclein aggregates are similar to pathologic findings in Gaucher disease patients who had Parkinsonism. However, such patients characteristically exhibit Lewy bodylike α-synuclein inclusions with gliosis in cortical layers 3 and 5 (deeper layers) and in the hippocampal neurons (CA2-4 regions) [15]. Similar aggregates were detected in 4L/PS-NA and 9H/PS-NA brain, but Lewy bodies were not observed in the current studies.
The relationships of α-synuclein accumulation and ubiquitin, astrocyte (GFAP), dopaminergic neuron (tyrosine hydroxylase), and microglial cells (CD68) were examined in brain sections. In 9H/PS-NA or 4L/PS-NA and PS-NA mice, ubiquitin partially colocalized with α-synuclein signals. In human samples, α-synuclein positive Lewy bodies were generally ubiquitin positive [31]. The differences in co-localization of α-synuclein and ubiquitin between human and mouse suggest variation in the initiation mechanisms between these species. However, the end-stage nature of autopsy specimens from patients with both Gaucher disease and PD patients does not provide insights into the time course of this process. In comparison, 9H/PS-NA mice showed a progressive α-synucleinopathy that was detected as early as 4-wks, whereas ubiquitin aggregates were seen almost exclusively after 10 wks. The precise role of α-synuclein in the Lewy body formation or in the neurodegenerative processes is unclear. Lewy bodies are complex structures and contain multiple proteins [52, 53]. However, GCase has recently been found in Lewy bodies in patients heterozygous for GBA1 mutations suggesting a role for the GCase in their formation [55]. In 9H/PS-NA or 4L/PS-NA brains, massive astrogliosis, and activated microglial cells were present, but signals for GFAP (astrocytes) or CD68 (microglial cells) colocalized with α-synuclein only occasionally. Interestingly, a few α-synuclein signals localize near the center of CD68 positive microglial cells (Fig. 10, CD68 positive), suggesting a microglial cell response to aggregated proteins in the brain. These observations indicate that α-synuclein was not expressed to a great extent in either astrocytes nor in microglial cells, but rather in neurons. Tyrosine hydroxylase expression in the mice used here was not significantly different from WT control mice.
Screening of several other LSD mouse models confirmed the presence of small α-synuclein aggregates in brains from NPC1 and MPS1 mice (data not shown), but not in LAL or individual saposin deficiency mice. NPC1 mice have a defect in a cholesterol efflux protein in the membrane of the lysosome [56-58]. However, glycosphingolipids, including glucosylceramide and gangliosides, also accumulate in NPC1 brain. MPS1 mice accumulate glycosaminoglycans heparan and dermatan sulfate, as well as GM2 and GM3 [59]. These observations support the contention that glycosphingolipids and gangoliosides contribute to the pathogenesis of these diseases and may increase the risk for developing α-synuclein accumulations. In Sandhoff mice, neuronal accumulations of α-synuclein and GM2 ganglioside were present throughout the brain [60]. α-Synuclein aggregates are also found in cathepsin D deficient mice, sheep, and humans. This is potentially due to the role of cathepsin D in α-synuclein processing. α-Synuclein and ubiquitin signals in NPC1 and MPS1 brains were significantly less than those in 9H/or 4L/PS-NA brains, some specificity for the α-synucleinopathy. Specific saposin A, B, C or D deficiencies in mice produced no α-synuclein and ubiquitin accumulation. The accumulated GSLs in each of these models are different, e.g., galactosylceramide and galactosylsphingosine with saposin A deficiency [61], sulfatide with saposin B deficiency [62], lactosylceramide with saposin C deficiency [63], and ceramide with saposin D deficiency [64]. In total saposin deficient mice, (i.e., prosaposin knockout) moderate increases of glucosylceramide and positive anti-ubiquitin antibody staining were observed in neurons [65]. These studies suggest that the respective sphingolipids or other saposin effects in the saposin deficient mice are not causally related to α-synuclein and ubiquitin accumulation. With the hypomorphic prosaposin-deficiency combined with low activity GCase mutations (4L/PS-NA or 9H/PS-NA), α-synuclein accumulation was observed as early as 4-wks. These studies suggest a more complicated relationship between glucosylceramide, GCase mutations, GSL accumulations, and the risk for α-synuclein/ubiquitin accumulations.
Many LSDs are associated with progressive and severe neurodegeneration. To date, the diverse biological pathways from lysosomal enzyme deficiency to neuron dysfunction and death have not been clarified [66]. In the mouse models of multiple sulfatase deficiency (MSD) and mucopolysaccharidosis type IIIA (MPSIIIA), defective autophagosome-lysosome fusion was observed with severe neurodegeneration [67]. In Huntington disease or familial of Parkinson disease, impaired autophagocytic pathways led to aggregation-prone proteins, which cause late-onset neurodegenerative conditions [68]. However, knockout of selected autophagy genes caused abnormal protein accumulation in ubiquitinated inclusions and neurodegeneration in mice [69, 70]. These phenotypic similarities suggest the presence of common pathogenic mechanisms for LSDs with neurodegeneration [71].
Clinically, earlier onset and more progressive PD patients had heterozygous mutations in GBA1 [5-13]. However, Parkinsonism manifestations [6-8, 11] and α-synuclein/ubiquitin aggregates [7, 15, 22] were found in only some GD type 1 or 3 patients. Thus, the connections between GCase mutations (gain or loss of function or both) and the development of Parkinsonism or PD remain unclear. The current findings in mice provide a biological system and potential clues to the pathogenesis of neuronopathic Gaucher disease including the α-synuclein distribution patterns in Gba1 mutant mice. Ongoing studies should provide insight into the underlying neuronopathic mechanisms in Gaucher disease and the relationship between GCase deficiency and/or GBA1 heterozygosity and the development of Parkinsonian manifestations.
Supplementary Material
Supplemental Fig 1: Distribution of α-synuclein in sagittal sections of brain cortex from 9H/PS-NA mice. The figure was composed from ∼70 images using 20×. Serial sagittal images were from 10-wk 9H/PS-NA mice and examined by immunofluorescence staining using anti-α-synuclein antibody conjugated with Alexa-610 (red). The abbreviations from left to right are as follows: VO, ventral orbital cortex; DTT, dorsal tenia tecta; Cg1, cingulate cortex area 1; PrL, prelimbic cortex; DP, dorsal peduncular cortex; gcc, genu of corpus callosum; Cpu, caudate putamen; M2, secondary motor cortex; LV, lateral ventricle; fi, fimbria of Hippocampus; cc, corpus callosum; CA2-3, hippocampus CA2-3 fields; RSA, retrosplenial agranular cortex; dentate gyrus (DG); LHb, lateral habenular nu; scc, splenium corpus callosum; RSGa, and retrosplenial granular cortex [43].
Acknowledgments
The authors thank Michelle Cooley for her clerical expertise, and Venette Inskeep for her excellent technical assistance; Drs. Hong Du and Dao Pan for providing LAL-/- and MPSI mouse brains, respectively. Lisa McMillin, Meredith Farmer and Sabina Sylvest for skilled tissue preparation; Irene Hofmann for EM preparation; Chris Woods for photograph processing. This work was supported by a grant from the NIH (DK 36729) to GAG.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beutler E, Grabowski GA. Gaucher Disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3635–3668. [Google Scholar]
- 2.Grabowski GA, Saal HM, Wenstrup RJ, Barton NW. Gaucher disease: a prototype for molecular medicine. Crit Rev Oncol Hematol. 1996;23:25–55. doi: 10.1016/1040-8428(96)00199-0. [DOI] [PubMed] [Google Scholar]
- 3.Sibille A, Eng CM, Kim SJ, Pastores G, Grabowski GA. Phenotype/genotype correlations in Gaucher disease type I: clinical and therapeutic implications. Am J Hum Genet. 1993;52:1094–1101. [PMC free article] [PubMed] [Google Scholar]
- 4.Zimran A, Kay A, Gelbart T, Garver P, Thurston D, Saven A, Beutler E. Gaucher disease. Clinical, laboratory, radiologic, and genetic features of 53 patients. Medicine (Baltimore) 1992;71:337–353. [PubMed] [Google Scholar]
- 5.McKeran RO, Bradbury P, Taylor D, Stern G. Neurological involvement in type 1 (adult) Gaucher's disease. J Neurol Neurosurg Psychiatry. 1985;48:172–175. doi: 10.1136/jnnp.48.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Machaczka M, Rucinska M, Skotnicki AB, Jurczak W. Parkinson's syndrome preceding clinical manifestation of Gaucher's disease. Am J Hematol. 1999;61:216–217. doi: 10.1002/(sici)1096-8652(199907)61:3<216::aid-ajh12>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 7.Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, Rosenbaum H, Schiffmann R, Bembi B, Sidransky E. Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab. 2003;79:104–109. doi: 10.1016/s1096-7192(03)00071-4. [DOI] [PubMed] [Google Scholar]
- 8.Varkonyi J, Rosenbaum H, Baumann N, MacKenzie JJ, Simon Z, Aharon-Peretz J, Walker JM, Tayebi N, Sidransky E. Gaucher disease associated with parkinsonism: four further case reports. Am J Med Genet A. 2003;116:348–351. doi: 10.1002/ajmg.a.10028. [DOI] [PubMed] [Google Scholar]
- 9.Halperin A, Elstein D, Zimran A. Increased incidence of Parkinson disease among relatives of patients with Gaucher disease. Blood Cells Mol Dis. 2006;36:426–428. doi: 10.1016/j.bcmd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Ziegler SG, Eblan MJ, Gutti U, Hruska KS, Stubblefield BK, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol Genet Metab. 2007;91:195–200. doi: 10.1016/j.ymgme.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Parkinsonism among Gaucher disease carriers. J Med Genet. 2004;41:937–940. doi: 10.1136/jmg.2004.024455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nichols WC, Pankratz N, Marek DK, Pauciulo MW, Elsaesser VE, Halter CA, Rudolph A, Wojcieszek J, Pfeiffer RF, Foroud T. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology. 2009;72:310–316. doi: 10.1212/01.wnl.0000327823.81237.d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab. 2004;81:70–73. doi: 10.1016/j.ymgme.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Wong K, Sidransky E, Verma A, Mixon T, Sandberg GD, Wakefield LK, Morrison A, Lwin A, Colegial C, Allman JM, Schiffmann R. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 2004;82:192–207. doi: 10.1016/j.ymgme.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 16.Adachi M, Wallace BJ, Schneck L, Volk BW. Fine structure of central nervous system in early infantile Gaucher's disease. Arch Pathol. 1967;83:513–526. [PubMed] [Google Scholar]
- 17.Lloyd OC, Norman RM, Urich H. The neuropathology of infantile Gaucher's disease. J Pathol Bacteriol. 1956;72:121–131. doi: 10.1002/path.1700720116. [DOI] [PubMed] [Google Scholar]
- 18.Kaye EM, Ullman MD, Wilson ER, Barranger JA. Type 2 and type 3 Gaucher disease: a morphological and biochemical study. Ann Neurol. 1986;20:223–230. doi: 10.1002/ana.410200208. [DOI] [PubMed] [Google Scholar]
- 19.Espinas OE, Faris AA. Acute infantile Gaucher's disease in identical twins. An account of clinical and neuropathologic observations. Neurology. 1969;19:133–140. doi: 10.1212/wnl.19.2.133. [DOI] [PubMed] [Google Scholar]
- 20.Conradi NG, Sourander P, Nilsson O, Svennerholm L, Erikson A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta Neuropathol (Berl) 1984;65:99–109. doi: 10.1007/BF00690463. [DOI] [PubMed] [Google Scholar]
- 21.Conradi N, Kyllerman M, Mansson JE, Percy AK, Svennerholm L. Late-infantile Gaucher disease in a child with myoclonus and bulbar signs: neuropathological and neurochemical findings. Acta Neuropathol (Berl) 1991;82:152–157. doi: 10.1007/BF00293959. [DOI] [PubMed] [Google Scholar]
- 22.Goker-Alpan O, Giasson BI, Eblan MJ, Nguyen J, Hurtig HI, Lee VM, Trojanowski JQ, Sidransky E. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology. 2006;67:908–910. doi: 10.1212/01.wnl.0000230215.41296.18. [DOI] [PubMed] [Google Scholar]
- 23.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- 24.Dev KK, Hofele K, Barbieri S, Buchman VL, van der Putten H. Part II: alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 25.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- 26.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 27.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 28.Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Schindzielorz A, Okochi M, Leimer U, van Der Putten H, Probst A, Kremmer E, Kretzschmar HA, Haass C. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 30.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci. 2002;22:2780–2791. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 32.Duda JE, Lee VM, Trojanowski JQ. Neuropathology of synuclein aggregates. J Neurosci Res. 2000;61:121–127. doi: 10.1002/1097-4547(20000715)61:2<121::AID-JNR1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Sun Y, Quinn B, Witte DP, Grabowski GA. Gaucher disease mouse models: point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J Lipid Res. 2005;46:2102–2113. doi: 10.1194/jlr.M500202-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Xu YH, Reboulet R, Quinn B, Huelsken J, Witte D, Grabowski GA. Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol Genet Metab. 2008;94:190–203. doi: 10.1016/j.ymgme.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandhoff K, Kolter T, Van Echten-Deckert G. Sphingolipid metabolism. Sphingoid analogs, sphingolipid activator proteins, and the pathology of the cell. Ann N Y Acad Sci. 1998;845:139–151. doi: 10.1111/j.1749-6632.1998.tb09667.x. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y, Qi X, Grabowski GA. Saposin C is required for normal resistance of acid beta-glucosidase to proteolytic degradation. J Biol Chem. 2003;278:31918–31923. doi: 10.1074/jbc.M302752200. [DOI] [PubMed] [Google Scholar]
- 37.Xu YH, Quinn B, Witte D, Grabowski GA. Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am J Pathol. 2003;163:2093–2101. doi: 10.1016/s0002-9440(10)63566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H, Suzuki K. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–725. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Qi X, Witte DP, Ponce E, Kondoh K, Quinn B, Grabowski GA. Prosaposin: threshold rescue and analysis of the “neuritogenic” region in transgenic mice. Mol Genet Metab. 2002;76:271–286. doi: 10.1016/s1096-7192(02)00114-2. [DOI] [PubMed] [Google Scholar]
- 40.te Vruchte D, Lloyd-Evans E, Veldman RJ, Neville DC, Dwek RA, Platt FM, van Blitterswijk WJ, Sillence DJ. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J Biol Chem. 2004;279:26167–26175. doi: 10.1074/jbc.M311591200. [DOI] [PubMed] [Google Scholar]
- 41.Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–511. doi: 10.1093/hmg/6.4.503. [DOI] [PubMed] [Google Scholar]
- 42.Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet. 1998;7:1347–1354. doi: 10.1093/hmg/7.9.1347. [DOI] [PubMed] [Google Scholar]
- 43.Paxinos G, Franklin KBJ. The Mouse Brain in stereotaxic Coordinates. Academic Press; San diego: 2001. [Google Scholar]
- 44.Kanfer JN, Legler G, Sullivan J, Raghavan SS, Mumford RA. The Gaucher mouse. Biochem Biophys Res Commun. 1975;67:85–90. doi: 10.1016/0006-291x(75)90286-7. [DOI] [PubMed] [Google Scholar]
- 45.Grabowski GA, Osiecki-Newman K, Dinur T, Fabbro D, Legler G, Gatt S, Desnick RJ. Human acid beta-glucosidase. Use of conduritol B epoxide derivatives to investigate the catalytically active normal and Gaucher disease enzymes. J Biol Chem. 1986;261:8263–8269. [PubMed] [Google Scholar]
- 46.Xu YH, Barnes S, Sun Y, Grabowski GA. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J Lipid Res. 2010 doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nicholl DJ, Vaughan JR, Khan NL, Ho SL, Aldous DE, Lincoln S, Farrer M, Gayton JD, Davis MB, Piccini P, Daniel SE, Lennox GG, Brooks DJ, Williams AC, Wood NW. Two large British kindreds with familial Parkinson's disease: a clinico-pathological and genetic study. Brain. 2002;125:44–57. doi: 10.1093/brain/awf013. [DOI] [PubMed] [Google Scholar]
- 48.Braak E, Braak H. Silver staining method for demonstrating Lewy bodies in Parkinson's disease and argyrophilic oligodendrocytes in multiple system atrophy. J Neurosci Methods. 1999;87:111–115. doi: 10.1016/s0165-0270(98)00173-3. [DOI] [PubMed] [Google Scholar]
- 49.Uchihara T, Nakamura A, Mochizuki Y, Hayashi M, Orimo S, Isozaki E, Mizutani T. Silver stainings distinguish Lewy bodies and glial cytoplasmic inclusions: comparison between Gallyas-Braak and Campbell-Switzer methods. Acta Neuropathol. 2005;110:255–260. doi: 10.1007/s00401-005-1044-2. [DOI] [PubMed] [Google Scholar]
- 50.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson's disease. J Biol Chem. 1999;274:19509–19512. doi: 10.1074/jbc.274.28.19509. [DOI] [PubMed] [Google Scholar]
- 52.Shults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olanow CW, Perl DP, DeMartino GN, McNaught KS. Lewy-body formation is an aggresome-related process: a hypothesis. Lancet Neurol. 2004;3:496–503. doi: 10.1016/S1474-4422(04)00827-0. [DOI] [PubMed] [Google Scholar]
- 54.Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 55.Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010;120:641–649. doi: 10.1007/s00401-010-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patterson M, Vanier M, Suzuki K, Morris J, Carstea E, Neufeld E, Blanchette-Mackie J, Pentchev P. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3611–3633. [Google Scholar]
- 57.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 58.Treiber-Held S, Distl R, Meske V, Albert F, Ohm TG. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J Pathol. 2003;200:95–103. doi: 10.1002/path.1345. [DOI] [PubMed] [Google Scholar]
- 59.Russell C, Hendson G, Jevon G, Matlock T, Yu J, Aklujkar M, Ng KY, Clarke LA. Murine MPS I: insights into the pathogenesis of Hurler syndrome. Clin Genet. 1998;53:349–361. doi: 10.1111/j.1399-0004.1998.tb02745.x. [DOI] [PubMed] [Google Scholar]
- 60.Suzuki K, Iseki E, Katsuse O, Yamaguchi A, Katsuyama K, Aoki I, Yamanaka S, Kosaka K. Neuronal accumulation of alpha- and beta-synucleins in the brain of a GM2 gangliosidosis mouse model. Neuroreport. 2003;14:551–554. doi: 10.1097/00001756-200303240-00004. [DOI] [PubMed] [Google Scholar]
- 61.Matsuda J, Vanier MT, Saito Y, Tohyama J, Suzuki K, Suzuki K. A mutation in the saposin A domain of the sphingolipid activator protein (prosaposin) gene results in a late-onset, chronic form of globoid cell leukodystrophy in the mouse. Hum Mol Genet. 2001;10:1191–1199. doi: 10.1093/hmg/10.11.1191. [DOI] [PubMed] [Google Scholar]
- 62.Sun Y, Witte DP, Ran H, Zamzow M, Barnes S, Cheng H, Han X, Williams MT, Skelton MR, Vorhees CV, Grabowski GA. Neurological deficits and glycosphingolipid accumulation in saposin B deficient mice. Hum Mol Genet. 2008;17:2345–2356. doi: 10.1093/hmg/ddn135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Y, Ran H, Zamzow M, Kitatani K, Skelton MR, Williams MT, Vorhees CV, Witte DP, Hannun YA, Grabowski GA. Specific saposin C deficiency: CNS impairment and acid {beta}-glucosidase effects in the mouse. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsuda J, Kido M, Tadano-Aritomi K, Ishizuka I, Tominaga K, Toida K, Takeda E, Suzuki K, Kuroda Y. Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse. Hum Mol Genet. 2004;13:2709–2723. doi: 10.1093/hmg/ddh281. [DOI] [PubMed] [Google Scholar]
- 65.Oya Y, Nakayasu H, Fujita N, Suzuki K, Suzuki K. Pathological study of mice with total deficiency of sphingolipid activator proteins (SAP knockout mice) Acta Neuropathol (Berl) 1998;96:29–40. doi: 10.1007/s004010050857. [DOI] [PubMed] [Google Scholar]
- 66.Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. doi: 10.1038/nrm1423. [DOI] [PubMed] [Google Scholar]
- 67.Settembre C, Fraldi A, Rubinsztein DC, Ballabio A. Lysosomal storage diseases as disorders of autophagy. Autophagy. 2008;4:113–114. doi: 10.4161/auto.5227. [DOI] [PubMed] [Google Scholar]
- 68.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 69.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 70.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 71.Jeyakumar M, Dwek RA, Butters TD, Platt FM. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6:713–725. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- 72.Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- 73.Pan D, Sciascia A, 2nd, Vorhees CV, Williams MT. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008;1188:241–253. doi: 10.1016/j.brainres.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig 1: Distribution of α-synuclein in sagittal sections of brain cortex from 9H/PS-NA mice. The figure was composed from ∼70 images using 20×. Serial sagittal images were from 10-wk 9H/PS-NA mice and examined by immunofluorescence staining using anti-α-synuclein antibody conjugated with Alexa-610 (red). The abbreviations from left to right are as follows: VO, ventral orbital cortex; DTT, dorsal tenia tecta; Cg1, cingulate cortex area 1; PrL, prelimbic cortex; DP, dorsal peduncular cortex; gcc, genu of corpus callosum; Cpu, caudate putamen; M2, secondary motor cortex; LV, lateral ventricle; fi, fimbria of Hippocampus; cc, corpus callosum; CA2-3, hippocampus CA2-3 fields; RSA, retrosplenial agranular cortex; dentate gyrus (DG); LHb, lateral habenular nu; scc, splenium corpus callosum; RSGa, and retrosplenial granular cortex [43].