

Abstract
Background
Junctophilin-2 (JPH2), a protein expressed in the junctional membrane complex, is necessary for proper intracellular calcium (Ca2+) signaling in cardiac myocytes. Down-regulation of JPH2 expression in a model of cardiac hypertrophy was recently associated with defective coupling between plasmalemmal L-type Ca2+ channels and sarcoplasmic reticular ryanodine receptors. However, it remains unclear whether JPH2 expression is altered in patients with hypertrophic cardiomyopathy (HCM). In addition, the effects of down-regulation of JPH2 expression on intracellular Ca2+-handling are presently poorly understood. We sought to determine whether loss of JPH2 expression is noted among patients with HCM and whether expression silencing might perturb Ca2+-handling in a pro-hypertrophic manner.
Methods and Results
JPH2 expression was reduced in flash frozen human cardiac tissue procured from patients with HCM compared to ostensibly healthy traumatic death victims. Partial silencing of JPH2 expression in HL-1 cells by a small interfering RNA probe targeted to murine JPH2 mRNA (shJPH2) resulted in myocyte hypertrophy and increased expression of known markers of cardiac hypertrophy. While expression levels of major Ca2+-handling proteins were unchanged, shJPH2 cells demonstrated depressed maximal Ca2+ transient amplitudes that were insensitive to LTCC activation with JPH2 knock-down. Further, reduced caffeine-triggered SR store Ca2+ levels were observed with potentially increased total Ca2+ stores. Spontaneous Ca2+ oscillations were elicited at a higher extracellular [Ca2+] and with decreased frequency in JPH2 knock-down cells.
Conclusions
Our results show that JPH2 levels are reduced in patients with HCM. Reduced JPH2 expression results in reduced excitation-contraction coupling gain as well as altered Ca2+ homeostasis which may be associated with pro-hypertrophic remodeling.
Keywords: JPH2, junctophilin, hypertrophic cardiomyopathy, calcium, ryanodine receptor
INTRODUCTION
Junctophilins are members of a family of proteins found in all excitable cells from striated muscle to neurons that bridge the subcellular space between the plasma membrane and the sarco/endoplasmic reticulum (SR/ER). In cardiac tissue, JPH2-encoded junctophilin type 2 (JPH2) plays a critical role in the maintaining effective calcium (Ca2+) flux1,2. In cardiocytes, voltage-gated L-type Ca2+ channels (LTCC) at the sarcolemma allow for an influx of extracellular Ca2+, which triggers Ca2+ release from the SR via intracellular Ca2+ release channels known as ryanodine receptors (RyR2). This process, known as Ca2+-induced Ca2+-release (CICR), regulates myocyte contraction. Contraction is terminated, and relaxation is initiated, by uptake of cytosolic Ca2+ back into the SR through the action of the SR Ca2+ ATPase (SERCA2) or transport into the extracellular space by the sodium-calcium exchanger (NCX1). A growing body of evidence suggests that defects in the communication between LTCC and RyR2 play a central role in the development of hypertrophic cardiomyopathy (HCM) and heart failure3–5.
HCM, defined as unexplained cardiac hypertrophy, affects approximately 1 in 500 persons and is one of the most common genetic cardiovascular diseases6. In addition, HCM is the most common cause of sudden cardiac arrest in young athletes and a significant cause of sudden death in the young in general7,8. Classically, HCM is viewed as a disease of the cardiac sarcomere whereby mutations in the genes encoding key sarcomeric proteins of the heart, such as MYH7-encoded β-myosin heavy chain (MYH7) and MYBPC3-encoded cardiac myosin binding protein C (MYBPC3), cause pathological cardiac hypertrophy. While the pathogenesis of HCM may be initiated with a genetic mutation, emerging evidence has associated gain-of-function disruptions in Ca2+ homeostasis which increase intracellular Ca2+ levels to initiation of a pro-hypertrophic response. Increased Ca2+ release from the SR, either through impaired inhibition of RyR2 function, or increased expression and function of SR inositol 1,4,5-trisphosphate receptors (IP3Rs), have been associated with cardiac hypertrophy9,10. Further, activation of IP3Rs in the nuclear envelope and increased store-operated Ca2+ entry through the transient receptor potential protein TRPC3 can similarly lead to cardiac enlargement in animal models11,12. In some patients with HCM, unabated disease progression results in further pathogenic remodeling ultimately leading to heart failure with loss of contractility13.
Heart failure is one of the major causes of morbidity and mortality in the US as well as the world14. Depressed contractility, particularly rate-dependent contractility reserve, is a common feature of a failing myocyte, and several lines of evidence point to an underlying defect in Ca2+-handling15. Compared to gain-of-function defects in HCM, failure generally demonstrates a loss-of-function defect in CICR, prolonged transient duration, and increased basal Ca2+ levels16. Loss of CICR is likely due to uncoupling of RyR2 from the LTCC, either functionally or physically, resulting in an orphaned RyR2 which has reduced sensitivity to Ca2+ entry from the LTCC4. This is reflected in a loss of excitation-contraction (e-c) coupling gain, such that the same influx of extracellular Ca2+ produces a reduced RyR2-mediated Ca2+ transient17,18. Despite the apparent dichotomous association of gain-of-function Ca2+ defects in HCM and loss-of-function Ca2+ defects in heart failure, there is evidence for overlapping Ca2+ perturbations between the disease states.
JPH2, the major cardiac junctophilin isoform, is necessary for the critical approximation of the LTCC with the RyR2 in the dyad19,20. Amino-terminal membrane occupation and recognition nexus (MORN) motifs interact with the sarcolemma, while a carboxy-terminal transmembrane domain tethers the opposite end to the SR. The critical role of this protein in maintaining the precise sub-sarcolemmal geometry needed for CICR is underscored by the embryonic lethality of JPH2-deficient mice stemming from altered dyadic ultrastructure and stochastic Ca2+ transients with reduced amplitude19. Loss of JPH2 expression was observed in rodent models of HCM and heart failure and is present during an early stage of dyadic “intermolecular failure”21,22. In skeletal muscle, JPH expression silencing resulted in loss of triadic ultrastructure and reduced CICR amplitude23. Recently, we identified three patients with HCM hosting mutations in JPH2, over-expression of which reduced CICR and disrupted cellular ultrastructure24.
To this end, we sought to determine whether a loss of JPH2 protein expression is found in human HCM, and to determine the effect of JPH2 gene silencing on Ca2+-handling within the cardiocyte. We demonstrate reduced JPH2 expression in patients with HCM as well as increased HL-1 cell size and up-regulation of the pro-hypertrophic gene program with expression silencing. Further, our observation that maximal Ca2+ transient, caffeine-stimulated store Ca2+-release, and spontaneous transients at low extracellular Ca2+ levels were each reduced supporting a conclusion that JPH2 knock-down may result in reduced e-c coupling through uncoupled RyR2 and LTCC.
MATERIALS AND METHODS
Human Cardiac Tissue
Human cardiac tissue was obtained from the left ventricle of traumatic death victims with no structural heart disease (BioChain Institute, CA; P1234139, Lot A506041; P1234138, Lot A710149; and CP-R01-T1234130, Lot B201244). Left ventricular myectomy tissue samples from patients with HCM were obtained according to protocols approved by the Mayo Foundation Institutional Review Board following informed consent. Immediately upon surgical resection, tissue samples were flash frozen in liquid nitrogen and stored at −80°C. Each HCM specimen was genotyped comprehensively for mutations in MYH7, MYBPC3, MYL2-encoded regulatory myosin light chain, MYL3-encoded essential myosin light chain, TNNI3-encoded troponin I, TPM1-encoded alpha-tropomyosin, TNNT2-encoded troponin T, and ACTC-encoded alpha-cardiac actin to determine the genetic substrate for HCM pathogenesis using previously outlined methods25. While not equivalent to the clinical genetic testing panel, these genes contain the majority of HCM-associated mutations.
Western Blot
Western blot analysis on whole protein lysates was conducted as previously described25. Briefly, reduced lysates were resolved by SDS-polyacrylamide gel electrophoresis (4–15% for JPH2, CAV3, GAPDH; 6% for RyR2; 7.5% for LTCC; 10% for NCX1, SERCA2; and 15% for PLN), transferred to a nitrocellulose membrane, and blotted with appropriate primary antibodies: anti-JPH2, anti-CAV3 (generous gift from Dr. Jonathan Makielski), anti-LTCC (Alomone Labs, Jerusalem), anti-RyR2 and -PLN (Thermo Fisher Scientific, IL), anti-NCX1 (Swant, Switzerland), anti-SERCA2 (Santa Cruz Biotechnology, CA), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Millipore, CA) antibodies. After application of appropriate secondary antibody, protein was detected using chemiluminescence reagent (Perkin Elmer, MA) or developed using Alexa-Fluor680-conjugated anti-mouse (Invitrogen Molecular Probes, CA) and/or IR800Dye-conjugated anti-rabbit secondary antibodies (Rockland Immunochemicals, PA), and scanned on an Odyssey infrared scanner (Li-Cor, NE). Band density was quantified using ImageJ software26. For human myectomy studies, samples were run unblinded to genetic mutation status and each replication Western blot utilized a different portion of the subject’s myectomy tissue.
Short Hairpin RNA Construct, Cell Culture, Transfection, Adenovirus Infection
The shRNA-encoded construct utilized for transient transfection experiments is described previously23. Briefly, screening experiments for RNA interfering probes to suppress JPH transcription identified a probe which targeted a conserved region of the mRNA encoding the second MORN motif domain of JPH2 (shJPH2) and JPH123. Subsequent cloning of the oligonucleotide into the pCMS-EGFP (BD Biosciences Clontech, CA) vector, in which the GFP reporter was exchanged with DsRED2, created the transient transfection construct pCMS-shRNA.
Lipofectamine-based transfection of pCMS-shRNA maximally achieved a 1% transfection rate in our studies. While suitable for single cell analyses (cell size, Ca2+ transient and store studies) high-efficiency shRNA oligo delivery was obtained through viral transduction/infection in studies requiring 100% shRNA delivery. Adenoviral delivery of shJPH2 and shLuc was achieved by insertion of the shRNA-encoding cassette into the Dual-Basic-RFP construct prior to ligation of the shRNA-RFP expression cassettes into a shuttle vector and in vitro recombinant to Ad5 adenoviral backbone with E1/E3 deletion (Vector Biolabs, PA) creating AdX-shRNA. Virus was packaged in HEK293 cells to produce viral lysates, viral titer was measured by Adeno-X Rapid Titer Kit (Clontech Laboratories, Inc., CA), and the viral lysate was stored in 1M sucrose, 5% tertiary amine beta cyclodextrin in PBS (in mM: 137 NaCl2, 10 NaH2PO4, 2.7 KCl2, 1 KH2PO4, pH 7.4)27.
Murine atrial tissue-derived HL-1 cells were maintained as described previously24. Briefly, low passage number HL-1 cells were grown on 0.02% gelatin, 1% fibronectin coated surfaces with Claycomb media (10% FBS (Sigma, Lot 116K8400), 100U/mL pen, 100ug/mL strep, 0.1mM norepinephrine, 2mM L-glutamine). For all confocal analysis, 5×104 cells were plated on 23mm diameter glass cover slip Delta TPG culture dishes (Bioptechs, PA). For Western blot analysis, 5×105 cells were plated on 60mm diameter plastic tissue culture dishes (Falcon). Cells were transiently transfected using GeneJammer transfection reagent (Stratagene) per manufacturer’s instructions with media replacement every 48 hours. Cells were assayed 96 hours post transfection or infection and shRNA delivery was confirmed through DsRED2 or RFP fluorescence respectively. Prior to phenylephrine (PE) treatment, cells were maintained in Claycomb media without norepinephrine for 96 hours to ensure absence of adrenergic receptor de-sensitization. Cells were then treated with Claycomb media containing 100μM PE (Sigma) or vehicle.
Cell Size and Calcium Measurements
Measurements of cell size and intracellular Ca2+ levels were performed using an Axiovert 200M-Apotome (Carl Zeiss Inc., CA) system with a rapid filter changer (Lambda DG-4, Sutter Instrument Co., CA), and analyzed using AxioVision v4.6 (Carl Zeiss Inc.) software. For all cell size experiments, the area of RFP-positive cells was measured by tracing of the cell periphery after scaling calibration. To confirm accurate identification of the sarcolemma and accurate cell size measurements, wheat germ agglutinin (WGA; emission 495nm, excitation 519nm; Invitrogen) diluted in Hank’s balanced salt solution (Invitrogen) was utilized to fluorescently label the sarcolemma, and cell size measurements were repeated in an independent assay. Prior to [Ca2+] measurements, cells were washed twice with Tyrode buffer (in mM: 140 NaCl, 5 KCl, 10 HEPES, 2.5 CaCl2, 2 MgCl2, 10 D-glucose; pH 7.4), followed by loading for 45 min with 2.5μM Fura-2 AM or 5μM Fluo-4 AM dye (Invitrogen) prior to 15 min de-esterification in Tyrode. For Fura-2 AM studies, regular spontaneous Ca2+ transients were measured via fluorescence (excitation 340nm and 380nm, emission 510nm) versus time with maximal Ca2+ transients (Fmax) measured as the peak fluorescence achieved relative to basal fluorescence (Fo). Ratiometric 340/380nm measurements allowed for correction of intracellular dye levels and estimation of rest/basal Ca2+ levels. For Fluo-4 AM studies, transients were measured via fluorescence (excitation 494nm, emission 526nm) versus time. Transient amplitude (Fmax/Fo), time to amplitude peak (Tpeak), and time to decay to 50% the maximal fluorescent intensity (TD50), and resting calcium levels (Fo) were measured. Pharmacologic modulation of LTCC activity was achieved by addition of two LTCC agonists: 10 μM (S)-(-)-Bay K 864428 (BayK; Tocris Bioscience, Ellisville, MO) and 500 nM FPL 6417629 (FPL; Tocris Bioscience) and an LTCC antagonist: 100 nM nifedipine30 (Tocris Bioscience) during Fluo-4 AM de-esterification. Alteration of extracellular Ca2+ levels ([Ca2+]o) was achieved by equilibrating cells in Tyrode buffer with no Ca2+ (0Ca2+) without EGTA buffering for 10 minutes, and successive increases in [Ca2+]o utilized similarly prepared Tyrode buffer with appropriate CaCl2 concentrations.
To ascertain SR-stored Ca2+ levels through activation of RyR2, 10mM caffeine (Sigma) was applied to the tyrode-immersed, Fura-2-loaded cells directly under real-time confocal analysis. At this concentration, caffeine triggers SR-stored Ca2+ release through a highly Ca2+ sensitive RyR2. To determine store Ca2+ levels through an SR-independent release mechanism, cells were treated with 10μM ionomycin (Sigma) which is a Ca2+ ionophore known to permeabilize membranes allowing for free movement of Ca2+ 31. Prior to ionomycin treatment, cells were rapidly switched to 0Na+/0Ca2+ tyrode (in mM: 140 NaCl, 5 KCl, 10 HEPES, 4.5 MgCl2, 10 EGTA, 10 D-glucose, pH 7.4) to prevent influx of extracellular Ca2+ with treatment as well as alterations in cytosolic Ca2+ due to the action of the LTCC and the NCX1.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was extracted from HL-1 cells 96 hours after infection with AdX-shRNA using the RNeasy Protect Mini Kit (Qiagen) prior to storage at −80°C. RNA integrity was confirmed by Agilent 2100 Bioanalyzer analysis prior to cDNA library genesis utilizing poly-T primer-based reverse transcription (Quanta Biosciences, MD). RNA expression levels of skeletal actin (ACTA, Mm00808218_g1), atrial natriuretic factor (ANF, Mm01255747_g1), brain natriuretic factor (BNP, Mm00435304_g1), MYH7 (Mm00600555_m1), and regulator of calcineurin 1-exon 4 splice isoform (RCAN1-4, Mm00627762_m1) were evaluated using quantitative real-time polymerase chain reaction (qRT-PCR) via TaqMan Gene Expression Assays (Applied Biosciences, CA). Detection of an interferon (IFN)-response by the cells was conducted by RNA expression levels of 2′-5′ oligoadenylate synthetase 1b (OAS1, Mm00449297_m1) and signal transducer and activator of transcription 1 (STAT1, Mm00439531_m1). The threshold cycle value, representing the number of PCR cycles (CT) at which a normalized reporter signal (Rn) first crosses a detection threshold, was determined. The inverse level of RNA template was calculated and normalized to endogenous GAPDH (4352932E) levels. Relative fold change in expression of AdX-shJPH was then calculated compared to AdX-shLuc treatment.
Statistics
ANOVA and t-test analysis was conducted for all studies, as appropriate, with the threshold of significance set to P < 0.05. All statistical analyses were done on at least three independent experiments and are expressed as mean ± standard error of the mean.
RESULTS
JPH2 is Down-Regulated in Human Hypertrophic Cardiomyopathy
Previous investigations have shown JPH2 down-regulation in rodent models of HCM, dilated cardiomyopathy, pressure-induced hypertrophy, and decompensated cardiac failure21,22. Despite these findings, little is known about the expression levels of JPH2 in human disease. Surgical myectomy to remove left ventricular septal myocardium, a treatment for obstructive HCM, provides a relatively unique means of acquiring diseased myocardium from human subjects. Given this resource, we sought to determine whether the down-regulation of JPH2 previously seen in rodent models of cardiac disease was also a characteristic of HCM in humans.
Western blot analysis of JPH2 expression levels from left ventricular protein lysates obtained from three otherwise-healthy traumatic death victims were compared to protein lysates obtained from 11 patients with HCM. Five were acquired from patients with mutations in MYH7 (hosting MYH7-G741R, -R663H, -I736T, -R403Q, and -M922K mutations from left to right lanes in Figure 1A) and three with mutations in MYBPC (MYBPC-E258K, -L527 fs/3, and -L1258 fs/71, respectively) which represent the two most common genetic subtypes of HCM. Further, three samples obtained from probands not hosting mutations in the analyzed genes were similarly assayed. While there was some variation in JPH2 expression among presumably “healthy” subjects, a reduction in expression was observed in all samples acquired from patients with HCM. Quantification of blots taken from independent portions of resected myocardium revealed that all individuals with HCM had reduced JPH2 expression (ranging from 0.011 ± 0.006 to 0.343 ± 0.089 normalized intensity units) compared to trauma samples (from 1.536 ± 0.331 to 0.700 ± 0.078) as depicted in (Figure 1B). When taken as a whole, HCM samples had a mean relative JPH2 expression of 0.120 ± 0.022 compared to ostensibly healthy tissue (1.079 + 0.638, P < 0.0001; Figure 1B, inset).
Figure 1. JPH2 expression is decreased in humans with hypertrophic cardiomyopathy.
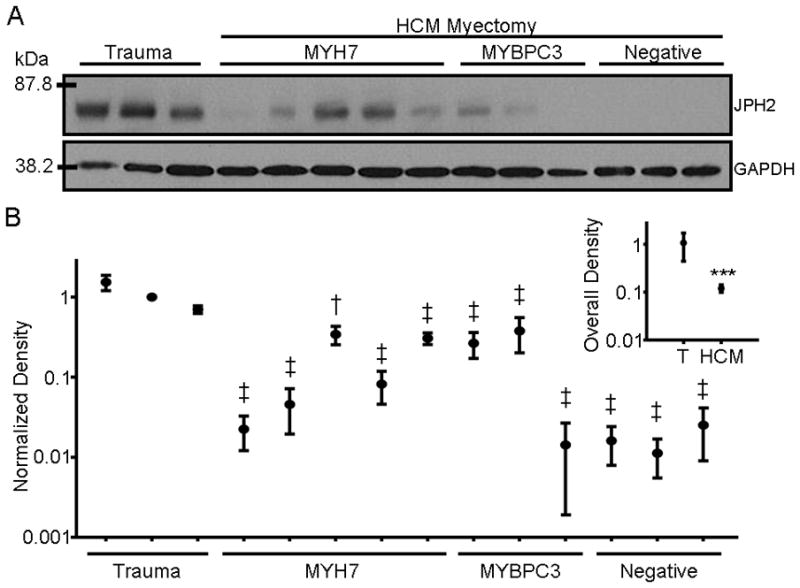
A. Representative Western blot of JPH2 expression levels in lysates prepared from left ventricular myocardium obtained from three healthy traumatic death victims and myectomy tissue from 11 patients with HCM genotype-positive for mutations in MYH7 (N = 5 individuals), MYBPC3 (3) or genotype-negative (3). From left to right, HCM patients hosted mutations of MYH7-G741R, -R663H, -I736T, -R403Q, -M922K and MYBPC-E258K, -L527 fs/3, and -1258 fs/71, respectively. GAPDH served as a loading control. B. Graph of relative mean band density of JPH2 expression of individuals with HCM (mean of 8 blots per individual) relative to control specimens (mean of 5 blots per individual). ‡, P < 0.05 versus each of the trauma samples; †, P < 0.05 versus two of the three trauma samples. Inset) Graph of the mean Western blot band density of all HCM specimens relative to mean control tissue band density. ***, P < 0.0001.
shJPH2 Achieves Durable Expression Knock-Down
Given the reduction of JPH2 protein expression in human HCM, we sought to determine whether this phenomenon was a compensatory physiologic response of the heart or whether it represents a potential initiating factor in hypertrophic remodeling. To address this, interfering RNA targeted to JPH2 mRNA was introduced into HL-1 cells which express endogenous JPH2 as well as all critical cellular components necessary for cardiac excitation contraction coupling, including RyR2, LTCC, SERCA2, NCX1, and the β-adrenergic receptor32. These cells demonstrate spontaneous CICR and have been used previously to characterize the effect of genetic mutations in RyR2 function33. Previous studies utilizing this shRNA probe have demonstrated a significant reduction in the expression levels of JPH1 as well as JPH2 in skeletal muscle23; however, HL-1 cells do not express JPH1 by qRT-PCR (Figure 2A), leaving JPH2 as the only junctophilin family member silenced by expression of this shRNA.
Figure 2. Adenovirus-mediated shRNA silencing of JPH2 in HL-1 cells.
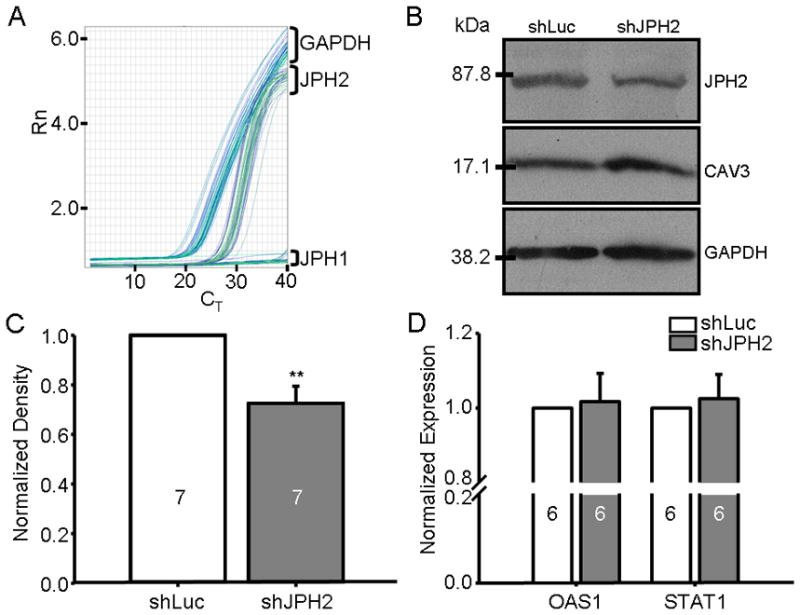
A. Representative qRT-PCR plot of normalized fluorescence (Rn) intensity versus PCR cycle number (CT) for three transcript-specific probe sets including JPH1, JPH2, and GAPDH targeted to un-treated HL-1 cDNA. B. Western blot of JPH2, CAV3, and GAPDH levels from HL-1 cells infected with AdX-shLuc and controls. C. Bar graph showing relative difference in band density of AdX-shJPH2 infected cells from independent Western blots relative to AdX-shLuc normalized to GAPDH (N = 7). **, P < 0.01. D. Bar graph demonstrating mean RNA expression levels of OAS1 and STAT1 by qRT-PCR of AdX-shJPH2 infected cells (dark gray bars) relative to controls (white bars, N = 6 runs).
Utilizing the RFP marker co-expressed by the adenovirus, we achieved nearly 100% infectivity of the HL-1 cells with a multiplicity of infection of 100IFU/cell (data not shown). As shown in Figure 2B & C, adenoviral delivery of the shJPH2 probe demonstrated a sustained JPH2 expression knock-down of 26.5% after 96 hours when normalized to GAPDH expression relative to shLuc (P < 0.01). The expression of caveolin-3 (CAV3), a JPH2 binding partner, was not affected by JPH2 silencing21.
Expression of double stranded RNA in mammalian cells may induce a type 1 IFN response that induces global alterations to multiple cellular processes which may confound the effect of JPH2 expression silencing34. To ensure that such a response was not present with adenoviral delivery of AdX-shJPH2 and shLuc, and to test this known cardiac remodeling signaling cascade, RNA expression levels of IFN-induced OAS1 and STAT1 were determined by qRT-PCR35. As demonstrated in Figure 2D, there was no alteration in OAS1 and STAT1 levels in HL-1 cells treated with AdX-shJPH2 relative to AdX-shLuc normalized to GAPDH expression.
JPH2 Knock-Down Increases Cell Size and Induces Pro-Hypertrophic Marker Expression
To determine whether an acute suppression of JPH2 expression is sufficient to initiate cardiac hypertrophic remodeling, HL-1 cell area was measured after both transient transfection and adenoviral infection of shJPH2. In pCMS-JPH2 transfected cells, partial JPH2 expression silencing resulted in a marked increase in HL-1 cellular size of 75.3 ± 7.5% (N = 68) when compared to control shLuc cells (N = 245; P < 0.0001). To confirm accuracy of cell size measurements, we fluorescently labeled the cell sarcolemma with WGA (Figure 3A) and confirmed an increase in cellular size of 57.0 ± 14.0% (N = 59) when compared to control (N = 80; P = 0.004) which was not statistically significantly different from the degree of hypertrophy seen without WGA-labeling (P = 0.2). Similarly, adenoviral delivery of shJPH2 resulted in an increase of 94.2 ± 7.3%, N = 346, when compared to AdX-shLuc (N = 469; P < 0.0001) which was not statistically significantly between transfection delivery of shJPH2 (P = 0.3; Figure 3B). As a positive control, a known pharmacologic inducer of hypertrophy in this cell line, PE, was applied (Figure 3B, gray bars)36. Treatment of these cells with an α1 adrenergic receptor agonist initiated a similar hypertrophic response (increase of 75.4 ± 7.9%, N = 485) relative to vehicle treated cells (N = 657; P < 0.0001) when compared to shJPH2 expression silencing.
Figure 3. JPH2 knock-down induces hypertrophy of HL-1 cells.
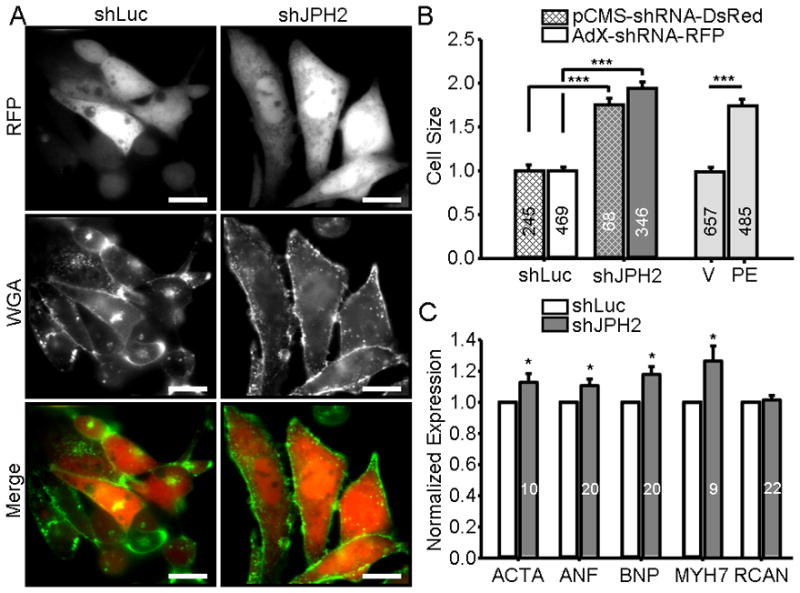
A. Representative image of RFP-positive HL-1 cells infected with control pCMS-shLuc or pCMS-shJPH2 and labeled with WGA. Scale bar, 100μm. B. Bar graph showing mean HL-1 cell size transiently transfected with pCMS-shRNA (cross-hatch) or adenovirally infected with AdX-shRNA (no cross-hatch). Delivery of shJPH2 by either transient transfection or adenoviral infection (dark gray bars) induced an increase in cell size compared to controls (white bars) that was not significantly different between oligo-delivery methodologies. Light gray bars, treatment with phenylephrine (PE), induced a similar increase in cell size compared to vehicle-treated controls (V). Numbers in bar graph indicate number of cells analyzed. ***, P < 0.0001. C. Relative RNA expression levels of ACTA, ANF, BNP, MYH7, RCAN1-4 in HL-1 cells infected with AdX-shJPH2-RFP normalized control cells. Numbers in bar graphs indicate number of assays. *, P < 0.05.
As many potential pathways could contribute to increased cellular size in vitro, we attempted to correlate this observation with molecular markers of the well-established cardiac hypertrophic remodeling process. Induction of four markers of this transcriptional process, ACTA, ANF, BNP, and MYH7, are consistently associated with a pathologic hypertrophic response. To determine whether the increase in HL-1 cell size is associated with a similar induction of expression of these markers, qRT-PCR was conducted on cells infected with AdX-shRNA. As shown in Figure 3C, a significant increase in ACTA (12.8 ± 5.6%), ANF (10.6 ± 4.4%), BNP (17.9 ± 5.0%), and MYH7 (26.4 ± 9.1%) was observed in AdX-shJPH2 cells compared to control cells (P < 0.05). Furthermore, we tested transcriptional expression of RCAN1-4, a marker of calcineurin-dependent signaling which is one potential mediator of remodeling in the cardiocyte. In AdX-shJPH2 infected cells, there was no increase in RCAN1-4 expression levels by qRT-PCR.
JPH2 Knock-Down Perturbs Calcium Flux and Homeostasis
Based on the observation of increased cellular area as well as transcriptional up-regulation of a pro-hypertrophic makers in the cells, we explored the possibility that Ca2+ dysregulation may be associated with JPH2 expression knock-down. Alterations in Ca2+ transients and basal Ca2+ levels have been linked to both the initiation of hypertrophy as well as arrhythmogenesis in the heart. HL-1 cells spontaneously and regularly oscillate Ca2+ through the action of excitation-contraction coupling proteins. We have previously utilized HL-1 cells to measure Ca2+-transient amplitude24, and the firm adherence of these cells to the growth surface is suitable for maintaining real-time confocal measurements during pharmacological treatment.
Maximal Ca2+ transients achievable were significantly reduced in cells transiently transfected with pCMS-shJPH2 (1.10 ± 0.09, N = 69) when compared to control cells (1.60 ± 0.03, N = 126; P < 0.0001) as demonstrated in Figure 4A & 4B. This reduction in transient amplitude was insensitive to pharmacological stimulation of the LTCC. While BayK and FPL, two LTCC stimulators, increased control cell transient amplitudes (1.75 ± 0.07, N = 43; P = 0.008 and 1.88 ± 0.07, N = 53; P < 0.0001 versus untreated control cells, respectively), this increase was not seen in shJPH2 cells (Bay K: 1.20 ± 0.04, N = 43 and FPL: 1.14 ± 0.03, N = 38) which demonstrated transient amplitudes that were not significantly different from untreated shJPH2 cells (Figure 4B). Conversely, application of nifedipine reduced Ca2+ transient amplitude in control cells (1.14 ± 0.03, N = 52; P < 0.0001 versus untreated control cells), while there was no statistically significant difference between nifedipine treated shJPH2 cells (1.06 ± 0.02, N = 28) and untreated cells (P = 0.5, Figure 4B).
Figure 4. JPH2 knock-down reduces intracellular Ca2+ transients.
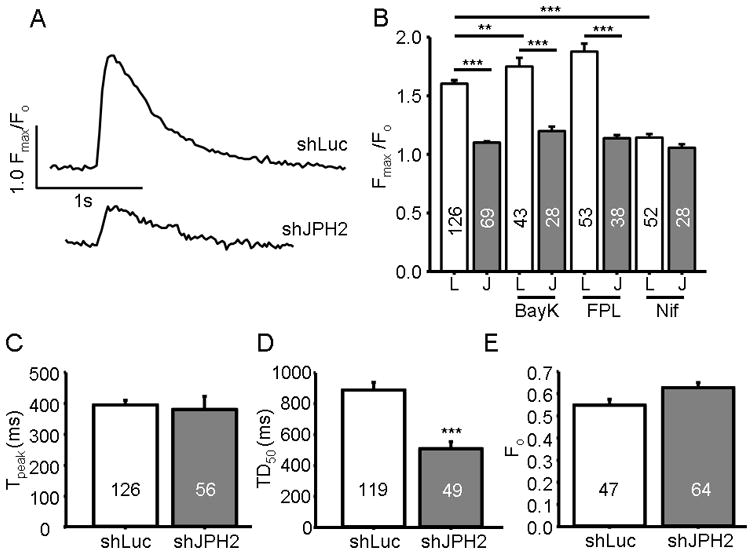
A. Representative Ca2+ transient tracing of an HL-1 cell transiently transfected with pCMS-shJPH2 or shLuc measuring relative fluorescent intensity (Fmax/Fo) versus time. B. Bar graph of the mean Ca2+ transient amplitude in HL-1 cells transiently transfected with pCMS-shJPH2 or control. Cells were treated with LTCC activators BayK or FPL or LTCC blocker nifedipine (Nif). Numbers in bar graphs indicate number of cells analyzed. **, P < 0.01; ***, P < 0.0001 C, and D. Bar graph of time to transient peak, and time to decay to half maximum intensity, respectively, in HL-1 cells transiently transfected with pCMS-shJPH2 or control. ***, P < 0.0001 E. Bar graph of the mean resting Ca2+ levels in cells transiently transfected with pCMS-shJPH2 or control. Numbers in bar graphs indicate number of cells analyzed.
In addition to reductions in transient amplitude, transient kinetics were altered with JPH2 knock-down. While the time to transient peak was unchanged, the time for the Ca2+-transient to decay was reduced in shJPH2 cells (508 + 46ms, N = 49) compared to control cells (888 ± 49ms, N = 119) as depicted in Figure 4C & 4D, respectively. Further, despite impaired Ca2+ transient amplitude, the resting Fura-2 AM fluorescence was higher in cells with shJPH2 (0.63 ± 0.03, N = 64) compared to control (0.55 ± 0.04, N = 47) as demonstrated in Figure 4E, although this finding was not statistically significant (P = 0.06).
JPH2 Knock-Down Alters Store Calcium Levels
To determine whether the reduction in Ca2+ transient and potential alteration in basal Ca2+ levels were due to alterations in store Ca2+, caffeine was applied to measure RyR2-dependent SR store Ca2+ levels. As demonstrated in Figure 5A & 5B, HL-1 cells transiently transfected with pCMS-shJPH2 had a significantly lower caffeine-induced Ca2+ release (0.82 ± 0.07, N = 25) compared to control (1.06 ± 0.05, N = 48; P < 0.0001). To determine whether this apparent loss of store Ca2+ was due to alterations in the SR Ca2+ store levels in isolation or whether other Ca2+ stores are similarly altered, ionomycin was applied31. As demonstrated in Figure 5C & 5D, under 0Na+/0Ca2+ buffer conditions, ionomycin treatment resulted in release of store Ca2+ which was greater in the pCMS-shJPH2 transfected cells (1.84 ± 0.10, N = 27) compared to controls (1.35 ± 0.06, N = 75; P < 0.0001). Thus, despite an overloaded total Ca2+ store in the shJPH2 cells, there was a significant reduction in caffeine-mediated SR store levels.
Figure 5. Knock-down of JPH2 increased store Ca2+ yet reduced RyR2-dependent caffeine-mediated store Ca2+ release.
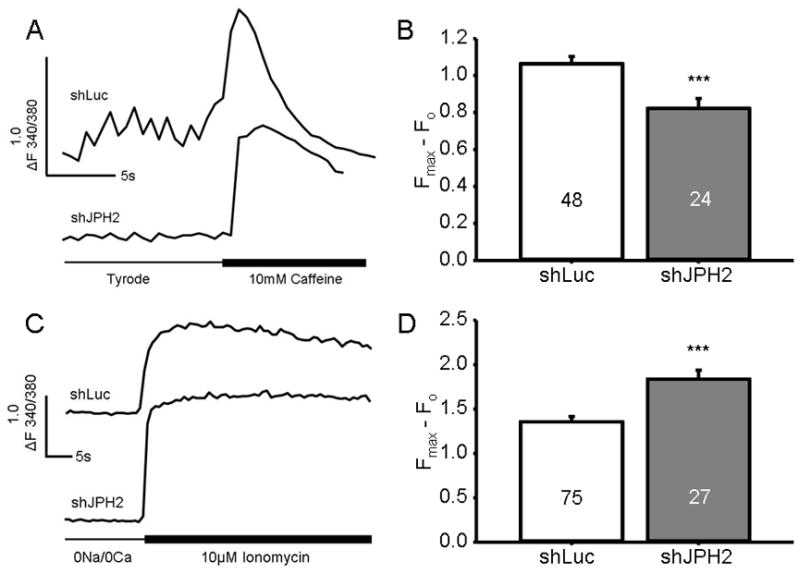
A. Representative Ca2+ tracing of HL-1 cells following caffeine application. B. Bar graph showing mean amplitude of Ca2+ efflux from the SR in pCMS-shRNA-treated cells exposed to caffeine. Numbers in bar graph indicate number of cells analyzed. ***, P < 0.0001. C. Representative Ca2+ tracing of HL-1 cells following ionomycin treatment. D. Bar graph showing mean amplitude of Ca2+ efflux from all stores in pCMS-shRNA-treated cells exposed to ionomycin. Numbers in bar graph indicate number of cells analyzed. ***, P < 0.0001.
JPH2 Knock-Down Suppresses Spontaneous Calcium Oscillations
The HL-1 cell line demonstrates regular spontaneous Ca2+ oscillations under normal extracellular [Ca2+]. Based on the previously observed loss of Ca2+ transient amplitude and insensitivity of the Ca2+ transient to LTCC stimulation with JPH2 expression silencing, we next tested the coupling of spontaneous Ca2+ transients generation with extracellular [Ca2+]. As demonstrated in Figure 6A, AdX-shLuc cells in 0Ca2+ buffer generate no regular Ca2+ oscillations and occasionally demonstrate stochastic transients which are presumably not LTCC-triggered Ca2+ release events. With addition of 0.1 and 0.25mM [Ca2+]o, the stochastic, non-LTCC-mediated transients begin to yield regular oscillations which are coordinated among the cells. Application of 1 and 2.5mM [Ca2+]o induced regular oscillations which are spontaneous and are simultaneously triggered in nearly every cell in each field. In contrast, knock-down AdX-shJPH2 infected HL-1 cells do not demonstrate Ca2+ oscillations until 1mM [Ca2+]o. This is quantified in Figure 6B, in which there is a blunting of Ca2+ oscillation generation with JPH2 silencing which occur at 0.1 and 0.25mM [Ca2+]o. This is reflected in a lower proportion of oscillating cells infected with AdX-shJPH2 (0.04 ± 0.01%, N = 12 fields) than control (0.13 ± 0.02%, N = 12; P = 0.002) at 0.1mM and 2.5mM (shJPH2: 0.05 ± 0.05%, N = 13 compared to control: 0.38 ± 0.01%, N = 11; P = 0.009). Further, while there is no difference in the number of field-wide Ca2+ oscillations per time at lower [Ca2+]o levels, there is a significant reduction in the oscillation frequency in cells infected with shJPH2 (7.7 ± 1.2 transients/30s, N = 15 fields) than shLuc (17.0 ± 4.3, N = 11 fields; P = 0.02) at 2.5mM [Ca2+]o (Figure 6C).
Figure 6. Knock-down of JPH2 blunts generation of spontaneous Ca2+ oscillations with modulation of extracellular Ca2+ levels.
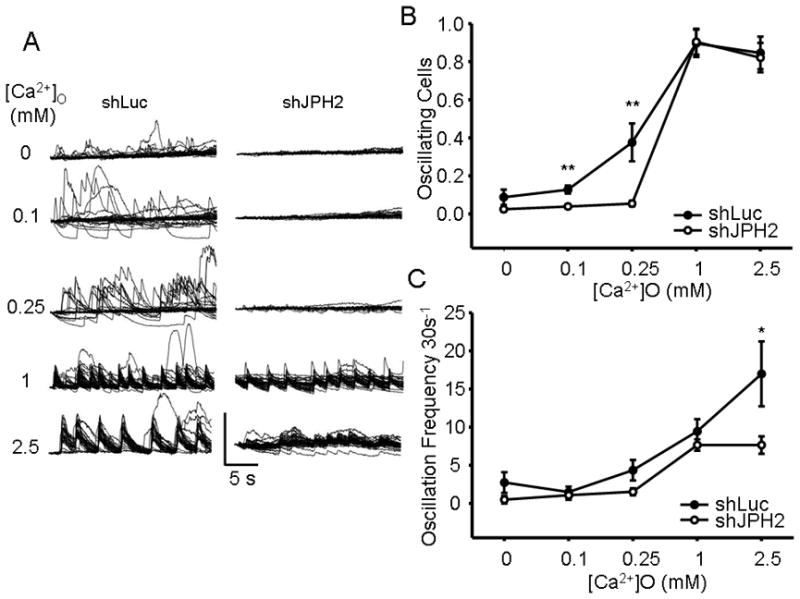
A. Representative individual Ca2+ tracings from a field of AdX-shJPH2 infected HL-1 cells with increasing levels of [Ca2+]o. B. A graph of proportion of spontaneously oscillating cells per field versus increasing [Ca2+]o for both AdX-shJPH2 infected cells and control. **, P < 0.01. C. A graph of the number of field-wide spontaneous oscillations per 30 seconds versus increasing [Ca2+]o for both AdX-shJPH2 infected cells and control. *, P < 0.05.
JPH2 Knock-Down Does Not Alter Calcium-Handling Protein Expression
Due to the observed reduction in Ca2+ transient amplitude, altered transient kinetics, and perturbed store Ca2+ levels, we next explore the expression levels of multiple proteins necessary for effective e-c coupling and transient formation (Figure 7). Despite the reduction of CICR amplitude, western blot analysis of LTCC and RyR2 consistently revealed no change in expression with AdX-shJPH2-treated cells. Further, there was not a statistically significant change in expression of NCX1, SERCA2 and PLN in JPH2 knock-down cells compared to controls.
Figure 7. Calcium-handling protein expression levels are unchanged with JPH2 knock-down.
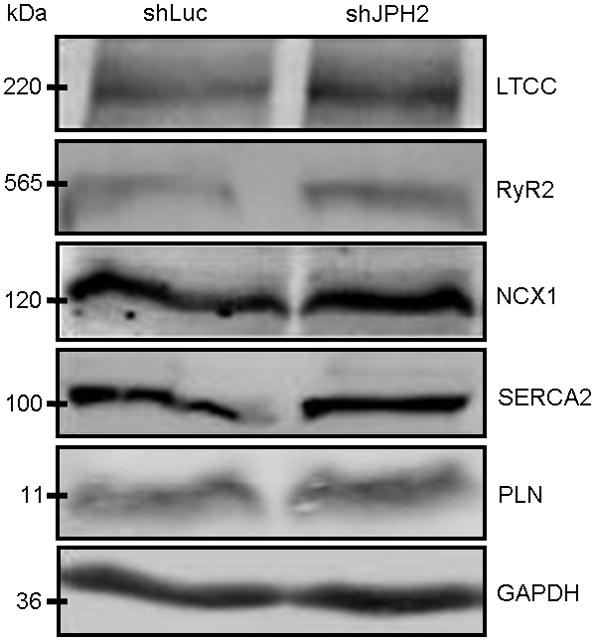
Representative Western blots of LTCC, RyR2, NCX1, SERCA2, and PLN expression levels in lysates prepared from cells treated with AdX-shRNA. GAPDH served as a loading control.
DISCUSSION
The junctophilin family of proteins are critical for proper Ca2+ flux and homeostasis in excitable cells including skeletal muscle (JPH1 and 237,38), cardiac muscle (JPH2), and neuronal tissue (JPH3 and 439–41). In concert with the recent insight into the cellular role of these proteins, genetic mutations which perturb the function of JPH2 and JPH3 have been associated with the pathogenesis of human disease. While its pathogenic mechanism remains elusive, a CAG/CTG nucleotide repeat expansion in a potential alternatively-spliced exon of JPH3-encoded junctophilin type 3 has been associated with Huntington disease type 2 in humans42. Further, we recently demonstrated three missense mutations in a cohort of 388 patients with HCM that were mutation negative for the traditional HCM-associated sarcomeric protein genes24. Based upon the emerging role of JPH dysregulation and dysfunction, we sought to understand how reduced expression of JPH2 might play a role in cardiomyopathy.
Herein, we demonstrated that myocardium from HCM patients demonstrate a significant reduction in JPH2 expression independent of underlying disease-causing genotype and may reflect a common feature in the cardiomyopathic remodeling of this disease. This fits well with the previous observations that reduction of JPH2 levels occurs across a relatively diverse range of rodent cardiomyopathic disease states. The generalizability of this phenomena in human heart failure, pressure (hypertension)-induced hypertrophy, or myopathic sequelae of organic disease states such as idiopathic cardiomyopathy of diabetes, is an unanswered question43. It is also possible that the down-regulation of JPH2 is not a property of other mechanisms of cardiac hypertrophy.
In order to test whether this reduction of JPH2 expression is sufficient to induce cardiac dysfunction and hypertrophy, we used a RNA silencing probe to knock-down JPH2 expression. Our observation that interfering RNA silencing of endogenous JPH2 results in a marked increase in cellular size, potentially through further induction of the fetal gene program, are evidence that even relatively small reductions in JPH2 expression can be an initiating factor in the pathogenic remodeling of the heart, and not simply an adaptive response with no pathophysiologic consequence. In experiments using native cardiac tissue, up-regulation of ACTA, ANF, BNP, and MYH7 as transcriptional markers of the pro-hypertrophic cellular response are on a fold-scale that is greater than the 10–30% increase we observe. As the HL-1 cells are immortalized and perpetually grow and divide, it is possible that there is a higher basal level of these transcripts within the transcriptional milieu of the cell, and we are observing a relative modest increase in a highly active pro-growth/hypertrophic gene program. Alternatively, it is possible that the observed increase in cell size seen may not be solely mediated through the traditional hypertrophic response of which these transcripts are markers.
Silencing of JPH2 in HL-1 cells resulted in significant alterations in both basal and dynamic Ca2+ signaling within the cell. Our observations that JPH2 expression silencing results in reduced CICR through a reduction in the maximal transient amplitude without alteration of LTCC, RyR2, SERCA2, PLN, or NCX1 expression supports a role for JPH2 in maintaining effective e-c coupling gain. This closely follows our previous work in which functional characterization of the three HCM-associated mutations demonstrated similar decrease in CICR amplitude when expressed in HL-1 cells24 and the reduced CICR seen in skeletal muscle with JPH1 and 2 knock-down23. When Ca2+ is titrated into the extracellular buffer of spontaneously oscillating HL-1 cells, JPH2 knock-down reduces the sensitivity of CICR to extracellular Ca2+. While RyR2-mediated Ca2+ transients are clearly present at 0.25mM [Ca2+]o with control shLuc-infected cells, no Ca2+ oscillations are seen in the shJPH2-infected cells. Since the cells did not demonstrate Ca2+ oscillations in 0Ca2+ extracellular buffer and at 2.5mM [Ca2+]o all cells regularly and uniformly oscillated in unison, extracellular Ca2+ entry through the voltage-sensitive LTCC is likely triggering the observed transients. Thus, it may be that the observed right shift in the dose-response curve with JPH2 knock-down is due to a loss of e-c coupling gain such that more Ca2+ entry into the sub-sarcolemmal space is needed to trigger sufficient RyR2 opening to cause CICR. This possibility is supported by the relative insensitivity of JPH2 knockdown cells Ca2+ transients to LTCC activation.
These loss-of-function defects in Ca2+-handling would generally reflect those seen in patients and experimental models of heart failure. Indeed, the loss of CICR amplitude, potential blunting of e-c coupling gain, and the observed reduction in SR store Ca2+ by caffeine-stimulation would fit with several previous models of heart failure such as rats4, dogs44, and humans18. Based on these previous studies, and the known role of JPH2 in maintaining critical dyadic geometry, one possibility is that this reduction in CICR may be due to increased distance of RyR2 from the sarcolemmal LTCC, or otherwise disrupted cellular ultrastructure. While not directly addressed herein, this physical separation may result in a relatively Ca2+-insensitive RyR2 which does not respond fully to Ca2+ influx from the LTCC, resulting in a loss of CICR. This possibility is supported by previous observation that over-expression of HCM-associated JPH2 mutations disrupted cellular ultrastructure24. In addition the observed defect in CICR, alterations in cardiac relaxation is often seen in cardiac hypertrophy and failure. Reduced SERCA2 expression is commonly seen in models of cardiac hypertrophy and failure resulting in reduced SR Ca2+ and increased basal cytosolic Ca2+ 45,46. Our observation that SERCA2 and PLN expression are unchanged is not without precedent and may reflect variation in the way this cellular model undergoes hypertrophy with JPH2 expression silencing. While the protein levels are unchanged, it is possible that the ATPase activity of SERCA2, and the inhibitory role of PLN, are nonetheless functionally impacted by post-translational modification resulting in the observed reduction in SR Ca2+.
The loss of JPH2 expression in rodent models of HCM, our previous association of JPH2 mutations in human HCM, and our observation of loss of JPH2 expression in patients with HCM would seemingly run counter to a solely loss-of-function defect in CICR activity. Indeed, the development of cardiac hypertrophy has largely been associated with increases in cytosolic Ca2+, whereby either elevation of global or dyadic Ca2+ levels induce the pro-hypertrophic fetal gene program through either a calmodulin kinase II- or calcineurin-mediated pathway, among others47,48. In a setting of reduced CICR amplitude, we observed a strong trend toward increased basal Ca2+ in JPH2 knock-down cells. It is unlikely that the increase in resting Ca2+ is directly due to alterations in the Ca2+ transient kinetics as the amplitude and TD50 are both reduced, it may indicate increased Ca2+ leakage of RyR2 at rest. This possibility is in agreement with previous work showing that RyR2 mutations which disrupt the calmodulin binding which normally reduces RyR2 Ca2+ release activity, lead to induction of hypertrophy as well as extra Ca2+ oscillations which elevate cytosolic Ca2+ levels9. While the ability of JPH2 to modulate the gating of RyR2 has not been directly demonstrated to date, skeletal muscle JPH1 modulates gating of RyR1 through a redox-sensitive direct interaction49. Any role, either direct or indirect, that JPH2 may have in modulating RyR2 channel function, as well as the involvement of pro-hypertrophic signaling pathways, will require future study.
There are several limitations to our study. While we observe a clear reduction in JPH2 expression in HCM myectomy specimens compared to the control samples, we are unable to evaluate the possibility of regional differences in cardiac JPH2 expression. In particular, we are unable to ensure that the control samples were obtained from the same region of the left ventricular myocardium as the HCM myectomy specimens. In addition, while the HL-1 cell line has proven useful for our analysis, the immortalized nature of these cells, and loss of native cardiocyte shape and t-tubular ultrastructure limits our analysis and prevents direct measurement of the intermolecular coupling of dyadic proteins. Further, we are unable to determine how knock-down of JPH2 expression might affect the beating heart in vivo and whether important hallmarks of cardiac remodeling, such as fibrous replacement of myocardium in heart failure, might be present. Future studies will need to be done to further elucidate any potential role JPH2 may have in modulating RyR2 function and how perturbations in this protein might predispose to Ca2+-mediated diseases of the heart.
Acknowledgments
FUNDING SOURCES
A.P.L. is supported by the American Heart Association Predoctoral Fellowship. R.J.v.O. is the recipient of the 2008–2010 American Physiological Society Postdoctoral Fellowship in Physiological Genomics. X.H.T.W. is a W.M. Keck Foundation Distinguished Young Scholar in Medical Research, and is supported by NIH/NHLBI grants R01-HL089598 and R01-HL091947; and Muscular Dystrophy Association grant #69238. M.J.A. is an Established Investigator of the American Heart Association and is supported by NIH grants R01-HD42569 and P01-HL94291, and Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program. M.J.A., and X.H.T.W. are supported by a Fondation Leducq Award to the ‘Alliance for Calmodulin Kinase Signaling in Heart Disease’.
Non-Standard Abbreviations and Acronyms
- CAV3
caveolin type 3
- CICR
calcium-induced calcium-release
- e-c
excitation-contraction
- HCM
hypertrophic cardiomyopathy
- IP3R
inositol 1,4,5-triphosphate receptor
- JPH
junctophilin
- LTCC
L-type calcium channel
- NCX1
sodium-calcium exchanger
- MORN
membrane occupation and recognition nexus
- MYBPC3
cardiac myosin binding protein
- MYH7
β myosin heavy chain
- RyR2
ryanodine receptor type 2
- SERCA2
sarcoplasmic reticulum calcium ATPase
- shRNA
short hairpin RNA
- SR
sarcoplasmic reticulum
- TRPC3
transient receptor potential protein type 3
Footnotes
CONFLICT OF INTEREST DISCLOSURES
MJA is a consultant for Medtronic, St. Jude Inc., Boston Scientific, and PGxHealth and serves on the Scientific Advisory Board of and receives royalties from PGxHealth.
References
- 1.Nishi M, Mizushima A, Nakagawara K, Takeshima H. Characterization of human junctophilin subtype genes. Biochem Biophys Res Commun. 2000;273:920–927. doi: 10.1006/bbrc.2000.3011. [DOI] [PubMed] [Google Scholar]
- 2.Garbino A, van Oort RJ, Dixit SS, Landstrom AP, Ackerman MJ, Wehrens XHT. Molecular evolution of the junctophilin gene family. Physiol Genomics. 2009;37:175–186. doi: 10.1152/physiolgenomics.00017.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen-Izu Y, Chen L, Banyasz T, McCulle SL, Norton B, Scharf SM, Agarwal A, Patwardhan A, Izu LT, Balke CW. Hypertension-induced remodeling of cardiac excitation-contraction coupling in ventricular myocytes occurs prior to hypertrophy development. Am J Physiol Heart Circ Physiol. 2007;293:H3301–3310. doi: 10.1152/ajpheart.00259.2007. [DOI] [PubMed] [Google Scholar]
- 4.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chantawansri C, Huynh N, Yamanaka J, Garfinkel A, Lamp ST, Inoue M, Bridge JHB, Goldhaber JI. Effect of metabolic inhibition on couplon behavior in rabbit ventricular myocytes. Biophys J. 2008;94:1656–1666. doi: 10.1529/biophysj.107.114892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of Hypertrophic Cardiomyopathy in a General Population of Young Adults: Echocardiographic Analysis of 4111 Subjects in the CARDIA Study. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 7.Maron B, Roberts W, McAllister H, Rosing D, Epstein S. Sudden death in young athletes. Circulation. 1980;62:218–229. doi: 10.1161/01.cir.62.2.218. [DOI] [PubMed] [Google Scholar]
- 8.Maron B, Epstein S, Roberts W. Causes of sudden death in competitive athletes. J Am Coll Cardiol. 1986;7:204–214. doi: 10.1016/s0735-1097(86)80283-2. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J Clin Invest. 2007;117:1344–1353. doi: 10.1172/JCI29515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harzheim D, Movassagh M, Foo RSY, Ritter O, Tashfeen A, Conway SJ, Bootman MD, Roderick HL. Increased InsP3Rs in the junctional sarcoplasmic reticulum augment Ca2+ transients and arrhythmias associated with cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:11406–11411. doi: 10.1073/pnas.0905485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakayama H, Wilkin BJ, Bodi I, Molkentin JD. Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J. 2006;20:1660–1670. doi: 10.1096/fj.05-5560com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna WJ, Borggrefe M, England D, Deanfield J, Oakley CM, Goodwin JF. The natural history of left ventricular hypertrophy in hypertrophic cardiomyopathy: an electrocardiographic study. Circulation. 1982;66:1233–1240. doi: 10.1161/01.cir.66.6.1233. [DOI] [PubMed] [Google Scholar]
- 14.Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KKL, Murabito JM, Vasan RS. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–1402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 15.Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 16.Gwathmey JK, Copelas L, MacKinnon R, Schoen FJ, Feldman MD, Grossman W, Morgan JP. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76. doi: 10.1161/01.res.61.1.70. [DOI] [PubMed] [Google Scholar]
- 17.Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ. Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science. 1997;276:800–806. doi: 10.1126/science.276.5313.800. [DOI] [PubMed] [Google Scholar]
- 18.Lindner M, Erdmann E, Beuckelmann DJ. Calcium Content of the Sarcoplasmic Reticulum in Isolated Ventricular Myocytes from Patients with Terminal Heart Failure. J Mol Cell Cardiol. 1998;30:743–749. doi: 10.1006/jmcc.1997.0626. [DOI] [PubMed] [Google Scholar]
- 19.Takeshima H, Komazaki S, Nishi M, Iino M, Kangawa K. Junctophilins: a novel family of junctional membrane complex proteins. Mol Cell. 2000;6:11–22. doi: 10.1016/s1097-2765(00)00003-4. [DOI] [PubMed] [Google Scholar]
- 20.Ziman AP, Gómez-Viquez NL, Bloch RJ, Lederer WJ. Excitation-contraction coupling changes during postnatal cardiac development. J Mol Cell Cardiol. 2010;48:379–386. doi: 10.1016/j.yjmcc.2009.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minamisawa S, Oshikawa J, Takeshima H, Hoshijima M, Wang Y, Chien KR, Ishikawa Y, Matsuoka R. Junctophilin type 2 is associated with caveolin-3 and is down-regulated in the hypertrophic and dilated cardiomyopathies. Biochem Biophys Res Commun. 2004;325:852–856. doi: 10.1016/j.bbrc.2004.10.107. [DOI] [PubMed] [Google Scholar]
- 22.Xu M, Zhou P, Xu S-M, Liu Y, Feng X, Bai S-H, Bai Y, Hao X-M, Han Q, Zhang Y, Wang S-Q. Intermolecular failure of L-type Ca2+ channel and ryanodine receptor signaling in hypertrophy. PLoS Biology. 2007;5:e21. doi: 10.1371/journal.pbio.0050021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirata Y, Brotto M, Weisleder N, Chu Y, Lin P, Zhao X, Thornton A, Komazaki S, Takeshima H, Ma J, Pan Z. Uncoupling store-operated Ca2+ entry and altered Ca2+ release from sarcoplasmic reticulum through silencing of junctophilin genes. Biophys J. 2006;90:4418–4427. doi: 10.1529/biophysj.105.076570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landstrom AP, Weisleder N, Batalden KB, Martijn Bos J, Tester DJ, Ommen SR, Wehrens XHT, Claycomb WC, Ko J-K, Hwang M, Pan Z, Ma J, Ackerman MJ. Mutations in JPH2-encoded junctophilin-2 associated with hypertrophic cardiomyopathy in humans. J Mol Cell Cardiol. 2007;42:1026–1035. doi: 10.1016/j.yjmcc.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theis J, Bos J, Theis J, Miller D, Dearani J, Schaff H, Gersh B, Ommen S, Moss R, Ackerman M. Expression Patterns of Cardiac Myofilament Proteins - Genomic and Protein Analysis of Surgical Myectomy Tissue from Patients with Obstructive Hypertrophic Cardiomyopathy. Circ Heart Fail. 2009;2:325–333. doi: 10.1161/CIRCHEARTFAILURE.108.789735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abramoff M, Magelhaes P, Ram S. Image Processing with ImageJ. Biophotonics International. 2004;11:36–42. [Google Scholar]
- 27.Croyle M, Cheng X, Wilson J. Development of formulations that enhance physical stability of viral vectors for gene therapy. Gene Ther. 2001;8:1281–1290. doi: 10.1038/sj.gt.3301527. [DOI] [PubMed] [Google Scholar]
- 28.Franckowiak G, Bechem M, Schramm M, Thomas G. The optical isomers of the 1,4-dihydropyridine BAY K 8644 show opposite effects on Ca channels. Eur J Pharmacol. 1985;114:223–226. doi: 10.1016/0014-2999(85)90631-4. [DOI] [PubMed] [Google Scholar]
- 29.Zheng W, Rampe D, Triggle DJ. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol Pharmacol. 1991;40:734–741. [PubMed] [Google Scholar]
- 30.Stork A, Cocks T. Pharmacological reactivity of human epicardial coronary arteries: phasic and tonic responses to vasoconstrictor agents differentiated by nifedipine. Br J Pharmacol. 1994;113:1093–1098. doi: 10.1111/j.1476-5381.1994.tb17108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu C, Hermann TE. Characterization of ionomycin as a calcium ionophore. J Biol Chem. 1978;253:5892–5894. [PubMed] [Google Scholar]
- 32.White SM, Constantin PE, Claycomb WC. Cardiac physiology at the cellular level: use of cultured HL-1 cardiomyocytes for studies of cardiac muscle cell structure and function. Am J Physiol Heart Circ Physiol. 2004;286:H823–829. doi: 10.1152/ajpheart.00986.2003. [DOI] [PubMed] [Google Scholar]
- 33.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93:531–540. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 34.Gil J, Esteban M. Induction of apoptosis by the dsRNA-dependent protein kinase (PKR): mechanism of action. Apoptosis. 2000;5:107–114. doi: 10.1023/a:1009664109241. [DOI] [PubMed] [Google Scholar]
- 35.Tsoutsman T, Kelly M, Ng DCH, Tan J-E, Tu E, Lam L, Bogoyevitch MA, Seidman CE, Seidman JG, Semsarian C. Severe Heart Failure and Early Mortality in a Double-Mutation Mouse Model of Familial Hypertrophic Cardiomyopathy. Circulation. 2008;117:1820–1831. doi: 10.1161/CIRCULATIONAHA.107.755777. [DOI] [PubMed] [Google Scholar]
- 36.McWhinney CD, Hansen C, Robishaw JD. Alpha-1 adrenergic signaling in a cardiac murine atrial myocyte (HL-1) cell line. Mol Cell Biochem. 2000;214:111–119. doi: 10.1023/a:1007129723949. [DOI] [PubMed] [Google Scholar]
- 37.Komazaki S, Ito K, Takeshima H, Nakamura H. Deficiency of triad formation in developing skeletal muscle cells lacking junctophilin type 1. FEBS Lett. 2002;524:225–229. doi: 10.1016/s0014-5793(02)03042-9. [DOI] [PubMed] [Google Scholar]
- 38.Ito K, Komazaki S, Sasamoto K, Yoshida M, Nishi M, Kitamura K, Takeshima H. Deficiency of triad junction and contraction in mutant skeletal muscle lacking junctophilin type 1. J Cell Biol. 2001;154:1059–1068. doi: 10.1083/jcb.200105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishi M, Sakagami H, Komazaki S, Kondo H, Takeshima H. Coexpression of junctophilin type 3 and type 4 in brain. Brain Res Mol Brain Res. 2003;118:102–110. doi: 10.1016/s0169-328x(03)00341-3. [DOI] [PubMed] [Google Scholar]
- 40.Kakizawa S, Kishimoto Y, Hashimoto K, Miyazaki T, Furutani K, Shimizu H, Fukaya M, Nishi M, Sakagami H, Ikeda A, Kondo H, Kano M, Watanabe M, Iino M, Takeshima H. Junctophilin-mediated channel crosstalk essential for cerebellar synaptic plasticity. EMBO J. 2007;26:1924–1933. doi: 10.1038/sj.emboj.7601639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishi M, Hashimoto K, Kuriyama K, Komazaki S, Kano M, Shibata S, Takeshima H. Motor Discoordination in Mutant Mice Lacking Junctophilin Type 3. Biochem Biophys Res Commun. 2002;292:318–324. doi: 10.1006/bbrc.2002.6649. [DOI] [PubMed] [Google Scholar]
- 42.Holmes SE, O’Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, Ross CA, Margolis RL. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet. 2001;29:377–378. doi: 10.1038/ng760. [DOI] [PubMed] [Google Scholar]
- 43.Rubler S, Dlugash J, Yuceoglu Y, Kumral T, Branwood A, AG New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 44.O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: Experimental studies. Circ Res. 1999;84:562–570. doi: 10.1161/01.res.84.5.562. [DOI] [PubMed] [Google Scholar]
- 45.Arai M, Alpert NR, MacLennan DH, Barton P, Periasamy M. Alterations in sarcoplasmic reticulum gene expression in human heart failure. A possible mechanism for alterations in systolic and diastolic properties of the failing myocardium. Circ Res. 1993;72:463–469. doi: 10.1161/01.res.72.2.463. [DOI] [PubMed] [Google Scholar]
- 46.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest. 2002;109:1013–1020. doi: 10.1172/JCI14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Little GH, Bai Y, Williams T, Poizat C. Nuclear Calcium/Calmodulin-dependent Protein Kinase II{delta} Preferentially Transmits Signals to Histone Deacetylase 4 in Cardiac Cells. J Biol Chem. 2007;282:7219–7231. doi: 10.1074/jbc.M604281200. [DOI] [PubMed] [Google Scholar]
- 48.Molkentin JD, Lu J-R, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A Calcineurin-Dependent Transcriptional Pathway for Cardiac Hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phimister AJ, Lango J, Lee EH, Ernst-Russell M, Takeshima H, Ma J, Allen PD, Pessah IN. Conformation-dependent stability of junctophilin 1 (JP1) and ryanodine receptor type 1 (RyR1) channel complex is mediated by their hyper-reactive thiols. J Biol Chem. 2007:M609936200. doi: 10.1074/jbc.M609936200. [DOI] [PubMed] [Google Scholar]
- 50.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BEC, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann B. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci USA. 2009;106:7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]