

Abstract
Though there have been many advances in providing access to linked and integrated biomedical data across repositories, developing methods which allow users to specify ambiguous and exploratory queries over disparate sources remains a challenge to extracting well-curated or diversely-supported biological information. In the following work, we discuss the concepts of data coverage and evidence in the context of integrated sources. We address diverse information retrieval via a simple framework for representing coverage and evidence that operates in parallel with an arbitrary schema, and a language upon which queries on the schema and framework may be executed. We show that this approach is capable of answering questions that require ranged levels of evidence or triangulation, and demonstrate that appropriately-formed queries can significantly improve the level of precision when retrieving well-supported biomedical data.
Keywords: Data integration, Database management, Information storage and retrieval, Bioinformatics, Ontology, Query language, Data diversity and provenance
1 Introduction
Analytical methods that rely on the integration of information from multiple sources are becoming increasingly common in biomedical research. For instance, determining the likelihood of a protein as a drug target or assessing the therapeutic value of a compound requires careful examination of experimental and derived data; information accumulated as a result of this may be cross-referenced across biomedical repositories whose contents collectively span the molecular, chemical, phenotypic and clinical spectrums [1, 2].
Though biomedical data is abundant and publicly-available in an ever-growing number of databases (over 1000 in molecular biology alone [3]), it is costly and effort-intensive to identify the most relevant information for any one task. Even starting from a single data source such as EntrezGene, one may easily find cross-references to other sources, potentially leading to an exponential number of possible choices. Retrieval of relevant data of high confidence in such a situation can take considerable time for a single gene – time that is compounded for high-throughput biomedical research.
In the following work, we present a simple, proof-of-concept framework and method for representing federation-based data provenance and evidence using a hierarchical ontology centered around membership. This hierarchy may be maintained separate from any common data model, and when given an ordering and mappings to query results represented in an external schema, can be used as a evidence cutoff function for information retrieval. To demonstrate utility of the added level of abstraction, we further present a query language as a means of obtaining data from multiple, interlinked repositories. This query language expressively accommodates retrieval of well-triangulated data based on source, data type and provenance, the specifics of which may be defined by the user using restrictions on the content of the data retrieved or means of retrieval.
2 Background
2.1 Linking over biomedical repositories
The interconnected nature of many biological data sources allows one to imagine the space of biomedical data as a directed graph where each vertex within the graph represents any individual record in any database, and the edges reference other records within or outside of that database (see Figure 1). Taking a mediated approach, we can further define a schema 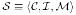 over this data;
over this data; 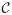 is the set of generalized entities within the schema with n : n correspondences to sources
is the set of generalized entities within the schema with n : n correspondences to sources 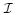 via
via 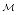 mappings.
mappings.
Figure 1.

References between six different biological databases. Arrows represent links from an individual record in each database to records in another.
Use of a graph-based representation allows queries to be expressed in terms of satisfying and valid paths. When supports inheritance, as in frames, queries can even be posed with a limited level of abstraction; if inter-source links, defined in , are annotated with additional descriptive information then the queries on 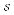 would effectively be done over a semantic network, the expressive benefits of which are numerous [4, 5]. Furthermore, adapting a common schema across any number of sources and materializing links between them not only allows one to query myriad sources in a uniform manner but also deterministically enumerate the possible paths from one source or schema entity to another, via other sources or schema entities [6].
would effectively be done over a semantic network, the expressive benefits of which are numerous [4, 5]. Furthermore, adapting a common schema across any number of sources and materializing links between them not only allows one to query myriad sources in a uniform manner but also deterministically enumerate the possible paths from one source or schema entity to another, via other sources or schema entities [6].
2.2 Path selection
However, all databases are not created equal and levels of scientific validation vary across and within sources (e.g., the contrast between TrEMBL and Swiss-Prot, the latter being manually curated). Thus, certain paths may be preferable to others, and selection of paths may be difficult based on the size and number of records and links; a method of filtering for or highlighting choice results, given some retrieval task, is necessary to navigate abundant data. Because these data record relationships may be represented as paths, one method is to employ regular expressions as a means of specifying restrictions going from one data type to another.
Even with the expressivity afforded by the use of regularly-described paths, some queries are by nature challenging to answer without additional information or overhead. Continuing within the context of genomic research, it is difficult and unwieldy to answer queries such as: “give me the proteins in my organism of study involved in biosynthesis whose functions were elucidated using at least manually-curated computational methods,” or “within a particular loci, return all functional SNPs associated with a gene whose involvement in a pathway is supported via multiple types of experiments and analyses.” Queries such as these express levels of uncertainty commonly encountered in active biomedical research, and implies that any information retrieved satisfies minimal coverage and evidence, in addition to the other requirements.
2.3 Query methods over disparate biomedical data
Given the disparate nature of many biomedical resources, there have been several query systems and methods specifically developed or adapted to deal with retrieving data from heterogeneous or remote scientific sources. XQuery, an XML query language, has been adapted for purposes of interrogating a variety of structured data such as XML files, ontologies and relational databases in a collaborative setting for the Human Brain Project [7]. Later work extended XQuery usability for non-experts by the development of a graphical interface for interactive query formation [8]. QIS (Query Integrator System) relies on a combination of SQL-like syntax and XML to allow a user to formulate a query [9]. The core of the system is a set of modules that mediate requests between a client and service in a scalable fashion. Notable features of QIS include automatic schema change determination (including a robust warning structure that notifies a user during querying if a source's schema may have changed) and a Web-based user interface.
More closely related to our work are query methods that rely on the paths and links formed between external references of databases. Such approaches can quickly generate large amounts of data of varying relevance, and provide a browsing paradigm that can be appropriate for exploratory research [10]. PQL, based on the StruQL [11] query language for Web content management, relies on path constraints placed on inter- and intra-databases references [12]. Similarly, BioNavigation builds upon this path-constraining approach by further ranking paths based on path cost and cardinality [22]. Notably, while these methods are capable of retrieving data from disparate data sources, they cannot explicitly query over minimal coverage and evidence.
2.4 Data quality and query methods over disparate biomedical data
Related to the challenge of querying biomedical data is ascertaining from where it came, how it has been processed and how much trust or belief to assign it. Capturing, recording and annotating biologic information with metadata regarding its origin, nature of alterations and quality has become an area of interest for many disciplines of science [13, 14, 15]; such tasks can include prospective examination of data generated for validation or comparison, and tracing the processes which transformed data to generate a replicable workflow [16, 17]. Methods of tracking provenance, or lineage, have been developed to address these and similar needs.
Systems which support one or more of these capabilities have the potential to help manage the varied data that is often collected from multiple repositories and experiments. Under the model implemented within the Karma provenance system, provenance information is recorded for data as it moves through a workflow [18]. Within this system, queries may be posed by a blend of both API calls and direct SQL queries to a back-end MySQL database. Similarly, the Taverna workbench supports the capture of provenance metadata during the execution of workflows for the myGrid project using RDF [19]. Other Semantic Web technologies have been examined as a means of representing provenance information for biomedical data management [15]. A strength of these systems is a traceable audit trail upon which queries regarding evidence may be posed to discern lineage, and the use of standardized representations encourage inter-system integration and data dissemination. At the same time, some of these technologies lack abilities desirable in a system where queries permit some level of ambiguity, e.g., SPARQL does not natively support regular path expressions, and queries using relational database-driven systems can become unwieldy as sources are added or the provenance model increases in complexity.
While related to workflow data capture, the problem we are most interested in for this work is to improve the ability of biomedical researchers to navigate data across heterogeneous resources, particularly for exploratory phases. The methods described below execute path-based queries with user-determined levels of ambiguity over distributed resources remotely, without the requirement of a database back-end to store or drive queries. Relying on a top-down approach that does not require a comprehensive system with end-to-end tracking, the implementation provides robust querying abilities in the context of evidence and coverage with comparatively little cost or overhead.
3 Methods
3.1 Modeling data diversity and evidence
To practically illustrate coverage and evidence as it pertains to our work and integrated data sources, refer to Figure 2 of a generalized query q against a set of databases A, B and C. Let A represent a semi-manually curated database of known structures and their functions, B a database of experimentally-derived data and C a database of genus-specific sequences. The target results of q are the records directly linked from A, B and C (among them x,y,z).
Figure 2.

Simple example graph showing how information that is supported through a greater diversity of sources or types may not be easily-discernable amongst a multitude of other data.
Examining cardinality alone,a possible conclusion is that x is a more preferable result to y as there is one more path from q → x than q → y, and this indeed may be true in the case of an investigator interested in finding targets whose structures are elucidated. On the other hand,y may be preferable as a well-triangulated result, as it is being linked via two different sources of information. Queries that are sensitive to coverage would allow a user to specify requirements on how diversely-supported their results are with respect to evidence without having to necessarily enumerate specific data sources or types.
From another perspective, an investigator interested primarily in specific genera of organisms and their protein structure may trust data from sources A, C more than B. Queries capable of execution over evidentiary knowledge would allow a user to express their preferred data origin needs, again without having to explicitly state the exact sources themselves.
3.2 Modeling a domain knowledge hierarchy
Being able to succinctly query on minimal requirements of evidence or coverage abstractly requires further metadata than normally provided in a traditional mediated schema such as the one defined formally in (Sec. 2). Formally, we introduce a domain hierarchy 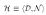 where each element (domain) d ∈
where each element (domain) d ∈ 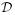 is an evidence type, such as information that was “derived from assay” or data ”elucidated from crystallization”;
is an evidence type, such as information that was “derived from assay” or data ”elucidated from crystallization”;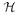 contains relations to any individual source or entity s ∈ via correspondences n : d ↦ s in
contains relations to any individual source or entity s ∈ via correspondences n : d ↦ s in 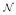 . In this way, any element of or from may then be claimed as a member of an element of . Multiple inheritance on would further allow us to state, for example, that structural information from TargetDB may be “elucidated from crystallization”, which is a type of “structural experiment” and, transitively, “experiment” and “manually-determined.”
. In this way, any element of or from may then be claimed as a member of an element of . Multiple inheritance on would further allow us to state, for example, that structural information from TargetDB may be “elucidated from crystallization”, which is a type of “structural experiment” and, transitively, “experiment” and “manually-determined.”
Mapping the domains of the hierarchy to actual sources or entities allows one to differentiate between the origin of various results, and pose questions that include restrictions on evidence coverage. Additional utility is added by imposition of an ordering upon the domains, entities and sources such that any given element may have precedence over another. (see Figure 3). This allows for various sources, records or links to have preference over others for any given task, as with the previous example of data sources A, B,C.
Figure 3.

Portion of the declarations of membership and rank order for a domain hierarchy oriented around exploratory sequence annotation. The first two entities (prefixed by the :dom command) create two domains, one based on short sequence alignments, and the other on pathway-related information; within each of these domains, various sources and classes native to the data integration engine's schema claim membership. The last entity (prefixed by :ord) defines the order for the domain, in increasing rank, as appropriate for the task at hand.
The content of the domain hierarchy can yield useful metadata regarding the results, and can be used to answer queries that may be ambiguous with respect to data coverage and evidence. Notably, we define these concepts as an external component to any schema, and rely on loose couplings from the domains to the schema. As a consequence, the order among the domains may be changed without having to disturb or alter any pre-existing class descriptions within a schema, maintaining scalability in how both the schema and the domain hierarchy are composed. This added layer of abstraction further allows for flexibility of ranking as requirements change, since the exact same data model may be used for any number of distinct tasks but return appropriately different results depending on the domain order imposed.
3.3 Supporting domain-aware queries
A primary objective of this research is to incorporate support for the concepts of data coverage and evidence within a query-capable framework. Recall that there have been many systems in the past to support structured queries; while some of these approaches, such as PQL and BioNavigation allow for somewhat ambiguous path queries (e.g., wildcard proxies for record elements), none of these approaches directly address the complementary issues of coverage and evidence. Subsequently, we developed a query language built to allow a user to easily write queries which directly consider information diversity and provenance, DaRQL (pronounced dar-kle and short for Domain-aware Regular Query Language).
Based loosely on SQL syntax for familiarity and usability purposes, and with similar regular expression capabilities as PQL and BioNavigation to traverse paths, DaRQL further supports user-defined restrictions on evidence and coverage for results. DaRQL queries have the following format:
TARGET <bindings for selected nodes>
FROM <bindings for start nodes>
RESTRICT <path and node constraints>
where TARGET specifies any number of nodes a user wishes to bind to a variable for later reference, FROM specifies nodes that serve as default path starts and RESTRICT the selection of predicate constraints to place upon the nodes and paths, represented via regular expressions.
As an example, consider the query in Figure 4. The variables ?t and ?g on line 1 are bound to all nodes in the graph that are either Terms or Genes, respectively (including any hyponyms, as defined by the schema and domain hierarchy); all paths, by default, originate at ?q, defined as any Query node within the graph. Variables may also be bound to specific data sources (enclosed by “), as well as evidence types defined in the domain hierarchy (prefixed with @), e.g., ?x∼'EntrezGene', ?x∼@StructureBased.
Figure 4.

Example showing a basic query in DaRQL.
The statements following the RESTRICT clause are the path and node restrictions that any path found in the graph must meet, and predicates contained in {} (e.g., line 4) represent a specific regular expression over a path. The path expression in Figure 4 would only be satisfied with a path that begins at a Query that transitively reaches a Gene, which has a direct link to a Term. Path expressions support most regular expression characters.1 For example, (*) is a transitive closure on a relation between specified terms, connected via the join (.) symbol; disjunction and conjunction among nodes are expressed using the ( ?x|?y) and ( ?x&?y) constructs.
Non-path restrictions in Figure 4 must also be met. The first specifies that the Gene identified in the path must be of human origin (where ?x:Y refers to any attribute Y of some record in the graph, ?x). The second references the order of the Term ?t using a reserved function order, and enforces a constraint on any result that a valid ?t must be supported by more than sequence-based motif evidence, as determined by the ordering in the domain hierarchy. Another reserved function, divcount, returns the number of distinct (as determined recursively via domain subsumption) types of evidence that lead to a result. Using path and node constraints, as well as functions for accessing a node's evidence and coverage characteristics, allows a user to formulate queries in DaRQL with a significant amount of ambiguity while taking into account data noise reduction.
3.4 Implementation of model
For feasibility testing of querying using coverage and evidence parameters, we developed and coupled a DaRQL interpreter with an integration engine to allow us to query over live results. Data integration across sources was handled using an open-source general-purpose data integration system2, as was our lexing and parsing module.3 Briefly, PyDI is a general-purpose data integration system that permits a user to pose a seeding query, such as an accession number, in the form of an entity-attribute-value tuple whose vocabulary is defined within the schema [20]. To illustrate, a user interested in seeding a query based on the human acyl-CoA protein related to long chain fatty acid mutation may submit the query via the following tuple: (Gene, #ID, “51”)), where “51” is the gene identifier of interest; conversely, a user, faced with an unknown protein, may choose to submit a sequence instead, with the appropriate parameters. In the case of the first query, the initial result will be a single node corresponding to the EntrezGene entry for the protein; in the latter case, the result would be any number of records with alignments to the sequence query that meet user-defined requirements.
When these parameters are submitted to the integration engine, any sources which support the specified entity and attribute, as specified by the schema, will be queried. Any data reachable from the seed across myriad repositories is then returned indiscriminately; such results may then be used to retrieve even further data. This paradigm of integrating data through browsing and daisy-chaining has been used in the past for exploring biologic data in disparate sources, and provide retrieved data to the user in the form of a query graph of interconnected results [21, 22, 23]. We implemented the DaRQL interpreter over PyDI, and defined loose couplings from the domain hierarchy to the system schema (refer to Figure 5 for system components).
Figure 5.

Above image is the schematic architecture of the querying and integration system; note that the query module and its subparts (lexer/parser, query planner) are modularized from the rest of the system.
A query is processed as follows: a user initiates an exploratory (non-DaRQL) query via the data integration system (refer to Fig. 5, step 1). Results from this initial, exploratory query are gathered via the integration engine by reformulating the query into the native format of the individual data sources (step 2). When sources respond with query results, the data is translated into the entity-attribute-value format of the integration engine (step 3).
The exploratory query can recursively re-query current results, thus expanding any query graph to a size limited only by the source-to-source mappings defined in the schema. To filter the results, the user may then submit a DaRQL query to the graph (step 4), which passes it on to the interpreter for processing. Bindings are resolved to entities, sources and domains and the validity of the terms in the query are checked against both the domain hierarchy and the data integration system's schema (step 5).
Prior to execution, a pathing plan is created to minimize traversal cost; paths that would not yield any valid answer, given the constraints on sources, classes and domains, would not be followed. At this point, the query engine inspects the terms expressed within the path and variable constraints. Any evidence and coverage requirements (per order and div keywords) within the query are cross-referenced with the domain hierarchy to ensure that any results satisfy the minimum levels of evidence or coverage requested, or fall within the defined range. For example, a terminal node of a path constraint which also requires a minimum coverage of three different, non-overlapping, domains would fail if its lineage can only be traced two or fewer domains.
Once a plan is ready, a series of deterministic finite state machines (DFSMs) are created to check the constraints over the live query graph, each representative of an expression in the RESTRICT clause. The DFSMs are evaluated individually using a non-greedy search, each navigating a singular path in the graph to determine the path's validity (step 6). If a path does not meet all of the defined restrictions, the end of the path is noted as invalid with respect to the query. Alternatively, any terminal node within the graph which contains all successful DFSMs is a valid result that meets all the restrictions defined in the RESTRICT clause. The results may then be serialized into XML form as a subgraph of the full query graph that contains only the nodes and edges that satisfy the query (step 7; see Fig. 6).
Figure 6.

Sample truncated output from graph serialization; the above query identifies paths to GO terms that run through genes, pathway-based data and have more than three different “types” of other data leading to them (i.e., two other domains in addition to pathway-based).
4 Results
4.1 System performance
Depending on the restrictiveness of the query formulated, use of the query planner increased execution speed on average 35%, a difference we found to be statistically significant (see Fig. 7). One limitation of relying on a DFSM, however, is that query evaluations over graphs that have a higher ratio of edges to nodes (i.e., are more well-connected) or queries whose paths are generally unrestrictive (e.g., { ?q.*.?t}, where ?t is not directly linked from ?q and no further constraints are expressed) are more time-expensive to execute. Conversely, even somewhat large graphs with relatively few well-connected nodes may be evaluated quickly.
Figure 7.

Query execution times for 99 randomly selected proteins from yeast, without query planning and with query planning for a query with many restrictions, and for a loosely-defined query; solid horizontal lines mark the means for each query, and dotted horizontal lines the standard deviations. Benchmarks were done on an Intel dual core 2.66GHz Linux machine with 3GB of RAM.
4.2 Model utility
Ultimately, the goal of our knowledge representation and querying model is to improve a researcher's ability to interrogate disparate data sources efficiently by facilitating queries that easily accommodate ambiguity in ranges and whose specific formulation is independent of data sources. To this end, we tested our implementation of DaRQL over a set of queries aimed at retrieving “high-quality” GO terms - that is, GO terms whose annotation is supported by experimental evidence. For this experiment, the actual evidence codes provided by the GO annotations revealed during the querying and retrieval process were not used as part of the evaluation. In doing so, we attempted to measure the utility of our approach in retrieving the most well-supported annotations a priori based on memberships to domains and entities, and levels of coverage only.
We randomly selected 500 proteins from the H. sapiens proteome from UniProt 4 which possessed one or more GO annotations supported by evidence at the experimental level, i.e., ‘EXP', ‘IDA’, ‘IPI’, ‘IMP’, ‘IGI’ and ‘IEP’ 5. These 500 proteins, and their experimental GO terms, serve as the test set against five DaRQL queries targeted at retrieving gene annotations at varying levels and ranges of stringency (see Table1, queries A-E).
Table 1.
Queries tested against the DaRQL implementation and their associated translations. GO terms retrieved from these queries used in a pairwise comparison without posed queries (cf. Figure 8).
| Name | Query | Description |
|---|---|---|
| A | 1: TARGET ?g∼‘AmiGO’ 2: FROM ? s∼ ProteinSequenceQuery 3: RESTRICT 4: {?s.*.@NonRepository.*.?g}, 5: divcount(?g) > 2, 6: order(?g) >= @CurationBased |
Retrieves GO terms linked from motif-, conserved domain- or protein family-based databases that are supported by more than two domain types and whose levels of evidence is equal to or greater than semi-manual curation. |
| B | 1: TARGET ?g∼‘AmiGO’ 2: FROM ?s∼ProteinSequenceQuery 3: RESTRICT 4: {?s.*.Gene.*.?g}, 5: divcount(?g) > 3 |
Retrieves GO terms whose path to the query passes through at least one Gene entity as defined in the schema, and which are also supported by more than three different domain types. |
| C | 1: TARGET ?g∼‘AmiGO’ 2: FROM ?s∼ProteinSequenceQuery 3: RESTRICT 4: {?s.*.Gene.*.?g}, 5: divcount(?g) > 2 |
Retrieves GO terms whose path to the query passes through at least one Gene entity, and which are also supported by more than two different domain types. |
| D | 1: TARGET ?g∼‘AmiGO’ 2: FROM ?s∼ProteinSequenceQuery 3: RESTRICT 4: {?s.*.‘InterPro’.*.?g}, 5: order(?g) >= @PathwayBased |
Retrieves GO terms linked from a specific data source (Inter Pro), and which is supported by a level of evidence equal to or greater than that of pathway support. |
| E | 1: TARGET ?g∼‘AmiGO’ 2: FROM ?s∼ProteinSequenceQuery 3: RESTRICT 4: order(?g) >= @ExperimentBased |
Retrieves GO terms supported by a level of evidence equal to or greater than that of experimental results. |
Annotations retrieved by the five queries were scored using two flavors of precision and recall: macro-average and micro-average. Macro-average precision was calculated by computing the precision for each protein individually, then averaging over all 500 queries. Let n be the number of proteins in the total test set, Px the set of experimental terms for protein x and Qx the set of terms in a query graph for protein x. The macro-average precision for one of the five test queries is then calculated by:
| (1) |
and macro-average recall with:
| (2) |
Micro-average precision was computed using:
| (3) |
and micro-average recall, likewise:
| (4) |
Using these four metrics, we were able to measure the overall precision and recall at the protein level (macro-average) as well as at the annotation term level (micro-average) for the test queries. For this test, only exact GO annotation matches were considered as correct; terms not listed in the gold standard, though they themselves may be parents to a correct annotation, were marked as incorrect.
We computed these macro- and micro-average values for each of the five test queries based on all 500 test proteins. The metrics were also computed for the full query graph for each of the test proteins, absent any DaRQL queries, thus allowing for comparison of our methodology against a baseline. Furthermore, we used two additional annotation sets generated from the full query graph, where 20% and 80% of the annotations for each protein were randomly selected as results. These two final sets, rand20 and rand80, were used as a control to ensure that by filtering the query graph via DaRQL we were not merely measuring any effects normally present when a subset of the query graph is returned.
The experiment was conducted thusly: the initial step involved generation of the baseline full query graphs from the data integration system (as described in Sec. 3.4); the sequences of each of the 500 test proteins were queried against a subset of the data sources currently supported by PyDI: EntrezProtein, BioCyc (proteins), KEGG (proteins), TIGRFAM, InterPro (motifs) and PDB (structures).From these initial sequence queries, further queries were recursively generated to return other components of these repositories (e.g., KEGG pathways) and other, external data sources (e.g., EntrezGene, AmiGO); recursive query calls were continued until the size of the graph did not increase, resulting in 500 full query graphs. Following this, each of the test queries was executed over the graphs to determine which terms within each query graph satisified the query criteria; for rand20 and rand80, 20% and 80% of the graphs' terms, respectively, were randomly selected. Finally, macro- and micro-average precision and recall metrics were collected for each test query and compared to the measures found for the baseline graphs and the random sets. Notably, UniProt is absent as a source, as we derived the gold standard experimental GO terms from this data source.
The findings of the query utility evaluation are shown in Table 2. As expected, the full graph – returning all terms without posing any queries - exhibited high recall and low precision; indiscriminate retrieval of GO annotation terms across data repositories successfully identified 98% of all experimental terms. However, only 5.9% of all retrieved terms were experimentally supported. Performances for both random sets were similar, with no appreciable increases in precision as recall fell.
Table 2.
Performance results of GO term data retrieved after queries A-E were executed (shown in Table 1) on the test set of human proteins; ‘Full graph’ corresponds to using all terms as available in the full graph, absent any querying. Findings are shown at the macro- and micro-level for both Pr and Re. Macro-average significance was measured using paired non-parametric rank sum tests, with correction; instances where the query results are not significantly different from the full graph (p > 0.01) or either of the random sets are denoted by the appropriate subscripts.
| Query | PrMacro | PrMicro | ReMacro | ReMicro |
|---|---|---|---|---|
| Full graph | 0.082 | 0.059 | 0.988 | 0.980 |
| rand20 | 0.075 | 0.057 | 0.165 | 0.180 |
| rand80 | 0.082 | 0.059 | 0.781 | 0.773 |
| Query A | 0.146 | 0.145 | 0.820≈r80 | 0.823 |
| Query B | 0.171 | 0.161 | 0.898 | 0.928 |
| Query C | 0.154 | 0.146 | 0.944 | 0.955 |
| Query D | 0.050≈r20 | 0.065 | 0.080 | 0.065 |
| Query E | 0.055≈r20 | 0.049 | 0.618 | 0.695 |
While the recall values for the five test queries were generally lower than those of the full graph, precision values were higher in cases than those of the full graph and random results. In one case, Query C, precision increased at the macro-level 46.7% over the full graph, with a recall decrease of only 4.4%. Three of the five test queries showed significantly better performance over the full graph and random controls, as determined by non-parametric tests. Nonetheless, utility for each individual query varied, and two queries displayed very poor results across both precision and recall. One of these queries declared a specific source as a path requirement while simultaneously constraining returned annotations to pathway-supported information (Query D); the other permitted return of only terms whose data types or resources were known to involve experimental findings (Query E).
Pairwise examination of individual protein annotation term scores further illustrates that precisions are generally higher for three of the test queries (A-C), and conversely lower for the remaining two (D, E; cf. Figure 8). In all cases, it is clear that the majority of instances where the query underperformed in comparison to the full graph occur where all experimental terms are omitted from the results, i.e., where points lie along the axis of ‘No query’. For the poorly-scoring queries, D and E, the vast majority of results exhibit this behavior, suggesting that for the purposes of retrieving experimental annotation terms these queries were likely too stringent.
Figure 8.

Pairwise protein-level comparison between the posed queries and the full query graph. Points which appear along the dotted line of each graph are cases where the performance of the query and of the full graph are equivalent; points above are results favorable for the posed query, and those below favorable for the full graph.
As the evaluation metrics of the various queries differed, so too did the features of the query graphs themselves. The plots in Figure 9 briefly illustrate some of the characteristics of the protein test set as well as the resulting query graphs. Notably, the vast majority of the test set is comprised of proteins associated with six or less experimental terms; indeed, proteins with five or less terms contribute 50% of all terms.
Figure 9.

The number of experimental terms per protein plotted as a cumulative fraction of all proteins in the test set (left), and frequency counts of terms within individual query graphs regardless of evidence (right).
Furthermore, we were interested in how the performance metrics were reflected within the log-normal distribution of the frequency of terms within the query graphs. The plots of Figure 10 show frequency curves for the query graphs, stratified by median: the lower half (red dotted) are the frequencies of the F1 score for graphs whose number of results fell below the median; the upper half (blue solid) are the frequencies for graphs whose number of results are larger than the median. Because of the nonlinearity of the distributions, we found no strong correlation between the size of the results returned and the F1 score. It is clear from a visual inspection, and confirmed with statistical testing, that larger query graphs (i.e., graphs with more returned results) tended to produce better metrics. The shifts in distribution ranges are most stark for the best-performing test queries, A-C, whereas for the full graph and the lower-performing queries, the upper and lower halves trended toward the same ranges. These findings suggest that the optimal use of our presented query model for researchers may be in highlighting well-supported data when faced with a large number of results; this utility may diminish with the number of records retrieved.
Figure 10.

Frequency curves of the full graph (‘No query’) and the five test queries, stratified by the median of the query result size; the blue, solid line represents the half of queries whose number of results rested above the median, and the red, dotted line those which fell below. For these plots, the (protein-level) measure is used as the x-axes (note the differences in scale for the y-axes). Differences between distributions per query are statistically significant by permutation test (p ≤ 0.001), save Query D.
5 Discussion
5.1 Design motivations
We have described a method of representing data coverage and evidence in the context of integrated, heterogeneous data sources. The result is a proof-of-concept artifact,6 feasible methodology, and performance and utility evaluations of a model for incorporating the concepts of range and minimality into queries for biomedical data integration. While our framework was kept intentionally simple and logically-based, we believe that it is flexible enough to accommodate more complex and nuanced additions as necessary. For example, in many fields of research probabilistic representation is a de facto standard for expressing uncertainty in evidence and provenance; the domain hierarchy can accommodate finer-grained annotations of relationships or orderings that reflect a user's belief in individual sources, similar to approaches taken by others [24, 25].
In introducing and implementing a model for dealing with ambiguous and ranged queries over integrated biomedical data, we have opted for a loose coupling between a hierarchy that manages how the data is internally represented (the schema) and a hierarchy that manages the evidence and diversity of data (domains). The primary motivating factor behind this design decision was to keep each individual component of the data integration and querying system simple and easily manageable. A beneficial side effect of this goal is that systems which rely on a basic data model would not require dramatic or extensive changes to support domain-aware queries; defining the domain hierarchy, its ordering and mappings to the entities in the schema could be done without disturbing the schema.
As a demonstration of this querying model, we opted for a top-down data integration approach that did not require any data source to offer Web services, a route taken by previous federated models of querying [12]. Furthermore, though our implementation permitted standardized output of results in the form of XML, our choice to limit the use of markup encoding was driven by a desire to minimize the overhead and footprint of the system implementation, and improve speed by relying primarily on internal data structures for representation. From the authors' experience, this was a fair tradeoff for an implementation whose primary purpose was not necessarily to form the backbone of a production system, but rather to explore a new method of querying disparate biomedical data using levels of ambiguity.
5.2 Limitations and future work
We were encouraged that the results of the evaluation validated the general approach, which found that overall DaRQL queries with consideration of evidence and coverage were able to succinctly improve system precision. At the same time, we note that performance naturally depended on the query crafted, a task often left to an individual researcher. Though this is a difficulty generally faced when pruning data, one possible solution with respect to the model we have outlined would be to provide more granularity when discriminating between data records. Our domain implementation focused primarily on sources (i.e., PDB) and classes (i.e., ‘Pathways’), but can be extended to include attributes or descriptors at a finer-grained level (e.g., expect values for alignments), thereby giving the user more control over the specificity of the query.
Indeed, an important limitation our findings illustrate is that manual, researcher interaction remains an important filter when dealing with biological data. While the queries improved precision over the baseline approach, scores still remained relatively low; the best precisions reported were between 0.14-0.17, and not nearly high enough to endorse automated application of queries for the purposes of identifying well-supported annotations. Identifying methods of improving the evaluation metrics is a key future challenge, though the analysis we have done here provides clues as to where to direct effort. For example, scores tended to be lower for query graphs that returned fewer results (see Figure 10), and as yet our methodology does not take advantage of knowledge concerning the state of the query graph overall. A smaller number of results imply a less well-known protein for which some terms within a query would be too restrictive, and where requiring too many different domains or specific data sources may be detrimental.
Even with further improvements, the purpose and goal of the above methods are not replacements to manual review, but instead should serve as aids to user-driven scientific research. In this regard, providing a way of prioritizing attention can be valuable, particularly when such methods permit scientists to pose ambiguous or specific queries over multiple biological sources as needed. To this end, in the future we plan to extend the system to support other data repositories, domains and attributes, thus expanding generalizability across tasks and resources. While this work demonstrates the technical practicality of implementing our proposed model, we have not yet done a study involving potential users in regards to the practicality of use and usability, which is one future direction of this research.
Also, though our preliminary work was implemented via a novel query language for feasibility, flexibility and relative simplicity in syntax, we envision the concepts of data diversity and coverage incorporated in future standardized languages such as SPARQL, either formally or as well-supported extensions; we expect that these technologies shall continue to mature, and their weaknesses addressed (such as regular path support [26]). Indeed, formal representations of data uncertainty is already under consideration by the World Wide Web Consortium [27], and desiderata and models in the form of the Open Provenance Model have gained traction from annual challenges and community discussion [28, 29]. Methods formally adopted for representing uncertainty and trust could be extended to handle task domains and data diversity such that navigation and integration becomes less onerous for users.
Independent of any method or approach taken, however, it is important that a user is able to formulate their questions and navigate their search results in a way that is optimized for their preference and task. This is an issue that is all the more important as the boundaries between medicine and biological research blur and repositories become more cross-disciplinary in usage and application. In this regard, methods of query formulation, such as that provided through DaRQL that give a searching user control over the level of ambiguity, specificity and trustability of their results can be a valuable aid.
Supplementary Material
Table 3.
Example of annotations found for the protein PNMT_HUMAN (P11086). Results are for the baseline and the five test queries; all terms shown were retrieved by the baseline method, and terms retrieved by any of the five test queries are denoted by an ‘X’ in the appropriate column. The experimentally-supported annotations, per the gold standard, are shown in bold. Notably, the GO terms most prevalent across all queries (GO:0016740, ‘transferase activity’; GO:0008168, ‘methyltransferase activity’) are parent terms to the gold standard term GO:0004603 (‘phenylethanolamine N-methyltransferase activity’).
| Baseline | A | B | C | D | E |
|---|---|---|---|---|---|
| GO:0003674 | X | X | |||
| GO:0004603 | X | X | X | ||
| GO:0005625 | X | X | |||
| GO:0005737 | X | X | X | ||
| GO:0005829 | X | X | X | ||
| GO:0008112 | X | X | X | ||
| GO:0008168 | X | X | X | X | X |
| GO:0008757 | X | ||||
| GO:0010243 | X | ||||
| GO:0016740 | X | X | X | X | |
| GO:0030748 | X | X | X | ||
| GO:0031100 | X | ||||
| GO:0042418 | X | ||||
| GO:0042423 | X | X | X | ||
| GO:0042493 | X | ||||
| GO:0046498 | X | ||||
| GO:0046500 | X |
Acknowledgments
The authors would like to thank Ira Kalet for his helpful suggestions, Jeffrey Dee for his editing assistance and the anonymous reviewers for their thoughtful comments and recommendations for improvement. This work was supported by National Library of Medicine grant T15LM07442 and National Science Foundation grant IIS-0513877.
A Appendix
Supplementary information, including executable code and usage instructions, is available at http://purl.oclc.org/eithon/supplemental.
Footnotes
UW Biomedical and Health Informatics, 1959 NE Pacific St. HSB I-264, Seattle, WA USA 98195-7240
?x.+.?y is unsupported, as it can be equivalently stated in schema form as ?x.Entity.?y.
A complete list of GO evidence codes is available at http://www.geneontology.org/GO.evidence.shtml
See supplementary information for software and step-by-step instructions on use.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Searls DB. Data integration: challenges for drug discovery. Nat Rev Drug Discov. 2005 Jan;4(1):45–58. doi: 10.1038/nrd1608. [DOI] [PubMed] [Google Scholar]
- 2.Bredel M, Jacoby E. Chemogenomics: an emerging strategy for rapid target and drug discovery. Nat Rev Genet. 2004 Apr;5(4):262–275. doi: 10.1038/nrg1317. [DOI] [PubMed] [Google Scholar]
- 3.Galperin MY. The molecular biology database collection: 2008 update. Nucleic Acids Res. 2008 Jan;36(Database issue):D2–4. doi: 10.1093/nar/gkm1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosse C, Mejino J. A reference ontology for biomedical informatics: the Foundational Model of Anatomy. J Biomed Inform. 2003 Dec;36(6):478–500. doi: 10.1016/j.jbi.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 5.Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004 Jan 1;32(Database issue):D267–70. doi: 10.1093/nar/gkh061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mork P, Halevy A, Tarczy-Hornoch P. A model for data integration systems of biomedical data applied to online genetic databases. Proc AMIA Symp. 2001:473–477. [PMC free article] [PubMed] [Google Scholar]
- 7.Bales N, Brinkley J, Lee ES, Mathur S, Re C, Suciu D. A framework for XML-based integration of data, visualization and analysis in a biomedical domain. In: Bressan S, Ceri S, Hunt E, Ives ZG, Bellahsene Z, Rys M, Unland R, editors. XSym. Vol. 3671. Lecture Notes in Computer Science; Springer: 2005. pp. 207–221. [Google Scholar]
- 8.Li X, Gennari JH, Brinkley JF. XGI: a graphical interface for XQuery creation. AMIA Annu Symp Proc. 2007:453–457. [PMC free article] [PubMed] [Google Scholar]
- 9.Marenco L, Wang TY, Shepherd G, Miller PL, Nadkarni P. QIS: A framework for biomedical database federation. J Am Med Inform Assoc. 2004 Nov-Dec;11(6):523–534. doi: 10.1197/jamia.M1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaker R, Mork P, Brockenbrough JS, Donelson L, Tarczy-Hornoch P. The BioMediator system as a tool for integrating biologic databases on the web. Proceedings of the 30th VLDB Conference. 2004 [Google Scholar]
- 11.Fernandez MF, Florescu D, Levy AY, Suciu D. A query language for a web-site management system. SIGMOD Record. 1997;26(3):4–11. [Google Scholar]
- 12.Mork P, Shaker R, Halevy A, Tarczy-Hornoch P. PQL: a declar-ative query language over dynamic biological schemata. Proc AMIA Symp. 2002:533–537. [PMC free article] [PubMed] [Google Scholar]
- 13.da Silva PP, McGuinness DL, McCool R. Knowledge provenance infrastructure. IEEE Data Eng Bull. 2003;26(4):26–32. [Google Scholar]
- 14.Zhao J, Wroe C, Goble CA, Stevens R, Quan D, Greenwood RM. Using semantic web technologies for representing e-science provenance. In: McIlraith SA, Plexousakis D, van Harmelen F, editors. International Semantic Web Conference. Vol. 3298. Lecture Notes in Computer Science; Springer: 2004. pp. 92–106. [Google Scholar]
- 15.Sahoo SS, Sheth AP, Henson C. Semantic provenance for escience: Managing the deluge of scientific data. IEEE Internet Computing. 2008;124:46–54. [Google Scholar]
- 16.Miles S, Groth PT, Branco M, Moreau L. The requirements of using provenance in e-science experiments. J Grid Comput. 2007;5(1):1–25. [Google Scholar]
- 17.Simmhan Y, Plale B, Gannon D. A survey of data provenance in e-science. SIGMOD Record. 2005;34(3):31–36. [Google Scholar]
- 18.Simmhan YL, Plale B, Gannon D. Query capabilities of the Karma provenance framework. Concurrency and Computation: Practice and Experience. 2008;20(5):441–451. [Google Scholar]
- 19.Zhao J, Goble CA, Stevens R, Turi D. Mining Taverna's semantic web of provenance. Concurrency and Computation: Practice and Experience. 2008;20(5):463–472. [Google Scholar]
- 20.Cadag E, Tarczy-Hornoch P, Myler PJ. On the reachability of trustworthy information from integrated exploratory biological queries. In: Paton NW, Missier P, Hedeler C, editors. DILS. Vol. 5647. Lecture Notes in Computer Science; Springer: 2009. pp. 55–70. [Google Scholar]
- 21.Donelson L, Tarczy-Hornoch P, Mork P, Dolan C, Mitchell JA, Barrier M, Mei H. The BioMediator system as a data integration tool to answer diverse biologic queries. Stud Health Technol Inform. 2004;107(Pt 2):768–772. [PubMed] [Google Scholar]
- 22.Cohen-Boulakia S, Davidson S, Froidevaux C, Lacroix Z, Vidal ME. Path-based systems to guide scientists in the maze of biological data sources. J Bioinform Comput Biol. 2006 Oct;4(5):1069–1095. doi: 10.1142/s0219720006002375. [DOI] [PubMed] [Google Scholar]
- 23.Boulakia SC, Masini K. Biobrowsing: Making the most of the data available in Entrez. In: Winslett M, editor. SSDBM. Vol. 5566. Lecture Notes in Computer Science; Springer: 2009. pp. 283–291. [Google Scholar]
- 24.Sevon P, Eronen L, Hintsanen P, Kulovesi K, Toivonen H. Link discovery in graphs derived from biological databases. In: Leser U, Naumann F, Eckman BA, editors. DILS. Vol. 4075. Lecture Notes in Computer Science; Springer: 2006. pp. 35–49. [Google Scholar]
- 25.Louie B, Detwiler L, Dalvi NN, Shaker R, Tarczy-Hornoch P, Suciu D. SSDBM. IEEE Computer Society; 2007. Incorporating uncertainty metrics into a general-purpose data integration system; p. 19. [Google Scholar]
- 26.Detwiler LT, Suciu D, Brinkley JF. Regular paths in SPARQL: querying the NCI Thesaurus. AMIA Annu Symp Proc. 2008:161–165. [PMC free article] [PubMed] [Google Scholar]
- 27.Uncertainty reasoning for the world wide web. 2008 March; [Google Scholar]
- 28.Moreau L, Freire J, Futrelle J, McGrath RE, Myers J, Paulson P. The Open Provenance Model: An overview. In: Freire J, Koop D, Moreau L, editors. IPAW. Vol. 5272. Lecture Notes in Computer Science; Springer: 2008. pp. 323–326. [Google Scholar]
- 29.Moreau L, Pale B, Miles S, Goble C, Missier P, Barga R, Simmhan Y, Futrelle J, McGrath R, Myers J, Paulson P, Bowers S, Ludaescher B, Kwasnikowska N, den Bussche JV, Ellkvist T, Freire J, Groth P. Tech rep. University of Southampton; 2008. The Open Provenance Model (v1.01) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.