

Abstract
Globozoospermia, characterized by round-headed spermatozoa, is a rare (< 0.1% in male infertile patients) and severe teratozoospermia consisting primarily of spermatozoa lacking an acrosome. Studying a Jordanian consanguineous family in which five brothers were diagnosed with complete globozoospermia, we showed that the four out of five analyzed infertile brothers carried a homozygous deletion of 200 kb on chromosome 12 encompassing only DPY19L2. Very similar deletions were found in three additional unrelated patients, suggesting that DPY19L2 deletion is a major cause of globozoospermia, given that 19% (4 of 21) of the analyzed patients had such deletion. The deletion is most probably due to a nonallelic homologous recombination (NAHR), because the gene is surrounded by two low copy repeats (LCRs). We found DPY19L2 deletion in patients from three different origins and two different breakpoints, strongly suggesting that the deletion results from recurrent events linked to the specific architectural feature of this locus rather than from a founder effect, without fully excluding a recent founder effect. DPY19L2 is associated with a complete form of globozoospermia, as is the case for the first two genes found to be associated with globozoospermia, SPATA16 or PICK1. However, in contrast to SPATA16, for which no pregnancy was reported, pregnancies were achieved, via intracytoplasmic sperm injection, for two patients with DPY19L2 deletion, who then fathered three children.
Main Text
In a previous work, we described the identification of a mutation in SPATA16 (MIM 609856) responsible for a complete form of globozoospermia (MIM 102530) in three affected brothers of an Ashkenazi Jewish family.1 We report here the involvement of DPY19L2 on chromosome 12 in cases of complete forms of globozoospermia. Our study started with the analysis of a Jordanian family of ten siblings, including two sisters and three brothers who were naturally fertile (all of them having 3–7 children) and five affected brothers with a complete form of globozoospermia (Figure 1A). Intracytoplasmic sperm injection (ICSI) was performed for the five brothers at the Farah Hospital, Amman, Jordan, and despite a total of 20 cycles there was only one single-term pregnancy, as well as two miscarriages.2 Because of the consanguineous marriage of the grandparents, who were first-degree cousins, we postulated that the genetic abnormality was transmitted as an autosomal-recessive disorder. Four of the five affected individuals, Globo1 to Globo4 (IV-1 to IV-4 in Figure 1A), and all three fertile individuals, brothers B1 to B3 (IV-5 to IV-7 in Figure 1A), consented to give blood samples for research (Figure 1A).
Figure 1.
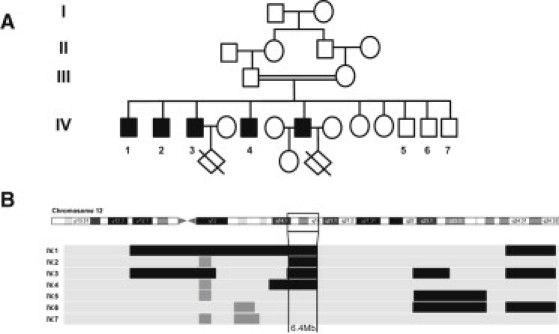
Pedigree and Linkage Analysis of the Jordanian Family
(A) Jordanian pedigree showing second-degree consanguinity.
(B) SNP array results of the four infertile and three fertile brothers for the region of chromosome12. Shared regions of homozygosity are visualized by the HomoSNP software, which displays one patient per line. The areas of homozygosity with 25 or more SNPs are black, whereas homozygosity regions defined by 15–25 consecutive SNPs are gray. Regions of heterozygosity are light gray. The four affected siblings, Globo1 to Globo4 (IV-1 to IV-4), share a region of homozygosity of 6.4 Mb on chromosome 12, which is not shared by the three fertile brothers, B1 to B3 (IV-5 to IV-7).
We performed a genome-wide scan analysis of all seven brothers, using 10K SNP arrays (Affymetrix Genechip). Regions of homozygosity were defined by the presence of at least 25 consecutive homozygous SNPs. We identified a unique region of 30 homozygous SNPs shared by the four affected brothers; the fertile brothers were heterozygous for this region (Figure 1B). This region of about 6.4 Mb on chromosome 12 (positions 63060074 to 69409722, on the GRCh37/hg19 version of the human genome, corresponding to SNPs rs345945 and rs2172989; see Table S1 available online) contains 101 genes, 40 of which are expressed in the testis, according to the UCSC Genome Browser3 (Table S2), and thus are potentially involved in spermatogenesis. We selected DPY19L2 as the most plausible candidate gene because of its predominant testis expression and its potential involvement in cellular polarization.4 The spermatozoon is a highly polarized cell, and the initial stages of spermiogenesis involve polarization of the round spermatid. Functional studies of DPY-19 (an ortholog of DPY19L2) in C. elegans highlighted its role in the establishment of polarity in the migrating neuroblasts.4 DPY19L2 is composed of 22 exons and belongs to the DPY19L family5 coding for proteins harboring 9–11 predicted transmembrane domains. This gene family, encompassing four genes and six pseudogenes, has a complex evolutionary history involving several duplications and pseudogenizations.5 In particular, DPY19L2 stems from an initial duplication of DPY19L1 on chromosome 7, followed by a recent relocalization on chromosome 12 in humans, leaving the initial copy on chromosome 7 as a pseudogene (DPY19L2P1). This evolutionary event may rely on the presence of low copy repeats (LCRs) surrounding the DPY19L2 locus (Figure 2A). Because of the high level of conservation between DPY19L2 and DPY19L2P1,5 the design of DPY19L2-specific oligonucleotides was complex. PCR conditions were optimized for exons 2, 7, 9, 10, 13, 17, and 21, resulting in specific amplifications in controls but never in the patients, suggesting a large deletion of the whole gene (Figure 2B). In order to better characterize the two described LCRs surrounding DPY19L2, we performed a Blastn analysis6 using a 100 kb region up- and downstream of the gene. This approach revealed a highly similar duplicated region (96.5% identity), corresponding to the two identified LCRs (Figure 2B). The telomeric LCR (named here LCR1) is 26,998 bp long and is located upstream of exon 1 (64,119,249–64,146,247), and the centromeric LCR (named here LCR2) is 28,200 bp long and starts ∼1000 bp downstream of exon 22 (63,923,419–63,951,619). In order to define as precisely as possible the breakpoints within the recombined LCRs, specific oligonucleotides were designed to walk on both sides of the deletion (Figures 2B–2D and Table S4). This allowed us to define a first target segment of 107 bp, which we named DPY19L2-BP1. Further refinement is not possible because of the high percentage of identity shared by the two LCRs (Figure 2E and Figure S1). The exact size of the deletion is 195,150 bp. All three healthy brothers B1, B2 and B3 are homozygous wild-type.
Figure 2.
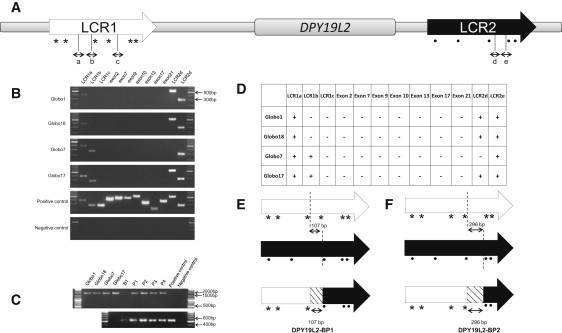
Analysis of DPY19L2
(A) Schematic representation of DPY19L2 flanked by two LCRs, LCR1 and LCR2. LCR1 is located upstream of the gene and LCR2 downstream. Different sequences were amplified on both sides of the gene in order to determine the breakpoints: “a,” “b,” and “c” sequences are localized in LCR1, whereas “d” and “e” sequences are localized in LCR2. Mismatches between the two LCRs sequences are represented by (∗) in the 5′ side and by (.) in the 3′ side.
(B) PCR results of DPY19L2 exons and flanking regions. Patients Globo1, Globo7, Globo17, and Globo18 are deleted for exons 2, 7, 9, 10, 13, 17, and 21, whereas the positive control showed an amplification of all of these exons. All patients showed specific amplifications for region “a” in LCR1 and region “e” in LCR2. Globo1 and Globo18 are missing region “b” in LCR1, which is not the case for Globo7 and Globo17. Globo7 is the only patient missing region “d” in LCR2. A DNA ladder was included on both sides of each gel for accurate determination of the PCR band size.
(C) PCR of DPY19L2 breakpoints. This PCR was performed with the use of the “LCR1a forward” and “DPY19L2-BP reverse” primers (Table S4). The four globozoospermic patients showed a fragment of 1700 bp. The fertile brother (B1, IV-5 in Figure 1A) is homozygous wild-type, because no fragment was amplified, whereas parents of Globo18 (P1 and P2) and Globo17 (P3 and P4) are heterozygous. A fertile man (positive control) was used as control of the PCR specificity. Amplification of exon 10 (lower panel) was used to control the presence or absence of DPY19L2.
(D) Table summarizing the PCR results of the four patients: (+) indicates presence of a PCR band at the corresponding size, and (-) indicates absence of a PCR band.
(E) Schematic representation of the breakpoint area determined for Globo1, Globo18, and the paternal allele of Globo17. The breakpoint area (DPY19L2-BP1) identified by PCR and sequencing is defined between the two discontinued lines and represented by a hatched box in the recombined LCR.
(F) Schematic representation of the homologous recombination that occurred in Globo7 and the maternal allele of Globo17. The breakpoint area (DPY19L2-BP2) identified by PCR and sequencing is defined between the two discontinued lines and represented by a hatched box in the recombined LCR.
The DPY19L2 deletion was then screened in 24 globozoospermic patients from 20 independent families, originating from Algeria, France, Iran, Italy, Lybia, Morocco, and Tunisia. Twelve of them had a complete form of globozoospermia, and six had a partial form (defined as between 20% and 90% of the spermatozoa having round heads7). For the remaining two, we did not have any detailed information (Table 1). We found a similar deletion of DPY19L2 in three unrelated cases presenting with a complete form of globozoospermia (Figures 2C and 2D), two cases originating from France (Globo17 and Globo18) and one familial case from Algeria comprising two brothers (Globo7 and Globo8). One of the French patients (Globo17) had two children, born after ICSI treatment (Table 1). We localized the breakpoints for all three additional patients. For Globo18, the deletion breakpoints are located, as suggested by the PCR walk (Figure 2D), in the same 107 bp LCR area (DPY19L2-BP1) as that of the Jordanian family. Both of his parents are heterozygous for the deletion, as demonstrated by the possibility of amplifying both the recombined region (i.e., absence of DPY19L2) and exon 10 (i.e., at least one copy of DPY19L2) (Figure 2C). The analysis of Globo7 and Globo8 allowed us to identify a different breakpoint, which we named DPY19L2-BP2, (Figure 2D), in an area of 296 bp, positioned 169 bp 3′ to the 107 bp area. Interestingly, Globo17 is a heterozygous composite, because the two different breakpoints (DPY19L2-BP1 and DPY19L2-BP2) could be observed (Figure 2D). The analysis of his parents showed their heterozygous status for the deletion and allowed us to detect the two different breakpoints on each allele. The maternal allele carries DPY19L2-BP2 and the paternal allele carries DPY19L2-BP1. For both parents we could amplify exon 10, demonstrating the presence of at least one copy of DPY19L2 (Figure 2C). The deletion of DPY19L2 was not found in the homozygous state in 105 European-descent males and 101 fertile Jordanian males.
Table 1.
List of Globozoospermic Patients
| Patient | Origin | SNP Screening | Sperm Concentration (Million/mL) | Progressive Motility (%) | Acrosome less Morphology (%) | Fertilization Attempts and Results |
|---|---|---|---|---|---|---|
| Complete Globozoospermia | ||||||
| Globo1 | Jordan | Yes | 52 | 22 | 100 | 6 ICSI failed |
| Globo2 | Jordan | Yes | 223 | 4 | 100 | 3 ICSI failed |
| Globo3 | Jordan | Yes | 118 | 4 | 100 | 4 ICSI failed |
| Globo4 | Jordan | Yes | 22 | 30 | 100 | 3 ICSI failed (1 miscarriage) |
| Globo5 | France | No | ND | ND | 100 | ND |
| Globo6 | France | No | ND | ND | 100 | ND |
| Globo7 | Algeria | No | 90 | 10 | 100 | ND |
| Globo8 | Algeria | No | 13.2 | 2 | 100 | ND |
| Globo9 | France | No | 0.35 | ND | 100 | 1 ICSI failed |
| Globo10 | France | Yes | 38 | ND | 100 | 1 ICSI succeeded |
| Globo11 | France | Yes | 109 | 40 | 100 | 2 ICSI failed |
| Globo12 | ND | No | ND | ND | 100 | DSI |
| Globo13 | Iran | No | 10.5 | ND | 100 | ND |
| Globo14 | Tunisia | No | 52.6 | 25 | 100 | ND |
| Globo15 | Tunisia | No | ND | ND | 100 | 1 ICSI failed |
| Globo16 | Libya | No | 4 | 8 | 100 | 3 ICSI failed |
| Globo17 | France | Yes | 78 | 45 | 100 | IMSI succeeded twice |
| Globo18 | France | Yes | 71 | 30 | 100 | ND |
| Partial Globozoospermia | ||||||
| Globo19 | Italy | No | 50 | ND | ND | 1 ICSI failed |
| Globo20 | France | Yes | 0.6 | ND | 84 | 1 IUI + 3 ICSI failed |
| Globo21 | France | Yes | 2 | ND | ND | 2 ICSI failed |
| Globo22 | Morocco | No | ND | ND | ND | ND |
| Globo23 | France | Yes | ND | ND | 95 | 1 ICSI succeeded |
| Globo24 | France | Yes | ND | ND | 98 | 1 ICSI succeeded |
| Globo25 | Tunisia | No | 59 | 50 | 88 | ND |
| Globo26 | Tunisia | No | 100 | 49 | 99 | ND |
| Globo27 | ND | No | ND | ND | ND | ND |
| Globo28 | ND | No | ND | ND | ND | ND |
Abbreviations are as follows: ND, not determined; ICSI, intracytoplasmic sperm injection; IUI, intrauterine insemination; IMSI, intracytoplasmic morphologically selected sperm injection; DSI, donor sperm insemination.
Several studies have reported copy-number variants (CNVs) at the genome-scale level that are fully available and searchable in databases such as the Database of Genomic Variants (DGV).8 DGV reports, in its latest version (November 2010), 66,741 CNVs from 42 studies. At the DPY19L2 locus, DGV references 29 CNVs that are described by 12 different studies encompassing different ethnic groups (Figure 3 and Table S3). Considering only the nonredundant CNVs identified in non-BAC (bacterial artificial chromosome) studies and those that are fully overlapping DPY19L2, only five studies can be considered.8–13 The detailed inventory of the CNVs revealed 64 duplications and 22 heterozygous deletions out of 4886 patients studied, whereas no homozygous deletion was reported (Table 2). Interestingly, about three times more duplications than deletions can be observed at this locus. This is surprising because in most of the cases studied, deletions are more frequent than duplications.14 We do not have a clear explanation for this, but it would be of interest to determine whether the heterozygous status is affecting male fertility, which would explain the selection against deletion. The frequency of heterozygous deletion in the surrounding region of DPY19L2 is 1/222, implying an overall disease risk of ∼1/200,000 (see Table 2). Examining the localization of the CNVs, one can observe that all deletion breakpoints recorded in DGV fall into the two LCRs, and this is also true for most of the duplications (Figure 3).
Figure 3.
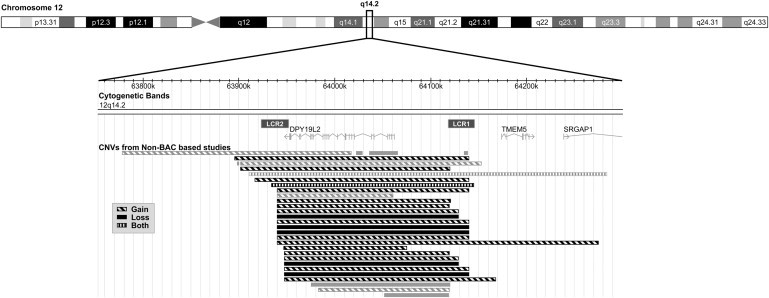
CNVs at the DPY19L2 Locus
CNVs are displayed as rectangles according to the following scheme: gains are drawn with hatched lines, losses with plain rectangles, and vertical lines are used when both gains and losses are observed. The CNVs excluded from the count are colored in gray. LCR1 and LCR2 are displayed as dark gray rectangles on top of the gene representation.
Table 2.
CNV Frequency of Individuals at the DPY19L2 Locus and Expected Prevalence of Homozygotes
Considering the presence of highly conserved LCRs surrounding DPY19L2, the most probable mechanism to explain the multiple occurrence of this deletion would be NAHR between highly similar sequences. Indeed, such a mechanism has been reported for a large number of disorders involving deletions, duplications, inversions, or gene fusions15–17 (reviewed in14). It is estimated that this is one of the most common mechanisms responsible for genomic disorders. So far, no obvious founder effect has been described for genetic disorders involving NAHR.14 That we found the DPY19L2 deletion in patients from three different origins (the two French cases are not known to be related), that two different breakpoints were observed, and that Globo18 is not sharing the same haplotype of the Jordanian brothers (Table S1) strongly suggest that the deletion results from recurrent events linked to the specific architectural feature of this locus rather than from a founder effect, without fully excluding a recent founder effect. It is worth noting that the two identified breakpoints are located near an Alu repeat (AluSq2), one being fully included in this repeated element (Figure S1).
Interestingly, the patients with DPY19L2 deletion present a complete globozoospermia yet have normal or, in one case (Globo8), near-normal concentration of sperm (Table 1). This contrasts with the case in which SPATA16 is mutated, suggesting that DPY19L2 may disrupt only spermiogenesis and not germ cell proliferation and meiosis. It should also be noted that, despite the testicular expression of the other three members of the DPY19L family,5 it does not appear that there is functional redundancy. No homozygosity was found for the loci of the other functional DPY19L genes.
We selected DPY19L2 as the primary candidate linked to globozoospermia in our Jordanian family because of its testicular expression and function of the DPY-19 ortholog in C. elegans, involved in the polarization of neuroblasts.4 This would suggest that DPY19L2 is not directly involved in the biogenesis of the acrosome, as for SPATA161 or PICK1 (MIM 605926),18,19 but rather in preceding steps associated with, for example, the polarity of the spermatid, therefore allowing the correct positioning of the different organelles such as the acrosome or the flagella. However, this hypothesis is not supported by the examination of the immature germ cells in the semen of three globozoospermic patients (Globo7, Globo17, and Globo18). For Globo7, all stages of spermatogenesis could be observed (Figure 4A) and there was no obvious abnormality in the earlier stages of germ cells. In contrast, no proacrosomal vesicle was observed in the round spermatids, yet they appeared correctly polarized. Indeed, the polarization of the spermatid was indicated by the correct positioning of the chromatoid body (in the area of the attachment of the flagellum to the spermatid nucleus) observed in elongating spermatids. In Globo17 and Globo18 semen samples, we observed elongating spermatids as well as spermatozoa that are comparable to that of Globo7 (Figures 4C–4E). In contrast to what is observed in C. elegans, the function of DPY19L2 does not seems to be involved in the polarization of the spermatozoa but, rather, in the formation of the acrosome.
Figure 4.
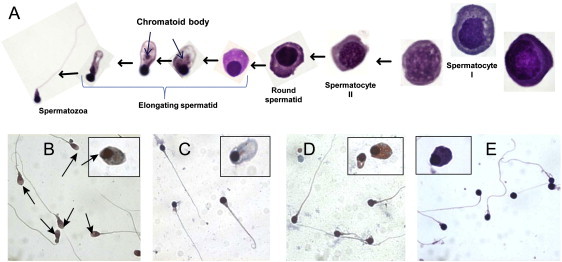
Seminal Germ Cells in Patients with DPY19L2 Deletion
(A) Detailed seminal germ cell of Globo7. There is no evidence of defective spermatogenesis in immature germ cells; the spermatid polarization and elongation is consistent with controls: e.g., the correct positioning of the chromatid body (blue arrows), except for the absence of the acrosomal vesicle.
(B) Spermatozoa and spermatids of fertile controls. The acrosome is clearly observed in spermatozoa and elongating spermatid (arrows).
(C–E) Spermatozoa and spermatid of Globo17, Globo18, and Globo7, respectively. The spermatozoa present the typical round head and an absence of acrosome. No evident polarization abnormality in seminal spermatids was observed in the globozoospermic patients.
DPY19L2 is the third gene identified as being associated with globozoospermia1,19 and, according to our data, the most frequently mutated in this phenotype. Indeed, in our series of 21 unrelated cases, we identified four individual cases, meaning that the DPY19L2 deletion is found in 19% of the globozoospermic cases. Looking at the frequency of heterozygous deletion in control populations, we would expect an incidence of 1/200,000 for homozygous individuals, whereas the incidence of globozoospermia is estimated at less than 0.1% in infertile males.7 Although we cannot fully exclude a positioning effect, we do not believe that this could explain the observed phenotype, because the four genes surrounding the DPY19L2 locus (two centromeric: AVPR1A and PPM1H; two telomeric: TMEM5 and SRGAP1) are not expressed in the testis and their function has no relation with cell polarity or acrosome formation.
Considering that round-headed spermatozoa do not have a higher incidence of chromosomal abnormalities20–22 and that pregnancy can be obtained through ICSI, albeit at a low frequency,2,23–26 it is reasonable to propose ICSI treatment for all globozoospermic patients. It will be interesting to enlarge the cohort of patients and establish which conditions are able to give rise to live births. Indeed, so far, no pregnancies have been obtained with patients carrying a SPATA16 mutation1 (data not shown), in contrast to patients with DPY19L2 deletion. Such an analysis may suggest that a genetic test would assist in determining the best option for treatment.
Our strategy has proven its interest and should be extended to other countries, given that family clustering of male infertility cases has been found in other studies and appears to be related to patterns of consanguineous marriage over generations.27,28
Acknowledgments
We are very grateful to our colleagues Manuel Mark, for his help in interpreting the gametogenesis of our patients, and Michel Koenig and Regen Drouin, for their critical reading of this manuscript. We would also like to thanks all of the clinicians—C. Jimenez (Dijon), F. Carré-Pigeon (Reims), J.M. Grillot (Marseille), F. Brugnon (Clermont Ferrand), D. De Briel (Colmar), and S. Declève-Paulhac (Limoges)—who have sent us samples from globozoospermic patients and for whom we still expect to find a mutation. We would like to recognize the intellectual input of Jean-Louis Mandel, Christelle Thibault-Carpentier, and Mirna Assoum and the technical help of Anne-Sophie Jaeger and Nathalie Drouot. We would like to thank Frederic Plewniak (plewniak@igbmc.fr) from the Bioinformatics Platform of Strasbourg for the development of the HomoSNP software. We are grateful to the Institute of Genetics and Molecular and Cellular Biology (IGBMC) services and the Department of Human Genetics for their invaluable assistance. This work was supported by the French Centre National de la Recherche Scientifique (CNRS), Institut National de la Santé et de la Recherche Médicale (INSERM), the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche, the University of Strasbourg, the University Hospital of Strasbourg, and the Agence de la BioMédecine.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Genomic Variants, http://projects.tcag.ca/variation/
UCSC Genome Browser Gateway, http://genome.ucsc.edu/cgi-bin/hgGateway
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
References
- 1.Dam A.H., Koscinski I., Kremer J.A., Moutou C., Jaeger A.S., Oudakker A.R., Tournaye H., Charlet N., Lagier-Tourenne C., van Bokhoven H., Viville S. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am. J. Hum. Genet. 2007;81:813–820. doi: 10.1086/521314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kilani Z., Ismail R., Ghunaim S., Mohamed H., Hughes D., Brewis I., Barratt C.L. Evaluation and treatment of familial globozoospermia in five brothers. Fertil. Steril. 2004;82:1436–1439. doi: 10.1016/j.fertnstert.2004.03.064. [DOI] [PubMed] [Google Scholar]
- 3.Rhead B., Karolchik D., Kuhn R.M., Hinrichs A.S., Zweig A.S., Fujita P.A., Diekhans M., Smith K.E., Rosenbloom K.R., Raney B.J. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38(Database issue):D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Honigberg L., Kenyon C. Establishment of left/right asymmetry in neuroblast migration by UNC-40/DCC, UNC-73/Trio and DPY-19 proteins in C. elegans. Development. 2000;127:4655–4668. doi: 10.1242/dev.127.21.4655. [DOI] [PubMed] [Google Scholar]
- 5.Carson A.R., Cheung J., Scherer S.W. Duplication and relocation of the functional DPY19L2 gene within low copy repeats. BMC Genomics. 2006;7:45. doi: 10.1186/1471-2164-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 7.Dam A.H., Feenstra I., Westphal J.R., Ramos L., van Golde R.J., Kremer J.A. Globozoospermia revisited. Hum. Reprod. Update. 2007;13:63–75. doi: 10.1093/humupd/dml047. [DOI] [PubMed] [Google Scholar]
- 8.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 9.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Smith A.J., Tsalenko A., Sampas N., Scheffer A., Yamada N.A., Tsang P., Ben-Dor A., Yakhini Z., Ellis R.J., Bruhn L. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum. Mol. Genet. 2007;16:2783–2794. doi: 10.1093/hmg/ddm208. [DOI] [PubMed] [Google Scholar]
- 11.Itsara A., Cooper G.M., Baker C., Girirajan S., Li J., Absher D., Krauss R.M., Myers R.M., Ridker P.M., Chasman D.I. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 2009;84:148–161. doi: 10.1016/j.ajhg.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto D., Marshall C., Feuk L., Scherer S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007;16(Spec. No. 2):R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- 13.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tézenas Du Montcel S., Mendizabai H., Aymé S., Lévy A., Philip N. Prevalence of 22q11 microdeletion. J. Med. Genet. 1996;33:719. doi: 10.1136/jmg.33.8.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoogendijk J.E., Hensels G.W., Gabreëls-Festen A.A., Gabreëls F.J., Janssen E.A., de Jonghe P., Martin J.J., van Broeckhoven C., Valentijn L.J., Baas F. De-novo mutation in hereditary motor and sensory neuropathy type I. Lancet. 1992;339:1081–1082. doi: 10.1016/0140-6736(92)90668-s. [DOI] [PubMed] [Google Scholar]
- 17.Lakich D., Kazazian H.H., Jr., Antonarakis S.E., Gitschier J. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat. Genet. 1993;5:236–241. doi: 10.1038/ng1193-236. [DOI] [PubMed] [Google Scholar]
- 18.Xiao N., Kam C., Shen C., Jin W., Wang J., Lee K.M., Jiang L., Xia J. PICK1 deficiency causes male infertility in mice by disrupting acrosome formation. J. Clin. Invest. 2009;119:802–812. doi: 10.1172/JCI36230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu G., Shi Q.W., Lu G.X. A newly discovered mutation in PICK1 in a human with globozoospermia. Asian J. Androl. 2010;12:556–560. doi: 10.1038/aja.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carrell D.T., Emery B.R., Liu L. Characterization of aneuploidy rates, protamine levels, ultrastructure, and functional ability of round-headed sperm from two siblings and implications for intracytoplasmic sperm injection. Fertil. Steril. 1999;71:511–516. doi: 10.1016/s0015-0282(98)00498-1. [DOI] [PubMed] [Google Scholar]
- 21.Machev N., Gosset P., Viville S. Chromosome abnormalities in sperm from infertile men with normal somatic karyotypes: teratozoospermia. Cytogenet. Genome Res. 2005;111:352–357. doi: 10.1159/000086910. [DOI] [PubMed] [Google Scholar]
- 22.Viville S., Mollard R., Bach M.L., Falquet C., Gerlinger P., Warter S. Do morphological anomalies reflect chromosomal aneuploidies?: case report. Hum. Reprod. 2000;15:2563–2566. doi: 10.1093/humrep/15.12.2563. [DOI] [PubMed] [Google Scholar]
- 23.Banker M.R., Patel P.M., Joshi B.V., Shah P.B., Goyal R. Successful pregnancies and a live birth after intracytoplasmic sperm injection in globozoospermia. J. Hum. Reprod. Sci. 2009;2:81–82. doi: 10.4103/0974-1208.57228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bechoua S., Chiron A., Delcleve-Paulhac S., Sagot P., Jimenez C. Fertilisation and pregnancy outcome after ICSI in globozoospermic patients without assisted oocyte activation. Andrologia. 2009;41:55–58. doi: 10.1111/j.1439-0272.2008.00884.x. [DOI] [PubMed] [Google Scholar]
- 25.Coetzee K., Windt M.L., Menkveld R., Kruger T.F., Kitshoff M. An intracytoplasmic sperm injection pregnancy with a globozoospermic male. J. Assist. Reprod. Genet. 2001;18:311–313. doi: 10.1023/A:1016678604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dirican E.K., Isik A., Vicdan K., Sozen E., Suludere Z. Clinical pregnancies and livebirths achieved by intracytoplasmic injection of round headed acrosomeless spermatozoa with and without oocyte activation in familial globozoospermia: case report. Asian J. Androl. 2008;10:332–336. doi: 10.1111/j.1745-7262.2008.00248.x. [DOI] [PubMed] [Google Scholar]
- 27.Inhorn M.C., Kobeissi L., Nassar Z., Lakkis D., Fakih M.H. Consanguinity and family clustering of male factor infertility in Lebanon. Fertil. Steril. 2009;91:1104–1109. doi: 10.1016/j.fertnstert.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Inhorn M.C., Kobeissi L., Nassar Z., Lakkis D., Fakih M.H. Consanguinity and Male Infertility in Lebanon: The Power of Ethnographic Epidemiology. In: Hahn R.A., Inhorn M.C., editors. Anthropology and Public Health: Bridging Differences in Culture and Society. Oxford University Press; New York: 2009. pp. 165–195. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.