

Abstract
Posterior microphthalmos (MCOP) is a rare isolated developmental anomaly of the eye characterized by extreme hyperopia due to short axial length. The population of the Faroe Islands shows a high prevalence of an autosomal-recessive form (arMCOP) of the disease. Based on published linkage data, we refined the position of the disease locus (MCOP6) in an interval of 250 kb in chromosome 2q37.1 in two large Faroese families. We detected three different mutations in PRSS56. Patients of the Faroese families were either homozygous for c.926G>C (p.Trp309Ser) or compound heterozygous for c.926G>C and c.526C>G (p.Arg176Gly), whereas a homozygous 1 bp duplication (c.1066dupC) was identified in five patients with arMCOP from a consanguineous Tunisian family. In one patient with MCOP from the Faroe Islands and in another one from Turkey, no PRSS56 mutation was detected, suggesting nonallelic heterogeneity of the trait. Using RT-PCR, PRSS56 transcripts were detected in samples derived from the human adult retina, cornea, sclera, and optic nerve. The expression of the mouse ortholog could be first detected in the eye at E17 and was maintained into adulthood. The predicted PRSS56 protein is a 603 amino acid long secreted trypsin-like serine peptidase. The c.1066dupC is likely to result in a functional null allele, whereas the two point mutations predict the replacement of evolutionary conserved and functionally important residues. Molecular modeling of the p.Trp309Ser mutant suggests that both the affinity and reactivity of the enzyme toward in vivo protein substrates are likely to be substantially reduced.
Main Text
Microphthalmos (MCO) is a developmental anomaly resulting from diverse pathomechanisms and characterized by small malformed eyes(s). When the condition occurs in the absence of other ocular defects, it is called isolated or pure microphthalmos. Autosomal-recessive isolated posterior microphthalmos (arMCOP) defines a rare distinct phenotype restricted to the posterior segment of the eye (MIM 613517). In adults, isolated arMCOP is clinically characterized by extreme hyperopia (from +7.5 to +21 diopters) due to short axial length (14–20 mm compared to normal >21 mm), with essentially normal anterior segment, steep corneal curvatures, shallow anterior chamber, thick lenses, and thickened scleral wall.1–3 Palpebral fissures appear narrow because of relatively deep-set eyes, visual acuity is mildly to moderately reduced, and anisometropic or strabismic amblyopia is common. The fundus of the eye shows crowded optical discs, tortuous vessels, and an abnormal foveal avascular zone.1,4,5 In addition, papillomacular folds are often reported.6–10 Morphometric features of the small eyes predispose to complications such as narrow-angle glaucoma and uveal effusion.7,11–14 Some interfamilial clinical heterogeneity is seen to affect, at a minimum, corneal curvature and papillomacular folds.2,15
The gene mutated in isolated arMCOP has not yet been identified. By a genome-wide linkage analysis (homozygosity mapping) in five consanguineous Tunisian families with isolated arMCOP, the disease locus (MCOP6) was mapped between D2S2340 and D2S331, corresponding to a genomic interval of about 2.65 Mb in chromosome 2q37.1.2 In two-point analysis, locus D2S2344 showed close linkage and no recombination with MCOP6 with a maximum LOD score of z = 8.85 at Θ = 0.00. Four of the five families were shown to share the same haplotype in the critical interval, suggesting the presence of a founder mutation in the families studied. The analysis of five positional candidate genes (SAG [MIM 181031], PDE6D [MIM 602676], CHRND [MIM 100720], CHRNG [MIM 100730], and KCNJ13 [MIM 603208]) did not reveal any disease-causing mutations in the Tunisian families.2
An unusually high prevalence of arMCOP has been reported in the Faroe Islands in the North Atlantic.1,3,15 The approximately 48,000 inhabitants represent a genetically isolated population, and in such cases rare genetic diseases are often due to founder mutations (identity by descent). We performed clinical studies and ascertained several Faroese families with arMCOP.1,3,15 In addition, we examined the medical records of approximately 30,000 outpatients visiting the Ophthalmology Department of the National Hospital in Tórshavn, Faroe Islands, between 1967 and 2006, as well as an earlier archive dating back to 1947–1966. The selection criterion of patients from the ophthalmological files was hypermetropia ≥ +10 diopters.1,3,15 In this way, we identified 23 additional patients with MCOP.
Based on the mapping information obtained in the Tunisian families,2 we genotyped all available members of two large Faroese families with arMCOP (HOP00201 and HOP00202; see Figure S1 available online) in an initial two-point linkage analysis for a total of nine microsatellite markers on 2q37.1 between D2S427 and D2S395 covering a region of about 25 cM. We assumed autosomal-recessive inheritance with complete penetrance, but, in order to avoid biasing the outcome, homozygosity due to identity by descent was not required at this stage. Close linkage without recombination was observed between MCOP6 and D2S2344 with maximum LOD scores of 4.79 at Θ = 0.00 (data not shown). Positive LOD scores with zmax of 1.74–2.76 at Θ = 0.05 were also obtained in two-point linkage analyses between MCOP6 and D2S427, between MCOP6 and D2S206, and between MCOP6 and D2S2348, loci known to map close to D2S2344. Analysis of multiple informative meioses in the two Faroese families assigned MCOP6 between D2S2193 and D2S206, a region spanning about 1.5 Mb (Figure S2).
Two attractive candidate genes are assigned to the interval between D2S2193 and D2S206: B3GNT7, encoding UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 7, an enzyme involved in the synthesis of corneal keratan sulfate glycosaminoglycans, and KCNJ13, a member of the subfamily J of inwardly rectifying potassium channel genes. Mutations in the latter gene are associated with autosomal-dominant snowflake vitreoretinal degeneration (MIM 193230). No pathogenic KCNJ13 mutation was found in the patients with arMCOP from the Tunisian cohort.2 No likely disease-causing variant was detected in the genes SNORA75 (small nucleolar RNA, H/ACA box 75), SNORD20 (small nucleolar RNA, C/D box 20), NMUR1 (neuromedin U receptor 1; MIM 604153), or PTMA (prothymosin, alpha; MIM 188390) in the DNA samples of the two patients from each of the Faroese families analyzed (data not shown). Comparison of individual genotypes of patients for intragenic polymorphisms detected in B3GNT7, NCL (MIM 164035), NMUR1, ALPP (MIM 171800), ECEL1 (MIM 605896), EFHD1 (MIM 611617), and KCNJ13 mapping within the 1.5 Mb region excluded the possibility that mutations of any of these genes are causative for the arMCOP in the Faroese families (data not shown and Figure S2).
In order to reduce the critical interval beyond that defined with the microsatellite markers used so far, we analyzed the segregation of six SNP alleles identified in genes mapped in the critical region (Table S1) in all members of the two families. The data obtained in a small nuclear family within pedigree HOP00201 are shown in Figure 1. The maternal haplotype of patient V.2 is a recombinant one that differs from that of his two affected sisters (V.5 and V.6) at the loci EFHD1, KCNJ13, and D2S206, suggesting that MCOP6 maps proximal to EFHD1. Individual VI.2, the unaffected daughter of V.2, carries the recombinant haplotype of her affected father, i.e., the one segregating with the MCOP phenotype in this family, as does the affected daughter (VI.1) of individual V.2. Individual VI.2 also carries a recombinant maternal chromosome with the same haplotype for the interval D2S427–ALPP as her affected sister VI.1, but different alleles for the genomic region D2S2344–D2S206. Because individual VI.2 is unaffected, these data suggest that MCOP6 maps distal to ALPP. Together, the data map MCOP6 in a genomic interval of about 250 kb between ALPP and EFHD1 (Figure S2). However, it is remarkable that both patients V.2 and VI.1 are heterozygous for the polymorphism at D2S2344, a locus mapped in the critical interval. This observation contradicts our initial hypothesis of an autosomal-recessive trait with a single founder mutation and instead suggests that—at least in some patients—two different mutations of the same gene may be responsible for the phenotype. Therefore, we selected a pool of 29 patients with arMCOP, two from each of the large families, 23 from small nuclear families from the Faroe Islands, one (SE) from a small consanguineous family from Turkey, and one (NB8) from a large consanguineous family from Tunisia, to screen for mutations in candidate genes in our subsequent studies.
Figure 1.
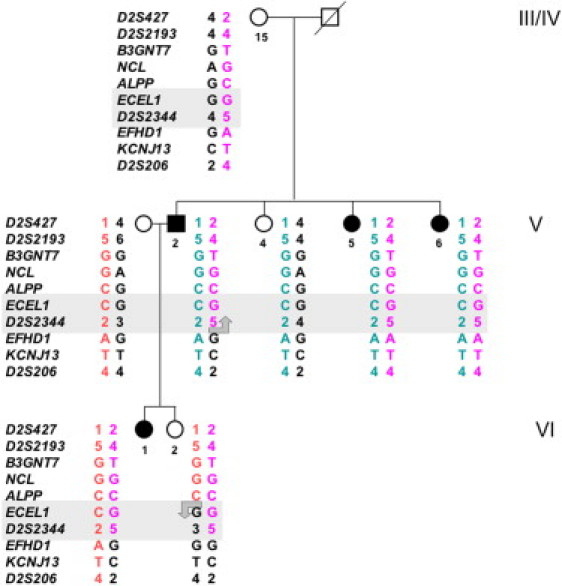
Mapping the MCOP6 Locus in a Branch of Family HOP000201 by the Analysis of Multiple Informative Meioses
Alleles of a total of ten DNA polymorphisms in the region between D2S427 and D2S206 on chromosome 2q37.1 are arranged as probable haplotypes. Individuals V.2 and VI.2 carry recombinant chromosomes, suggesting that the MCOP6 locus maps between ALPP and EFHD1. Generation and individual numbers shown in the figure are the same as in Figure S1.
PRSS56 (protease serine 56; also known as LOC646960) encodes a protein of 603 amino acids (GenBank RefSeq: NM_001195129 and NP_001182058) showing similarity to trypsin-like serine proteases. Expressed sequence tag data suggest that PRSS56 is expressed in embryonic tissue, testis, brain, and eye. We PCR amplified the 13 coding exons with short adjacent intronic sequences of PRSS56 (see Table S2) and found, in addition to known SNPs, three very likely disease-causing mutations. In patient NB8 from Tunisia, a homozygous 1 bp duplication (c.1066dupC) was identified that results in a shift in the reading frame, inclusion of PRSS56-unrelated residues, a premature stop codon (p.Gln356ProfsX152), and, most likely, a functional null allele (Figure 2B). The mutation was shown to cosegregate with the disease phenotype in the family consisting of healthy consanguineous parents and a total of 11 children, five of them affected. The ophthalmological picture in the family consists of nanophthalmos with microcornea (9–10 mm, mean of 9.75 mm), shallow anterior chamber and angle, extreme hyperopia (+10 to +18 diopters; mean of 14 diopters), short axial length (15.4–17.6 mm; mean of 16.15 mm), high lens/eye volume ratio (mean of 29%), thick sclera and choroid, and absence of overt structural defects (unlike microphthalmia). Best corrected vision ranged between 0.05 and 0.5 (mean of 0.27). Fundus abnormality was seen in five eyes (papillomacular fold [n = 3], a choroidal neovascular membrane [n = 1], and a central venous occlusion secondary to angle-closure glaucoma [n = 1]). Phenotypes in the Tunisian families reported by Hmani-Aifa et al.2 differ from the phenotype in this family with respect to normal corneal diameter and steepness.
Figure 2.
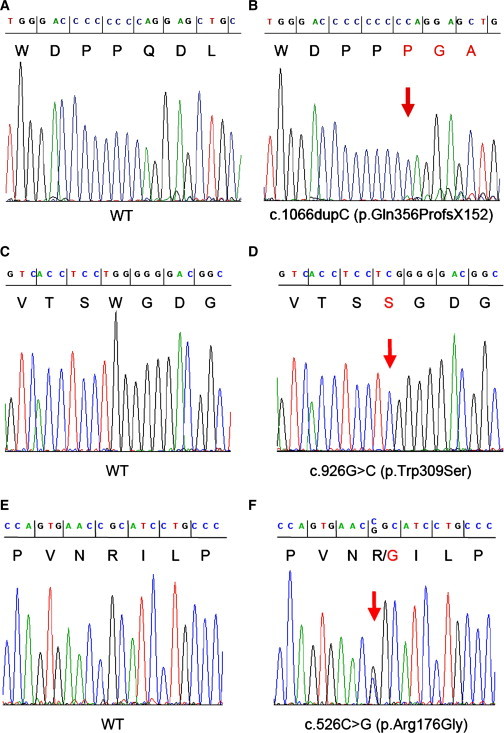
Electropherograms of DNA Sequences of PCR Amplicons
(A–F) The panels show the homozygous c.1066dupC and c.926G>C mutations of PRSS56 in patients NB8 (B) and III.5/HOP000202 (D) and the heterozygous c.526C>G mutation in V.2/HOP000201 (F), as well as the corresponding regions in controls (wild-type, WT; A, C, and E, respectively). DNA and predicted amino acid (one letter code) sequences are shown on the top of the figure.
All but one of the patients of the Faroese cohort (n = 27; two from each of the large families and 23 “sporadic” cases) carried a PRSS56 mutation: a c.926G>C change in exon 8 (p.Trp309Ser; Figure 2D) in homozygous form (n = 20) or compound heterozygous form (n = 6) with c.526C>G (p.Arg176Gly) in exon 5 (Figure 2F). We also genotyped all members of the two families for the c.526C>G and c.926G>C mutations (Figure S1) and showed cosegregation of the variants with the disease phenotype. Consistent with the 1 bp duplication in the Tunisian family that should result in a loss-of-function allele, the two Faroese point mutations result in the replacement of evolutionarily conserved and functionally important residues and are likely to lead to reduced catalytic activity of the protein. The phenotype of the Faroese patients is very similar to that seen in the Tunisian family of NB8, except for the corneal diameter, which is described as microcornea in the Tunisian family, whereas corneal diameters in the Faroese subjects are normal, albeit in the lower range. The modest differences among, and the partial overlap of, the phenotypes of the patients in the Faroese families and the NB8 Tunisian family suggest that a distinction between posterior microphthalmos and nanophthalmos might be artificial, thus leading to the view that these two conditions represent a continuum of phenotypes.
We genotyped selected patients for polymorphisms of the genes mapped within the 250 kb interval. Of the 20 patients carrying a homozygous c.926G>C PRSS56 mutation, one was heterozygous for the G-to-C change of the rs6750085 SNP of ECEL1. This observation can be explained by a putative recombination between ECEL1 and PRSS56, confirms that MCOP6 maps between ECEL1 and EFHD1, and thus reduces the critical interval to about 150 kb (Figure S2). In addition to PRSS56, this interval contains four other positional candidate genes: CHRND, CHRNG, TIGD1 (tigger transposable element derived 1; MIM 612972), and EIF4E2 (eukaryotic translation initiation factor 4E, family member 2; MIM 605895; Figure S2). In the DNA sample of a Faroese patient homozygous for the PRSS56 c.926G>C mutation, only very likely nonpathogenic variants were found by sequencing all coding exons of TIGD1 (c.252G>A [rs4973540] and c.822G>A [rs6752614]) and EIF4E2 (c.213+48delT). Mutation analysis of CHRND and CHRNG was performed by Hmani-Aifa et al. in patients from the Tunisian families with arMCOP and did not reveal any disease-causing changes.2
Genealogical studies identified a married couple (born in 1598 and 1631) as ancestors of the parents of all arMCOP patients from the Faroe Islands carrying the c.926G>C mutation in homozygous or heterozygous state genotyped in this study. None of the three PRSS56 sequence variants found were detected on 100 chromosomes of unaffected German controls. However, three out of 94 anonymous unaffected Faroese controls genotyped in this study were heterozygous for the c.926G>C mutation, corresponding to a heterozygote frequency of 3.2%. This finding is in line with the incidence of heterozygotes for other disorders in the Faroe Islands caused by founder mutations, e.g., mitochondrial encephalomyopathy caused by SUCLA2 (MIM 603921) mutations16 and retinitis pigmentosa due to MERTK (MIM 604705) mutations.17 The c.526C>G mutation was not found in the 94 controls. Genealogical studies identified a male born in 1827 as the ancestor of all Faroese arMCOP patients carrying the c.526C>G mutation in compound heterozygous state. On the one hand, the likely younger age of the c.526C>G change explains the predominance of the c.926G>C variant in both patients and controls. On the other hand, the high number of patients carrying the c.526C>G mutation in heterozygous state in family HOP00201 is unexpected. However, this observation can easily be explained by assuming that one of the parents (1 or 1a) in generation I carried the c.526C>G variant and transmitted it to both his/her children, whereas the c.926G>C change was contributed by one of the other parents (2 or 2a) in generation I (Figure S1).
In two patients, one from the Faroe Islands and the other, patient SE, from Turkey, we sequenced all exons of PRSS56 and found that they did not carry any of the mutations identified in other patients of this study or any other disease-related changes. Similarly, exon-spanning PCR amplicons did not reveal any size difference following nondenaturing electrophoretic separation compared to control amplicons, making the presence of a larger, exon-spanning deletion or insertion unlikely and suggesting nonallelic heterogeneity of MCOP. This assumption is further substantiated by the fact that patient SE from a consanguineous family was heterozygous for two different SNPs in PRSS56 (data not shown).
We evaluated the expression of PRSS56 via RT-PCR with gene-specific primers (Table S3) to amplify cDNAs generated from equal amounts of total RNAs isolated from human donor eye tissues. PRSS56 transcripts were abundant in the samples derived from the neural retina, cornea, sclera, and optic nerve (Figure 3A). To evaluate expression during development, we PCR amplified Prss56 (also called 1700027L20Rik, the mouse ortholog of the human PRSS56) from cDNAs generated from total RNAs isolated from mouse eyes (embryonic day 12 through postnatal day 1) and mouse retinas (postnatal day 5 through adult). Prss56 transcripts were first detected at embryonic day 17, with expression maintained into adulthood (Figure 3B). The expression of Prss56 was also evaluated by in situ hybridization (Figure 4). Images of nonpigmented mouse eyes in cross-section (top panels) show that specific (antisense) signals were obtained in the retina and in the optic nerve (seen as punctate staining), whereas nonspecific staining artifacts were observed in lens fragments. At higher magnification (bottom panels), strong reactivity was seen in the retinal ganglion cells, moderate reactivity in the inner nuclear layer, and low-level reactivity in the outer nuclear layer and other eye tissues (Figure 4). No reactivity was detected in E16 mouse eye tissue (data not shown). Taken together, these data suggest a unique requirement for PRSS56 in the physiology of the neural retina.
Figure 3.
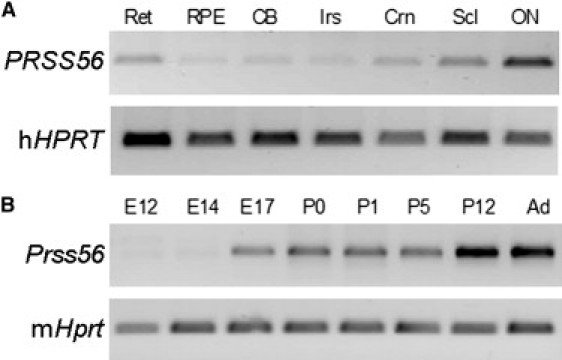
Expression Analysis
(A) RT-PCR products from human eye tissues. The following abbreviations are used: Ret, retina; RPE, retinal pigment epithelium/choroid; CB, ciliary body; Irs, iris; Crn, cornea; Scl, sclera; ON, optic nerve.
(B) RT-PCR products from mouse eye and retinas at the ages shown.
Total RNA was isolated from human donor (male, 60 yr) eye tissues obtained within 20 hr postmortem. Mouse total RNA was isolated from whole eyes of C57Bl/6 at embryonic day ages E12, E14, and E17 and postnatal day ages P0 and P1, as well as from dissected retinas at ages P5, P12, and adult (Ad). RNA concentrations and integrity were evaluated with Agilent Bioanalysis. First-strand cDNAs were prepared from 1 μg RNA with Superscript II, followed by treatment with RNase H. DNA sequences were PCR amplified with primers for PRSS56, Prss56, and the hypoxanthine phosphoribosyltransferase gene (HPRT; Table S3) by using 35 cycles of 94°C for 1 min, 55°C for 1 min, and 68°C for 2 min. The products were visualized on agarose gels stained with SyberGreen.
Figure 4.
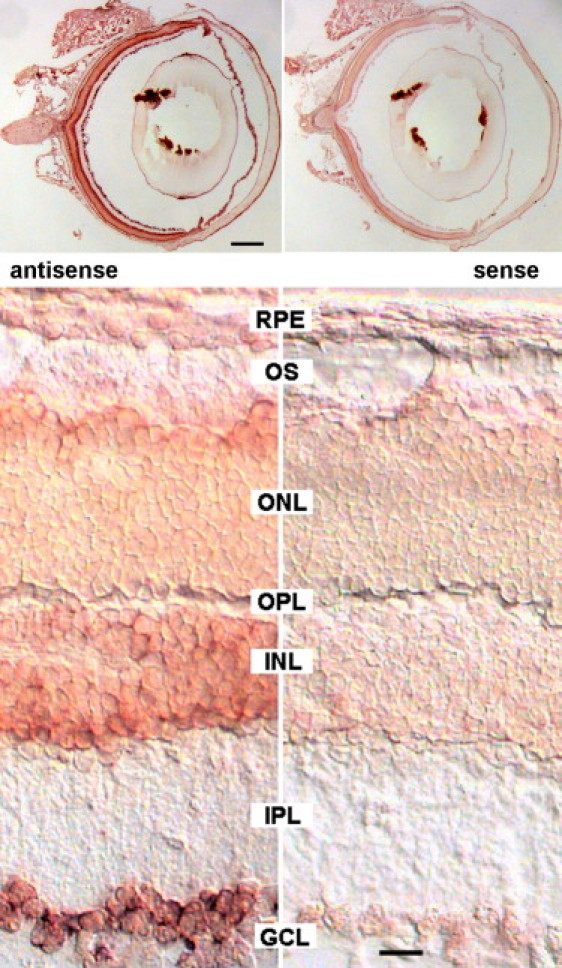
In Situ Hybridization of Sections of a 3-Month-Old Balb/c Mouse Eye with Prss56 RNA Probes
Sagittal sections were hybridized with digoxygenin-labeled antisense and sense RNA probes transcribed from a 411 bp fragment of the Prss56 cDNA (accession number JF323950; amplified with primer pair 2 from P12 mouse retina RNA; see Table S3 for primers). Probes were detected with alkaline phosphatase-coupled anti-DIG antibodies, followed by staining in Nitro-Blue Tetrazolium/5-Bromo-4-Chloro-3′-Indolyphosphate (NBT/BCIP; Roche) for 4 hr at 37°C. Sections were viewed with a Zeiss Axiophot microscope equipped with differential interference contrast optics and a Kappa camera. Scale bars represent 0.5 mm (top panels) and 20 μm (bottom panels).
Based on the little data available to date, Prss56 shows a limited expression in the mouse nervous system. Coulpier et al.18 reported that 1700027L20Rik (Prss56) is a marker for embryonic boundary cap (BC) cells, a transient population of cells of neural crest origin. BC cells form clusters at the surface of the neural tube, at entry and exit points of peripheral nerve roots (i.e., at the boundary between central and peripheral nervous system), and give rise to neuron and glia cells. We found that Prss56 is highly expressed in retinal ganglion cells of adult animals. Although the functional relation between retinal ganglion cells and BC cells is unknown, we conclude that this putative protease is expressed in different populations of highly specialized neural cells.
Analysis of the predicted PRSS56 protein sequence showed that it may be classified as a serine peptidase of the chymotrypsin family (clan PA, family S1 according to Merops Peptidase Database).19 We performed molecular modeling with the SWISS-MODEL server in automated mode and based on the best possible alignment toward proteins present in the Research Collaboratory for Structural Bioinformatics protein database (pdb). The PRSS56 protein sequence (both wild-type and the p.Trp309Ser mutant) exhibited the highest similarity to the catalytic domain of enteropeptidase (pdb entry 1ekbB). However, enteropeptidase does not carry tryptophan, but instead carries phenylalanine in position 263 (corresponding to position 309 in PRSS56), and thus it may not be the best model to analyze the effect of the p.Trp309Ser mutation. Therefore, modeling was based on trypsin (2agg) that carries tryptophan at this position (Trp215 in trypsin). The enteropeptidase- and trypsin-based models were largely comparable, because in both cases Trp309 adopted a similar position lining part of the substrate-binding pocket. As shown in Figure 5, several features of the PRSS56 sequence clearly support its assignment to the chymotrypsin family. (1) Like other members of this family, PRSS56 is predicted to be a secreted protein with an N-terminal signal peptide that directs the protein into the secretory pathway (Figure 5A). (2) Members of this family are secreted as catalytically inactive proenzymes that require proteolytic cleavage at a highly conserved position to convert the enzyme to the active form. The sequence around the cleavage site (residues 104–105) is conserved in PRSS56. (3) PRSS56 carries a charge relay system (also termed catalytic triad) consisting of Asp191-His145-Ser286 (similar to Asp102-His57-Ser195 in trypsin), which is required for the nucleophilic activity of the catalytic Ser286.19 (4) Amino acids involved in substrate binding are conserved in PRSS56 (Figure 5B). Asp280 (corresponding to Asp189 in trypsin) is located at the bottom of the rather deep pocket of the predicted active site (blue in Figure 5C) in a position to stabilize positively charged Arg/Lys side chains of the substrate through its negative charge.20,21 In addition, residues Ser308-Trp309-Gly310 are in a position to form a secondary binding pocket that can contribute to specificity by accommodating the side chains of residues N terminal to this Arg/Lys residue.
Figure 5.
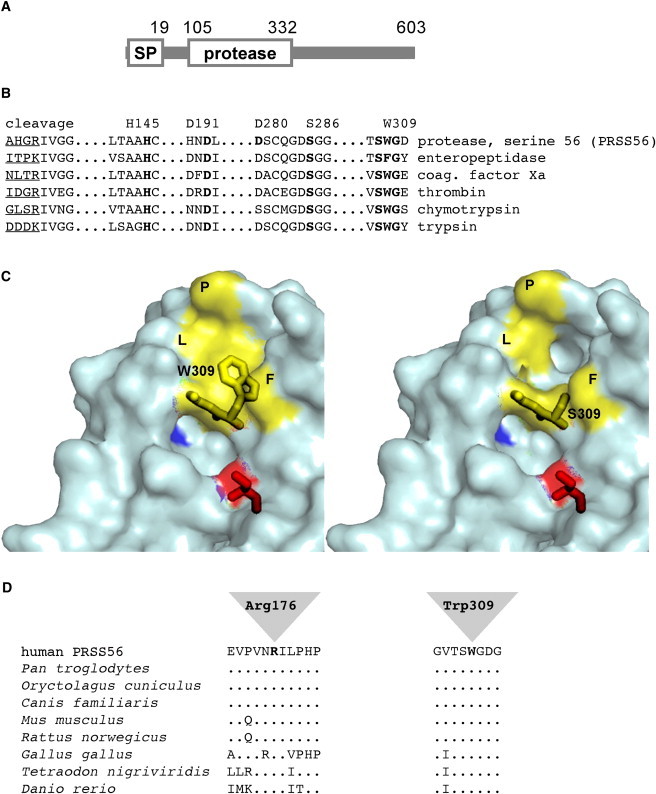
Trypsin-Like Serine Protease PRSS56
(A) Schematic structure of PRSS56. Positions of the signal peptide (SP, cleaved between residues 19 and 20), the cleavage site of the proprotein (residues104–105), and the protease domain are indicated. The C-terminal domain of the protein does not exhibit similarity to any known protein.
(B) Alignment of functionally relevant sequence elements of members of the serine-protease family. The position of the cleavage site for conversion of the proenzymes into the active enzymes is indicated (trypsinogen to trypsin, prothrombin to thrombin, etc.); the sequence N-terminal to the cleavage site is underlined. Elements of the charge relay system or catalytic triad (Asp191/His145/Ser286 in PRSS56), as well as an aspartate residue localized in the P1 specificity pocket (Asp280 in PRSS56), and the sequence surrounding Trp309 are indicated by bold print.
(C) 3D model of wild-type (left) and p.Trp309Ser mutant (right) PRSS56 based on the atomic structure of trypsin. Molecular modeling was performed with the SWISS-MODEL server in automated mode. PDB data sets obtained by the modeling procedure were visualized with the help of the polyview 3D visualization server. Residues Trp309-Gly310 and the catalytic Ser286 (red) are indicated by stick models, and the negatively charged Asp280 at the bottom of the P1 active-site pocket is shown in blue. In the wild-type, Trp309 together with residues Phe188 (F), Leu260 (L), and Pro262 (P) contributes to the formation of a shallow hydrophobic cavity (yellow surface), which is disrupted in the p.Trp309Ser mutant.
(D) Sequence alignment of vertebrate homologs of PRSS56. Sequences flanking Arg176 and Trp309 are shown. Identical amino acids are indicated by dots.
Proteases of the chymotrypsin family exhibit variable degrees of substrate specificity. On the one hand, trypsin shows little specificity, because the presence of a positively charged Arg or Lys immediately C-terminal to the cleavage site is sufficient for substrate recognition. On the other hand, catalytic activity of other members of this protein family, such as enteropeptidase or thrombin, is highly sequence specific. In most cases, residues N-terminal to the Arg/Lys residue contribute to specificity as their side chains are accommodated by a shallow pocket adjacent to the active site. The sequence around Trp215 (Trp309 in PRSS56) is highly conserved among trypsin-like enzymes and is also predicted to contribute to formation of this pocket in PRSS56. In the case of thrombin, the corresponding Trp215 residue has been shown to be essential for efficient binding of hydrophobic residues of the substrate, and analysis of mutant proteins generated by in vitro mutagenesis indicated that this residue is the primary determinant of substrate selectivity.22,23
Molecular modeling of the wild-type and p.Trp309Ser mutant forms of PRSS56 suggests that p.Trp309, together with Phe188, Leu260, and Pro262, forms a hydrophobic cavity that may be required to accommodate hydrophobic residues of the substrate. Modeling of the mutant form suggests that a serine at position 309 leads to a much wider pocket and a disruption of the hydrophobic surface as a result of insertion of the small and hydrophilic serine side chain (Figure 5C). These changes are likely to disrupt binding of one or more in vivo substrates of PRSS56, suggesting that affinity and reactivity toward the (so far unknown) natural substrates of PRSS56 are likely to be reduced substantially. The second missense mutation, p.Arg176Gly, affects a residue that is evolutionarily highly conserved in all vertebrate orthologs of PRSS56 (Figure 5D). In 3D models, it is distant from the active site and thus is not likely to directly participate in substrate binding. Arg176 may interact with a helical segment at the C-terminal end of the protease domain. Loss of this interaction in the mutant leads to very subtle conformational changes in a number of amino acids in the vicinity of the catalytic Ser286 (data not shown). This suggests a possible effect of the p.Arg176Gly mutation on catalytic efficiency, which can only be experimentally ascertained once the in vivo substrate or substrates of the PRSS56 protease are identified. Alternatively, loss of Arg176 might affect the solubility of the protein, leading to aggregation.
Sequence comparisons show that orthologs of PRSS56 exist in all vertebrate species analyzed (Figure 5 and data not shown). Similarity is high between the human protein and its mammalian counterparts. In particular, all sequence elements required for processing of the protein and all residues likely to be involved in catalysis are highly conserved (see Figure 5). In addition, the C-terminal region, which does not contain any obvious functional motifs, is very similar in different mammalian species, suggesting that it is also relevant for the function of the PRSS56 protein. In other vertebrates such as zebrafish, the catalytic domain is more highly conserved than the C-terminal domain. No PRSS56 orthologs in nonvertebrate species could be detected, suggesting a function unique to the development of the vertebrate visual system.
The mouse homolog of PRSS56 has been described as a protein of unknown function with high similarity to the transmembrane serine protease 9.18,24 This protein, also known as polyserase-I, consists of an N-terminal cytoplasmic segment followed by a type II transmembrane domain and three independent trypsin-like serine protease domains. However, the similarity between PRSS56 and poyserase-I is limited to the protease domains, whereas the overall domain structure is different. As mentioned above, the PRSS56 protein is predicted to be processed in the secretory pathway because of a cleavable N-terminal signal peptide, whereas the transmembrane serine protease was shown to be an integral membrane protein localized on the cell surface.25
Postnatal development of the ocular refractive components is a tightly regulated adaptive process by which optical defocus leads to changes in axial length moving the retina toward the image plane. This emmetropization process involves a retinal feedback mechanism. The concept of such mechanism is underscored by evidence from experimental myopia. The postnatal increase in axial length of the eye is mainly due to an elongation of the posterior segment from 10–11 mm at birth to 15–16 mm at the age of 13.26 The pathophysiology involved in MCOP may theoretically be explained by a postnatal growth retardation of the posterior segment (axial length of the vitreous), in accordance with the expression pattern of PRSS56. The corneal changes, however, may indicate the presence of a prenatal action. Very little is known about the molecular pathways involved in this remodeling. It is, however, tempting to anticipate the involvement of secreted peptidases in catalytic processes in the vitreous and/or the sclera. We propose that the proteolytic activity provided by the protein encoded by PRSS56 is essential for this process during development.
Acknowledgments
The study was performed in collaboration with the Genetic Resource Center, Ílegusavnið, and approved by the National Scientific Ethics Committee, Vísindasiðsemisnevnd Føroya; it also conformed to the guidelines of the University of Michigan, Laboratory of Animal Medicine. The study was supported by grants of the Alcon Research Institute (ARI Award 2006 to A.G.), the Danish Society of the Blind, the Swiss National Science Foundation (grant 320030-127558 to D.F.S. and F.L.M.), the National Eye Institute (P30-EY09003), and the Unitè de Recherche en Oculo-Génétique UR 17/04 de l'Institut Hedi Rais d'Ophtalmologie de Tunis. D.A.T. is the recipient of a Research to Prevent Blindness Senior Investigator award. Mouse RNAs were prepared by Marina Zieger and Afaf Absood. Human donor eyes were obtained from the Midwest Eye Banks and Transplantation centers.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Merops Peptidase Database, http://merops.sanger.ac.uk/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Polyview 3D visualization server, http://polyview.cchmc.org/polyview3d.html/
SWISS-MODEL server, http://swissmodel.expasy.org
Accession Number
The GenBank accession number for the mouse cDNA sequence reported in this paper is JF323950.
References
- 1.Fuchs J., Holm K., Vilhelmsen K., Rosenberg T., Scherfig E., Fledelius H.C. Hereditary high hypermetropia in the Faroe Islands. Ophthalmic Genet. 2005;26:9–15. doi: 10.1080/13816810590918406. [DOI] [PubMed] [Google Scholar]
- 2.Hmani-Aifa M., Ben Salem S., Benzina Z., Bouassida W., Messaoud R., Turki K., Khairallah M., Rebaï A., Fakhfekh F., Söderkvist P., Ayadi H. A genome-wide linkage scan in Tunisian families identifies a novel locus for non-syndromic posterior microphthalmia to chromosome 2q37.1. Hum. Genet. 2009;126:575–587. doi: 10.1007/s00439-009-0688-8. [DOI] [PubMed] [Google Scholar]
- 3.Fledelius H.C., Fuchs H.J., Rosenberg T. Oculometric characteristics of extreme hypermetropia in two faroese families. Optom. Vis. Sci. 2004;81:762–768. doi: 10.1097/00006324-200410000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Serrano J.C., Hodgkins P.R., Taylor D.S., Gole G.A., Kriss A. The nanophthalmic macula. Br. J. Ophthalmol. 1998;82:276–279. doi: 10.1136/bjo.82.3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh M.K., Goldberg M.F. Abnormal foveal avascular zone in nanophthalmos. Am. J. Ophthalmol. 2007;143:1067–1068. doi: 10.1016/j.ajo.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 6.Spitznas M., Gerke E., Bateman J.B. Hereditary posterior microphthalmos with papillomacular fold and high hyperopia. Arch. Ophthalmol. 1983;101:413–417. doi: 10.1001/archopht.1983.01040010413014. [DOI] [PubMed] [Google Scholar]
- 7.Khairallah M., Messaoud R., Zaouali S., Ben Yahia S., Ladjimi A., Jenzri S. Posterior segment changes associated with posterior microphthalmos. Ophthalmology. 2002;109:569–574. doi: 10.1016/s0161-6420(01)00996-4. [DOI] [PubMed] [Google Scholar]
- 8.Aras C., Ozdamar A., Ustundag C., Ozkan S. Optical coherence tomographic features of papillomacular fold in posterior microphthalmos. Retina. 2005;25:665–667. doi: 10.1097/00006982-200507000-00021. [DOI] [PubMed] [Google Scholar]
- 9.Erdol H., Kola M., Turk A., Akyol N. Ultrasound biomicroscopy and OCT findings in posterior microphthalmos. Eur. J. Ophthalmol. 2008;18:479–482. doi: 10.1177/112067210801800331. [DOI] [PubMed] [Google Scholar]
- 10.Buyukyildiz H.Z., Demirci G., Gulkilik G. Optical coherence tomography in posterior microphthalmos with papillomacular fold and high hyperopia in two siblings. Ann. Ophthalmol. (Skokie) 2008;40:45–47. [PubMed] [Google Scholar]
- 11.Lesnoni G., Rossi T., Nistri A., Boccassini B. Nanophthalmic uveal effusion syndrome after prophylactic laser treatment. Eur. J. Ophthalmol. 1999;9:315–318. doi: 10.1177/112067219900900411. [DOI] [PubMed] [Google Scholar]
- 12.Yalvac I.S., Satana B., Ozkan G., Eksioglu U., Duman S. Management of glaucoma in patients with nanophthalmos. Eye (Lond.) 2008;22:838–843. doi: 10.1038/sj.eye.6702742. [DOI] [PubMed] [Google Scholar]
- 13.Thapa S.S., Paudyal G. Choroidal effusion following laser peripheral iridotomy for the treatment of angle closure glaucoma in a patient with nanophthalmos. Nepal Med. Coll. J. 2005;7:81–82. [PubMed] [Google Scholar]
- 14.Burgoyne C., Tello C., Katz L.J. Nanophthalmia and chronic angle-closure glaucoma. J. Glaucoma. 2002;11:525–528. doi: 10.1097/00061198-200212000-00013. [DOI] [PubMed] [Google Scholar]
- 15.Fledelius H.C., Rosenberg T. Extreme hypermetropia and posterior microphthalmos in three siblings. An oculometric study. In: Ossoinig K.C., editor. Ophthalmic Echography. Martinus Nijhoff/Junk; Dordrecht, The Netherlands: 1987. pp. 87–91. [Google Scholar]
- 16.Ostergaard E., Hansen F.J., Sorensen N., Duno M., Vissing J., Larsen P.L., Faeroe O., Thorgrimsson S., Wibrand F., Christensen E., Schwartz M. Mitochondrial encephalomyopathy with elevated methylmalonic acid is caused by SUCLA2 mutations. Brain. 2007;130:853–861. doi: 10.1093/brain/awl383. [DOI] [PubMed] [Google Scholar]
- 17.Ostergaard E., Duno M., Batbayli M., Vilhelmsen K., Rosenberg T. A novel MERTK deletion is a common founder mutation in the Faroe Islands that is responsible for a high proportion of retinitis pigmentosa cases. Mol. Vis. 2011 in press. [PMC free article] [PubMed] [Google Scholar]
- 18.Coulpier F., Le Crom S., Maro G.S., Manent J., Giovannini M., Maciorowski Z., Fischer A., Gessler M., Charnay P., Topilko P. Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia. 2009;57:1450–1457. doi: 10.1002/glia.20862. [DOI] [PubMed] [Google Scholar]
- 19.Di Cera E. Serine proteases. IUBMB Life. 2009;61:510–515. doi: 10.1002/iub.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perona J.J., Hedstrom L., Rutter W.J., Fletterick R.J. Structural origins of substrate discrimination in trypsin and chymotrypsin. Biochemistry. 1995;34:1489–1499. doi: 10.1021/bi00005a004. [DOI] [PubMed] [Google Scholar]
- 21.Radisky E.S., Lee J.M., Lu C.J., Koshland D.E., Jr. Insights into the serine protease mechanism from atomic resolution structures of trypsin reaction intermediates. Proc. Natl. Acad. Sci. USA. 2006;103:6835–6840. doi: 10.1073/pnas.0601910103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cantwell A.M., Di Cera E. Rational design of a potent anticoagulant thrombin. J. Biol. Chem. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- 23.Marino F., Pelc L.A., Vogt A., Gandhi P.S., Di Cera E. Engineering thrombin for selective specificity toward protein C and PAR1. J. Biol. Chem. 2010;285:19145–19152. doi: 10.1074/jbc.M110.119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hooper J.D., Clements J.A., Quigley J.P., Antalis T.M. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 2001;276:857–860. doi: 10.1074/jbc.R000020200. [DOI] [PubMed] [Google Scholar]
- 25.Cal S., Quesada V., Garabaya C., Lopez-Otin C. Polyserase-I, a human polyprotease with the ability to generate independent serine protease domains from a single translation product. Proc. Natl. Acad. Sci. USA. 2003;100:9185–9190. doi: 10.1073/pnas.1633392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larsen J.S. The sagittal growth of the eye. 3. Ultrasonic measurement of the posterior segment (axial length of the vitreous) from birth to puberty. Acta Ophthalmol. (Copenh.) 1971;49:441–453. doi: 10.1111/j.1755-3768.1971.tb00969.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.