

Abstract
Dilated cardiomyopathy commonly causes heart failure and is the most frequent precipitating cause of heart transplantation. Familial dilated cardiomyopathy has been shown to be caused by rare variant mutations in more than 30 genes but only ∼35% of its genetic cause has been identified, principally by using linkage-based or candidate gene discovery approaches. In a multigenerational family with autosomal dominant transmission, we employed whole-exome sequencing in a proband and three of his affected family members, and genome-wide copy number variation in the proband and his affected father and unaffected mother. Exome sequencing identified 428 single point variants resulting in missense, nonsense, or splice site changes. Genome-wide copy number analysis identified 51 insertion deletions and 440 copy number variants > 1 kb. Of these, a 8733 bp deletion, encompassing exon 4 of the heat shock protein cochaperone BCL2-associated athanogene 3 (BAG3), was found in seven affected family members and was absent in 355 controls. To establish the relevance of variants in this protein class in genetic DCM, we sequenced the coding exons in BAG3 in 311 other unrelated DCM probands and identified one frameshift, two nonsense, and four missense rare variants absent in 355 control DNAs, four of which were familial and segregated with disease. Knockdown of bag3 in a zebrafish model recapitulated DCM and heart failure. We conclude that new comprehensive genomic approaches have identified rare variants in BAG3 as causative of DCM.
Introduction
Dilated cardiomyopathy, (DCM [MIM 115200]), a common primary myocardial disease, causes systolic dysfunction and heart failure, a major public health problem. Familial dilated cardiomyopathy has been shown to be caused by rare variant mutations in more than 30 genes, most of which encode sarcomeric or sarcomeric-associated proteins.1,2 However, mutations in the known genes only account for ∼35% of genetic causes, leaving the majority of them unknown. This highlights two observations: (1) additional rare variants in other genes remain to be found and (2) rare copy number variants (large deletions and duplications > 1 kb) have been largely ignored in DCM genetics. These types of genetic variation would be missed by traditional sequencing approaches to coding regions because of the background presence of the wild-type allele. At this time, no systematic study of the contribution of copy number variation to DCM has been performed.
On these bases, we simultaneously performed exome sequencing and a high-resolution genome-wide study of copy number variation on a large multiplex family (Figure 1) with DCM. The family was previously screened for coding mutations in 15 known DCM genes and was mutation negative.3–7 Exome sequencing was performed with NimbleGen solution-based capture and Illumina 76 bp read next-generation sequencing. Copy number variation was assessed with NimbleGen 2.1M catalog arrays, followed by NimbleGen 135K custom arrays, allowing detection of deletions and duplications at the resolution of a single exon. Our approach identified a deletion of exon 4 of BCL2-associated athanogene 3 (BAG3) (MIM 603883) as a rare variant causative of DCM. Furthermore, post hoc analyses of our genomic data suggest that both point mutations and rare copy number variants can be identified from a single genomic platform (Figures S3A and S3B, available online), further development of which will have a significant impact on the discovery of unknown genetic causes of cardiovascular disease.
Figure 1.
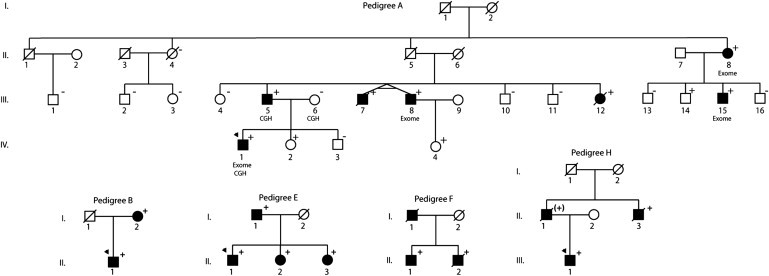
Pedigrees of BAG3-Associated Dilated Cardiomyopathy
Pedigrees have been labeled by letter, which correspond to their respective mutation as shown in Table 3. Squares represent males and circles represent females. An arrowhead denotes the proband. A diagonal line marks deceased individuals. Solid symbols denote dilated cardiomyopathy with other causes except genetic ruled out. Open symbols denoteunaffected individuals. The presence or absence of the pedigree's BAG3 mutation is indicated by a + or − symbol, respectively; obligate carriers are noted in parenthesis, (+). Individuals who underwent exome sequencing are denoted (exome), as are those who underwent comparative genomic hybridization (CGH).
Material and Methods
Subjects
Written, informed consent was obtained from all subjects, and the institutional review boards at the Oregon Health and Science University and the University of Miami approved the study. The investigation included 311 probands with DCM as previously described.5,6,8 The investigation also included a total of 355 population controls: 45% male, 97% white, and 3.0% African American. Genomic DNA was extracted from whole blood according to a standard salting-out procedure. Prior to the current study, we sequenced the coding and flanking intronic regions of 15 DCM genes in genomic DNA from these probands.3–7
Exome Sequencing
Three to five micrograms of DNA from four family members affected with DCM (pedigree A, Figure 1) were sent to the University of Washington Genome Sciences for exome capture and sequencing. These samples were sent as a batch, (a total of 20 samples across five DCM families). The remaining exome data are under investigation and will be published elsewhere. Sequence capture was performed in solution with a NimbleGen probe library that included a 26.4 Mb target sequence (152,917 exons from 16,515 genes). Exome sequencing was performed on the Illumina GAIIx platform with paired-end 76 base reads.
Fastq sequence files were aligned against the human reference sequence (hg18) with the Burrows-Wheeler Aligner (BWA).9 Duplicate paired-end reads were removed from the merged data sets. SNP and indel calling was performed with the GATK Unified Genotyper and annotated with SeattleSeq SNP annotation. To insure sample identity at each stage in the process, we genotyped all samples (Illumina BeadXpress) prior to exome sequencing for 96 high-allele-frequency, exome-specific polymorphisms and compared them to polymorphisms derived from both lane and merged exome data. Variants with either quality scores ≤ 50, allelic imbalance ≥ 0.75 (number reads a reference allele as a proportion of total number of reads), homopolymer runs > 3 of the variant allele, or read depth < 5 were filtered out. We further filtered out those variants with a heterozygous genotype in 20 out of 20 exomes from five DCM families (all sequenced in the same batch at the University of Washington) assuming these to be false positives.
We assumed an autosomal dominant model and prioritized nonsynonymous, splice site, and insertion/deletion variants that were heterozygous and shared across all four affected individuals; not present in dbSNP (build 131), 1000 Genomes Project, and also not present in a further eight exome sequences from HapMap individuals;10 and also not present in eight exome sequences from a pedigree with spastic paraplegia.11 The remaining variants were further prioritized by transcriptome data generated in our laboratory from left ventricular myocardium of two control samples and two unrelated DCM samples. We determined variants with reads per kilobase of exon model per million mapped reads (RPKM) > 3 in any of the four transcriptome data sets as being expressed in left ventricular myocardium. Of the remaining variants, we further prioritized those that were unique to pedigree A. Remaining variants that were also present in the exome sequences of four additional DCM families (16 additional exome sequences sequenced in the same batch at the University of Washington), and that did not segregate with disease in these families, were assumed to be polymorphisms.
Comparative Genomic Hybridization
Three micrograms of DNA from the proband of pedigree A were sent to NimbleGen to perform comparative genomic hybridization (CGH) on the NimbleGen 2.1M HD2 catalog microarray, median probe spacing 1.2 kb. Microarrays were scanned with the NimbleGen MS200 scanner at 2 μ resolution. We used three different algorithms to score copy number variants (CNVs): NimbleGen segMNT, Nexus copy number (BioDiscovery), and wuHMM.12 Copy number variants were viewed in the Nexus Copy Number software (version 4.1) and scored with a log2 ratio ±0.2. CNV regions called with any one of the three algorithms with less than 50% overlap with variants in the database of genomic variants13 were designed onto a custom NimbleGen 135K array with average probe spacing of ∼156 bp, including 3 kb flanking sequence and backbone probe spacing every ∼100 kb across each autosome. Validation of putative CNV's not present in the database of genomic variants was performed with custom array CGH on the proband and also in both parental DNA samples at the Florida State University core microarray facility following standard NimbleGen protocols. We prioritized the validated CNV's as those inherited from the affected parent and overlapping exons as putative disease causing.
Transcriptome Sequencing
RNA was extracted from frozen left ventricular myocardium of two normal and two DCM hearts with Trizol reagent and purified with QIAGEN RNeasy columns and treated with 20 U DNase with RNase inhibitor. RNA yield and purity was assessed with the RNA 6000 Nano Kit RNA and Agilent 2100 Bioanalyzer. RNAs with integrity number (RIN) > 7.8 were used for cDNA synthesis. Poly-A containing mRNA was isolated by poly-T oligo-attached magnetic beads. cDNA libraries were prepared with Illumina's mRNA-Seq Sample Prep kit according to the manufacturer's instructions. Briefly, mRNA was fragmented into small pieces (∼200 bp) with divalent cations in the fragmentation buffer at 94°C for 5 min. Cleaved RNA fragments were transcribed into first-strand cDNA with SuperScript II (Invitrogen) reverse transcriptase and random primers. This was followed by second-strand cDNA synthesis with DNA Polymerase I and RNase H. The designated adaptor oligos were ligated to the ends of the DNA fragments with T4 DNA ligase. Gel isolation was used to extract expected cDNA fragments. Limited PCR amplification (15 cycles) with two primers annealed to the ends of the adaptors was performed to enrich the purified cDNA templates. Paired-end sequencing was performed at the University of Miami Hussman Institute of Human Genomics on the GAII Illumina platform with mRNA-Seq Cluster Generation kits (76 cycles). Data collection, base calling, and preliminary data analysis were performed with standard Illumina software. Sequence alignment to human reference sequences and expression quantification in RPKM were performed with the NextGENe software package (SoftGenetics, State College, PA). RPKM is the number of reads that map per kb of exon model per million mapped reads for each gene.14 We used a RPKM > 3 as criteria for expression in the heart. Despite this low threshold, it is possible that some cardiac genes would not be identified by this criteria because of the small number of samples in the data set (n = 4).
Long-Range PCR across BAG3, ATE1, and DOCK1 Deletions
PCR assays were used to genotype putative disease causing deletions in the remaining family members to assess segregation with disease. We used the Phusion PCR amplification kit (Finnzymes, New England Biolabs) under the manufacturer's instructions at annealing temperatures of 68°C for BAG3 and DOCK1 (MIM 601403) and 70°C for ATE1 (MIM 607103). Assays were designed with forward and reverse primers set outside of the deleted regions and an additional forward primer inside of the deletion breakpoint to simultaneously amplify the wild-type fragment. Primer sequences are shown Table S1.
Sanger Sequencing of BAG3 Coding Region in 311 Probands and 180 Population Controls
Bidirectional Sanger sequencing of the BAG3 coding region (NM_004281.3), in 311 DCM probands was run on a 3130xl as previously published.3–7 We were unable to consistently amplify exon 1. Primer sequences are shown in Table S1. Exons 2, 3, and 4 were also sequenced in 180 population controls.
Screening for Missense, Nonsense, and Frameshift Mutations in an Additional 175 Population Controls
DNA pools of n = 7 or n = 8 population controls (14–16 chromosomes) were constructed. We used SNaPshot primer extension (Applied Biosystems) to test for the presence of BAG3 missense, nonsense, and frameshift mutations in population controls from 22 DNA pools. Primer extension reactions were run on a 3130xl DNA sequencer (Applied Biosystems). Prior to pool construction, each DNA sample was placed on a rotator in a cold room for one week to ensure DNA homogeneity, then PCR amplified by long-range PCR to test for sample quality and amplification. Subdilutions of approximately 30 ng/μl were made for each sample based on the initial reading by spectrophotometer. Each subdilution was then quantified four times by nanodrop. All samples were quantified on the same day. On the basis of the average reading, a total of 600 ng of each DNA sample was pooled. The final DNA pools were diluted to a concentration of 20 ng/μl. Validation of the DNA pools is described in Supplemental Data.
Zebrafish Bag3 Knockdown
Zebrafish Lines
Wild-type TüAB zebrafish were maintained and raised as described previously.15 Dechorionated embryos were kept at 28.5°C in E3 solution with or without 0.003% 1-phenyl-2-thiourea (PTU; Sigma) to suppress pigmentation and were staged according to somite number (som) or hours postfertilization (hpf).15
Morpholino Antisense Oligonucleotide Injections
To study the effects of altered expression of bag3 in zebrafish, two morpholino oligonucleotides directed against the translation start codon (5′-CAGAGGATCGTTAGTTGCCATCGTT) and splice donor sites of exon 2 (5′-GCTTTCTCATGATCTTACCTCAGGC) were purchased from Gene Tools (Philomath, OR) and injected into one- to two-cell-stage zebrafish embryos as previously described.16 The cardiac morphology and function as well as the general development of fish were evaluated at 24, 48, and 72 hpf.
PCR primers were designed in flanking exon sequences to evaluate the efficacy of the bag3 exon2 splice-blocking morpholino oligonucleotide and to characterize mRNA splicing products in cDNA isolated from both wild-type and morphant embryos. The following nested PCR primer sets were designed to amplify both wild-type and morphant mRNA fragments: outF-5′-ATCAAAATTGACCCGCAGAC-3′, outR-5′-CCCGGAGACCTGTATTGCT-3′ and inF-5′-ATCACAACAATCGCACAACG-3′, inR-5′-GGTGAAGGCCACTGATTTGT-3′. RT-PCR analysis of wild-type embryos produced an amplification product of 408 bp, whereas two products of 408 and 99 bp were generated from the bag3 exon2 morpholino-treated embyros showing the alternate splicing of exon 2 (Figure S2). These RT-PCR amplicons were gel extracted and sequenced with standard Sanger sequencing protocols. The 408 bp fragment from both wild-type and morphant embyros contained exon 2 sequence, whereas the 99 bp fragment contained sequence from only exon 1 and exon 3. The resulting deletion of exon 2 yielded an in-frame sequence for the remainder of the protein.
Zebrafish Image Analysis and Cardiac Measures
Approximately 1200 frames were recorded for each ventricular measurement of fractional shortening (FS) and peak flow velocity. FS movies were recorded at 120 frames per s and peak flow velocities were recorded at 240 frames per s with a Grasshopper high-speed video camera (Point Grey Research, Vancouver). All measurements were performed with ImageJ software. FS was determined as the ventricle area (in pixels) at diastole minus the area at systole as a proportion of the area at diastole × 100. FS was calculated for three heart cycles in each fish. Peak flow measurements were taken by mapping the coordinates of a single blood cell in ImageJ every eight frames (equivalent to every 0.033 s). A correction factor to convert pixels to distance was determined as per Shin et al.,17 allowing the velocity to be calculated as distance divided by time. We measured the velocity of three blood cells (each in a different heart cycle) per fish in a total of six wild-type and six ATG morphant embryos. Significance was determined by an unpaired t test with unequal variance.
Results
Exome-Sequencing Quality Control
Exome data were generated on the Illumina GAIIx with paired-end 76 base reads and multilane data merged. We generated an average of 75 M total reads, with 55 M mapping to the exome target for analysis. All duplicate read pairs were removed. All samples had a 104-fold average coverage [range 52–172] and an average of 86% of the exome target region at >20-fold coverage. Concordance with presequencing fingerprint variant set was >99% across all samples. On average 16,407 variants positions were identified across these samples.
We previously sequenced our DCM cohort in 15 known DCM genes with bidirectional Sanger sequencing.3–7 Exome data were generated for five unrelated individuals (including the proband from pedigree A) with Sanger sequencing data available, and of the 15 genes screened by Sanger sequencing, 11 were also present on the exome capture design and within these genes. Notably, 35 out of 35 heterozygous coding variants identified by Sanger sequencing in these 11 genes were also identified in the exome-sequence data, suggesting both an extremely low false negative rate and high genotyping accuracy by the GATK software.
Exome Sequencing Did Not Identify Genetic Cause in a Multiplex DCM Family
The number of shared variants observed with exome sequencing in four affected pedigree A individuals are shown at each filtering stage (Table 1). At the final filtering stage, a single missense variant (c.626G>A [p.Arg209His]), position chr14: 75,243,154 (hg18) in tubulin tyrosine ligase-like family, member 5, (TTLL5, NM_015072.4), (MIM 612268) remained. Genotyping in the extended family showed that it was absent from 1 of the 7 affected members and was also present in 4 of 12 unaffected family members, suggesting it was unlikely to be relevant for DCM.
Table 1.
Heterozygous Variants Identified by Exome Sequencing and Shared between Four Affected Individuals in pedigree A
| Pedigree A: Exome Variants (Single Base and Small Insertion Deletions) | |
|---|---|
| Missense, nonsense, splice, indel | 479 |
| Not present in dbSNP build 131 or 1000 Genomes Project | 13 |
| Expressed in heart | TTLL5, TUBGCP6, FRG1, C1orf163 |
| Unique to DCM pedigree A | TTLL5 |
In addition to TTLL5, a second variant passing all filtering criteria with the exception that it was not unique to pedigree A was found in the gene TUBGCP6, (MIM 610053). This missense variant was also present in one of two affected exome data sets from a second DCM family. We screened additional DNA samples in pedigree A for the TUBGCP6 variant and found this to be nonsegregating. The variant was also present in 3 out of 88 population controls and was thus considered a polymorphism. Two missense variants in the genes FRG1 (MIM 601278) and C1orf163, expressed in the heart, were present in all four exome sequences from pedigree A. These variants were present in a total of 19 out of 20 of our affected DCM exome data sets and were not further considered.
High-Density Genome-wide Assessment of Copy Number Variation
Genome-wide copy number analysis with NimbleGen 2.1M CGH arrays on the proband in pedigree A is summarized in Table 2. A total of three CNVs were inherited from the affected father and spanned exons and genes expressed in heart: BAG3 (8.7 kb), ATE1 (15.6 kb), and DOCK1 (67.1 kb). Deletions in BAG3 and ATE1 spanned the last exon of each gene with breakpoints in the final intron and 3′ flanking regions. We designed PCR assays across the BAG3, ATE1, and DOCK1 deleted regions, including a third primer inside of the deletion, and we genotyped all available DNA samples in pedigree A. All seven of the affected members were heterozygous for the BAG3 deletion (Figure 1), which was not present in 355 population control DNA specimens. The ATE1 deletion was also heterozygous but was absent from two of seven affected members. Both deletions map to within 2 Mb of each other on chromosome 10q26, suggesting recombination between them. The DOCK1 deletion was present only in the proband and father.
Table 2.
Iterative Approach Identifying CNV with Comparative Genomic Hybridization in Pedigree A
| Pedigree A: CNV Variants | No. CNVs | Loss | Mean Sizea | Median Sizea | Gain | Mean Sizea | Median Sizea |
|---|---|---|---|---|---|---|---|
| Total (2.1M array) | 440 | 180 | 48,074 | 13,573 | 260 | 62,662 | 12,868 |
| <50% overlap with DGV | 81 | 21 | 14,433 | 5,699 | 60 | 12,864 | 9958 |
| Designed onto 135K custom array | 41 | 7 | 23,841 | 13,223 | 34 | 16,395 | 9252 |
| Validated by custom array | 9 | 6 | 24,017 | 11,930 | 3 | 10,835 | 8264 |
| Inherited from affected father | 6 | 5 | 27,517 | 15,004 | 1 | 8264 | 8264 |
| Spanning exons | 5 | 4 | 32,182 | 26,511 | 1 | 8264 | 8264 |
| Expressed in heart | BAG3, ATE1, DOCK1 | 3 | 30,312 | 15,615 | 0 | NA | NA |
| Segregation with DCM | BAG3 | 1 | 8,733 |
DGV is an abbreviation for database of genomic variants and NA is an abbreviation for not applicable.
CNV sizes are given in base pairs.
Identification of BAG3 Point Mutations in Additional DCM Families
BAG3 is highly expressed in the heart, as shown by both transcriptome data from our laboratory and the literature.18 A point mutation in BAG3 is already known to cause fulminant skeletal myopathy and early lethality in knockout mice18 and myofibrillar myopathies (MIM 612954) with restrictive (RCM [MIM 115210]) or hypertrophic (HCM [MIM 192600]) cardiomyopathy in humans.19,20 To seek further evidence that mutations in BAG3 cause DCM, we resequenced exons 2, 3, and 4 in 311 additional DCM probands previously resequenced for 15 other genes.3–7 We identified point mutations in seven unrelated probands, including two premature stop codons and one frameshift (Table 3, Figure 2) that were absent in 355 in-house population controls and over 967 exomes sequenced at Seattle Seq (a total of 2644 chromosomes). One stop codon and a frameshift mutation were identified in sporadic cases, suggesting, as with the myofibrillar mutations observed in this gene,20 that they may result from de novo mutations or from germ-line mosaicism in a parent. The remaining mutations were observed in familial cases. For four mutations (c.211C>T [p.Arg71Trp], c.367C>T [p.Arg123stop], c.784G>A [p.Ala262Thr], and c.1430G>A [p.Arg477His]), DNA was available from additional affected family members, and all typed positive for these mutations (Figure 1, Table 4). A single missense variant, p.Val298Met showing very low conservation scores, was identified in the control population alone (Table 3).
Table 3.
Variants Identified by Sequencing BAG3
| Exon | UCSC hg18 | Nucleotide Change | Amino Acid Change | Phastcon Score | GERP Score | Ped ID | Present in Controls |
|---|---|---|---|---|---|---|---|
| 2 | chr10:121419383 | c.211C>T | p.Arg71Trp | 0.067 | 2.50 | B | no |
| 2 | chr10:121419440 | c.268C>T | p.Arg90stop | 0.996 | 5.05 | C | no |
| 2 | chr10:121419498 | c.326A>G | p.His109Arg | 0.585 | 4.23 | D | no |
| 2 | chr10:121419539 | c.367C>T | p.Arg123stop | 0.479 | 2.36 | E | no |
| 3 | chr10:121421901 | c.652 delC | p.Arg218GlyfsX89 | NA | NA | G | no |
| 3 | chr10:121422033 | c.784G>A | p.Ala262Thr | 0 | 2.56 | F | no |
| 4 | chr10:121426486 | c.1430G>A | p.Arg477His | 1 | 5.46 | H | no |
| 2 | chr10:121419452 | c.280A>T | p.Ile94Phe | 1 | 5.05 | - | yes (2) |
| 2 | chr10:121419602 | c.430A>G | p.Thr144Ala | 0.001 | 1.14 | - | yes (1) |
| 3 | chr10:121421902 | c.653G>A | p.Arg218Gln | 0.168 | 4.93 | - | yes (1) |
| 3 | chr10:121422141 | c.892G>A | p.Val298Met | 0.001 | 0.08 | - | yes (1) |
Conservation measured by Phastcons and GERP scores. Nucleotide positions based on reference sequence NM_004281.3. NA is an abbreviation for not applicable.
Figure 2.
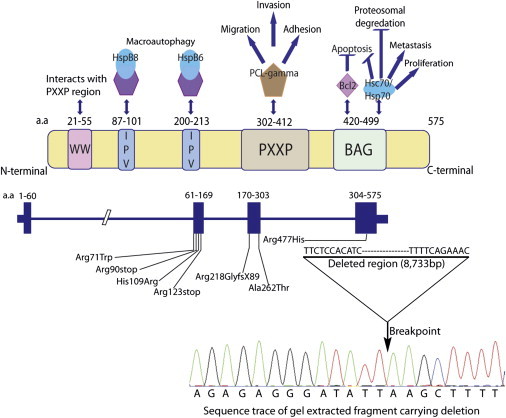
Summary of BAG3 Function
Adapted from McCollum et al.22 and annotated with mutations identified in DCM families.
Table 4.
Clinical Characteristics
| Subject | Age at Diagnosis or Screening | Gender | DCM | ECG/Arrhythmia | LVEDD (mm), Z Score | Ejection Fraction | LV Septum/ Posterior Wall Thickness (mm) | Mutation Status (Yes, No, Unknown) | Comment |
|---|---|---|---|---|---|---|---|---|---|
| Pedigree A: 8.7 kb Deletion | |||||||||
| II.4 | 75 | F | no | 40, NA | 60 | 8/11 | no | ||
| II.8 | 64 | F | yes | NSSTT, Q's V1-V2 |
55, 2.91 | 15 | 8/14 | yes | coronary angiogram negative |
| III.1 | 51 | M | no | 39, −3.24 |
55 | 9/11 | no | ||
| III.2 | 62 | M | no | 54.6, −1.14 |
63 | 11/12 | no | ||
| III.3 | 60 | F | no | 41.5, −1.21 |
50 | 11/8 | no | ||
| III.4 | 57 | F | no | sinus bradycardia; minimal voltage criteria for LVH | 51.2, 1.89 | 63 | 11/8 | no | |
| III.5 | 48 | M | yes | RBBB | 64, 3.61 | 30 | 9/11 | yes | |
| III.7 | 44 | M | yes | Q's V1–V3, LAE | NA | 15 | NA | yes | echocardiogram: LV severely dilated with global hypokinesis, coronary angiogram negative; died of advanced heart failure at age 44 |
| III.8 | 54 | M | yes | Q's III, F | 68, 4.34 | 33 | yes | no known coronary disease | |
| III.10 | 52 | M | no | 50, −0.24 |
68 | no | |||
| III.11 | 39 | M | no | 52, 0.47 | 60 | no | |||
| III.12 | 35 | F | yes | LVH | 77, 8.15 | 17 | 6/9 | yes | died of advanced heart failure at age 38 |
| III.13 | 58 | M | no | 46, −1.39 |
58 | no | |||
| III.14 | 57 | M | no | 46, −1.42 |
58 | 11/13 | yes | ||
| III.15 | 54 | M | yes | NSSTT | 60, 2.30 | 48 | 14/13 | yes | |
| III.16 | 47 | M | no | NSR | 39, −3.80 |
68 | no | ||
| IV.1 | 23 | M | yes | Q's V1-V2, PVCs, LAD | 75, 6.18 | 7 | 8/7 | yes | heart transplant, age 23; no coronary disease |
| IV.2 | 22 | F | no | 50, 1.32 | 60 | NA | yes | ||
| IV.3 | 32 | M | no | LVH | 54,1.13 | 60 | 11/7 | no | |
| IV.4 | 24 | F | no | low voltage QRS | 50, 1.09 | 58 | 9/9 | yes | |
| Pedigree B: p.Arg71Trp | |||||||||
| I.2 | 59 | F | yes | Q's V1-V2, PVCs | NA | 20 | NA | yes | heart transplant, age 65; coronary angiogram negative |
| II.1 | 41 | M | yes | iLBBB, AF, LAD, RVR | 71.2, 4.46 | 24 | 7/9 | yes | coronary angiogram negative |
| Pedigree C: p.Arg90Stop | |||||||||
| I.1 | 44 | M | yes | Q's III, F, V1; LAE | 70, 4.98 | 24 | 8/8 | yes | no coronary disease, idiopathic dilated cardiomyopathy by medical records |
| Pedigree D: p.His109Arg | |||||||||
| I.1 | 21 | M | yes | nonspecific IVCD | 79, 6.32 | 25 | 7/7 | yes | no coronary disease, idiopathic dilated cardiomyopathy by medical records |
| Pedigree E: p.Arg123Stop | |||||||||
| I.1 | 36 | M | unknown | NA | NA | NA | NA | yes | heart transplant, age 40; medical records unavailable to categorize cardiomyopathy |
| II.1 | 25 | M | yes | NA | 74, 5.14 | 25 | 8/8 | yes | ventricular assist device, heart transplant, age 26 |
| II.2 | 34 | F | Systolic dysfunction | NA | 49.5, 1.31 | 48.3 | 10/9 | yes | asymptomatic with mild LV dysfunction; coronary angiogram negative |
| II.3 | 31 | F | Yes | NA | 49.5, 1.44 | 37.5 | 9/9 | yes | asymptomatic |
| Pedigree F: p.Ala262Thr | |||||||||
| I.1 | 65 | M | Yes | NA | NA | NA | NA | unknown | DCM, no coronary disease by medical records |
| II.1 | 42 | M | Yes | 1AVB, Q's V1-V3, LAE | NA (severely dilated) | 15 | NA | yes | ICD, heart transplant, age 43; coronary angiogram negative |
| II.2 | 44 | M | Yes | 1AVB, Q's V1-V2, LAFB, LAD | 68, NA | 28 | NA | yes | heart transplant, age 45 |
| Pedigree G: p.Arg218GlyFsX89 | |||||||||
| 1.1 | 47 | M | Yes | NSSTT | 60, 2.90 | 25 | NA | yes | died, age 54; coronary angiogram negative |
| Pedigree H: p.Arg477His | |||||||||
| II.3 | 47 | M | Yes | NSCD, NSSTT | 80, 6.52 | 15 | 9/9 | yes | heart transplant, age 57; coronary angiogram negative |
| III.1 | 50 | M | Yes | iBBB, LPFB, NSSTT | 76, 5.88 | 5 | NA | yes | ventricular assist device, heart transplant, 50 years; coronary angiogram negative |
The following abbreviations are used: 1AVB, first-degree atrioventricular block; AF, atrial fibrillation; DCM, dilated cardiomyopathy; iBBB, incomplete bundle branch block; ICD, implantable cardiac defibrillator; iLBBB, incomplete left bundle branch block; IVCD, interventricular conduction delay; LAD, left axis deviation; LAE, left atrial enlargement; LAFB, left anterior fasicular block; LV, left ventricle; LVEDD, left ventricular end-diastolic dimension; LVH, left ventricular hypertrophy; LPFB, left posterior fasicular block; MI, myocardial infarction pattern; NA, not available; NSCD, nonspecific conduction delay; NSR, normal sinus rhythm; NSSTT, nonspecific ST-T changes; PVC, premature ventricular contraction; RBBB, right bundle branch block; RVH, right ventricular hypertrophy; RVR, rapid ventricular response.
The age at onset or diagnosis of the DCM phenotype for 17 subjects harboring one of the eight BAG3 rare variants (Table 4) ranged from 21 to 64 years (median = 44). Disease severity varied considerably. Although 8 of the 17 mutation carriers with DCM underwent heart transplant or died with advanced heart failure, a few mutation carriers had minimal or no disease, including three deletion carriers in pedigree A, ages 22, 24, and 57 years who showed lack of pentrance. Such phenotypic variability, including that related to age-dependent penetrance, has been widely seen in pedigrees with many different mutations in genes causing DCM.1,2
bag3 Knockdown in a Zebrafish Model
Because a point mutation in BAG3 had been previously associated with HCM and RCM, we sought to develop a model of DCM and heart failure. To accomplish this, we employed a knockdown approach by using morpholino oligonucleotides in the zebrafish. Two different morpholinos were designed to target zebrafish bag3, one blocking bag3 translation initiation and one blocking the splicing donor site at exon 2. The translation initiation-blocking morpholino specifically introduced heart failure phenotypes with pericardial effusion and decreased fractional shortening by 14%, p = 0.003, in measurements from five wild-type fish and six morphants, (Figure 3D), (see Movies S1 and S2). Peak blood cell flow velocity was also significantly decreased, p = 0.046, with a mean maximum velocity of 1287 μM per second in wild-type fish (n = 5) compared to 1045 μM per second in morphants (n = 6) (Figure 3E). The second morpholino introduced a dose-dependent phenotype of both heart failure (Figure 3C and 3D) and axis curvature (Figure S4) with pericardial effusion and significantly reduced fractional shortening, p = 0.0001.
Figure 3.
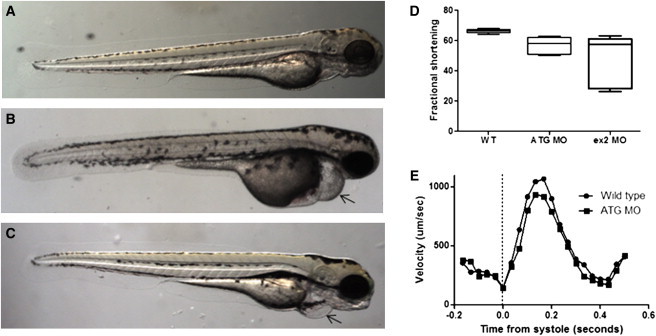
Morpholino Knockdown of bag3 in Zebrafish Causes Cardiac Phenotypes
Whole-mount brightfield images of 72hpf zebrafish embros. (A) wild-type, (B) bag3ATG morpholino-injected, (C) exon 2 splice donor-blocking morpholino-injected embryos. Measurement of cardiac performance comparing wild-type and morphant embryos for (D) fractional shortening and (E) peak flow velocity averaged across all data points from onset of systole. ATG morpholino injected at 0.5 mM and exon 2 morpholino injected at 0.3 mM. Arrows mark pericardial effusion. The following abbreviations are used: WT, wild-type; MO, morpholino; ATG, translation initiation blocking. Bars in (D) correspond to minimum and maximum values. Boundaries of each box correspond to 25th and 75th percentiles and horizontal lines within boxes correspond to the median value.
Discussion
Collectively, the genetic and pedigree data, showing that every subject with DCM carried one of the eight BAG3 rare variants, and the zebrafish heart failure data, recapitulating DCM phenotype, provide compelling evidence for a relevant cause of human adult-onset DCM. Further indication of BAG3 relevance to DCM is demonstrated by the observed excess of rare variants in the DCM cohort relative to our control sample (eight rare variants in cases including two nonsense and one frameshift compared to three missense variants in both cases and controls and one missense in the control population only). Given the often late onset and phenotypic heterogeneity of DCM, it is possible that some of our control group could go on to develop DCM or could be asymptomatic carriers of rare variants. If conservation scores are reliable in disease prediction, the c.280A>T (p.Ile94Phe) variant could also be disease causing, as both Phastcon and GERP scores are high (1 and 5.05, respectively). All other variants observed in controls show relatively low conservation compared to variants identified in DCM cases only, suggesting they are likely polymorphisms of low frequency.
Childhood-onset muscular dystrophy, termed myofibrillar myopathy, is dramatically different from the human DCM; it is caused by a BAG3 point mutation (c.626C>T [p.Pro209Leu]) accompanied in all cases with RCM or HCM. Three cases from one Italian and two British families19 and four cases in three US families20 showed onset ranging from 5 to 13 years old, with involvement of skeletal, respiratory, and cardiac muscle. Of the seven cases, three underwent cardiac transplantation (ages 13, 13, and 14) and two suffered sudden death (ages 9 and 20).19,20 Allelic heterogeneity common to genes disrupted in DCM has occasionally been associated with muscular dystrophy,1 most notably with mutations in LMNA (MIM 150330).21 However, the striking phenotypic difference between the childhood RCM or HCM with myofibrillar myopathy associated with BAG3 p.Pro209Leu and the adult-onset DCM observed in association with the eight BAG3 rare variants in this report suggests fundamental differences in mechanisms of disease.
BAG3, a member of the Bcl-2-associated athanogene family, encodes a 535 amino acid protein that has been classified as a cochaperone of the heat shock proteins (HSPs). The HSPs function as molecular chaperones, assisting with folding, stabilization, trafficking, and degradation of cellular proteins, and their disruption can lead to misfolded proteins and diseases that stem from abnormal protein aggregation,22 including expansion-repeat neurodegenerative disease proteins.23 Cochaperones such as BAG3 bind to chaperone proteins, modulating their function and providing specificity of protein binding or targeting to a cellular compartment. Proteins containing the BAG motif are thought to function primarily in protein degradation through both the proteasome and autophagy pathways.22 In addition to the BAG domain that binds to Bcl-2 and the heat shock proteins Hsp70 and Hsc70, BAG3 has several other motifs, including Ile-Pro-Val (IPV) domains that regulate oligomerization.22 The p.Pro209Leu BAG3 point mutation, modifying the proline in the IPV domain (Figure 2) and recently shown to interact with HspB8 and HspB6,23 is thought to provide the specificity of the childhood-onset phenotype of skeletal and cardiac myopathy.22,23
Although BAG3 or its binding partners have not yet been crystallized, extensive prior work with BAG3 biology suggests that a mutant BAG3 protein could disrupt binding to several key proteins to cause disease. One is Bcl-2, a protein that protects cells from apoptotic death. The amount of Bcl-2 has been shown to affect DCM course and survival in a murine model generated from a mutant small HSP, HspB5 (CRYAB [MIM 123590], also known as αB-crystallin).24 αB-crystallin-associated DCM in humans has also been reported25 and is considered part of the desmin-related myopathies (DRM [MIM 601419]).24 Both HspB5 and BAG3 colocalize to the Z disk,18,26 a well established DCM gene ontogeny including CSRP3, MYPN (MIM 600824, 608517) and others.1,2 Alternatively, it has been proposed that BAG3 and Hsc70 interaction is needed to preserve cardiac myofibrillar integrity under contractile stress,26 which could also be relevant for the development of adult-onset DCM. BAG3 has also been shown to modulate protein degradation via the proteasomal or autophagic pathways with aging,27 which might also contribute to the age of onset and severity of BAG3-mediated DCM.
The observed phenotypic variability in the BAG3 mutation spectrum may also be reflected in our zebrafish model. Translation initiation-blocking of the whole gene gave a single phenotype of heart failure. In contrast, a second morpholino that specifically splices out exon 2 resulted in axis curvature and may be analogous to skeletal myopathy (Figure S4).
Our study did not include functional assays to determine loss or gain of function for specific mutations. The large deletion observed in pedigree A probably results in a loss of function or haploinsufficiency, but specific disease mechanisms for the point mutations are uncertain. In the case of the nonsense and frameshift mutations, nonsense-mediated decay could lead to a loss of function, or alternatively, the presence of a truncated protein could lead to a gain of function with a more severe phenotype. Given that DCM is a late onset disease and significantly less severe than the childhood-onset myofibrillar myopathy, one could predict that the DCM mutations are the result of a loss of function. Further experimental work will be needed to determine the contribution of these BAG3 mutations to disease pathology, which may open a new window to disease treatment.
Acknowledgments
We thank all the family members who participated, without whom this study would not be a success. This work was supported by National Institutes of Health awards HL58626 (R.E.H.) and HL094976 (D.A.N., SeattleSeq), and the Florida Heart Research Institute (N.N.). We also thank and recognize the following ongoing studies that produced and provided exome variant calls for filtering purposes: National Heart Lung and Blood Institute Lung Cohort Sequencing Project (HL 1029230), NHLBI Women's Health Initiative Sequencing Project (HL 102924), and the Northwest Genomics Center (HL 102926).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes database, http://www.1000genomes.org/page.php
Database of Genomic Variants, http://projects.tcag.ca/variation/
dbSNP homepage, http://www.ncbi.nlm.nih.gov/SNP/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
SeattleSeq Annotation, http://gvs.gs.washington.edu/SeattleSeqAnnotation/
University of California Santa Cruz Human Genome Browser, http://genome.cse.ucsc.edu/cgi-bin/hgGateway
References
- 1.Hershberger R.E., Cowan J., Morales A., Siegfried J.D. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2:253–261. doi: 10.1161/CIRCHEARTFAILURE.108.817346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershberger R.E., Morales A., Siegfried J.D. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet. Med. 2010;12:655–667. doi: 10.1097/GIM.0b013e3181f2481f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li D., Parks S.B., Kushner J.D., Nauman D., Burgess D., Ludwigsen S., Partain J., Nixon R.R., Allen C.N., Irwin R.P. Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am. J. Hum. Genet. 2006;79:1030–1039. doi: 10.1086/509900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parks S.B., Kushner J.D., Nauman D., Burgess D., Ludwigsen S., Peterson A., Li D., Jakobs P., Litt M., Porter C.B. Lamin A/C mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am. Heart J. 2008;156:161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hershberger R.E., Parks S.B., Kushner J.D., Li D., Ludwigsen S., Jakobs P., Nauman D., Burgess D., Partain J., Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. doi: 10.1111/j.1752-8062.2008.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershberger R.E., Norton N., Morales A., Li D., Siegfried J.D., Gonzalez-Quintana J. Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ Cardiovasc Genet. 2010;3:155–161. doi: 10.1161/CIRCGENETICS.109.912345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D., Morales A., Gonzalez-Quintana J., Norton N., Siegfried J.D., Hofmeyer M., Hershberger R.E. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kushner J.D., Nauman D., Burgess D., Ludwigsen S., Parks S.B., Pantely G., Burkett E.L., Hershberger R.E. Clinical characteristics of 304 kindreds evaluated for familial dilated cardiomyopathy. J. Card. Fail. 2006;12:422–429. doi: 10.1016/j.cardfail.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hedges D.J., Burges D., Powell E., Almonte C., Huang J., Young S., Boese B., Schmidt M., Pericak-Vance M.A., Martin E. Exome sequencing of a multigenerational human pedigree. PLoS ONE. 2009;4:e8232. doi: 10.1371/journal.pone.0008232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahan P., Godfrey L.E., Eis P.S., Richmond T.A., Selzer R.R., Brent M., McLeod H.L., Ley T.J., Graubert T.A. wuHMM: a robust algorithm to detect DNA copy number variation using long oligonucleotide microarray data. Nucleic Acids Res. 2008;36:e41. doi: 10.1093/nar/gkn110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 14.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 15.Westerfield M. University of Oregon Press; Eugene, OR: 1995. The zebrafish book. [Google Scholar]
- 16.Mangos S., Lam P.Y., Zhao A., Liu Y., Mudumana S., Vasilyev A., Liu A., Drummond I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis Model Mech. 2010;3:354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shin J.T., Pomerantsev E.V., Mably J.D., MacRae C.A. High-resolution cardiovascular function confirms functional orthology of myocardial contractility pathways in zebrafish. Physiol. Genomics. 2010;42:300–309. doi: 10.1152/physiolgenomics.00206.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Homma S., Iwasaki M., Shelton G.D., Engvall E., Reed J.C., Takayama S. BAG3 deficiency results in fulminant myopathy and early lethality. Am. J. Pathol. 2006;169:761–773. doi: 10.2353/ajpath.2006.060250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Selcen D., Muntoni F., Burton B.K., Pegoraro E., Sewry C., Bite A.V., Engel A.G. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann. Neurol. 2009;65:83–89. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Odgerel Z., Sarkozy A., Lee H.S., McKenna C., Rankin J., Straub V., Lochmüller H., Paola F., D'Amico A., Bertini E. Inheritance patterns and phenotypic features of myofibrillar myopathy associated with a BAG3 mutation. Neuromuscul. Disord. 2010;20:438–442. doi: 10.1016/j.nmd.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Worman H.J., Courvalin J.C. How do mutations in lamins A and C cause disease? J. Clin. Invest. 2004;113:349–351. doi: 10.1172/JCI20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCollum A.K., Casagrande G., Kohn E.C. Caught in the middle: the role of Bag3 in disease. Biochem. J. 2010;425:e1–e3. doi: 10.1042/BJ20091739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuchs M., Poirier D.J., Seguin S.J., Lambert H., Carra S., Charette S.J., Landry J. Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction. Biochem. J. 2010;425:245–255. doi: 10.1042/BJ20090907. [DOI] [PubMed] [Google Scholar]
- 24.Maloyan A., Sayegh J., Osinska H., Chua B.H., Robbins J. Manipulation of death pathways in desmin-related cardiomyopathy. Circ. Res. 2010;106:1524–1532. doi: 10.1161/CIRCRESAHA.109.212639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inagaki N., Hayashi T., Arimura T., Koga Y., Takahashi M., Shibata H., Teraoka K., Chikamori T., Yamashina A., Kimura A. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 26.Hishiya A., Kitazawa T., Takayama S. BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ. Res. 2010;107:1220–1231. doi: 10.1161/CIRCRESAHA.110.225649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gamerdinger M., Hajieva P., Kaya A.M., Wolfrum U., Hartl F.U., Behl C. Protein quality control during aging involves recruitment of the macroautophagy pathway by BAG3. EMBO J. 2009;28:889–901. doi: 10.1038/emboj.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.