

Abstract
An increasing number of couples require medical assistance to achieve a pregnancy, and more than 2% of the births in Western countries now result from assisted reproductive technologies. To identify genetic variants responsible for male infertility, we performed a whole-genome SNP scan on patients presenting with total globozoospermia, a primary infertility phenotype characterized by the presence of 100% round acrosomeless spermatozoa in the ejaculate. This strategy allowed us to identify in most patients (15/20) a 200 kb homozygous deletion encompassing only DPY19L2, which is highly expressed in the testis. Although there was no known function for DPY19L2 in humans, previous work indicated that its ortholog in C. elegans is involved in cell polarity. In man, the DPY19L2 region has been described as a copy-number variant (CNV) found to be duplicated and heterozygously deleted in healthy individuals. We show here that the breakpoints of the deletions are located on a highly homologous 28 kb low copy repeat (LCR) sequence present on each side of DPY19L2, indicating that the identified deletions were probably produced by nonallelic homologous recombination (NAHR) between these two regions. We demonstrate that patients with globozoospermia have a homozygous deletion of DPY19L2, thus indicating that DPY19L2 is necessary in men for sperm head elongation and acrosome formation. A molecular diagnosis can now be proposed to affected men; the presence of the deletion confirms the diagnosis of globozoospermia and assigns a poor prognosis for the success of in vitro fertilization.
Main Text
The increasing incidence of infertile couples, potentially caused by a general deterioration of sperm parameters, is becoming a major concern worldwide.1 Although environmental or infectious causes play an important role in infertility, genetic defects are also believed to be frequently involved in the pathological process.2 Several hundred genes are believed to be involved in spermatogenesis, yet very few have so far been directly connected with male infertility in human. A better understanding of gametogenesis through the identification of genes involved in infertility can help to decrease this worrying trend. We investigated patients with globozoospermia (MIM 102530) to identify genes involved in this infertility syndrome. Globozoospermia is a rare phenotype of primary male infertility characterized by the production of a majority of round-headed spermatozoa without acrosome (Figure 1, for review see 3). The phenotype was described over 30 years ago, and familial cases pointed to a genetic component for this defect.4–7 Three brothers affected with total globozoospermia were recently analyzed and found to carry a homozygous mutation of SPATA16 (MIM 609856).8 The testicular expression of SPATA16 and its intracellular localization in the acrosome-building Golgi vesicles in the spermatids correlated well with the observed phenotype.9,10 However, no SPATA16 mutations were detected in 29 other affected men, thus suggesting that SPATA16 was not the main locus associated with globozoospermia.8 We recently demonstrated that the strategy of using whole-genome homozygosity mapping applied to infertile patients from the same ethno-geographical background presenting with a specific morphologic anomaly of the sperm could lead to the localization and identification of genes involved in spermatogenesis.11 We demonstrated that AURKC (MIM 603495) deficiency led to male infertility due to the production of large-headed, multiflagellar, polyploid spermatozoa (MIM 24306).12,13 Here, we applied the same genetic strategy to a cohort of mainly Tunisian patients presenting with total globozoospermia, and we were able to demonstrate that 15 of the 20 patients that we analyzed had a homozygous deletion encompassing only DPY19L2, therefore indicating that its absence induces the investigated phenotype. This work was previously presented orally,14 and a patent describing processes for the diagnosis of globozoospermia and the use of DPY19L2 inhibitors to achieve male contraception has been filed.15
Figure 1.
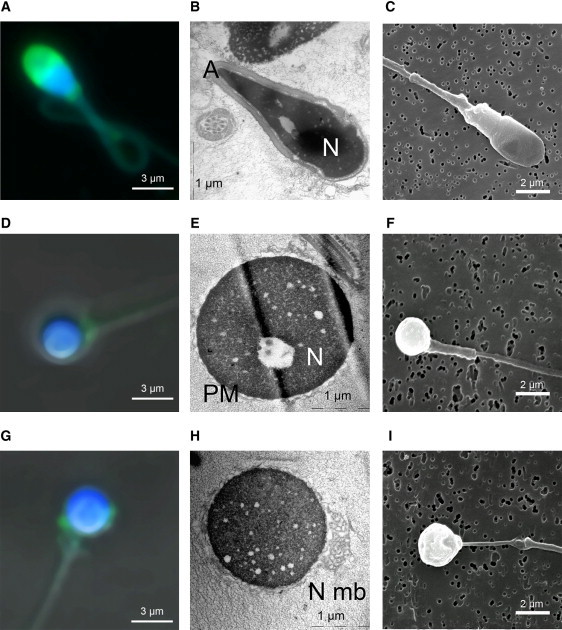
Sperm Head Is Round and the Acrosome Is Absent or Atrophied and Misplaced in Globozoocephalic Spermatozoa
Panels (A–C) show control spermatozoa, (D–F) show globozoocephalic spermatozoa with no acrosome, and (G–I) show globozoocephalic spermatozoa with atrophied and misplaced acrosome.
(A, D, G) Confocal microscopy. Sperm were double stained with TopRo3 (blue), for evidencing the nucleus, and with lectins from Pisum sativum conjugated to fluorescein isothiocyanate (PSA-FITC, Sigma Aldrich, France) (green), for evidencing the acrosome. In control sperm, the acrosome is displayed as a bell surrounding the tip of the sperm head (A). In globozoocephalic sperm, the acrosome is absent (D) or atrophied and misplaced (G). Lectin from PSA-FITC was used to label the acrosomal matrix, and TopRo3 was used to label the nucleus. Slide cells were washed in PBS and fixed in 4% PFA for 30 min on ice. Fixed spermatozoa were washed in PBS for 3–5 min and incubated with PSA-FITC (10 μg/ml in PBS). Slides were analyzed on a Leica TCS-SP2 (Mannheim, France) confocal laser scanning microscope.
(B, E, H) Electronic microscopy. The different organites, acrosome (A), and nucleus (N) of control sperm are clearly identified (B). In globozoocephalic sperm, the acrosome is absent and the plasma membrane (PM) surrounds the nucleus (E). In some cells, redundant nuclear membrane (N Mb) is clearly evidenced (H). Sperm cells were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer of pH 7.4 for 2 hr at room temperature. Cells were washed with buffer and postfixed with 1% osmium tetroxyde in the same buffer for 1 hr at 4°C. Ultrathin sections of the cell pellet were cut with an ultramicrotome (Leica, Nanterre, France) after dehydration and epon inclusion. Sections were poststained with 4% uranile acetate and 1% lead citrate before being observed in a 80 kV electron microscope (JEOL 1200EX).
(C, F, I) Scanning microscopy. The surface of control sperm appears smooth (C), whereas that of globozoocephalic spermatozoa appears wrinkled (F) and with a probable remnant of atrophic acrosome (I). Transmission electron microscopy was performed as detailed previously.12
A total of 20 globozoospermic patients were recruited. All sperm analyses were performed at least twice, in accordance with the World Health Organization recommendations.16 All subjects showed typical globozoospermia (Figure 1, Table 1) with close to 100% globozoospermic cells. The study was approved by local ethics committee, all patients gave written informed consent, and national laws and regulations were respected. All subjects had a normal somatic karyotype. None of the patients were related apart from patient 6 (P6) and P6b, who were brothers. Most patients originated from Tunisia, and only two patients did not originate from North Africa (Table 1). The parents of nine patients were related; most of them were first cousins. The homogeneity of the origin of the patients and their frequent consanguinity increased the chances of success of this homozygosity mapping study. Whole-genome analysis was carried out on nine patients (P1–P9) with the use of Affymetrix 250K StyI SNP arrays. Common regions of homozygosity were searched for with the homoSNP software. Seven patients shared a homozygous region ranging from 49 Mb to 1 Mb, centered on 12q14.2 (Figure 2A). The other regions of shared homozygosity identified elsewhere in the genome involved smaller genomic regions and concerned a maximum of four patients. We thus focused on the 12q14.2 region and analyzed the smallest common region of homozygosity defined by patient 7 (Figure 2A and Table 1). Four genes were localized in this region (Figure 2B), of which only one, DPY19L2, was described to be highly expressed in the testes. Furthermore, although there was no known function for DPY19L2 in human, previous work indicated that DPY-19, its ortholog in C. elegans, was involved in the establishment of cell polarity in the worm,17 a function coherent with the failure to achieve sperm head elongation that was observed in our patients. PCR amplification of three DPY19L2 exons was carried out for nine genotyped patients plus P6b, P6's affected brother who had not been genotyped. The results showed that all patients except P7 and P8 had a homozygous deletion of DPY19L2 (Figure 3A). PCR primers and the specific annealing temperatures used to amplify the different exons and loci are listed in Table S1 (available online). Ironically, P7, who presented the smallest region of homozygosity used to localize (and identify) the gene defect, was without the deletion and might present a “by chance” region of homozygosity centered on DPY19L2. Ten additional patients were analyzed, and seven of them also showed a homozygous deletion (Table 1). Subsequent amplifications on each side of the gene allowed us to pinpoint the two breakpoint zones to a 15 kb region included in two 28 kb low copy repeat (LCR) sequences with 97% identity, located on each side of DPY19L2 (Figure 3B). The deletion encompasses approximately 200 kb, harboring only DPY19L2. The same breakpoint localization could be observed for all 15 patients with the deletion. Agilent 180K comparative genomic hybridization (CGH) was carried out on P1. The analysis confirmed the presence of the homozygous deletion including DPY19L2 (Figure 4). The minimal deleted region spanned 22.3 kb, corresponding to a breakpoint ranging from 62,342,196 to 62,364,496 (NCBI36/hg18). Analyzing copy-number variant (CNV) databases, we saw that the DPY19L2 region was referenced as a CNV and that duplications and heterozygous deletions had been detected in all tested ethnic groups (Table 2 and Figure S1). Surprisingly, Affymetrix array genotyping results were obtained for all patients from most of the SNPs localized in the deleted region. Only rs11175121 gave three no-calls and three aberrant heterozygous calls (Figure 5). Performing a BLASTN search of these five SNPs, we see that all have a very homologous sequence present on chromosome 7 and that rs11175121 is the only one with a chromosome 12–specific nucleotide directly adjacent to the polymorphic nucleotide (Table S2). This illustrates that some genotypes provided by SNP arrays should be considered with caution, especially those located in CNVs or LCRs. The use of higher-coverage arrays such as the Affymetrix 6.0 in combination with a quantitative analysis of the results would have likely overcome the problem and might have enabled us to directly visualize the deletion. In fact, the deletion could be detected by CGH array despite a low number of probes (3) localized in the sequence of interest (Figure 4). Also, a quantitative analysis of the locus was achieved with the use of the Illumina Infinium II HumanHap 550 BeadChip SNP arrays in the CHOP project, which allowed the identification of the deleted region.18
Table 1.
Patient Information According to Genetic Status
| Patients | Region of Homoz. |
Sperm Parameters |
Country of Origin | Parents' Relation | Affected Siblings | DPY19l2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Volume (ml) | Conc. (M/ml) | % Globo | Cells (M/ml) | Motility | MAI | ||||||
| P1 | 13,344–62,370 | 4 | 100 | 100 | 3,8 | 35 | 2,56 | Tunisia | first cousins | 0/1 | deleted |
| P2 | 53,622–77,064 | 7 | 95 | 100 | 2 | 30 | 2,58 | Tunisia | first cousins | 3/6 | deleted |
| P3 | 51,030–67,271 | 4,5 | 89 | 100 | 16 | 40 | NA | Tunisia | first cousins | 0/4 | deleted |
| P4 | 57,021–63,398 | 3,5 | 52 | 100 | 6 | 23 | NA | Tunisia | third cousins | 0/0 | deleted |
| P5 | 60,521–68,314 | 4,5 | 0,02 | 100 | 0,1 | 1 | NA | Algeria | third cousins | 3/3 | deleted |
| P6 | 60,521–62,637 | 2,7 | 154 | 100 | 3 | 25 | 2,46 | Tunisia | none | 2/4 | deleted |
| P6b | NA | 7,5 | 126 | 100 | 3 | 25 | NA | Tunisia | none | 2/4 | deleted |
| P9 | none | 3,2 | 64 | 100 | 4 | 30 | 2,62 | Tunisia | first cousins | 0/2 | deleted |
| P10 | NA | 2,3 | 32 | 98 | NA | 40 | 2,06 | Morocco | NA | NA | deleted |
| P11 | NA | 3,5 | 36,5 | 100 | 0,6 | 20 | 3 | Tunisia | none | 0/3 | deleted |
| P12 | NA | 1,7 | 0,6 | 100 | 1,7 | 3 | 3 | Tunisia | none | 0/2 | deleted |
| P13 | NA | 1,4 | 108 | 100 | 1,2 | 23 | 3 | Tunisia | first cousins | 0/2 | deleted |
| P14 | NA | 2.9 | 18 | 100 | NA | 38 | NA | Morocco | NA | NA | deleted |
| P15 | NA | 2,7 | 25,2 | 94 | NA | 30 | NA | Tunisia | NA | NA | deleted |
| P16 | NA | 2 | 41 | 100 | NA | 20 | NA | Tunisia | NA | NA | deleted |
| average | 3,6 | 62,8 | 99,5 | 3,8 | 25,5 | 2,7 | |||||
| P7 | 61,770–62,815 | 6 | 1,4 | 99 | 1,5 | 5 | 2,91 | Turkey | first cousins | 1/1 | nondel. |
| P8 | none | 3 | 0,04 | 100 | 0,5 | 1 | NA | Tunisia | first cousins | 1/2 | nondel. |
| P17 | NA | 3 | 14 | 100 | 0,4 | 50 | 3,1 | Tunisia | NA | NA | nondel. |
| P18 | NA | 2,7 | 25,2 | 86 | NA | 30 | 1,88 | Tunisia | NA | NA | nondel. |
| P19 | NA | 4,7 | 8 | 64 | NA | 20 | 2,31 | Slovenia | NA | NA | nondel. |
| average | 3,9 | 9,7 | 89,8 | 0,8 | 21,2 | 2,6 | |||||
MAI, multiple anomalies index. Genetic status: DPY19L2 deleted (n = 15) or not deleted (nondel.; n = 5). Region of Homoz. indicates, for the patients analyzed by microarray (P1–P9), the extent of the region of homozygosity around DPY19L2 on chromosome 12 (NCBI36/hg18). Vol. indicates the volume of the ejaculate; Conc. indicates the number of spermatozoa in million per ml (M/ml); % Globo indicates the percentage of globozoosperm in the ejaculate; Cells indicates the concentration of nonsperm cells in the ejaculate (leukocytes or nondifferentiated germ cells); Motility is the percentage of motile spermatozoa with rapid (a) or slow (b) progression, 1 hr after ejaculation. Affected Siblings indicates the number of affected brothers (excluding the patient investigated) out of the total number of brothers. NA indicated values that were not availaible.
Figure 2.
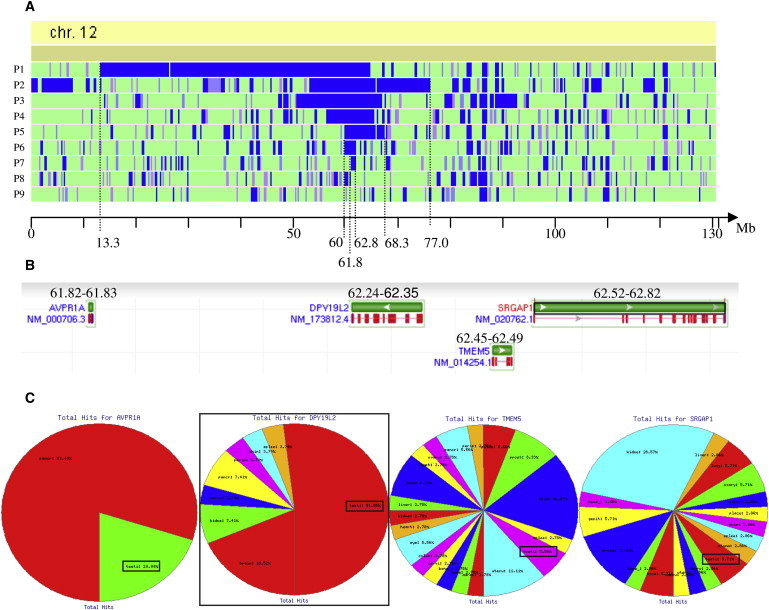
Identification of the Best Candidate Gene by Homozygosity Mapping
(A) Schematic representation of regions of homozygosity on chromosome 12 for globozoospermic patients P1–P9. Data were obtained with a GeneChip Mapping 250K StyI SNP Array from Affymetrix (Santa Clara, CA) in accordance with the manufacturer's recommendations. The analyses were carried out at the IGBMC (Strasbourg) Microarray and Sequencing Platform. The graphic representation is a view from homoSNP, developed by F. Plewniak of IGBMC, Strasbourg (software available on request from plewniak@igbmc.u-strasbg.fr). Regions of homozygosity greater than 45 SNPs are shown in blue. The entire chromosome 12 is represented, with the physical localization indicated at the bottom (NCBI36/hg18). Seven of nine patients have a region of homozygosity > 1 million bp centered around 62 Mb on chromosome 12.
(B) Identification of all the genes localized in the smallest common region of homozygosity of P7, between 61.8 Mb and 62.8 Mb . Representation from the NCBI Nucleotide database.
(C) Expression profile of the four genes present in the candidate region as obtained from the geneHub-GEPIS database, showing that DPY19L2 is expressed preferentially in the testis and that testis expression is marginal in the three other genes.
Figure 3.
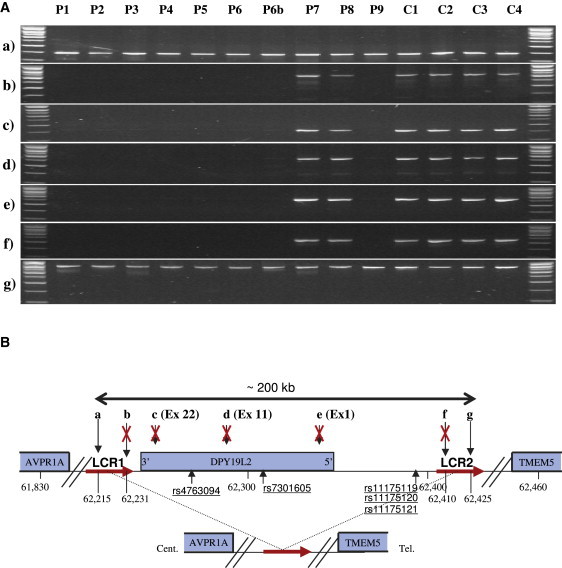
Characterization of a Large Homozygous Deletion Encompassing Only DPY19L2 in Eight of Ten Patients
(A) Seven loci (a–g) localized on and around DPY19L2 were amplified from ten patients with globozoospermia (P1–P9, including P6b) and four fertile controls (C1–C4). All of the tested loci yielded good PCR amplification from fertile individuals, whereas in eight of the ten globozoospermic patients there was no amplification of any of the intragenic loci (exons 1, 11, and 22) or of extragenic loci (b and f).
Genomic DNA was extracted either from peripheral blood leucocytes with the use of a guanidium chloride extraction procedure or from saliva via an Oragene DNA Self-Collection Kit (DNAgenotech, Ottawa, Canada). PCR primers with their specific annealing temperatures and exact genomic localization are listed in Table S1. Thirty-five cycles of PCR amplification were carried out with the use of Taq DNA polymerase (QIAGEN, Courtaboeuf, France).
(B) Schematic representation of the analyzed region with the position of the amplified loci (in kb). (a and b) Loci are localized approximately 25 kb and 9 kb 3′ of DPY19L2, respectively; (c), (d), and (e) show the position of DPY19L2 exons 22, 11, and 1, respectively; and (f) and (g) loci are localized approximately 62 and 77 kb 5′ of DPY19L2. LCRs 1 and 2 (red arrows) represent two 28 kb duplicated sequences showing 97% sequence identities located on each side of DPY19L2. PCR results in (A) indicate that the centromeric breakpoint is located between loci (a) and (b) (16 kb) within LCR 1 and the telomeric breakpoint between loci (f) and (g) (15 kb) within LCR 2. The deleted region encompasses a maximum of 210 kb, containing only one gene: DPY19L2.
Figure 4.
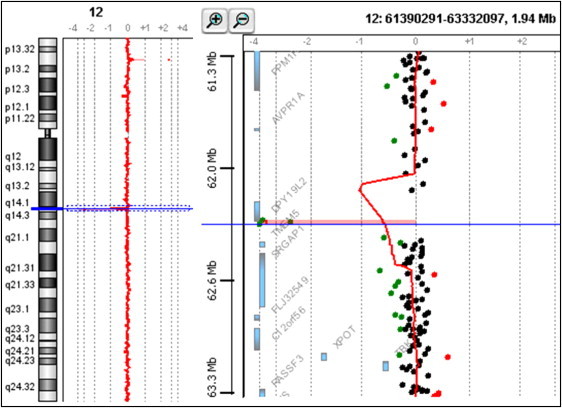
Confirmation of the Presence of a Homozygous Deletion in P1
Oligonucleotide array CGH was performed with the Agilent 180K Human Genome CGH Microarray (Agilent Technologies, Santa Clara, CA, USA) (Hospices Civils de Lyon, CGH Plateform). Graphical overview and analysis of the data were obtained with the Agilent DNA Analytics software version 4.0.81 (statistical algorithm: Z-score, sensitivity threshold: 2.5, window: 0.5). A graphical overview and analysis of the data were obtained by using the Agilent DNA Analytics software. The value of zero represents equal fluorescence intensity ratio between sample and reference DNA. Copy-number losses shift the ratio to the left (< −1), and copy-number gains shift it to the right (> 0.58).Three adjacent probes located on the telomeric side of DPY19L2 are homozygously deleted in the analyzed patient.
Table 2.
Frequency of Individuals Presenting with a Duplication or a Heterozygous Deletion at the DPY19L2 Locus and the Expected Prevalence of Individuals with Homozygous Deletion
| Number of Subjects | Number of Dup. | Frequency of Dup. (%) | Number of Het. Del. | Frequency of Het. Del. (%) | Expected Frequency of Hom. Del. | |
|---|---|---|---|---|---|---|
| Africans | 693 | 23 | 3.3 | 2 | 0.3 | 1/ 481 636 |
| Europeans | 1321 | 14 | 1.1 | 13 | 1.0 | 1/ 41 616 |
| Asians | 12 | 0 | 0 | 1 | 8.3 | 1/ 576 |
| TOTAL | 2026 | 37 | 1.8 | 16 | 0.8 | 1/ 64 516 |
Abbreviations are as follows: Dup., duplication; Het. Del., heterozygous deletion; Hom. Del., homozygous deletion. Data are from the CNV CHOP database.
Figure 5.
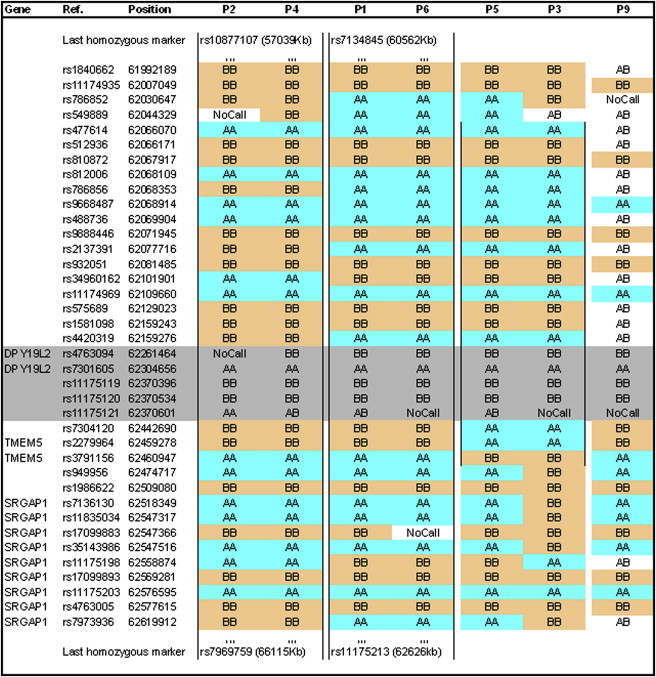
Haplotype of Seven Patients with the Deletion
Genotype of seven patients with the deletion centered around DPY19L2, as indicated by the microarray analysis. P2 and P4 share the longest identical haplotype, of approximately 9 Mb, ranging from rs10877107 to rs7969759. P1 and P6 have an identical haplotype of 2 MB, and P5 and P3 have a haplotype of 0.4 Mb. Markers in gray are located in the deleted region. The genotypes indicated for deleted markers very likely come from hybridization of homozygous sequences from chromosome 7 (see Table S2).
Because of the presence of duplicated DPY-19 sequences and, in particular, of DPY19L2P1 on chromosome 7, which has > 95% sequence identity with DPY19L2, it has been extremely difficult and tedious to identify primers and amplification conditions specific to DPY19L2 and we have not yet succeeded in sequencing the gene in totality. Five of our patients do not have a homozygous deletion, and we cannot exclude the possibility that some of these patients may harbor either two point mutations or a heterozygous deletion and a smaller mutation (in particular patient 7, who presents with the 1Mb homozygous region). LCRs 1 and 2 are also very homologous (> 97%), and this also prevented us from developing a long-range PCR across the deletion. This would prove useful for detecting heterozygotes and would characterize more precisely the deletion breakpoints. In the absence of this test, we carried out PCR amplification of two exons (1 and 11) on 200 fertile North African men and 100 fertile European men. All patient exons amplified, indicating that none were homozygous for the DPY19L2 deletion. For better ascertainment of the frequency of the deletion, additional data could be analyzed from the Copy Number Variation project at the Children's Hospital of Philadelphia (CHOP project), which presents Illumina HumanHap 550 BeadChip data obtained from 2026 disease-free individuals.18 Interrogation of the CHOP web server indicated that the overall frequency of DPY19L2 heterozygous deletion was 1 in 127 (Table 2), implying an estimated disease incidence of 1 in 64,516, which would be concordant with the rarity of the phenotype. A very high carrier prevalence was detected in the Asian cohort, but only 12 individuals were tested, and this result is probably due to the small size of the population tested. Data from the CHOP project strongly suggests that male globozoospermia due to a homozygous DPY19L2 deletion can be expected in all ethnic groups. The frequency of the deletion has not been determined in the North African population, which might be different from the African population—mainly African American—used in the CHOP study (Table 2). The higher incidence of globozoospermia observed in Maghrebian men is, however, not necessarily correlated with an increased allelic frequency. It is probably due to the high rate of consanguineous marriages specific to this population (as was the case for almost half of the patients tested here), which strongly favors the emergence of recessive traits. This was confirmed by our microarray data, which show that six of seven patients with the deletion were homozygous for a large region (ranging from 2–49 Mb) encompassing the deleted region (Table 1). The analysis of the SNP microarray data suggests the presence of three distinct haplotypes (Figure 5). This concurs with data from the CNV database and confirms that this deletion is not a due to a unique founder effect, even in our (rather homogeneous) population. Altogether, theses elements strongly suggest that DPY19L2 CNV is caused by nonallelic homologous recombination (NAHR) between LCRs 1 and 2. We could not identify any remnant of the LCR 1,2 sequences near the other DPY19L paralogs or pseudogenes, suggesting that the duplications that led to the creation of the DPY19L family might have been driven by a different mechanism.
Nucleotide changes have long been believed to be the major force in genomic evolution. It is now estimated that LCRs account for approximately 5% of the human genome19 and that the average rate of gene duplication is similar to the mutation rate per nucleotide site.20 Mutations in duplicated genes can cause (1) pseudogenization (function loss), (2) subfunctionalization, a process whereby both genes are required to perform the ancestral functions, or (3) acquisition of a new function.21,22 DPY19L2 is a member of the DPY19L family, which comprises four transmembrane proteins of unknown function: DPY19L1–DPY19L4. Genes encoding DPY19L proteins have undergone multiple duplications through evolution, leading to the recent creation of a family with LCR comprising DPY19L1 and six pseudogenized copies on chromosome 7 and DPY19L2 on chromosome 12.23 We can safely postulate that the duplication of DPY-19 eventually led to the acquisition of a new specialized function in spermiogenesis, in what could be seen as a paradigm for neofunctionalization through gene duplication. DPY19L1 was shown to be expressed in rat neural stem cell prior to their differentiation into GABAergic neurons,24 and DPY19L3, which is expressed mainly in the brain and the peripheral nervous system, was very recently described to be have a strong association with bipolar disorder.25 We can postulate that the presence of mutations in DPY19L1 or DPY19L3—potentially large deletions—could have severe pathological consequences caused by an abnormal neurological development. DPY19L2 is, however, the only of the four paralogs or of the other pseudogenes to have been described as a CNV and found to be duplicated or deleted, and we have not detected the 28 kb LCR near any of its paralogs or pseudogenes through extensive BLASTN search. Data from the CHOP database indicates that the breakpoints of a vast majority of the described CNV (101/108) are localized in LCRs 1 and 2 (Figure S1). This indicates that the observed deletion, like the described gain/loss CNV at the DPY19L2 locus, is indeed very likely caused by NAHR occurring between LCRs 1 and 2. NAHR usually takes place during meiosis between highly homologous sequences, usually LCRs, located (1) on the same chromatid, (2) on sister chromatids, or 3) on chromatids from paired bivalent chromosomes. Interchromosomal and interchromadid NAHR (2 and 3) between LCRs in direct orientation result in the production of two recombined chromosomes: one duplicated and one deleted, whereas intrachromatid NAHR (1) produces a deleted chromosome and a small, nontransmissible (thus lost), circularized chromatid portion (for review see 26). NAHRs are thus expected to produce a higher proportion of deletions than duplications. This was indeed confirmed through measuring the de novo meiotic deletions and duplications at four NAHR hotspots on sperm DNA.27 Surprisingly, the frequency of duplications at the DPY19L2 locus (1.8%) is more than twice that of deletions (0.8%) (Table 2). This could result from the negative selection against the deleted alleles leading to infertility in homozygous males, as demonstrated here. Alternatively, this could result from a positive selection of the duplicated alleles. At the moment, we have no data concerning the potential effect of the deletion in females. However, considering the sperm-specific function of DPY19L2, we do not anticipate any phenotype in females.
Antibodies were raised against human and mouse DPY19L2. We could demonstrate that the proteins are present in human and mouse testis but absent from ejaculated human sperm or epididymal mouse sperm (Figure 6). These results are concordant with a role of DPY19L2 during spermiogenesis. We performed flow cytometry analysis on spermatozoa from P1's spermatozoa (data not shown) and observed a single haploid peak. Others have carried out fluorescence in situ hybridization (FISH) analyses on sperm from globozoospermic patients and have also observed that the tested spermatozoa were euploid, albeit with a slight increase in aneuploidy rate.28–30 Altogether, these data demonstrate that globozoospermatozoa have undertaken both meiotic divisions and have reached the round spermatid stage but that the spermiogenesis is altered beyond this stage. One of the major complexes involved in acrosome formation and head elongation is the acroplaxome, a subacrosomal cytoskeletal plate toward which Golgi-derived vesicles fuse.31 Very little is known about DPY19L2 function. In the human, the only data come from the analysis of cDNA libraries, and they indicate that DPY19L2 transcripts are expressed mainly in the testes (Figure 2C). Our protein work confirmed that both human and mouse DPY19L2 are indeed present in the testis but that they are absent from mature sperm (Figure 6). We also observed in the mouse that the protein is predominantly expressed in the testis compared to muscle, spleen, kidney, or liver (data not shown). These results are concordant with a function of the protein during spermiogenesis. At the moment, the only key to its function comes from the study of DPY-19, the ancestor of DPY19L2 in C. elegans. Both genes encode multipass membrane proteins likely to contain 6–11 transmembrane domains. DPY-19 was shown to be necessary for the correct polarization of C. elegans neuroblasts and their subsequent migration along the anterior-posterior axis.17 We saw in our patients that without DPY19L2, head elongation and acrosome formation are not possible. We can postulate that DPY19L2 might be necessary to indicate the anterior pole of spermatozoa and might be involved in the acroplaxome positioning and fixation. The dpy19l2 mice knockout has just been produced in a recent effort to produce animal models for transmembrane proteins.32 The described phenotype concurs perfectly with what is observed in the human and was briefly described as “infertility, failure of the spermatid nuclei to elongate”32. We have no doubt that the utilization of this animal model will contribute to the elucidation of the function and mode of action of DPY19L2.
Figure 6.
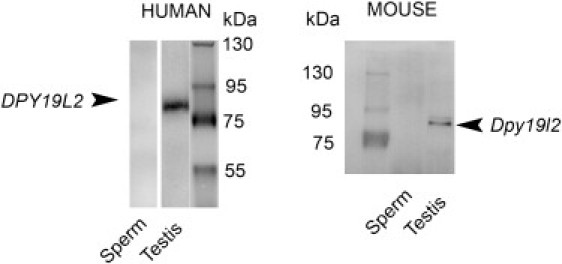
Immunoblots Showing the Presence of DPY19L2 in Testis and Its Absence in Sperm
Left; a unique band in the expected area of the gel is marked with human anti-DPY19L2 antibody with human testis extract but not with ejaculated sperm extract. A similar result is obtained with mouse anti-Dpy19l2 antibody, in which a band is marked with mouse testis extract but not with epididymal sperm extract . Protein loadings in each lane were similar and were checked by the Bradford protein assay.
Proteins were separated on 8% polyacrylamide denaturing gels and electrotransferred for 120 min at 350 mA to Immobilon P transfer membrane (Millipore). The membranes were then blocked for 60 min with 5% nonfat dry milk (Biorad) in PBS Tween 0.1%. The primary antibody was added and incubated overnight at 4°C. After washing in PBS Tween 0.1%, the secondary antibody (anti-rabbit, Jackson Laboratory) was added at a dilution of 1:10,000 for 1 hr at room temperature. The membrane was washed and incubated for 1 min in HRP substrate (Western Lightning, Perkin Elmer Life Science). The reactive proteins were detected with G-Box chemi XL (Syngene, England). Polyclonal antibodies against peptides from the N terminus of DPY19L2 (RSKLREGSSDRPQSSC for mouse Dpy19l2 and RSQSKGRRGASLAREPEC for human DPY19L2) were raised in rabbit. Antibodies were not purified, and serums were used at a dilution of 1:1000. All animal procedures were run according to the French guidelines on the use of living animals in scientific investigations with the approval of the local ethical review committee of Grenoble Neurosciences Institute. Sperm were obtained from 16-week-old Oncins France 1 strain (OF1) mice (obtained from Charles River, Macon, France) by manual trituration of caudae epididymis. Human testis tissue (from an 80-year-old donor) was obtained by surgery. Sperm were obtained by ejaculation (from a 30-year-old donor). Human tissues were obtained after approval by the local ethical committee and informed consent from the patients.
None of the patients with the deletion declared any medical impairment apart from their primary infertility. DPY19L2 inhibition could therefore provide an interesting target to achieve male contraception and a patent was filed to that effect.15 Many have attempted intracytoplasmic sperm injection (ICSI) with globozoospermic spermatozoa. Overall fertilization and pregnancy rate is low but successful pregnancies have been reported.3,33–35 In most of these reports, the distinction between type I (total globozoospermia) and type II (partial globozoospermia) patients is not clear. It is also very likely that not all of the more homogeneous type I patients have the same molecular etiology. Twenty ICSI cycles were carried out on five brothers with typical type I globozoospermia, resulting in a single birth.36 In the past 5 years, at the Clinique des Jasmin in Tunis, we have attempted seven ICSI cycles for six of the patients with DPY19L2 deletion who are described here. Normal fertilization rate was very low (13%), but one pregnancy could be obtained, illustrating the fact that a pregnancy could be obtained for type I globozoospermia caused by DPY19L2 deletion. Recent work suggests that the low fertilization rate observed with globozoospermatozoa is at least partially caused by a decrease or a defect in PLCζ, a protein involved in the induction of calcium oscillations triggering oocyte activation.37,38 Our report suggests that defects in PLCζ signaling would be secondary to the defect in head maturation caused by the absence of DPY19L2. It is now important to genotype a larger series of patients, including some with a positive ICSI outcome, to establish whether the globozoospermic patients without DPY19L2 deletion have a better prognosis. If this were the case, the realization of a molecular diagnosis for globozoospermic men would permit providers to adopt the best course of treatment for these patients.
Acknowledgments
We thank all affected individuals and family members for their participation. We thank Frederic Plewniak (fred@igbmc.fr) from the IGBMC of Strasbourg for having developed homoSNP and for the help he provided for utilization of the software. We thank Gaelle Vieville from Grenoble CHU and Damien Sanlaville from the Hospices Civils de Lyon for their help with the CGH array. We thank Peter White from the Children's Hospital of Philadelphia for helpful discussion on CNV and the use of the CHOP database. We thank Jean Luc Cracowski and the Centre d'Investigation Clinique (CIC) of Grenoble CHU for their help regarding the ethical and legal aspects of patient management.
This work was part of the project Identification and Characterization of Genes Involved in Infertility (ICG2I) funded by the program GENOPAT 2009 from the French Research Agency (ANR). This work was also funded in part by program CIBLE 2009 from the Rhône Alpes Région.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
The Copy Number Variation project at the Children's Hospital of Philadelphia (CHOP), http://cnv.chop.edu/
GENEHUB GEPIS, http://www.cgl.ucsf.edu/Research/genentech/genehub-gepis/
NCBI Nucleotide database, http://www.ncbi.nlm.nih.gov/nuccore
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Auger J., Kunstmann J.M., Czyglik F., Jouannet P. Decline in semen quality among fertile men in Paris during the past 20 years. N. Engl. J. Med. 1995;332:281–285. doi: 10.1056/NEJM199502023320501. [DOI] [PubMed] [Google Scholar]
- 2.Matzuk M.M., Lamb D.J. The biology of infertility: research advances and clinical challenges. Nat. Med. 2008;14:1197–1213. doi: 10.1038/nm.f.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dam A.H., Feenstra I., Westphal J.R., Ramos L., van Golde R.J., Kremer J.A. Globozoospermia revisited. Hum. Reprod. Update. 2007;13:63–75. doi: 10.1093/humupd/dml047. [DOI] [PubMed] [Google Scholar]
- 4.Kullander S., Rausing A. On round-headed human spermatozoa. Int. J. Fertil. 1975;20:33–40. [PubMed] [Google Scholar]
- 5.Flörke-Gerloff S., Töpfer-Petersen E., Müller-Esterl W., Mansouri A., Schatz R., Schirren C., Schill W., Engel W. Biochemical and genetic investigation of round-headed spermatozoa in infertile men including two brothers and their father. Andrologia. 1984;16:187–202. doi: 10.1111/j.1439-0272.1984.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 6.Nistal M., Herruzo A., Sanchez-Corral F. Absolute teratozoospermia in a family. Irregular microcephalic spermatozoa without acrosome. Andrologia. 1978;10:234–240. [PubMed] [Google Scholar]
- 7.Escalier D. Failure of differentiation of the nuclear-perinuclear skeletal complex in the round-headed human spermatozoa. Int. J. Dev. Biol. 1990;34:287–297. [PubMed] [Google Scholar]
- 8.Dam A.H., Koscinski I., Kremer J.A., Moutou C., Jaeger A.S., Oudakker A.R., Tournaye H., Charlet N., Lagier-Tourenne C., van Bokhoven H., Viville S. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am. J. Hum. Genet. 2007;81:813–820. doi: 10.1086/521314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu M., Xiao J., Chen J., Li J., Yin L., Zhu H., Zhou Z., Sha J. Identification and characterization of a novel human testis-specific Golgi protein, NYD-SP12. Mol. Hum. Reprod. 2003;9:9–17. doi: 10.1093/molehr/gag005. [DOI] [PubMed] [Google Scholar]
- 10.Lu L., Lin M., Xu M., Zhou Z.M., Sha J.H. Gene functional research using polyethylenimine-mediated in vivo gene transfection into mouse spermatogenic cells. Asian J. Androl. 2006;8:53–59. doi: 10.1111/j.1745-7262.2006.00089.x. [DOI] [PubMed] [Google Scholar]
- 11.Dieterich K., Soto Rifo R., Faure A.K., Hennebicq S., Ben Amar B., Zahi M., Perrin J., Martinez D., Sèle B., Jouk P.S. Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat. Genet. 2007;39:661–665. doi: 10.1038/ng2027. [DOI] [PubMed] [Google Scholar]
- 12.Dieterich K., Zouari R., Harbuz R., Vialard F., Martinez D., Bellayou H., Prisant N., Zoghmar A., Guichaoua M.R., Koscinski I. The Aurora Kinase C c.144delC mutation causes meiosis I arrest in men and is frequent in the North African population. Hum. Mol. Genet. 2009;18:1301–1309. doi: 10.1093/hmg/ddp029. [DOI] [PubMed] [Google Scholar]
- 13.Harbuz R., Zouari R., Dieterich K., Nikas Y., Lunardi J., Hennebicq S., Ray P.F. Function of aurora kinase C (AURKC) in human reproduction. Gynecol. Obstet. Fertil. 2009;37:546–551. doi: 10.1016/j.gyobfe.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 14.Ray P.F., Zouari R., Harbuz R., Ben Khelifa M., Kharouf M., Nikas Y., Hennebicq S., Koscinski I., Viville S., Escoffier J. Identification of the molecular defect responsible for most cases of globozoospermia. Hum. Reprod. 2010;25(Suppl. 1) I106–I106. [Google Scholar]
- 15.Ray, P.F. and Arnoult, C. (2009). Contrôle de la fertilité humaine via DPY19L2. Patent FR0957963. November 12, 2009.
- 16.World Health Organization . Cambridge University Press; Cambridge, UK: 1999. WHO Laboratory Manual for the Examination of Human Semen and Sperm–Cervical Mucus Interaction. [Google Scholar]
- 17.Honigberg L., Kenyon C. Establishment of left/right asymmetry in neuroblast migration by UNC-40/DCC, UNC-73/Trio and DPY-19 proteins in C. elegans. Development. 2000;127:4655–4668. doi: 10.1242/dev.127.21.4655. [DOI] [PubMed] [Google Scholar]
- 18.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 20.Lynch M., Conery J.S. The evolutionary demography of duplicate genes. J. Struct. Funct. Genomics. 2003;3:35–44. [PubMed] [Google Scholar]
- 21.Force A., Lynch M., Pickett F.B., Amores A., Yan Y.L., Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohta T. Simulating evolution by gene duplication. Genetics. 1987;115:207–213. doi: 10.1093/genetics/115.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carson A.R., Cheung J., Scherer S.W. Duplication and relocation of the functional DPY19L2 gene within low copy repeats. BMC Genomics. 2006;7:45. doi: 10.1186/1471-2164-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurokawa K., Tamura Y., Kataoka Y., Yamada H., Nakamura T., Taki K., Kudo M. A mammalian dpy-19 homologue is expressed in GABAergic neurons. Med. Mol. Morphol. 2005;38:79–83. doi: 10.1007/s00795-004-0280-1. [DOI] [PubMed] [Google Scholar]
- 25.Smith E.N., Bloss C.S., Badner J.A., Barrett T., Belmonte P.L., Berrettini W., Byerley W., Coryell W., Craig D., Edenberg H.J. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry. 2009;14:755–763. doi: 10.1038/mp.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu W., Zhang F., Lupski J.R. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1:4. doi: 10.1186/1755-8417-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner D.J., Miretti M., Rajan D., Fiegler H., Carter N.P., Blayney M.L., Beck S., Hurles M.E. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 2008;40:90–95. doi: 10.1038/ng.2007.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viville S., Mollard R., Bach M.L., Falquet C., Gerlinger P., Warter S. Do morphological anomalies reflect chromosomal aneuploidies?: case report. Hum. Reprod. 2000;15:2563–2566. doi: 10.1093/humrep/15.12.2563. [DOI] [PubMed] [Google Scholar]
- 29.Morel F., Douet-Guilbert N., Moerman A., Duban B., Marchetti C., Delobel B., Le Bris M.J., Amice V., De Braekeleer M. Chromosome aneuploidy in the spermatozoa of two men with globozoospermia. Mol. Hum. Reprod. 2004;10:835–838. doi: 10.1093/molehr/gah111. [DOI] [PubMed] [Google Scholar]
- 30.Moretti E., Collodel G., Scapigliati G., Cosci I., Sartini B., Baccetti B. ‘Round head’ sperm defect. Ultrastructural and meiotic segregation study. J. Submicrosc. Cytol. Pathol. 2005;37:297–303. [PubMed] [Google Scholar]
- 31.Kierszenbaum A.L., Tres L.L. The acrosome-acroplaxome-manchette complex and the shaping of the spermatid head. Arch. Histol. Cytol. 2004;67:271–284. doi: 10.1679/aohc.67.271. [DOI] [PubMed] [Google Scholar]
- 32.Tang T., Li L., Tang J., Li Y., Lin W.Y., Martin F., Grant D., Solloway M., Parker L., Ye W. A mouse knockout library for secreted and transmembrane proteins. Nat. Biotechnol. 2010;28:749–755. doi: 10.1038/nbt.1644. [DOI] [PubMed] [Google Scholar]
- 33.Liu J., Nagy Z., Joris H., Tournaye H., Devroey P., Van Steirteghem A. Successful fertilization and establishment of pregnancies after intracytoplasmic sperm injection in patients with globozoospermia. Hum. Reprod. 1995;10:626–629. doi: 10.1093/oxfordjournals.humrep.a136000. [DOI] [PubMed] [Google Scholar]
- 34.Battaglia D.E., Koehler J.K., Klein N.A., Tucker M.J. Failure of oocyte activation after intracytoplasmic sperm injection using round-headed sperm. Fertil. Steril. 1997;68:118–122. doi: 10.1016/s0015-0282(97)81486-0. [DOI] [PubMed] [Google Scholar]
- 35.Bechoua S., Chiron A., Delcleve-Paulhac S., Sagot P., Jimenez C. Fertilisation and pregnancy outcome after ICSI in globozoospermic patients without assisted oocyte activation. Andrologia. 2009;41:55–58. doi: 10.1111/j.1439-0272.2008.00884.x. [DOI] [PubMed] [Google Scholar]
- 36.Kilani Z.M., Shaban M.A., Ghunaim S.D., Keilani S.S., Dakkak A.I. Triplet pregnancy and delivery after intracytoplasmic injection of round-headed spermatozoa. Hum. Reprod. 1998;13:2177–2179. doi: 10.1093/humrep/13.8.2177. [DOI] [PubMed] [Google Scholar]
- 37.Yoon S.Y., Jellerette T., Salicioni A.M., Lee H.C., Yoo M.S., Coward K., Parrington J., Grow D., Cibelli J.B., Visconti P.E. Human sperm devoid of PLC, zeta 1 fail to induce Ca(2+) release and are unable to initiate the first step of embryo development. J. Clin. Invest. 2008;118:3671–3681. doi: 10.1172/JCI36942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heytens E., Parrington J., Coward K., Young C., Lambrecht S., Yoon S.Y., Fissore R.A., Hamer R., Deane C.M., Ruas M. Reduced amounts and abnormal forms of phospholipase C zeta (PLCzeta) in spermatozoa from infertile men. Hum. Reprod. 2009;24:2417–2428. doi: 10.1093/humrep/dep207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.