

Main Text
To the Editor: We read with great interest the article entitled “Pelizaeus-Merzbacher-Like Disease Caused by AIMP1/p43 Homozygous Mutation” by Feinstein et al, published in the December 2010 issue of The Journal.1
Although this report highlights the potential deleterious effects of one AIMP1/p43 (MIM 603605) mutation, we think that the relationship to a Pelizaeus-Merzbacher-like disease is erroneous and misleading.
The patients described by Feinstein et al. did not present the core clinical and neuroradiological symptoms that define a hypomyelinating leukodystrophy (HLD) corresponding to either Pelizaeus-Merzbacher disease (PMD, HLD1 [MIM 312080]) or Pelizaeus-Merzbacher-like disease 1 (PMLD1, HLD2 [MIM 608804]).2,3 In addition, those patients presented additional features that did not conform to diseases because of a primary central nervous system (CNS) myelin formation defect.
Patients described in this single Israeli Bedouin family presented with rapid neurologic deterioration, early microcephaly, dysmorphia, spastic paraparesis, limb deformities, and kyphoscoliosis. Although they presented horizontal or rotatory nystagmus, which is often present in patients with PMD or PMLD, the clinical symptoms presented by the patients clearly differ from those observed in patients with PMD or PMLD: microcephaly is never present, hypotonia is prominent, spasticity and/or dystonia appear usually after 2 years of age, and patients with PMD or PMLD do not have dysmorphia, even mild. In addition, the magnetic resonance imaging (MRI) data provided in this report are insufficient to state that the patients possibly have a severe hypomyelinating leukodystrophy. T2-weighted and FLAIR hypersignal of the white matter is observed in both hypomyelinating and demyelinating leukodystrophies, whereas both diseases have distinct T1-weighted signal of the white matter.4,5 In demyelinating forms, white matter T1 is always highly hypointense (relative to the cortex), whereas it ranges from normally hyperintense in moderate hypomyelination to mildly hypointense in severe hypomyelinating forms (Figure 1). Unlike what is mentioned by Feinstein et al. in the legend of Figure 2D, the signal of white matter on T1 sequence in the 14-month-old patient appears normally hyperintense with respect to cerebral cortex, suggesting an amount of myelin not compatible with a severe form of PMD or PMLD (Figure 1B).
Figure 1.
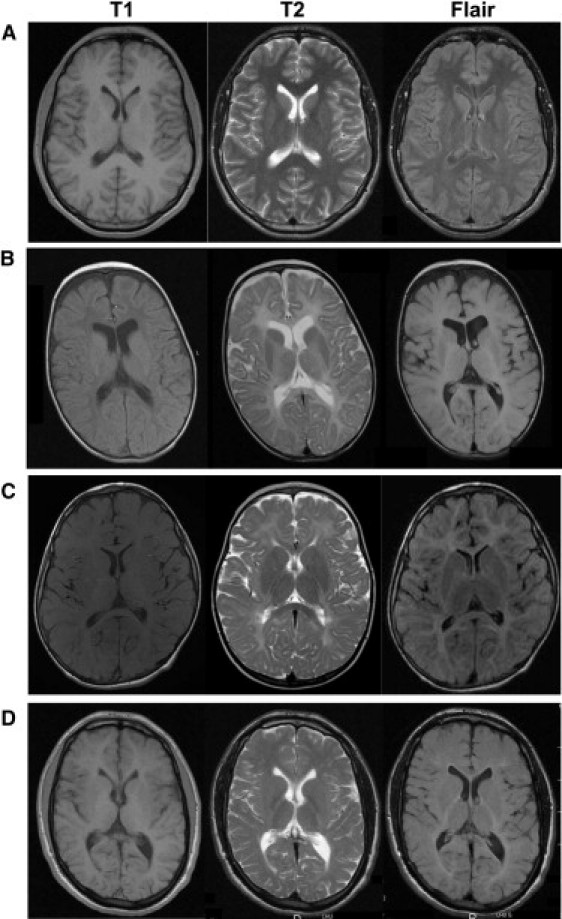
Magnetic Resonance Imaging of White Matter Changes in Hypomyelinating Leukodystrophies
(A–D) In a normal brain (A), when the myelination is achieved, the white matter signal is hyperintense relative to gray matter on T1-weighted images and hypointense relative to gray matter on T2-weighted and FLAIR images. In patients affected by hypomyelinating leukodystrophies, T1 white matter signal is hypointense relative to gray matter in severe form 0 related to proteolipid potein 1 (PLP1 [MIM 300401]) mutation (patient B, 2 years old), isointense in moderate form 3 related to PLP1 duplication (patient C, 7 years old), and normally hyperintense in mild form 4 related to gap junction protein gamma-2 (GJC2 [MIM 608803]) mutation (patient D, 20 years old).9 In patients B and C, the white matter signal on T2-weighted and FLAIR images appears hyperintense relative to gray matter. In patient D, the white matter tends to be isointense relative to gray matter on T2-weighted images, except in posterior internal capsules, and is hyperintense in FLAIR images.
Even with stringent criteria, the deciphering of genetic causes of CNS hypomyelinating diseases has remained a challenge. The patients described by Feinstein et al. present a complex phenotype, and the clinical and neuroimaging abnormalities suggest that they suffer from a primitive severe axonal disease. It must be noticed that the phenotype of patients with recessive mutations in the gene encoding for heat shock 60 kDa protein 1 (HSPD1 [MIM 118190]), classified as hypomyelinating leukodystrophy type 4 (HLD4 [MIM 612233]), also suggests an early and severe axonal involvment.6 Therefore, those phenotypes may rather correspond to infantile forms of neuronal degenerative disorders. It must be noticed that abnormal myelination is classically observed in such early-onset forms of neurodegenerative diseases.7 In this respect, it would have been interesting if the authors had provided neurophysiological analyses, particularly on the peripheral nerve functions of their patients.
The clinical and MRI presentation of the AIMP1 mutated patients reported in this paper is in agreement with the important role of AIMP1 in regulating neurofilament and maintaining axon-cytoskeleton integrity, and not as a primitive disorder of the myelination. Nevertheless, in such a highly inbred population, we cannot rule out the possibility of another mutated gene implicated in the etiology of the complex phenotype.8
In conclusion, the genetician and neurologist cannot consider AIMP1 as a candidate gene for PMLD mutations. Patients with early severe neurodegenerative disorder involving axonal functions and myelin development seem presently more appropriate for AIMP1 mutation screening.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Feinstein M., Markus B., Noyman I., Shalev H., Flusser H., Shelef I., Liani-Leibson K., Shorer Z., Cohen I., Khateeb S. Pelizaeus-Merzbacher-like disease caused by AIMP1/p43 homozygous mutation. Am. J. Hum. Genet. 2010;87:820–828. doi: 10.1016/j.ajhg.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mimault C., Giraud G., Courtois V., Cailloux F., Boire J.Y., Dastugue B., Boespflug-Tanguy O., The Clinical European Network on Brain Dysmyelinating Disease Proteolipoprotein gene analysis in 82 patients with sporadic Pelizaeus-Merzbacher Disease: Duplications, the major cause of the disease, originate more frequently in male germ cells, but point mutations do not. Am. J. Hum. Genet. 1999;65:360–369. doi: 10.1086/302483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henneke M., Combes P., Diekmann S., Bertini E., Brockmann K., Burlina A.P., Kaiser J., Ohlenbusch A., Plecko B., Rodriguez D. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748–754. doi: 10.1212/01.wnl.0000284828.84464.35. [DOI] [PubMed] [Google Scholar]
- 4.Boespflug-Tanguy O., Labauge P., Fogli A., Vaurs-Barriere C. Genes involved in leukodystrophies: A glance at glial functions. Curr. Neurol. Neurosci. Rep. 2008;8:217–229. doi: 10.1007/s11910-008-0034-x. [DOI] [PubMed] [Google Scholar]
- 5.Schiffmann R., van der Knaap M.S. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magen D., Georgopoulos C., Bross P., Ang D., Segev Y., Goldsher D., Nemirovski A., Shahar E., Ravid S., Luder A. Mitochondrial hsp60 chaperonopathy causes an autosomal-recessive neurodegenerative disorder linked to brain hypomyelination and leukodystrophy. Am. J. Hum. Genet. 2008;83:30–42. doi: 10.1016/j.ajhg.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steenweg M.E., Vanderver A., Blaser S., Bizzi A., de Koning T.J., Mancini G.M., van Wieringen W.N., Barkhof F., Wolf N.I., van der Knaap M.S. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain. 2010;133:2971–2982. doi: 10.1093/brain/awq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zlotogora J. The molecular basis of autosomal recessive diseases among the Arabs and Druze in Israel. Hum. Genet. 2010;128:473–479. doi: 10.1007/s00439-010-0890-8. [DOI] [PubMed] [Google Scholar]
- 9.Cailloux F., Gauthier-Barichard F., Mimault C., Isabelle V., Courtois V., Giraud G., Dastugue B., Boespflug-Tanguy O., Clinical European Network on Brain Dysmyelinating Disease Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Eur. J. Hum. Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]