

Abstract
High-affinity extrasynaptic GABAA receptors (GABAARs) are a prominent feature of cerebellar granule neurons and thalamic relay neurons. In both cell types, the presence of synaptic glomeruli would be expected to promote activation of these GABAARs, contributing to phasic spillover-mediated currents and tonic inhibition. However, the precise role of different receptor subtypes in these two phenomena is unclear. To address this question, we made recordings from neurons in acute brain slices from mice, and from tsA201 cells expressing recombinant GABAARs. We found that δ subunit-containing GABAARs of both cerebellar granule neurons and thalamic relay neurons of the lateral geniculate nucleus contributed to tonic conductance caused by ambient GABA but not to spillover-mediated currents. In the presence of a low “ambient” GABA concentration, recombinant “extrasynaptic” δ subunit-containing GABAARs exhibited profound desensitization, rendering them insensitive to brief synaptic- or spillover-like GABA transients. Together, our results demonstrate that phasic spillover and tonic inhibition reflect the activation of distinct receptor populations.
Introduction
Signaling through GABAA receptors (GABAARs) displays great diversity, reflecting both the heterogeneity of GABAAR subtypes and differences in the nature of their activation (Farrant and Nusser, 2005; Olsen and Sieghart, 2009). Synaptically released GABA transiently activates postsynaptic γ subunit-containing GABAARs opposite the release site, producing a brief increase in membrane conductance that underlies the “phasic” IPSC. By contrast, ambient GABA in the extracellular space activates predominantly δ or α5 subunit-containing extrasynaptic GABAARs to produce a persistent “tonic” conductance (Stell et al., 2003; Caraiscos et al., 2004; Glykys and Mody, 2006; Glykys et al., 2008). Between these extremes is a third form of signaling—“spillover”—where diffusion of GABA allows phasic activation of GABAARs other than those in the immediate postsynaptic density, either beneath the same bouton (Telgkamp et al., 2004), in extrasynaptic membrane (Brickley et al., 2001), in presynaptic structures (Trigo et al., 2008; Ruiz et al., 2010) or at adjacent synapses (Wei et al., 2003).
In cerebellar granule neurons (CGNs), extrasynaptic GABAARs, formed from α6, β2/3 and δ subunits, exist within synaptic glomeruli (Nusser et al., 1998), structures that are thought to promote spillover (Brickley et al., 1996; Wall and Usowicz, 1997; Rossi and Hamann, 1998). Thalamic relay neurons of the dorsal lateral geniculate nucleus (dLGN) express analogous high-affinity GABAARs formed from α4, β2 and δ subunits (Pirker et al., 2000; Peng et al., 2002). The high affinity and modest desensitization of δ-containing GABAARs (δ-GABAARs) (Saxena and Macdonald, 1994; Wohlfarth et al., 2002; Feng et al., 2009) are considered key to the generation of a tonic conductance in these cells (Brickley et al., 2001; Stell et al., 2003; Cope et al., 2005; Bright et al., 2007). Whether these same features also enable δ-GABAARs to contribute to phasic signaling via slow spillover currents remains unclear (Hamann et al., 2002; Wei et al., 2003; Peng et al., 2004; Bright et al., 2007).
Here we reexamined this question in morphologically identified thalamic relay neurons and in CGNs from wild-type, α6−/− and δ−/− mice. Thalamic neurons showed a clear mismatch between tonic and spillover currents. First, in X- and Y-type dLGN neurons, differences in the prevalence of “spillover-like” IPSCs were not mirrored by differences in δ-GABAAR-mediated tonic conductance. Second, putative spillover currents were present in neurons of the ventral LGN (vLGN) that lack δ-GABAARs (Pirker et al., 2000; Peng et al., 2002; Bright et al., 2007). Likewise, in CGNs, synaptic current components thought to reflect spillover of GABA within glomeruli did not involve δ-GABAAR activation. To understand why δ-GABAARs appear not to contribute to spillover currents but function solely as sensors of ambient GABA, we examined the responses of recombinant α1β2γ2S (“synaptic”) and α4β2δ (“extrasynaptic”) GABAARs. Unexpectedly, steady-state desensitization of δ-GABAARs by a low “ambient” GABA concentration rendered them insensitive to transient changes in GABA concentration. Together, our observations demonstrate that, in thalamus and cerebellum, spillover and tonic currents are separate phenomena, reflecting the activation of distinct receptor populations.
Materials and Methods
Heterologous expression.
Murine GABAAR α1, β2, and γ2S subunits were a gift from Trevor Smart (University College London, London, UK). The murine α4 and α6subunit coding sequences were amplified from a Clontech adult brain cDNA library with Phusion polymerase (New England Biolabs) and subcloned into the expression vector pRK5. A full-length rat δ cDNA was obtained (Open Biosystems clone 7317154) and subcloned into pRK5 as an EcoRI-SalI fragment. To conjugate a fluorescent epitope with the δ subunit, a XhoI site was introduced between the fourth and fifth codons of the predicted mature form (QuikChange mutagenesis, Stratagene) so that super ecliptic phluorin could be inserted. Expression of the δ subunit was then assessed by surface fluorescence, altered zinc sensitivity (Stórustovu and Ebert, 2006) and GABA EC50 values relative to αβ expression alone. The coding region of all clones was sequenced in full. tsA201 cells were grown according to standard protocols and transfected with cDNA encoding GABAAR subunits (2.4 μg of total DNA per dish) using Lipofectamine 2000 (Invitrogen) or the calcium phosphate method. Cotransfection with enhanced green fluorescent protein (EGFP) was performed for α1β2γ2S, α1β2, and α4β2 GABAARs. The cDNA ratio for transfections was 1:1 (αxβ2), 1:1:1 (αx:β2:δ) and 1:1:1:0.5 (αx:β2:γ2S:EGFP). Cells were split and plated on glass coverslips 6–24 h after transfection and used for recording after a further 16–72 h.
Acute slice preparations.
Brain slices were obtained from mature (>1 month postnatal) male wild-type (C57BL/6J), α6−/− (Jones et al., 1997) and δ−/− (Mihalek et al., 1999) mice, in accordance with the Animals (Scientific Procedures) Act 1986. In brief, the brain was rapidly removed and placed in ice-cold slicing solution composed of (in mm): 85 NaCl, 2.5 KCl, 1 CaCl2, 4 MgCl, 1.25 NaH2PO4, 26 NaHCO3, 75 sucrose, 25 glucose, pH 7.4 when bubbled with 95% O2 and 5% CO2. A moving blade microtome (DSK Super Zero 1 or DTK-1000; Dosaka EM) was used to prepare coronal thalamic slices (250 μm thick) and parasagittal cerebellar slices (150–250 μm thick). Slices were incubated at 37°C for 30 min. The high sucrose slicing solution was then gradually replaced with normal recording solution containing (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl, 1.25 NaH2PO4, 26 NaHCO3, 25 glucose, pH 7.4 when bubbled with 95% O2 and 5% CO2 over a period of 30 min.
Electrophysiology—recombinant receptors.
Cells were perfused with “external” recording solution, containing (in mm): 140 NaCl, 2.5 CaCl2, 1.2 MgCl2, 4.7 KCl, 5 HEPES, 11 glucose; the pH was adjusted to 7.4 with NaOH. The temperature of the solution in the recording chamber was monitored throughout, and maintained in physiological range (∼36°C). The “internal” (pipette) solution contained the following (in mm): 140 CsCl, 4 NaCl, 0.5 CaCl2, 10 HEPES, 5 EGTA, 2 Mg-ATP; the pH was adjusted to 7.3 with CsOH. Pipettes for whole-cell and outside-out patch recording were pulled from thick-walled borosilicate glass [1.5 mm outer diameter (o.d.), 0.86 mm inner diameter; Harvard Apparatus], coated with Sylgard resin (Dow-Corning 184) and fire-polished to a final resistance of 4–12 MΩ. Cells were viewed using a fixed stage upright microscope (SliceScope, Scientifica, or Olympus BX51 WI). Currents were recorded from fluorescent cells using an Axopatch 200A or 200B amplifier (Molecular Devices).
During whole-cell recording, GABA was bath applied at a flow rate of ∼6 ml/min, resulting in an exchange time of ∼1 min. This method was used to estimate the steady-state response of δ-GABAARs to a range of GABA concentrations, following at least 2 min of stable recording. To measure whole-cell peak responses GABA was instead applied locally through an 8-chamber manifold device (ALA Scientific Instruments) placed ∼150 μm from the recorded cell. Solutions were gravity fed via a solenoid valve system controlled using WinEDR software (John Dempster, University of Strathclyde, Glasgow, UK). Liquid junction currents, recorded in response to application of a 5% diluted solution across the open tip of a pipette, were used to estimate the solution exchange times (10–90% rise-time 308 ± 39 ms, n = 22).
Ultra-fast (1 ms duration) applications of GABA onto outside-out patches were achieved using a double-barreled application tool, made from theta glass (2 mm o.d.; Hilgenberg GmbH) pulled to a tip opening of ∼200 μm, and mounted on a piezoelectric translator (P-265.00, Physik Instrumente). “Control” and agonist solutions flowed continuously through the two barrels and solution exchange occurred when movement of the translator was triggered by a voltage step (pClamp10, Molecular Devices). To enable visualization of the solution interface and allow measurement of solution exchange, 2.5 mg/ml sucrose was added to the agonist solution and the control solution was diluted by 5%. The speed and duration of the solution exchange were estimated, respectively, from the 10–90% rise-time and half-height duration of the liquid junction current recorded from the open tip of the pipette at the end of each recording, after destroying the membrane patch (50–500 sweeps for each patch). Ultra-fast jumps at physiological temperature had a mean 10–90% rise-time of 202 ± 14 μs and a mean duration (at half-height) of 1.25 ± 0.04 ms (range: 0.94–1.84 ms, n = 30).
To generate a slower GABA transient, a GABA application pipette (tip diameter ∼1 μm) was moved across the outside-out patch at a speed of 2 mm/s using a stepper motor (MP-225, Sutter Instruments). When the application and recording pipette passed within <5 μm of each other the liquid junction currents were well described by a single Gaussian. The estimated mean duration of these liquid junction currents was 7.3 ± 1.7 ms (n = 5) with a 10–90% rise-time of 6.8 ± 1.3 ms, reaching a peak amplitude that was 55 ± 9% of the full amplitude recorded at steady state. Thus, with 1 mm GABA in the application pipette, a slowly rising and decaying GABA transient reaching a peak concentration of ∼500 μm was produced.
Temperature control for local GABA application or excised patches.
For whole-cell and excised patch recording with local GABA application at physiological temperature, warmed in-flowing bath solution was used to heat the GABA application pipettes or theta glass tools, the tips of which passed through the bath solution at a shallow angle for ∼0.75 cm. A miniature temperature probe was placed in the center of the chamber, or on the microscope objective, near the recording electrode. In recordings from excised patches, for example, a stable temperature of 36.3 ± 0.2°C (n = 30) was measured.
Electrophysiology—acute slice preparations.
Thalamic relay neurons in the dLGN were visually identified using a Zeiss Axioskop FS microscope (Carl Zeiss Ltd.) equipped with DIC-IR optics. Whole-cell recordings were made under voltage-clamp using a Multiclamp 700B amplifier (Molecular Devices) at physiological temperature (33–37°C). Recording pipettes were pulled from thick-walled borosilicate glass (GC-150F-10; Harvard Apparatus) and filled with “intracellular” solution, containing (in mm): 140 CsCl, 4 NaCl, 0.5 CaCl2, 10 HEPES, 5 EGTA, 2 Mg-ATP; the pH was adjusted to 7.3 with CsOH. Whole-cell voltage-clamp recordings were only included if the cell-attached seal resistance was >1 GΩ and the whole-cell series resistance was <25 MΩ. A fluorescent dye, Lucifer yellow or Alexa 488 (Sigma), was included in the intracellular solution (0.5 mg/ml) to allow for later confocal imaging of filled neurons. Thalamic relay neurons were identified in the dLGN based upon their soma size and input resistance; other small cells with high input resistance were considered as local interneurons (Bright et al., 2007; Bright and Brickley, 2008). During whole-cell recording, GABAAR-mediated responses were pharmacologically isolated by inclusion of the ionotropic glutamate receptor blocker kynurenic acid (0.5 mm; Sigma) in the recording solution.
Cerebellar granule neurons were identified from their position within the internal granule cell layer and their characteristically small input capacitance (∼3pF). Whole-cell recordings were made under voltage-clamp using an Axopatch 200A amplifier. Recordings in slices from wild-type mice, in which sIPSCs and mIPSCs were compared, were performed at physiological temperature. To maximize the yield of successful recordings from the limited numbers of animals available, experiments on slices from genetically modified mice were performed at room temperature (22–25°C). Recording pipettes and intracellular solutions were as for recordings from thalamic neurons. The following drugs were added to the external solution as indicated: 10 μm d-AP-5, 10 μm SR95531 (Research Biochemicals), 0.5 μm strychnine (Sigma), 5 μm CNQX (Tocris Bioscience) and 1 μm TTX (Tocris Bioscience).
Data analysis.
Data acquisition was performed using pClamp7, pClamp10, Axograph 4.2 (Molecular Devices) or WinEDR/WinWCP (John Dempster, University of Strathclyde, Glasgow, UK). Current records were filtered at 2 or 10 kHz and digitized at 10, 20 or 50 kHz using either a National Instruments (NI-DAQmx, PCI-6221) or a Digidata 1440A (Molecular Devices). Recordings from ultra-fast jump experiments were further filtered at 5 kHz (8-pole low-pass digital Bessel, Clampfit 10) and analyzed using Igor Pro 6.10 (WaveMetrics Inc.) with NeuroMatic 2.02 (http://www.neuromatic.thinkrandom.com). The decay of averaged ultra-fast jump currents (2–11 sweeps per patch) was described by one or, more often, two exponential functions. When fitted with two exponentials, the weighted time constant of decay (τw, decay) was calculated as the sum of the fast and slow time constants weighted by their fractional amplitudes. For simplicity, throughout the article this weighted time constant (as well as an equivalent fit-independent measure of decay applied to synaptic currents; see below, Analysis of neuronal recording) is referred to as τdecay. To generate the global average currents (shown below in Fig. 5), while preserving onset variability from different patches, averaged GABA-evoked currents from each patch were appended to the averaged liquid junction current (“step”) from the same patch and the concatenated traces from all patches realigned to the initial rising phase of the step before generating the global average. The single-channel amplitude from outside-out patches was determined from Gaussian fits to all-point histograms constructed from a section of the current record that did not contain superimposition of channel openings. This single channel amplitude was then used to estimate the maximum total charge transfer possible during this time period and the steady-state Popen was calculated by integrating the current record and dividing the measured charge by the estimated maximum total charge.
Figure 5.

Ultra-fast GABA applications onto α4β2δ and α1β2γ2S GABAARs at physiological temperature. A, Representative currents (superimposed) evoked by 1 ms application of 1 mm GABA to outside-out patches (−30 mV) taken from two tsA201 cells expressing α4β2δ or α1β2γ2S GABAARs. The vertical bar denotes the time of agonist application. B, Global averages illustrating the different current onset and decay in α4β2δ and α1β2γ2S GABAARs patches (n = 16 and 14, respectively, gray shaded areas denote SEM). The global average of the “open tip” liquid junction currents recorded at the end of each experiment is shown above the average traces (gray shaded areas denoting SEM are hidden by the average trace). C, Pooled data illustrating the slower rise-time and decay for currents mediated by α4β2δ (black, 16 patches) compared with α1β2γ2S receptors (gray, 14 patches; *p < 0.0001). Error bars indicate SEM. D, Top traces show representative currents (concatenated) evoked by ultra-fast GABA applications to an outside-out patch expressing α4β2δ GABAARs. Concentration jumps were repeated every 10 s, first from a control solution (left-hand set of currents), then from a solution containing 250 nm GABA (middle, gray bar) that was perfused for 3 min before resuming the jumps. Note that the peak current was reduced by >80% in this patch, recovering to 88% of control peak current after removal of 250 nm GABA. The bottom shows averaged traces obtained from the currents shown in the top. The vertical bar denotes the time of agonist application and the gray bar represents the 250 nm GABA treatments. Overall, pre-equilibration with 250 nm GABA did not change the 10–90% rise-time (from 2.2 ± 0.4 ms to 2.5 ± 0.4 ms; p = 0.2, n = 7) but accelerated the decay (from 76.2 ± 7.8 ms to 55.9 ± 5.2 ms; p = 0.02, n = 7). E, Same as D, for a patch from a cell expressing α1β2γ2S GABAARs. In the patch depicted, the peak current was reduced by only 32%, with 92% recovery. Pre-equilibration with GABA did not change the 10–90% rise-time or decay (from 0.30 ± 0.05 ms and 11.8 ± 0.8 ms to 0.30 ± 0.05 ms and 12.9 ± 0.6 ms; p = 0.98 and 0.45, respectively, n = 4).
Analysis of neuronal recordings.
For thalamic neurons, the series resistance, input resistance and capacitance were calculated from current responses to 10 mV hyperpolarizing voltage steps. For CGNs, series resistance and cell capacitance were read directly from the amplifier settings used to minimize the current responses to 5 mV hyperpolarizing voltage steps.
Synaptic events were analyzed using Axograph, Igor Pro, WinEDR/WinWCP or custom software (EVAN, courtesy of Istvan Mody, University of California, Los Angeles, CA). Event detection was performed using amplitude threshold crossing or scaled template matching. Events were aligned on their initial rising phases and averaged synaptic waveforms were constructed from IPSCs that exhibited monotonic rises and an uninterrupted decay phase. Average baseline current levels were calculated during a 10 ms epoch immediately before each detected event and the peak amplitude was determined relative to this value. The 10–90% rise-time was calculated between the start and the peak location with interpolation between samples. The decay constant of individual IPSCs was calculated as the charge transfer during the baseline corrected IPSC divided by the IPSC peak amplitude. This allowed a fit-independent estimate of the decay that could be determined for individual or averaged sIPSCs. To distinguish between fast- and slow-sIPSCs in thalamic neurons, histograms were constructed of 10–90% rise-time, peak amplitude and τdecay. These distributions were best described by multiple Gaussian functions (see Results). Average waveforms were constructed from either fast-rising/fast-decaying events or slow-rising/slow-decaying events based upon the cutoff values defined by the intersection between the Gaussian fits. These selected events were also used for peak-scaled nonstationary fluctuation analysis (ps-NSFA).
For both thalamic and cerebellar recordings, each averaged sIPSC (constructed from a minimum of 50 events) was scaled to the peak of individual contributing sIPSCs and the waveforms subtracted to determine the variance associated with the decaying phase of each event. Plots of mean current versus peak-scaled variance were fitted with a parabolic function of the form,
where σPS2 is the peak-scaled variance, Ī is the mean current, i is the weighted mean single-channel current, NP is the number of channels open at the peak of the IPSC, and σB2 is the background variance. However, the goal here was not to determine the single-channel current, but to examine deviations from the expected symmetrical parabolic relationship between mean current and current variance. To this end, each current-variance relationship was integrated, and the ratio of the areas either side of the median current was used to provide a fit-independent measure of skew.
For acute slice experiments, the GABA-mediated tonic conductance was calculated from the difference between the baseline current amplitude recorded before and after GABAA receptor block. SR-95531 and picrotoxin were used interchangeably at saturating concentrations (50–100 μm) to block all GABAA receptors. The baseline current amplitude was determined from a single Gaussian fit to the all-point histogram constructed from a 500 ms epoch that did not contain sIPSCs. To account for variability in cell size, the tonic conductance was normalized to input capacitance (pS/pF).
Concentration–response data were fitted with a Hill equation:
 |
where I is the normalized mean baseline current, Imax is the maximum response, [GABA] is the GABA concentration, and nH is the slope of the curve. For steady-state dose–response curves, data from each experiment were normalized to the response to a 10 μm GABA concentration. For peak dose–response curves data were normalized to the response with 1 mm GABA. Fits to this Hill equation were applied to data collected from individual cells and the mean EC20, EC50, and EC80 values calculated.
Morphological analysis of thalamic neurons.
Following electrophysiological recording, slices were fixed in PBS containing 4% paraformaldehyde for 24 h before being washed in PBS and mounted in Vectashield mounting medium (Vector Labs). Optical sections were obtained using a Zeiss LSM 510 upright confocal microscope, equipped with a 40× oil-immersion objective. Fluorescence was visualized using the 458 nm line of an argon laser, with emitted light passing through a 475 nm long-pass filter. Neurons were z-sectioned into 1 μm optical sections. The signal-to-noise ratio was improved by averaging at least two scans for each z-section. Morphological analysis of neurons was performed using the software program Reconstruct (version 1.0.9.6) (Fiala, 2005). For each optical section, the x, y, z coordinates and section radius were recorded enabling parameters, such as dendritic length and surface area to be measured (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Statistical analysis.
Statistical tests were performed using STATISTICA (StatSoft) or Prism (GraphPad Software). Unless stated otherwise, differences between groups were examined using the appropriate paired or unpaired Student's t test. When data were not normally distributed (Shapiro–Wilk test), we used a Mann–Whitney U test or Wilcoxon signed-rank test (as indicated). Differences were considered significant at p < 0.05.
Results
We made whole-cell voltage-clamp recordings at physiological temperature (37–38°C) from 125 dLGN and 35 vLGN relay neurons in slices prepared from adult (68 ± 3-d-old) C57BL/6J mice. Full morphological reconstruction followed by quantitative analysis of soma size, polarity scores and spine density, resulted in the identification of 10 X-type and 8 Y-type relay neurons in the dLGN and 5 X-type relay neurons in the vLGN. X-type neurons were distinguished by the significantly more polar arrangement of their dendrites (e.g., average polar score for dLGN X-type cells, 4.1 ± 0.6 vs 1.0 ± 0.1 for radial Y-type neurons; p = 0.0004) (see supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In X-type relay neurons of both vLGN and dLGN, we observed not only conventional fast-rising and fast-decaying spontaneous IPSCs (“fast-sIPSCs”) but also a population of slowly rising and slowly decaying IPSCs (“slow-sIPSCs”) (Fig. 1A,B). Multiple Gaussian fits to these rise- and decay-time distributions were used to define criteria for isolation of fast- and slow-sIPSCs, as shown in Figure 1A for an X-type cell of the vLGN. Similar slow-sIPSCs were seen in X-type relay neurons of the dLGN (Fig. 1B), where they represented 19 ± 4% of sIPSCs. These slow-sIPSCs occurred at a frequency of 0.46 ± 0.12 Hz and each event produced a significantly greater charge transfer than did fast-sIPSCs (79.7 ± 9.6 fC vs 31.3 ± 4.0 fC, p = 0.0002). Some sIPSCs with a fast rise exhibited a slowly decaying component, but slow-rising and fast-decaying sIPSCs were rarely observed in X-type relay neurons of the dLGN (3.7 ± 1.1% of all events) or vLGN (4.2 ± 0.5%). Furthermore, the proportion of fast-rising and slow-decaying IPSCs was, on average, not significantly different between dLGN and vLGN X-type relay neurons (40.0 ± 7.2% vs 44.8 ± 4.9%, respectively, p = 0.6). Therefore, the absence of δ-GABAARs from X-type relay neurons of the vLGN does not influence sIPSC properties in this preparation. Applying identical selection criteria to sIPSCs from Y-type relay neurons of the dLGN (that lack glomerular synapses) (Fig. 1C) showed the presence on average of threefold fewer slow-sIPSCs (6 ± 2% of total, p = 0.02; Mann–Whitney U test) but, no significant difference in the frequency of fast-sIPSCs (2.5 ± 1.3 Hz in X-type cells vs 1.2 ± 0.5 Hz in Y-type cells, p = 0.6, Mann–Whitney U test).
Figure 1.

Comparison of sIPSCs recorded from X- and Y-type relay neurons at physiological temperature. A, Coronal view of a reconstructed thalamic relay neuron with X-like morphology recorded from the vLGN. Note the clear polar distribution of dendrites (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Below are representative fast-rising, fast-decaying sIPSCs (black) and slower-rising and -decaying sIPSCs (gray) recorded from this cell (−60 mV). Plots of 10–90% rise-time against τdecay and peak amplitude for all sIPSCs recorded from this cell are shown on the right. The dashed lines denote the cutoff values for event grouping that were determined from the histograms of rise-time, τdecay, and peak amplitude (see Materials and Methods) (note, in B and C, the histograms themselves are omitted for clarity). The slow-rising and -decaying sIPSCs are represented in the upper right-hand quadrant. From comparison with the peak versus rise-time plot beneath, it is clear that these slow-sIPSCs tended to be small in amplitude. On average, the slow-sIPSCs in this cell had a mean 10–90% rise-time of 1.12 ± 0.06 ms and a τdecay of 7.01 ± 0.13 ms. Corresponding values for the fast-sIPSCs were 0.305 ± 0.002 ms and 4.06 ± 0.08 ms. B, Corresponding reconstruction and sIPSC analysis from a representative thalamic relay neuron with X-like morphology recorded from the dLGN, also showing both fast- and slow-sIPSCs. On average, the slow-sIPSCs in this cell had a mean 10–90% rise-time of 0.9 ± 0.1 ms and a τdecay of 3.89 ± 1.55 ms. Corresponding values for the fast-sIPSCs were 0.25 ± 0.001 ms and 1.68 ± 1.09 ms. C, Coronal view of a reconstructed thalamic relay neuron with clear Y-like morphology recorded from the dLGN; in this example, the dendrites emerge from the central soma with a radial distribution (see also supplemental Fig. 1, available at www.jneurosci.org as supplemental material). In this cell, all sIPSCs were fast-rising and -decaying (mean 10–90% rise-time of 0.213 ± 0.002 ms and a τdecay of 1.98 ± 0.04 ms). D, Continuous current recordings (−60 mV) from a representative X-type vLGN neuron and X- and Y-type dLGN neurons, showing the effects of 100 μm SR-95531. Current levels before and after antagonist application were determined from Gaussian fits to all-point histograms (gray). Dashed lines indicate the control current levels. In the vLGN cell, block of GABAARs resulted in loss of all sIPSCs, with no change in the holding current, indicating the absence of a GABA-mediated tonic conductance. By contrast, clear GABAAR-mediated tonic currents were seen in both neurons of the dLGN.
Although entrapment of GABA within glomeruli of X-type relay neurons might be expected to favor tonic GABAA receptor activation, blocking GABAARs with SR-95531 (or picrotoxin, data not shown) revealed no significant difference in the magnitude of the tonic conductance between X- and Y-type relay neurons of the dLGN (Fig. 1D). The tonic GABAA receptor conductance was 16.4 ± 3.8 pS/pF (n = 10) in the X-type relay neurons compared with 19.9 ± 9.2 pS/pF (n = 6) in the Y-type relay neurons (p = 0.8). Therefore, even though X-type relay neurons exhibited a greater proportion of slow-sIPSCs this was not associated with enhanced δ-GABAAR activation, as assayed by presence of tonic conductance. As expected from our previous studies (Bright et al., 2007), no tonic conductance was seen in cells of the vLGN (1.6 ± 1.7 pS/pF; n = 4) that lack δ-GABAARs (Fig. 1D). Again, consistent with previous observations (Bright et al., 2007), there was no relationship between the instantaneous sIPSC frequency and the magnitude of the tonic conductance in dLGN relay neurons and pooling data from 125 relay neuron recordings (regardless of morphological classification) also revealed no correlation between the average sIPSC decay and the magnitude of the tonic conductance (data not shown). These data support the view that δ-GABAARs do not shape the synaptic response of dLGN relay neurons and suggest that conventional low affinity γ subunit-containing GABAARs mediate the slow spillover-like currents seen in thalamic neurons of both the dLGN and vLGN.
Tonic and spillover-like currents in adult CGNs at physiological temperatures
Do the conclusions drawn from thalamic relay neurons extend to other neuronal types that also express high-affinity δ-GABAARs? To answer this question we examined the contribution of these receptors to the tonic conductance and spillover currents in CGNs of adult mice at physiological temperature. There was no relationship between the basal frequency of sIPSCs (0.7 ± 0.2 Hz, n = 29) and the magnitude of the tonic conductance (229 ± 25 pS/pF) across cells (Fig. 2A). However, block of action potential-dependent GABA release by bath application of TTX (500 nm) caused a large reduction in IPSC frequency (from 5.7 ± 0.9 Hz to 0.2 ± 0.1 Hz; p = 0.0002), which was associated with a significant reduction in the tonic conductance (from 321.8 ± 59.8 pS/pF to 148.2 ± 27.4 pS/pF, n = 7, p = 0.005) (Fig. 2B). A similar reduction in the tonic conductance was observed following bath perfusion of nominally Ca2+-free solution (175.3 ± 52.9 pS/pF vs 98.9 ± 49.9 pS/pF, n = 5, p = 0.01). Analysis of IPSC kinetics further established that blocking action potential-dependent GABA release onto CGNs significantly reduced the τdecay (Fig. 2C), from 7.9 ± 1.2 ms for sIPSCs (n = 11) to 4.8 ± 0.4 ms for mIPSCs recorded in the presence of TTX (n = 10, p = 0.03). These results are consistent with the established view that the slow component of sIPSCs reflects spillover of GABA that is not seen with quantal events (e.g., dentate gyrus granule cells) (Wei et al., 2003).
Figure 2.
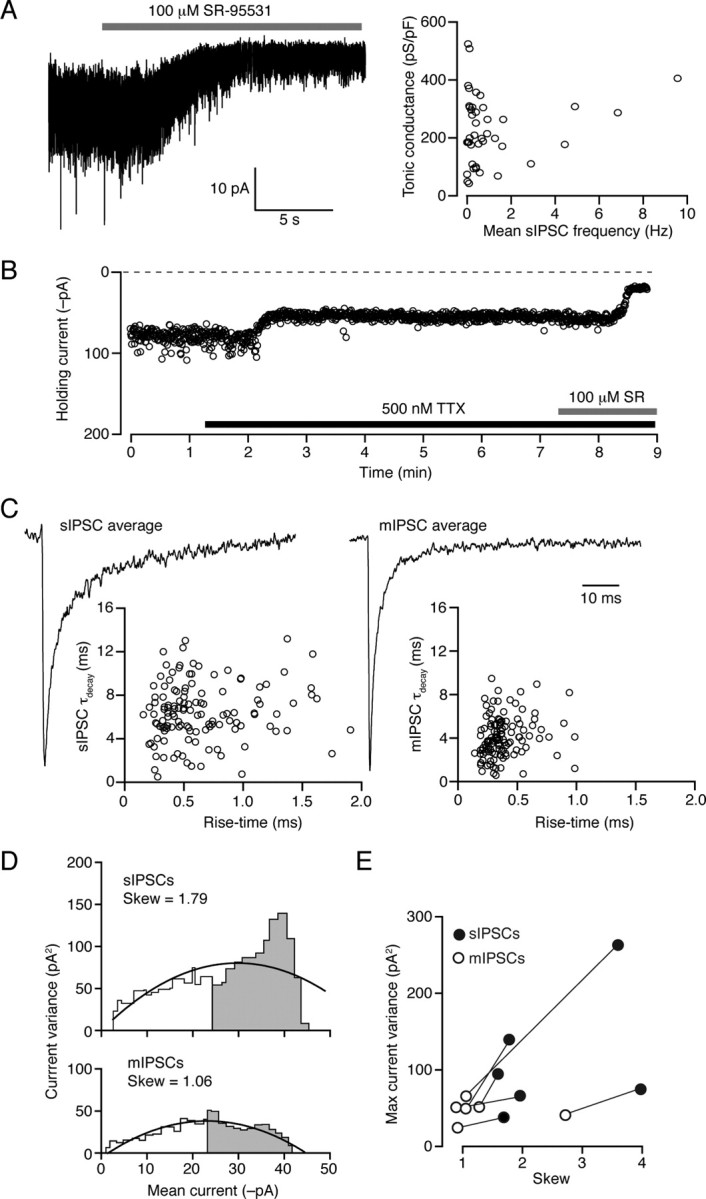
Functional properties of sIPSCs in CGNs from adult mice at physiological temperature. A, Continuous current record (−70 mV) during application of SR-95531 (100 μm). The change in holding current was calculated as shown in Figure 1D, and used to calculate the tonic GABAA receptor-mediated conductance. Right-hand plot shows lack of correlation between normalized tonic conductance (pS/pF) and sIPSC frequency (correlation coefficient 0.002). B, Representative plot from an adult CGN showing changes in holding current produced by bath application of 500 nm TTX; block of action potential-dependent GABA release resulted in a reduction in the holding current, which was reduced further by coapplication of 100 μm SR-95531. C, Scaled average IPSC waveforms from a different cell, before (sIPSC) and after (mIPSC) the application of 500 nm TTX. As illustrated in the scatter plots (below), the faster decay of the averaged mIPSC reflects the absence of slow action potential-dependent events. D, Plots of current variance against mean current for the sIPSCs and mIPSCs shown in C. In this cell, the plot from mIPSCs exhibits a clear reduction in skew (gray area/white area; see Materials and Methods) and maximum current variance compared with that from sIPSCs. E, Pooled data from six CGNs in which current-variance plots were produced for both sIPSCs and mIPSCs. TTX reduced the skew and the maximum variance in all cells. TTX also reduced the mean peak amplitude from −75.5 ± 7.0 pA to −49.7 ± 4.9 pA (n = 7; p = 0.0005).
Slow-sIPSCs in CGNs and thalamic relay neurons are consistent with spillover
If the slow component of sIPSCs reflects spillover of GABA from the synaptic cleft, causing activation of receptors distant from the local release site, GABAARs contributing to this component would be expected to open for the first time after the peak of the sIPSC. We found support for this possibility in the results of peak-scaled nonstationary fluctuation analysis (ps-NSFA), a method commonly used to determine synaptic channel conductance based upon the analysis of current variance arising from stochastic channel closure after the peak of the synaptic event. When ps-NSFA (see Materials and Methods) was applied to sIPSCs from six wild-type CGNs at physiological temperature, the relationship between mean current and current variance was skewed (skew = 2.2 ± 0.4; see Materials and Methods for calculation), with a maximum current variance of 82.5 ± 33.1 pA2. However, a less skewed relationship was observed when the same analysis was applied to mIPSCs from the same cells (skew = 1.3 ± 0.3; p = 0.017) and the maximum current variance was reduced in all cells, with a mean value of 47.0 ± 5.5 pA2 (Fig. 2D,E). Although deviations from the expected symmetrical parabolic relationship between mean current and current variance can arise through various mechanisms (for discussion see Hartveit and Veruki, 2006), a skewed relationship and increased current variance would be at least consistent with spillover of GABA contributing to delayed channel opening (see also supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
We next applied the same analysis to data collected from thalamic relay neurons, recorded at physiological temperatures, to determine whether similar characteristics were also associated with slow-sIPSCs in these cells. The relationship between current variance and mean current was calculated for the decay of fast- and slow-sIPSCs in seven X-type thalamic relay neurons. For slow-sIPSCs, where we speculate that spillover influences the decay kinetics, the plots showed conspicuous skew of 5.5 ± 1.4, with a maximum current variance of 54.1 ± 12.6 pA2. These values were significantly greater than those seen with fast-sIPSCs (1.8 ± 0.2 and 26.7 ± 4.7 pA2; p = 0.025 and 0.017). For the fast-sIPSCs, these parameters were comparable to those found in Y-type thalamic relay neurons that lack slow-sIPSCs (2.0 ± 0.2 and 44.2 ± 10.6 pA2; p = 0.56 and 0.20). Therefore, in both CGNs and thalamic relay neurons the current variance associated with the IPSC decay was reduced when the putative spillover current was absent and the relationship between mean current and current variance was less skewed.
Extrasynaptic δ-GABAARs do not mediate spillover currents in CGNs
We next analyzed sIPSCs from CGNs of genetically modified mouse strains to determine the subunit composition of the GABAARs underlying the spillover current in CGNs. We have demonstrated previously that the tonic conductance recorded at room temperature is absent from adult CGNs in both δ−/− mice (Stell et al., 2003) and α6−/− mice (Brickley et al., 2001). In the latter case, this occurs because CGNs lose surface expression of both the α6 and the δ subunit, which is degraded in the absence of the α6 protein (Jones et al., 1997). We now show that the sIPSCs recorded from CGNs of mature α6−/− mice at room temperature decay significantly faster than those in wild-type mice (15.0 ± 1.3 ms, n = 11 in wild-type vs 8.7 ± 1.0 ms, n = 6 in α6−/−, p = 0.002 Fig. 3A). Subtraction of the global average sIPSC waveforms revealed a difference current with a slow rise and decay (10–90% rise ∼7 ms; τdecay ∼50 ms). By contrast, there was no difference between the kinetics of sIPSCs recorded from CGNs of wild-type and δ−/− mice (13.2 ± 1.4 ms, n = 12 in wild-type vs 12.6 ± 1.4 ms n = 11 in δ−/−, p = 0.8 Fig. 3B). These results suggest that α6-, but not δ-containing receptors underlie a slow spillover-mediated component of IPSC decay. An alternative interpretation of the data would be that the difference current reflects the kinetic behavior of synaptic α6-containing receptors. If this were true, the absence or selective block of α6-containing receptors would be expected to speed the decay of IPSCs. This was not the case. For mIPSCs (that lack a spillover-mediated component) there was no significant difference between decay kinetics of events recorded from wild-type and α6−/− CGNs (τdecay 6.0 ± 1.2 ms, n = 6 for wild-type vs 7.9 ± 0.6 ms, n = 9 for α6−/−; p = 0.1). Similarly, blocking α6-containing receptors with the selective antagonist furosemide had no effect on mIPSC decay kinetics (τdecay 6.3 ± 0.7 ms; n = 4, p = 0.9) (Fig. 3C). Importantly, the effectiveness of furosemide was confirmed in the same recordings, where, as expected (Brickley et al., 2001; Wall, 2002; Crowley et al., 2009), it reduced both the tonic conductance and the peak synaptic current (Fig. 3C). Overall, our results are consistent with the view that transmitter spillover leads to the delayed activation of GABAARs. However, contrary to expectations, high-affinity δ-GABAARs do not appear to be involved in the generation of these slow spillover currents within thalamic or cerebellar glomeruli.
Figure 3.

Functional properties of sIPSCs in CGNs from adult mice at room temperature. A, Global average waveforms of sIPSCs from wild-type (black; n = 11) and α6−/− (gray; n = 6) CGNs. The two averages have been subtracted to give the difference current (dark gray). The inset shows a plot of τdecay (normalized to the mean of the control data) for all sIPSCs recorded from these cells. Note the faster τdecay for α6−/− CGNs. Asterisk indicates p < 0.05. B, The same as in A, but for recordings from δ−/− CGNs. Note the superimposition of the traces. The inset shows normalized τdecay for sIPSCs recorded from wild-type (n = 12) and δ−/− CGNs (n = 11). C, Current record from a representative CGN (dashed line shows zero current level). Application of 500 nm TTX reduced the tonic conductance and baseline current variance, allowing resolution of mIPSCs that were smaller than sIPSCs (−19.9 ± 4.6 pA vs −30.3 ± 4.4 pA; n = 5, p = 0.05) and occurred at a lower frequency (∼0.1 vs ∼0.8 Hz). The application of 200 μm furosemide reduced mIPSC amplitude, with no significant effect on decay. Average IPSC waveforms from this cell are shown below. In 5 paired experiments the mIPSC peak amplitude decreased from −19.5 ± 3.2 pA to −16.5 ± 3.0 pA (p = 0.03) in the presence of furosemide, with no effect on τdecay (6.0 ± 1.2 ms to 5.9 ± 0.7 ms, p = 0.8). Importantly, as would be predicted if the action of furosemide was mediated solely by α6-containing GABAA receptors, this effect on mIPSC amplitude was absent in α6−/− mice (−12.4 ± 2.3 pA vs −13.1 ± 3.1 pA, p = 0.3).
δ-GABAARs are insensitive to brief GABA transients in the presence of ambient GABA
For ligand-gated ion channels, such as glutamate or GABAA receptors, pre-equilibration with a low concentration of agonist can promote receptor desensitization and reduce the amplitude of the current evoked by brief exposure to a high concentration of agonist designed to mimic synaptic release (Colquhoun et al., 1992; Overstreet et al., 2000; Lagrange et al., 2007). Although plausible estimates of ambient GABA concentrations in the brain are available (Santhakumar et al., 2006; Wu et al., 2007), there are no data regarding the potency of such GABA concentrations on γ- or δ-GABAARs at physiological temperatures. To address this, we coexpressed α1, α4 or α6 subunits with β2 and δ or γ2S subunits in tsA201 cells to yield recombinant α4β2δ, α6β2δ or α1β2γ2S receptors, and recorded GABA-evoked currents at ∼37°C. We found that δ-GABAARs receptors exhibited considerable desensitization. Thus, with α4β2δ receptors, the initial peak of the whole-cell response to bath application of 1 μm GABA was reduced by 87 ± 6% (n = 8) at steady state (Fig. 4A). As little as 10 nm GABA produced measurable whole-cell steady-state currents, and the steady-state concentration–response relationship was well described by a Hill equation (see Materials and Methods) with an EC50 of 243 ± 5 nm (EC20 92 ± 3 nm, EC80 645 ± 40 nm) (Fig. 4A, inset).
Figure 4.
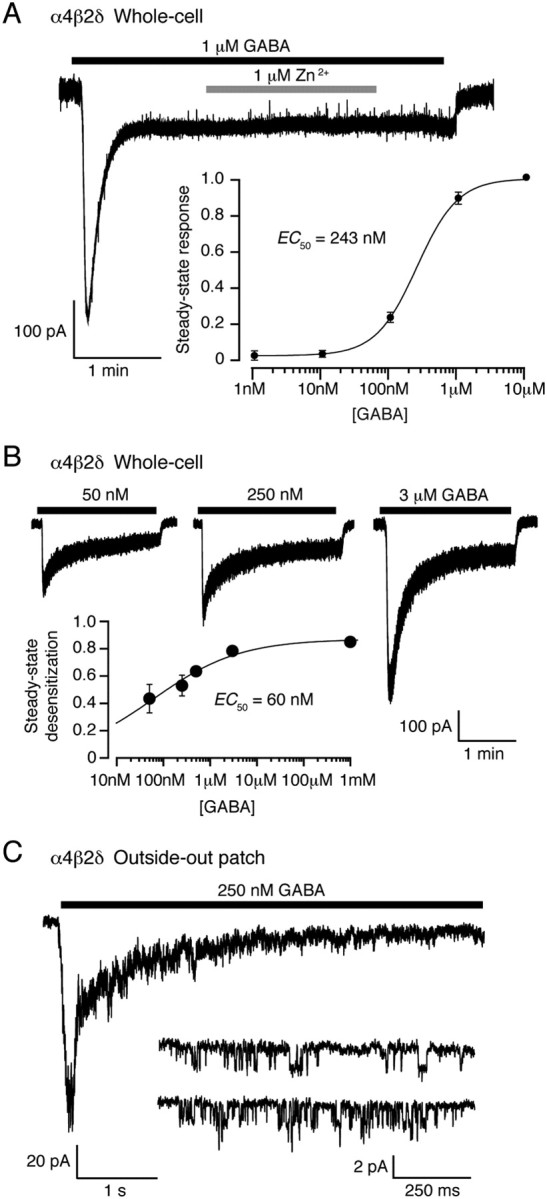
Steady-state desensitization of α4β2δ GABAARs at physiological temperature. A, Whole-cell record from a tsA201 cell expressing α4β2δ GABAARs (−30 mV, 37°C) during bath application of 1 μm GABA. Note the rapid desensitization to a steady-state response. Application of 1 μm Zn2+, which at low concentrations blocks αβ but not αβδ GABAARs (see Materials and Methods), did not affect the steady-state current, consistent with incorporation of the δ subunit into the GABAAR assembly. The inset shows the steady-state concentration–response curve. The solid line is a fit of the Hill equation, giving an EC50 of ∼250 nm. B, Whole-cell responses from a single tsA201 cell (−30 mV) in response to local GABA application. The peak and steady-state currents were used to construct the steady-state desensitization plot (inset). The solid line is a fit of the Hill equation, giving a desensitization EC50 of 60 nm. C, Current from an outside-out patch excised from a tsA201 cell expressing α4β2δ GABAARs (−30 mV) in response to application of 250 nm GABA. The inset shows single-channel openings recorded from the same patch, at steady state.
To more accurately delineate peak responses, we made additional whole-cell recordings with local application of GABA (see Materials and Methods for details) (Fig. 4B). For α4β2δ receptors, the ratio of steady-state to peak currents gave an EC50 for desensitization of only 60 nm (Fig. 4B, inset). Extensive desensitization was also seen in outside-out patches, where application of 250 nm GABA gave rise to currents with an amplitude at steady state that was only 10% of the peak response (Fig. 4C). Analysis of single-channel openings in the continued presence of 250 nm GABA (∼EC50 for steady-state current activation) (Fig. 4C, inset) indicated a main conductance level of 46.4 ± 5.6 pS and a steady-state Popen of 0.02 ± 0.01 (n = 8). Although this Popen value is low, maintained channel activity in the continued presence of 250 nm GABA is consistent with the idea that persistent channel opening underlies the tonic conductance in neurons. Similar results were obtained for α6β2δ receptors (data not shown). Together, these results suggest that steady-state desensitization of δ-GABAARs during exposure to ambient GABA concentrations would limit the ability of these high-affinity receptors to respond to brief changes in GABA concentration.
We tested this prediction for “synaptic-like” GABA transients, using ultra-fast application of GABA (1 mm, 1 ms) to outside-out patches containing α1β2γ2S or α4β2δ receptors (see Materials and Methods). GABA-evoked currents were characterized by monotonic rises and slower exponential decays (Fig. 5A,B). For α1β2γ2S GABAARs, the mean 10–90% rise-time was 0.48 ± 0.07 ms and the τdecay (see Materials and Methods for details) was 13 ± 2 ms (n = 14). For α4β2δ GABAARs, both the rise and decay times were significantly slower (2.5 ± 0.3 ms and 77 ± 4 ms, respectively; n = 16, both p < 0.0001) (Fig. 5C). To determine the effect of ambient GABA, we applied repeated 1 mm jumps (0.1 Hz) either from (and back to) control solution or 250 nm GABA. In the latter case this followed a 3 min equilibration in 250 nm GABA (Fig. 5D,E). In line with our observations from whole-cell recordings, continuous exposure to this low concentration of GABA induced a substantial desensitization of α4β2δ receptors, whose peak response to ultra-fast application of 1 mm GABA was significantly reduced (87 ± 2% reduction, n = 10, p = 0.031 Wilcoxon signed-rank test) (Fig. 5D). In 8 cells, where tested, 1 min after removal of 250 nm GABA this block was largely reversed (74 ± 7% recovery p = 0.125 vs control, Wilcoxon signed-rank test). We next repeated this series of experiments with α1β2γ2S receptors. Consistent with the relatively low-affinity of these γ-GABAARs, and in marked contrast to δ-GABAARs, the same low concentration of GABA had no significant effect (24 ± 4% reduction, n = 6, p = 0.125; 102 ± 6% after washout of ambient GABA, n = 5, p = 0.75 vs control, both Wilcoxon signed-rank test). These experiments show that when exposed to a low background GABA concentration at physiological temperatures, the high-affinity and the conspicuous desensitization of δ-GABAARs severely limit their ability to respond to synaptic-like GABA transients.
We also exposed outside-out patches to a slowly rising and decaying GABA transient (duration 7 ± 2 ms, peak concentration ∼500 μm) (Fig. 6) (see Materials and Methods for details). This was not designed to mimic a specific synapse, but simply to complement the ultra-fast jumps by providing a less abrupt GABA exposure, as might be experienced by extrasynaptic receptors during spillover from the synaptic cleft. Outside-out patches containing α1β2γ2S or α4β2δ receptors responded to these spillover-like GABA transients with currents that had slower monotonic rises and decays than those elicited by ultra-fast GABA applications (Fig. 6A). For α1β2γ2S receptors the average 10–90% rise-time was 4.7 ± 0.6 ms and the average τdecay was 73.2 ± 15.1 ms (n = 7). The corresponding values for α4β2δ receptors were 10.4 ± 8.9 ms and 305.9 ± 53.1 ms (n = 11). Following a 3 min equilibration with 250 nm GABA, responses from α4β2δ receptors to GABA transients were reduced by 91 ± 1% (n = 10, p = 0.005, Wilcoxon signed-rank test) (Fig. 6B) while responses from α1β2γ2S receptors were unaltered, being reduced by only 10 ± 10% (n = 3, p = 0.1, Wilcoxon signed-rank test) (Fig. 6C). These experiments suggest that in the presence of ambient GABA at physiological temperatures, the desensitization of δ-GABAARs severely limits their ability to detect spillover-like GABA transients.
Figure 6.
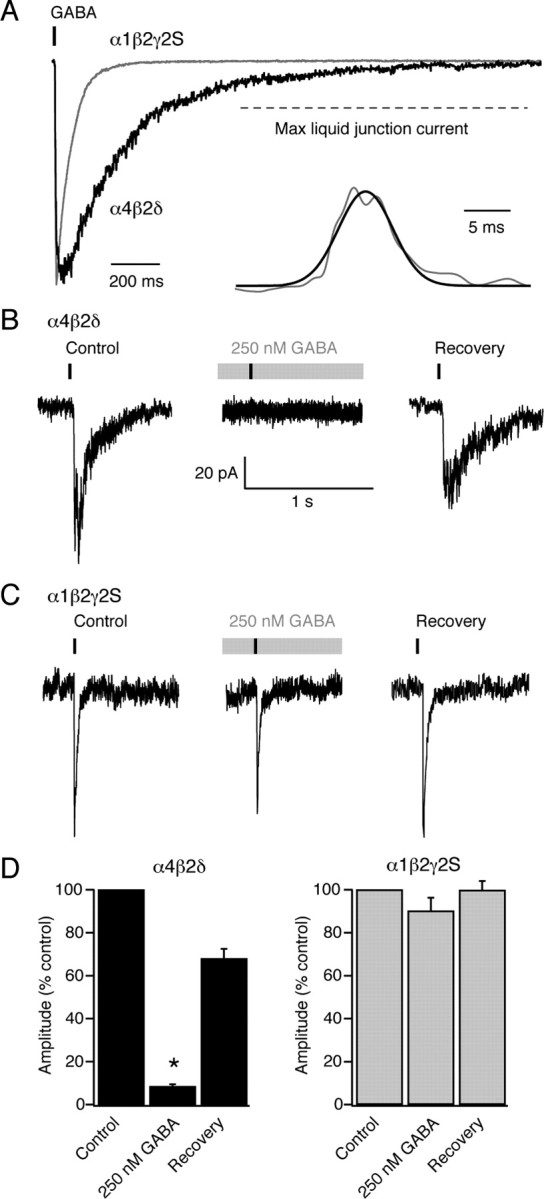
Response of recombinant α4β2δ and α1β2γ2S GABAARs to spillover-like GABA transients recorded at physiological temperature. A, Superimposed examples of patch responses (−30 mV) during the slow (7 ms) application of ∼500 μm GABA. Application of GABA was achieved by moving a GABA-containing pipette across the mouth of the recording pipette at a speed of 2 mm/s. Inset shows a liquid junction current recorded from the open tip of a recording pipette. The superimposed Gaussian fit had a half-width of 7 ms and reached a peak amplitude that was 55% of the maximum current. B, Currents from a patch expressing α4β2δ GABAARs that was exposed to a series of GABA transients, first from a control solution (left-hand trace), then from a solution containing 250 nm GABA (middle, gray bar) that was perfused for 3 min before resuming the jumps. There was no response in the presence of 250 nm GABA, but recovery was observed on washout of 250 nm GABA. C, The same experimental protocol as in B, but with α1β2γ2S GABAARs. Note the maintained responses in the presence of 250 nm GABA. D, Pooled data showing the effect of exposure to 250 nm GABA on the amplitude of currents evoked from α4β2δ receptors (n = 5) and α1β2γ2S receptors (n = 3). Error bars denote SEM. Asterisk denotes p < 0.05.
Discussion
Early in situ hybridization studies led to the suggestion that δ-GABAAR expression is a common feature of cells with glomerular synapses, including cells in the olfactory bulb, cerebellar cortex, and thalamus (Shivers et al., 1989). It was later suggested that ambient GABA concentrations were raised within glomeruli, leading to the persistent activation of δ-GABAARs and the generation of a novel form of tonic conductance (Brickley et al., 1996, 2001; Wall and Usowicz, 1997; Rossi and Hamann, 1998). Whether or not specific receptor populations are able to support spillover-mediated transmission is determined by their distance from the release site, affinity for GABA, and gating behavior. GABA spillover is often discussed in relation to the activation of high-affinity extrasynaptic δ-GABAARs within glomerular structures, leading to the widely held view that δ-GABAARs mediate both tonic and spillover currents (Rossi and Hamann, 1998; Wei et al., 2003; Farrant and Nusser, 2005; Glykys and Mody, 2007). Our findings challenge this assumption, and suggest that ambient GABA and synaptic spillover are sensed by distinct GABAAR populations.
In CGNs, the fact that neurosteroids did not modulate the slowly decaying component of IPSCs (Hamann et al., 2002), was originally taken as evidence in support of δ-GABAAR activation following transmitter spillover. This reasoning was based on a supposed insensitivity of δ-GABAARs to neurosteroids. It is now widely accepted that δ-GABAARs are, in fact, highly sensitive to neurosteroid modulation (Hosie et al., 2009) and our results from CGNs of δ−/− mice indicate that δ-GABAARs are not involved in synaptic spillover. In the case of thalamic relay neurons, the greater prevalence of slow-sIPSCs in X-type neurons, compared with adjacent Y-type cells, strengthens the suggestion that GABA spillover is accentuated within glomeruli. However, these data also argue against a role for δ-GABAAR activation during GABA spillover. Consistent with our previous observations (Bright and Brickley, 2008), the prevalence of spillover-like IPSCs shows little correlation with the magnitude of the tonic conductance and we find that slow-sIPSCs are also present in neurons of the vLGN, cells that lack δ-GABAARs. Due to a lack of published anatomical data, we cannot comment on the location of δ-GABAARs within thalamic glomeruli. However, in CGNs it is clear that δ-GABAARs are located at perisynaptic locations within glomerular structures (Nusser et al., 1998). Therefore, the inability of δ-GABAARs to sense GABA spillover within CGNs is best explained by the desensitization of these receptors in the presence of ambient GABA.
Our heterologous expression studies demonstrate that in the presence of low ambient GABA concentrations δ-GABAARs are insensitive to brief GABA transients. These experiments were performed using the EC50 concentration for steady-state activation (250 nm), a concentration that is within the range of expected ambient GABA concentrations (Kékesi et al., 1997; Santhakumar et al., 2006; Wu et al., 2007). For γ-GABAARs, the effective “removal,” by slow desensitization, of a fraction of synaptic receptors has been discussed previously (Overstreet et al., 2000; Lagrange et al., 2007), but this phenomenon has been largely ignored when considering δ-GABAARs, as these receptors were considered to exhibit only modest desensitization. Consistent with recent reports from studies performed at room temperature (Feng et al., 2009; Mortensen et al., 2010), we now show that at physiological temperatures δ-GABAARs are largely desensitized in the presence of steady-state EC50 GABA concentrations.
This finding, together with our data from different thalamic relay neurons, leads us to conclude that δ-GABAARs do not contribute to any aspect of synaptic transmission in the thalamus. Other evidence in support of this view comes from experiments using the δ-GABAAR selective agonist gaboxadol, which can increase the tonic conductance in thalamic relay neurons without affecting the kinetics or amplitude of sIPSCs (Cope et al., 2005). This is indeed what would be expected if slow-sIPSCs reflect activation of conventional γ-GABAARs within thalamic glomeruli. A similar conclusion can be drawn from our experiments with CGNs, where knock-out data suggest that α6-containing/δ-lacking GABAARs contribute to the spillover component of the IPSC.
Although we have shown that δ-GABAARs do not sense synaptic spillover, they are clearly implicated in the generation of the tonic conductance. For example, in CGNs (Brickley et al., 2001; Stell et al., 2003), thalamic relay neurons (Porcello et al., 2003; Herd et al., 2009) and dentate granule neurons of the hippocampus (Stell et al., 2003; Wei et al., 2003), removal of the δ subunit results in the complete loss of the tonic conductance. Likewise, alteration of δ-GABAAR number influences the magnitude of the tonic conductance(Maguire et al., 2005; Maguire and Mody, 2008). The tonic activation of these receptors under physiological conditions results from their high affinity for GABA and the presence of ambient GABA. In turn, the extracellular concentration of GABA is thought to reflect the equilibrium state of GABA transporters, and thus the intracellular GABA concentration, the membrane potential and the transmembrane distribution of Na+ and Cl− (Attwell et al., 1993; Richerson and Wu, 2003; Wu et al., 2006). On this basis, the theoretical ambient GABA concentration is predicted to be in the range 10–400 nm (Wu et al., 2007). These estimates are consistent with microdialysis studies that report a range of ambient GABA concentrations from 30 nm (de Groote and Linthorst, 2007) to 300 nm (Richter et al., 1999; Kennedy et al., 2002; Bianchi et al., 2003; Xi et al., 2003). These GABA concentrations are sufficient to activate and desensitize high-affinity extrasynaptic δ-GABAARs but will have little influence on conventional low affinity γ-GABAARs.
Tonic activation of GABAA receptors produces powerful shunting inhibition (Mitchell and Silver, 2003) that can modulate cell or network excitability in various ways (Cope et al., 2005; Vida et al., 2006; Bright and Brickley, 2008; Mann and Mody, 2010). In thalamic relay neurons, for example, the tonic current generates a persistent inhibition, modulation of which affects the balance between regular and burst firing (Cope et al., 2005). Such changes have been implicated in transitions between sleep and wake states (Belelli et al., 2005; Wafford and Ebert, 2006; Harrison, 2007) and in the generation of absence seizures (Cope et al., 2009). The importance of δ-GABAARs in regulating states of consciousness is further highlighted by the potency with which certain anesthetics enhance the tonic conductance in both thalamic and neocortical neurons (Orser, 2006). While our data confirm a role for δ-GABAARs in the generation of tonic conductance, our findings demonstrate that these receptors are poorly suited to the detection of GABA spillover and suggest that activation of δ-GABAARs should be considered functionally isolated from synaptic release. In this context, it is interesting to note the recent suggestion that a sizeable fraction of tonic conductance in CGNs arises from the activity of receptors activated by GABA released from glial cells through Bestrophin 1 anion channels (Lee et al., 2010). Finally, our study highlights why the effectiveness of drugs such as alcohol, anesthetics, and neurosteroids, that might modulate the tonic conductance through a specific action on δ-GABAARs, needs to be readdressed at physiological temperatures using relevant steady-state GABA concentrations.
Footnotes
This work is dedicated to the memory of Alastair M. Hosie, a dear friend and valued colleague. This work was supported by a Wellcome Trust Project Grant (S.G.B.), a Medical Research Council (MRC) project grant (S.G.B.), a Wellcome Trust Programme grant (M.F.), and an MRC New Investigator Award (A.H.). D.P.B., S.G.B., and M.F. performed, and analyzed data from, acute slice recordings. A.H., M.R., G.M., J.B., and T.P.M. performed, and analyzed data from, recombinant expression studies. All authors contributed to the writing of the manuscript. We thank Prof. William Wisden (Imperial College London) and Prof. Istvan Mody (University of California, Los Angeles, CA) for providing access to the α6−/− and δ−/− mouse strains. We also thank Prof. William Wisden for his comments on the manuscript.
References
- Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- Belelli D, Peden DR, Rosahl TW, Wafford KA, Lambert JJ. Extrasynaptic GABAA receptors of thalamocortical neurons: a molecular target for hypnotics. J Neurosci. 2005;25:11513–11520. doi: 10.1523/JNEUROSCI.2679-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi L, Ballini C, Colivicchi MA, Della Corte L, Giovannini MG, Pepeu G. Investigation on acetylcholine, aspartate, glutamate and GABA extracellular levels from ventral hippocampus during repeated exploratory activity in the rat. Neurochem Res. 2003;28:565–573. doi: 10.1023/a:1022881625378. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABA(A) receptors. J Physiol Lond. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Wisden W, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Bright DP, Brickley SG. Acting locally but sensing globally: impact of GABAergic synaptic plasticity on phasic and tonic inhibition in the thalamus. J Physiol. 2008;586:5091–5099. doi: 10.1113/jphysiol.2008.158576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Aller MI, Brickley SG. Synaptic release generates a tonic GABA(A) receptor-mediated conductance that modulates burst precision in thalamic relay neurons. J Neurosci. 2007;27:2560–2569. doi: 10.1523/JNEUROSCI.5100-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA. Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:3662–3667. doi: 10.1073/pnas.0307231101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol. 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Hughes SW, Crunelli V. GABAA receptor-mediated tonic inhibition in thalamic neurons. J Neurosci. 2005;25:11553–11563. doi: 10.1523/JNEUROSCI.3362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Di Giovanni G, Fyson SJ, Orbán G, Errington AC, Lorincz ML, Gould TM, Carter DA, Crunelli V. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–1398. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Fioravante D, Regehr WG. Dynamics of fast and slow inhibition from cerebellar Golgi cells allow flexible control of synaptic integration. Neuron. 2009;63:843–853. doi: 10.1016/j.neuron.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groote L, Linthorst AC. Exposure to novelty and forced swimming evoke stressor-dependent changes in extracellular GABA in the rat hippocampus. Neuroscience. 2007;148:794–805. doi: 10.1016/j.neuroscience.2007.06.030. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Botzolakis EJ, Macdonald RL. Context-dependent modulation of alphabetagamma and alphabetadelta GABA A receptors by penicillin: implications for phasic and tonic inhibition. Neuropharmacology. 2009;56:161–173. doi: 10.1016/j.neuropharm.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Hippocampal network hyperactivity after selective reduction of tonic inhibition in GABA A receptor alpha5 subunit-deficient mice. J Neurophysiol. 2006;95:2796–2807. doi: 10.1152/jn.01122.2005. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. Activation of GABAA receptors: views from outside the synaptic cleft. Neuron. 2007;56:763–770. doi: 10.1016/j.neuron.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mann EO, Mody I. Which GABA(A) receptor subunits are necessary for tonic inhibition in the hippocampus? J Neurosci. 2008;28:1421–1426. doi: 10.1523/JNEUROSCI.4751-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M, Rossi DJ, Attwell D. Tonic and spillover inhibition of granule cells control information flow through cerebellar cortex. Neuron. 2002;33:625–633. doi: 10.1016/s0896-6273(02)00593-7. [DOI] [PubMed] [Google Scholar]
- Harrison NL. Mechanisms of sleep induction by GABA(A) receptor agonists. J Clin Psychiatry. 2007;68(Suppl 5):6–12. [PubMed] [Google Scholar]
- Hartveit E, Veruki ML. Studying properties of neurotransmitter receptors by non-stationary noise analysis of spontaneous synaptic currents. J Physiol. 2006;574:751–785. doi: 10.1113/jphysiol.2006.111856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herd MB, Foister N, Chandra D, Peden DR, Homanics GE, Brown VJ, Balfour DJ, Lambert JJ, Belelli D. Inhibition of thalamic excitability by 4,5,6,7-tetrahydroisoxazolo[4,5-c]pyridine-3-ol: a selective role for delta-GABA(A) receptors. Eur J Neurosci. 2009;29:1177–1187. doi: 10.1111/j.1460-9568.2009.06680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG. Conserved site for neurosteroid modulation of GABA A receptors. Neuropharmacology. 2009;56:149–154. doi: 10.1016/j.neuropharm.2008.07.050. [DOI] [PubMed] [Google Scholar]
- Jones A, Korpi ER, McKernan RM, Pelz R, Nusser Z, Mäkelä R, Mellor JR, Pollard S, Bahn S, Stephenson FA, Randall AD, Sieghart W, Somogyi P, Smith AJ, Wisden W. Ligand-gated ion channel subunit partnerships: GABAA receptor alpha6 subunit gene inactivation inhibits delta subunit expression. J Neurosci. 1997;17:1350–1362. doi: 10.1523/JNEUROSCI.17-04-01350.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kékesi KA, Dobolyi A, Salfay O, Nyitrai G, Juhász G. Slow wave sleep is accompanied by release of certain amino acids in the thalamus of cats. Neuroreport. 1997;8:1183–1186. doi: 10.1097/00001756-199703240-00025. [DOI] [PubMed] [Google Scholar]
- Kennedy RT, Thompson JE, Vickroy TW. In vivo monitoring of amino acids by direct sampling of brain extracellular fluid at ultralow flow rates and capillary electrophoresis. J Neurosci Methods. 2002;114:39–49. doi: 10.1016/s0165-0270(01)00506-4. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Botzolakis EJ, Macdonald RL. Enhanced macroscopic desensitization shapes the response of alpha4 subtype-containing GABAA receptors to synaptic and extrasynaptic GABA. J Physiol. 2007;578:655–676. doi: 10.1113/jphysiol.2006.122135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee J. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire JL, Stell BM, Rafizadeh M, Mody I. Ovarian cycle-linked changes in GABA(A) receptors mediating tonic inhibition alter seizure susceptibility and anxiety. Nat Neurosci. 2005;8:797–804. doi: 10.1038/nn1469. [DOI] [PubMed] [Google Scholar]
- Mann EO, Mody I. Control of hippocampal gamma oscillation frequency by tonic inhibition and excitation of interneurons. Nat Neurosci. 2010;13:205–212. doi: 10.1038/nn.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihalek RM, Banerjee PK, Korpi ER, Quinlan JJ, Firestone LL, Mi ZP, Lagenaur C, Tretter V, Sieghart W, Anagnostaras SG, Sage JR, Fanselow MS, Guidotti A, Spigelman I, Li Z, DeLorey TM, Olsen RW, Homanics GE. Attenuated sensitivity to neuroactive steroids in gamma-aminobutyrate type A receptor delta subunit knockout mice. Proc Natl Acad Sci U S A. 1999;96:12905–12910. doi: 10.1073/pnas.96.22.12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Ebert B, Wafford K, Smart TG. Distinct activities of GABA agonists at synaptic- and extrasynaptic-type GABAA receptors. J Physiol. 2010;588:1251–1268. doi: 10.1113/jphysiol.2009.182444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orser BA. Extrasynaptic GABAA receptors are critical targets for sedative-hypnotic drugs. J Clin Sleep Med. 2006;2:S12–18. [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABA(A) receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z, Hauer B, Mihalek RM, Homanics GE, Sieghart W, Olsen RW, Houser CR. GABA(A) receptor changes in delta subunit-deficient mice: altered expression of alpha4 and gamma2 subunits in the forebrain. J Comp Neurol. 2002;446:179–197. doi: 10.1002/cne.10210. [DOI] [PubMed] [Google Scholar]
- Peng Z, Huang CS, Stell BM, Mody I, Houser CR. Altered expression of the delta subunit of the GABAA receptor in a mouse model of temporal lobe epilepsy. J Neurosci. 2004;24:8629–8639. doi: 10.1523/JNEUROSCI.2877-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Porcello DM, Huntsman MM, Mihalek RM, Homanics GE, Huguenard JR. Intact synaptic GABAergic inhibition and altered neurosteroid modulation of thalamic relay neurons in mice lacking delta subunit. J Neurophysiol. 2003;89:1378–1386. doi: 10.1152/jn.00899.2002. [DOI] [PubMed] [Google Scholar]
- Richerson GB, Wu Y. Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol. 2003;90:1363–1374. doi: 10.1152/jn.00317.2003. [DOI] [PubMed] [Google Scholar]
- Richter DW, Schmidt-Garcon P, Pierrefiche O, Bischoff AM, Lalley PM. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. J Physiol. 1999;514:567–578. doi: 10.1111/j.1469-7793.1999.567ae.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Hamann M. Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(A) receptors and glomerular geometry. Neuron. 1998;20:783–795. doi: 10.1016/s0896-6273(00)81016-8. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Campanac E, Scott RS, Rusakov DA, Kullmann DM. Presynaptic GABA(A) receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat Neurosci. 2010;13:431–438. doi: 10.1038/nn.2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhakumar V, Hanchar HJ, Wallner M, Olsen RW, Otis TS. Contributions of the GABAA receptor alpha6 subunit to phasic and tonic inhibition revealed by a naturally occurring polymorphism in the alpha6 gene. J Neurosci. 2006;26:3357–3364. doi: 10.1523/JNEUROSCI.4799-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Assembly of GABAA receptor subunits: role of the delta subunit. J Neurosci. 1994;14:7077–7086. doi: 10.1523/JNEUROSCI.14-11-07077.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers BD, Killisch I, Sprengel R, Sontheimer H, Köhler M, Schofield PR, Seeburg PH. Two novel GABAA receptor subunits exist in distinct neuronal subpopulations. Neuron. 1989;3:327–337. doi: 10.1016/0896-6273(89)90257-2. [DOI] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stórustovu SI, Ebert B. Pharmacological characterization of agonists at delta-containing GABAA receptors: Functional selectivity for extrasynaptic receptors is dependent on the absence of gamma2. J Pharmacol Exp Ther. 2006;316:1351–1359. doi: 10.1124/jpet.105.092403. [DOI] [PubMed] [Google Scholar]
- Telgkamp P, Padgett DE, Ledoux VA, Woolley CS, Raman IM. Maintenance of high-frequency transmission at Purkinje to cerebellar nuclear synapses by spillover from boutons with multiple release sites. Neuron. 2004;41:113–126. doi: 10.1016/s0896-6273(03)00802-x. [DOI] [PubMed] [Google Scholar]
- Trigo FF, Marty A, Stell BM. Axonal GABAA receptors. Eur J Neurosci. 2008;28:841–848. doi: 10.1111/j.1460-9568.2008.06404.x. [DOI] [PubMed] [Google Scholar]
- Vida I, Bartos M, Jonas P. Shunting inhibition improves robustness of gamma oscillations in hippocampal interneuron networks by homogenizing firing rates. Neuron. 2006;49:107–117. doi: 10.1016/j.neuron.2005.11.036. [DOI] [PubMed] [Google Scholar]
- Wafford KA, Ebert B. Gaboxadol—a new awakening in sleep. Curr Opin Pharmacol. 2006;6:30–36. doi: 10.1016/j.coph.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Wall MJ. Furosemide reveals heterogeneous GABA(A) receptor expression at adult rat Golgi cell to granule cell synapses. Neuropharmacology. 2002;43:737–749. doi: 10.1016/s0028-3908(02)00085-0. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci. 1997;9:533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x. [DOI] [PubMed] [Google Scholar]
- Wei W, Zhang N, Peng Z, Houser CR, Mody I. Perisynaptic localization of delta subunit-containing GABA(A) receptors and their activation by GABA spillover in the mouse dentate gyrus. J Neurosci. 2003;23:10650–10661. doi: 10.1523/JNEUROSCI.23-33-10650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlfarth KM, Bianchi MT, Macdonald RL. Enhanced neurosteroid potentiation of ternary GABA(A) receptors containing the delta subunit. J Neurosci. 2002;22:1541–1549. doi: 10.1523/JNEUROSCI.22-05-01541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wang W, Richerson GB. The transmembrane sodium gradient influences ambient GABA concentration by altering the equilibrium of GABA transporters. J Neurophysiol. 2006;96:2425–2436. doi: 10.1152/jn.00545.2006. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wang W, Díez-Sampedro A, Richerson GB. Nonvesicular inhibitory neurotransmission via reversal of the GABA transporter GAT-1. Neuron. 2007;56:851–865. doi: 10.1016/j.neuron.2007.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Ramamoorthy S, Shen H, Lake R, Samuvel DJ, Kalivas PW. GABA transmission in the nucleus accumbens is altered after withdrawal from repeated cocaine. J Neurosci. 2003;23:3498–3505. doi: 10.1523/JNEUROSCI.23-08-03498.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]