

Abstract
It is well known that electrostatic interactions play important roles in determining the thermodynamic stability of proteins. However, the investigation into the role of electrostatic interactions in mechanical unfolding of proteins has just begun. Here we used single molecule atomic force microscopy techniques to directly evaluate the effect of electrostatic interactions on the mechanical stability of a small protein GB1. We engineered a bi-histidine motif into the force-bearing region of GB1. By varying the pH, histidine residues can switch between protonated and deprotonated states, leading to the change of the electrostatic interactions between the two histidine residues. We found that the mechanical unfolding force of the engineered protein decreased by ∼34% (from 115 pN to 76 pN) on changing the pH from 8.5 to 3, due to the increased electrostatic repulsion between the two positively charged histidines at acidic pH. Our results demonstrated that electrostatic interactions can significantly affect the mechanical stability of elastomeric proteins, and modulating the electrostatic interactions of key charged residues can become a promising method for regulating the mechanical stability of elastomeric proteins.
Introduction
Elastomeric proteins are subject to stretching forces under their physiological conditions and play important roles in many biological processes, such as muscle contraction and cell adhesion (1–3). Desirable mechanical stability is critical for the working of elastomeric proteins. Thus, understanding the molecular determinants of mechanical stability and development of approaches to modulate the mechanical stability of proteins has been one of the focuses in protein mechanics. The importance of hydrogen bonds and hydrophobic interactions for protein mechanical stability has been well recognized and documented (4–7). However, investigation into the importance of electrostatic interactions on the mechanical stability of proteins has just begun (8,9).
Electrostatic interactions are important for thermodynamic stability of proteins and have been exploited extensively in nature (10–14). Electrostatic interactions between side chains of amino acid residues can be modulated by changing the protonation/deprotonation state of charged residues via the change of environmental pH. Thus, thermodynamic stability of proteins often depends on pH values (15,16). The effect of pH on thermodynamic stability of proteins has been investigated in great detail (17–19). However, mechanical stability, which is determined by the free energy difference between the native state and mechanical unfolding transition state, is kinetic stability of a protein along its mechanical unfolding pathway. Hence, mechanical stability is different from thermodynamic stability, which is the free energy difference between the unfolded and native states (20,21), and the influence of electrostatic interactions on mechanical stability of proteins needs to be examined independently. Single molecule atomic force microscopy (AFM) experiments on ubiquitin showed that the mechanical stability of ubiquitin decreases in acidic pH range (8), providing the first glimpse of the effect of electrostatic interactions on the mechanical unfolding of proteins. However, this study reported the average effect of all charged residues of ubiquitin on the mechanical stability, and no residue-specific information was obtained (8). Molecular dynamics (MD) simulations on the unfolding of the tenth type III domain from fibronectin (10FnIII) predicted that the mechanical stability of 10FnIII domain increases significantly due to the protonation of a few key acidic residues in the force-bearing AB β strands on lowering the pH from 7 to 4.7, pointing out the importance of residue-specific electrostatic interactions on mechanical stability (9). However, experimental study to test the MD prediction found that lowering the pH did not result in the increase of mechanical stability (22). Like ubiquitin, 10FnIII domain contains many charged residues, and changing pH will inevitably affect the protonation/deprotonation states of these charged residues. The specific effect caused by the protonation of the key acidic residues as predicted in the MD simulations was thus not directly examined in the experiment (22).
To specifically examine the effect of electrostatic interactions on mechanical stability of proteins, one has to carefully separate the effect of electrostatic interactions due to the protonation/deprotonation of the designated residues versus that originating from other charged residues. In this study, we used protein GB1 (the first B1 IgG binding domain of protein G from Streptococcus), which is thermodynamically stable in a broad range of pH (18,23), as a model system and engineered a pH sensitive bi-histidine motif into GB1 to investigate the effect of pH on the mechanical stability of GB1.
Material and Methods
Protein engineering
Bi-histidine mutant G6-53 and its polyprotein (G6-53)8 were constructed as reported (24).The genes of single-histidine mutants GI6H and GT53H were constructed using standard site directed mutagenesis method, and their sequences were verified by direct DNA sequencing. The genes of polyproteins (GI6H)8 and (GT53H)8 were constructed using a well-established method based on the identity of the sticky ends generated by BamHI and KpnI restriction digestion (4). Polyproteins were overexpressed in Escherichia coli DH5α strain, and purified by Co2+-affinity chromatography. Proteins were eluted with phosphate buffered saline with 300 mM imidazole at room temperature. Residual Co2+ was removed by adding EDTA (20 mM) in the eluted fraction. The protein was further dialyzed extensively against Tris-HCl buffer (10 mM, pH 7.4, containing 100 mM NaCl) to completely remove imidazole and EDTA before AFM experiments.
Single molecule AFM experiments
Single molecule AFM experiments were carried out on a custom-built AFM as described previously (24). The spring constant of each individual cantilever (MLCT type Si3N4 cantilever from Vecco (Plainview, NY) with a typical spring constant of 40 pN/nm) was calibrated in solution using the equipartition theorem in each individual experiment. The force-extension measurements were carried out in a two-component buffer with Na2HPO4 and citric acid under pH 3, 4, 5, 6, 7, and 8.5 with an ionic strength of 20 mM, 500 mM, and 2 M, respectively. Ionic strength of 2 M was achieved by adding NaCl to the two-component buffer. In a typical experiment, (G6-53)8 polyprotein solution (∼1 μL) was deposited onto a clean glass coverslip covered by ∼50 μL buffer at desired pH and the mixture was allowed to equilibrate for ∼15 min before the AFM experiments. For each experiment, force-extension measurements were carried out at different pH (at least three) using the same cantilever to eliminate errors associated with cantilever calibration. All experiments were carried out at a pulling speed of 400 nm s−1.
The mechanical unfolding of G6-53 can be modeled as a two-state unfolding process with force-dependent rate constants. The unfolding rate constant α0 at zero force at different pH was estimated using Monte Carlo simulations according to published procedures (25).
pKa determination of the engineered histidine residues by one-dimension 1H-NMR
One-dimensional 1H-NMR spectra were recorded on a 0.5 mM sample of G6-53 in 10 mM phosphate buffered saline, 8% D2O on a 500 MHz Bruker (Milton, Ontario, Canada) Avance spectrometer, at 298 K. The 1D spectra were acquired with presaturation for suppression of the water signal, over the pH range of 7.5–3.
Results and Discussion
Design of the model protein G6-53
GB1 is a small α+β protein that is composed of a 4-stranded β-sheet packing against a α helix (Fig. 1) (26). Its mechanical properties have been characterized extensively by using single molecule AFM and are mainly determined by the interaction between β strands 1 and 4 (27,28). MD simulations showed that the major barrier for the mechanical unfolding of GB1 corresponds to the sliding of β strand 1 against 4 and the rupturing of noncovalent interactions between these two force-bearing strands (29,30). GB1 is thermodynamically stable over a broad range of pH from 3 to 11 (23). Although it contains 16 charged residues, only three (Lys4, Lys50, and Glu56) are located in the force-bearing β strands 1 and 4. Hence GB1 is an ideal model system for protein mechanics studies. To investigate the potential of using GB1 as a template protein to investigate the effect of electrostatic interactions on the mechanical stability, we first carried out single molecule AFM experiments on GB1 at different pH. We found that the mechanical stability of GB1 shows a very weak dependence on pH: the unfolding force of GB1 changed from 182 pN at pH 8.5 to 172 pN at pH 4 (Fig. S1 in the Supporting Material), suggesting that the electrostatic interactions due to endogenous charged residues of GB1 do not have a major effect on the mechanical stability of GB1 in the pH range of 4–8.5.
Figure 1.
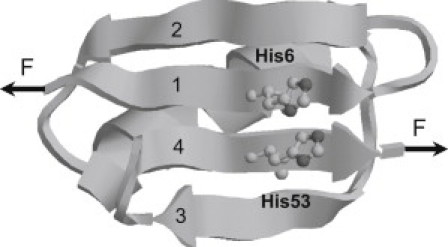
Three-dimensional structure of bi-histidine mutant G6-53. Residues 6 and 53 in the force-bearding β strands 1 and 4 were mutated to histidines (highlighted in ball and stick representation) to introduce pH sensitive motif to investigate the effect of electrostatic interactions on the mechanical stability of proteins. The 3D structure of G6-53 was obtained by homology modeling (http://swissmodel.expasy.org/).
To investigate whether electrostatic interactions can affect the mechanical stability of GB1, we mutated residues Ile6 and Thr53 to histidines in the force-bearing strands 1 and 4 to introduce an engineered pair of charged residues (Fig. 1) (24). Because the major barrier for the mechanical unfolding of GB1 corresponds to the sliding of β strand 1 against 4 and the rupturing of noncovalent interactions between these two force-bearing strands (29,30), introducing a bi-histidine site into this key region is essential for delineating the effect of electrostatic interactions on the mechanical stability. The average pKa of the imidazole side chain of histidine in proteins is ∼6.6 (31–33). Therefore, bi-histidines can be readily switched between deprotonated and protonated forms by varying the pH value. When the pH is below the pKa, the two histidine residues are protonated and carry positive charges, leading to electrostatic interactions. Electrostatic interactions diminish with distance r between two charges as (1/r) × exp(−r/D), where D is the Debye screening length. When the distance between two charges is smaller than the Debye screening length, electrostatic screening effect can be neglected. Otherwise, electrostatic interactions will be screened. Because the distance between the centroids of two imidazole rings of histidine residues 6 and 53 is ∼4 Å, which is smaller than the Debye length (20 Å at an ionic strength of 20 mM and ∼4.3 Å at an ionic strength of 0.5 M), electrostatic interactions between the two histidines will be present. Thus, interactions between the two histidine residues can be readily modulated by varying the pH: when the pH is lower than its pKa, histidine residues tend to be protonated and carry more positive charges, resulting in repulsive interactions between the two positively charged histidines; when the pH is higher, histidine residues tend to be deprontonated and electrostatically neutral. Hence, G6-53 serves as a good model system to examine the effect of electrostatic interactions on protein mechanical stability.
The mechanical stability of G6-53 decreases with the decrease of pH
We used single molecule AFM to examine the effect of changing pH values on the mechanical stability of G6-53. To elucidate the effect of pH on the mechanical stability of G6-53, we constructed polyprotein (G6-53)8, which consists of eight G6-53 domains arranged in tandem. We first studied the mechanical stability of (G6-53)8 in its deprotonated form in buffers of ionic strength of 20 mM under pH 8.5. Based on the average pKa of the imidazole side chain of isolated histidines, ∼99.7% of histidine residues are deprotonated. Therefore, electrostatic interactions between the two neutral histidine residues are negligible. Stretching polyprotein (G6-53)8 at pH 8.5 resulted in sawtooth-like force-extension curves, where individual sawtooth peak corresponds to the mechanical unfolding of individual G6-53 domains, and the last peak corresponds to the stretching and subsequent detachment of the unfolded polyprotein chain. Fitting worm-like chain (WLC) model of polymer elasticity (34) to consecutive unfolding force peaks measured contour length increment (ΔLc) of ∼18.0 nm for the unfolding of G6-53 (Fig. 2). The average unfolding force is 115 ± 30 pN (average ± standard deviation), similar to the unfolding force of 119 pN measured in our prior studies on G6-53 at pH7.4 in Tris buffer (24).
Figure 2.
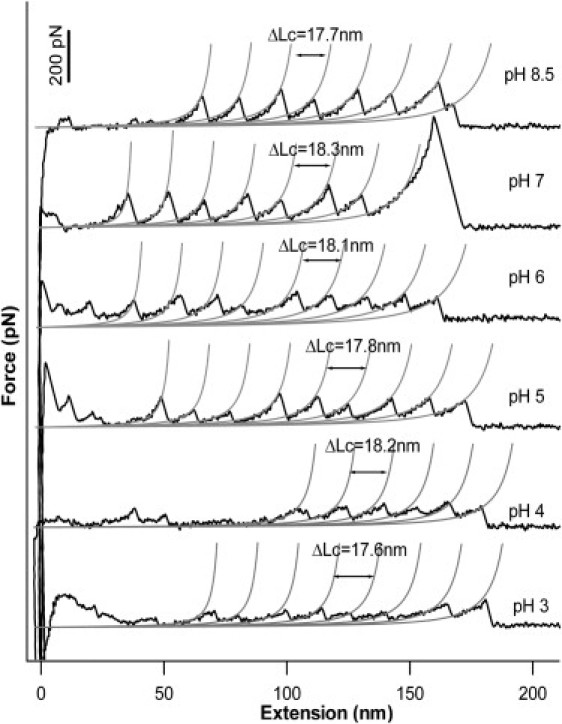
Typical force-extension curves of G6-53 at different pH measured at a pulling speed of 400 nm/s. Stretching polyproteins of (G6-53)8 results in characteristic sawtooth-like force-extension curve, in which the individual force peaks correspond to the mechanical unfolding of the individual domain. The last peak corresponds to the detachment of the unfolded polyprotein chain from either the AFM tip or substrate. Gray lines correspond to the WLC fits to the experimental data. On decreasing the pH, the mechanical unfolding force of G6-53 showed significant decrease.
We then measured the mechanical stability of (G6-53)8 under conditions that histidines are in their protonated form. At pH 3, ∼99.9% of histidines are protonated. Two protonated and positively charged histidines result in electrostatic repulsive interactions in the force-bearing region of G6-53. Stretching polyprotein (G6-53)8 at pH 3 resulted in force-extension curves with similar sawtooth-like appearance with ΔLc of ∼18 nm, suggesting that G6-53 domains remain folded at pH 3 and protonation of histidine residues does not cause dramatic change to its structure. However, the unfolding force of G6-53 decreased significantly by ∼34% to 76 ± 30 pN, indicating that the increased electrostatic repulsive interactions mechanically weakened G6-53. It is of note that the width of the unfolding force distribution remains largely unchanged.
To investigate how unfolding force of (G6-53)8 changes as a function of pH, we carried out force-extension measurements at pH 4, 5, 6, and 7. As shown in Figs. 2 and 3, the unfolding force of G6-53 decreases monotonically with the decreasing of pH from 7 to 4, but the width of the unfolding force distribution remains largely unchanged. Because the width of the unfolding force distribution is largely determined by the unfolding distance Δxu (20), which is the distance between the native state and the mechanical unfolding transition state, our results suggest that the unfolding distance Δxu remains the same at different pH, and the decreasing in unfolding force largely results from the decreasing in the unfolding energy barrier ΔGT−N. We also carried single molecule AFM experiments in buffers of ionic strength of 500 mM, under which condition the Debye screening length is ∼4 Å, and the results were indistinguishable from that at lower ionic strength (15).
Figure 3.
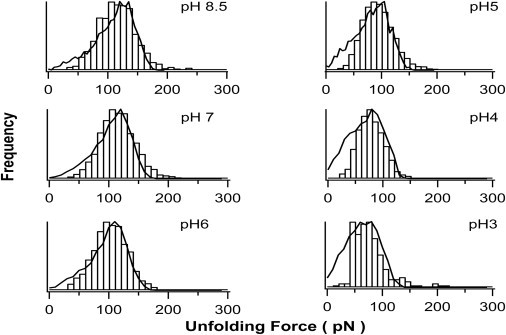
Histogram of the mechanical unfolding force for G6-53 at different pH. It is evident that the average unfolding force of G6-53 decreases on lowering the pH, whereas the width of the unfolding force distribution remained largely unchanged. The average unfolding forces of G6-53 are 115± 33 pN at pH 8.5 (n = 1328), 115 ± 33 pN at pH 7 (n = 2761), 101 ± 28 pN at pH 6 (n = 1036), 93 ± 30 pN at pH 5 (n = 2394), 78 ± 25 pN at pH 4 (n = 845) and 76 ± 32 pN at pH 3 (n = 197), respectively. Solid lines correspond to the unfolding force distribution generated using Monte Carlo simulations. In the Monte Carlo simulations, the same unfolding distance Δxu of 0.23 nm was used for all pH values. The following unfolding rate constant α0 values were found to be adequate to reproduce the unfolding force histograms obtained in AFM experiments: 0.10 s−1 for pH 8.5; 0.10 s−1 for pH 7; 0.12 s−1 for pH 6; 0.21 s−1 for pH 5; 0.38 s−1 for pH 4 and 0.45 s−1 for pH 3.
Using Monte Carlo simulation techniques (25), we reproduced the force-extension curves of G6-53 and estimated the two key parameters characterizing the mechanical unfolding of proteins at different pH, i.e., the spontaneous unfolding rate constant α0 at zero force and unfolding distance Δxu (Fig. 3).
Decreased mechanical stability of G6-53 at acidic pH is due to repulsive electrostatic interactions between bi-histidines, which can be screened by high ionic strength
Our results on the engineered bi-histidine mutant G6-53 clearly indicated that the mechanical unfolding force of G6-53 depends on pH. The mechanical unfolding force of G6-53 at pH 8.5 is ∼65 pN lower than that for wt GB1. This reduced mechanical stability of G6-53 reflects the destabilization effect of bi-histidine mutation on the mechanical stability of GB1. Because the mechanical unfolding force of wt GB1 only shows very weak dependence on pH, the observed pH-dependent unfolding force of G6-53 thus should originate from the electrostatic interactions of the two introduced histidine residues. To further confirm this point, we carried out 1D 1H-NMR experiments to determine the pKa of the engineered histidine residues. We found that one histidine titrates well showing a pKa of ∼4.6 (Fig. 4), which is much lower than the average pKa value of histidine residues in proteins (31). However, the chemical shift of the second histidine that titrates around pH 6 overlapped with other resonances, some of which showed surprising pH dependence. Thus, it was difficult to unambiguously track the change of the chemical shift on changing of pH to determine the pKa of the second engineered His residue. To accurately determine the second pKa, we attempted 2D NMR experiments using 15N-labeled protein. However, the 15N-labled G6-53 precipitated on changing of pH. Efforts to accurately determine the pKa of the second His residue are needed in our future endeavors but are beyond the scope of this work. Nonetheless, NMR results are consistent with the fact that in acidic pH range, histidine residues are protonated and positively charged, leading to destabilizing electrostatic interactions between the two engineered histidine residues. In addition, the measured effective pKa value of histidine (pKa 4.6) is consistent with our AFM experimental results that the mechanical stability of G6-53 showed a sharp decrease near pH 5 (Fig. 5 A).
Figure 4.
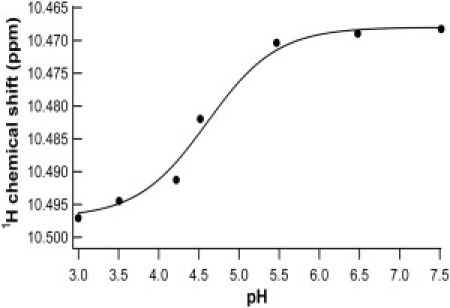
Chemical shift titration curves for proton resonances of histidine in the pH range of 3–7.5. The solid line is the least-square fit of the data to the following equation: , where δ1 and δ2 represent the chemical shift values at the low and high extremes of pH, respectively. The measured pKa of one of the engineered histidine residue is 4.6, which shifted to a lower value compared with the average value of histidine in proteins.
Figure 5.
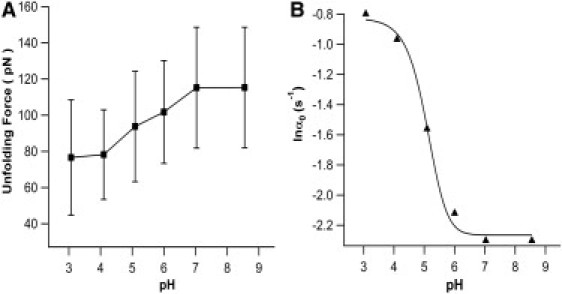
(A) Unfolding force of G6-53 decreases on lowering the pH. Error bars indicate the standard deviation of the experimental data. (B) Spontaneous unfolding rate constant α0 of G6-53 increases on the decreasing of pH. Gray line corresponds to the fit to the experimental data using Eq. 4.
To corroborate that the mechanical destabilization of G6-53 in acidic pH is mainly due to electrostatic interactions between histidine residues 6 and 53, we carried out single molecule AFM experiments on G6-53 in aqueous solution of high ionic strength 2 M. Under an ionic strength of 2 M, the Debye screening length (∼2 Å) is smaller than the distance between the two charged histidine residues. Thus, electrostatic interactions between the two charged histidines should be screened, leading to effective abolishment of the effect of electrostatic interactions on the mechanical stability of G6-53. Indeed, the mechanical unfolding force of G6-53 remained largely unchanged in the pH range of 4–8.5 (Fig. 6), strongly indicating that the origin of the pH-dependent mechanical unfolding force of G6-53 under lower ionic strength is electrostatic repulsion in nature between the two positively charged histidine residues at acidic pH.
Figure 6.
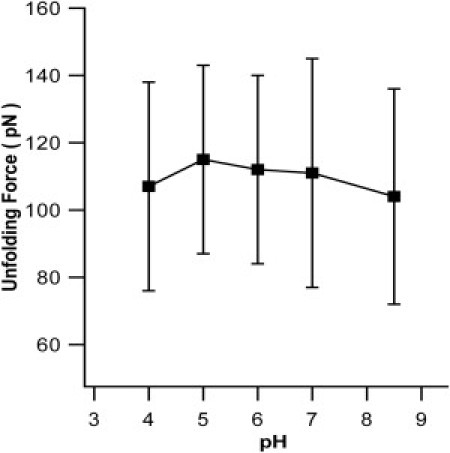
Unfolding force of G6-53 does not change with pH at ionic strength of 2 M. The average unfolding forces of G6-53 are 104 ± 32 pN at pH 8.5(n = 266), 111 ± 34 pN at pH 7 (n = 431), 112 ± 28 pN at pH 6 (n =166), 115 ± 28 pN at pH 5 (n = 131), 107 ± 31 pN at pH 4 (n = 375), respectively. At higher ionic strength, the electrostatic interactions between the two histidine residues are screened and the mechanical unfolding force of G6-53 no longer shows pH-dependence.
To further confirm that the decreased mechanical stability of G6-53 is due to the electrostatic interactions between His6 and His53 rather than interactions between either histidine with its adjacent charged residues Lys4 and Glu15, we engineered two single histidine mutants GI6H and GT53H. Single molecule AFM experiments showed that the mechanical unfolding forces of GI6H and GT53H remained largely constant within the pH range of 4–8.5 at ionic strength of 0.5 M (Fig. S2). These results clearly indicated that the decreased mechanical unfolding force of G6-53 at acidic pH indeed originates from the interactions between the two histidine residues, which are electrostatic in nature.
The observed change of mechanical unfolding force of G6-53 between pH 8.5 and 3 is 39 pN, which amounts to a change of ∼34%. Therefore, our results indicate that modulating the electrostatic interactions in the key region of proteins can significantly affect the mechanical stability of proteins, consistent with the MD prediction based on 10FnIII domain (9). Different from prior single molecule AFM studies (22), our results clearly separated the global effect of endogenous charged residues on the mechanical stability and allowed for pinpointing down the role of electrostatic interactions originating from specific charged residues.
Using a thermodynamic cycle analysis, previously we demonstrated that it is critical to preferentially stabilize (or destabilize) the native state over the mechanical unfolding transition state to increase (or decrease) the mechanical stability of proteins (24). Our results that repulsive electrostatic interactions destabilize G6-53 suggest that the repulsive interactions preferentially destabilize the native state of G6-53 over the mechanical unfolding transition state. In other words, the electrostatic interactions originating from residues 6 and 53 mainly take effect in the native state, and at the mechanical unfolding transition state the electrostatic interactions are largely diminished due to the separating of residues 6 and 53. This result is consistent with the prediction by MD simulations (29,30) that the major barrier to the mechanical unfolding of GB1 corresponds to the sliding of two force-bearing strands 1 and 4 and the rupture of interactions between these two strands, corroborating the fact that placing stabilizing or destabilizing interactions in the key region of proteins can have a major impact on the mechanical stability of proteins.
Electrostatic interactions modulate the mechanical stability of proteins in a continuous fashion
It is important to note that the unfolding force of G6-53 changes monotonically with the change of pH (Fig. 5 A), and the unfolding forces show unimodal distribution at different pH (Fig. 3). Similar continuous decrease in mechanical unfolding force with pH was also observed for ubiquitin (8). These results are in sharp contrast with the effect of protein-protein interaction and ligand binding on the mechanical stability of proteins (24). For example, our previous studies on the effect of metal chelation on the mechanical stability of G6-53 showed that the mechanical unfolding force of G6-53 shows two distinct unfolding forces, which correspond to that of apo- and metal-bound G6-53. Under nonsaturating concentration of metal ions, the unfolding force shows a clear bimodal distribution (35). Such contrast may reflect interesting mechanistic difference in the way electrostatic interactions and ligand binding affect the mechanical unfolding.
This difference may be explained by the ultrafast switch of histidine residues between its protonated and deprotonated forms. The hydrogen exchange rate for histidine (imidazole) is ∼1010 s−1, which is much faster than the diffusion coefficient of protein in solution (∼106 s−1) and the typical time window for AFM experiment (subsecond) (36). Therefore, such a fast exchange results in equal distribution of charges on all imidazole rings of histidine, as depicted in Fig. 7. The partial charge on each imidazole ring can be considered as a probability and calculated based on pKa of histidine and the pH of the solution
| (1) |
where q is the partial charge on the histidine, e is unit charge and pKa is the average acid dissociation constant for the imidazole ring on the two histidine residues.
Figure 7.
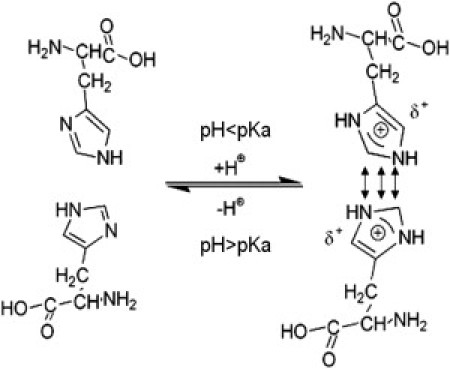
Schematic of the interconversion of histidine between protonated and deprotonated forms. When pH < pKa, histidine residues tend to be protonated, resulting in electrostatic repulsion between the two histidine residues in G6-53.
The decrease in mechanical unfolding force largely results from the preferential destabilization of the native state over the transition state caused by the repulsive electrostatic interactions. As an approximation, we assumed that the electrostatic interactions mainly affect the native state and have little effect on the transition state. Therefore, the mechanical unfolding energy barrier at acidic pH will be reduced by ΔGele, and the spontaneous unfolding rate constant at a given pH can be written as
| (2) |
where ΔGT−N(pH) is the unfolding energy barrier at a given pH, ΔGT−N (pH 8.5) is the unfolding energy barrier at pH 8.5, kBT is thermal energy. ΔGele can be calculated solely based on electrostatic interactions between histidine residues (37):
| (3) |
where ɛ0 is the dielectric constant, r is the distance between two histidine residues, D is the Debye–Hückel radius, and b = exp(−r/D)/4πɛ0r.
Combining Eqs. 1–3, we now have
| (4) |
Equation 4 can now be used to quantitatively describe the change of the spontaneous unfolding rate constant as a function of pH. Fitting Eq. 4 to the experimental data (Fig. 5 B) measures a pKa of 5.3. From the experimental data, it is clear that when pH is within 3 to 7, 0.01 < (1/1+10(pH-pKa)) < 0.99, we have the largest tunable range of mechanical stability of G6-53, in which the unfolding force can vary by ∼39 pN. Thus, this is the most suitable and sensitive range of pH for tuning the mechanical stability of G6-53. These results suggest that by carefully modulating the electrostatic interactions between charged residues, it is feasible to design pH-sensitive elastomeric proteins that can change their mechanical stability in response to the change of pH in a controlled fashion. Thus, modulating the electrostatic interactions in engineered charged residues may provide a new approach to rationally modulating the mechanical stability of elastomeric proteins using external stimuli.
In addition, the pH dependence of the unfolding force and unfolding rate constant provides insights into proton uptake of G6-53 at the mechanical unfolding transition state versus native state. The change in free energy as a function of pH can be related to ΔQA−B, the change of bound protons between two distinct states A and B (38–40):
| (5) |
Similarly, the change in mechanical unfolding energy barrier (ΔGT−N) with pH can be expressed in term of the change of bound protons between the native and mechanical unfolding transition state (8):
| (6) |
where ΔQN−T is the difference of number of proton bound to the protein between the native state and transition state. Substituting Eq. 2 into Eq. 6 we obtain
| (7) |
As shown in the Fig. 4 B, the unfolding rate increases as pH deceases between pH 7 and 3, indicating G6-53 uptakes protons to reach its mechanical unfolding transition state. We calculated that in the pH range of 4–6, ∼0.2 extra charge is uptaken at the transition state. In the pH range of 6–7 and 3–4, no significant change was observed for proton uptake.
Conclusion
Using a carefully designed bi-histidine mutant of GB1, we have demonstrated that the mechanical unfolding force of proteins can be affected by electrostatic interactions between charged residues, which can be controlled by changing the pH to control the charged states of specific amino acid residues. Such pH-dependent mechanical stability provides insights into the mechanical unfolding process of proteins, and offers a potential approach by modulating electrostatic interactions to regulate the mechanical stability of elastomeric proteins to achieve desirable mechanical properties.
Acknowledgments
This work is supported by the Natural Sciences and Engineering Research Council of Canada, Canada Foundation for Innovation, Canada Research Chairs program. H.L. is a Michael Smith Foundation for Health Research Career Investigator.
Footnotes
Yi Cao's present address is Department of Physics, Nanjing University, Nanjing, Jiangsu, People's Republic of China.
Supporting Material
References
- 1.Oberhauser A.F., Marszalek P.E., Fernandez J.M. The molecular elasticity of the extracellular matrix protein tenascin. Nature. 1998;393:181–185. doi: 10.1038/30270. [DOI] [PubMed] [Google Scholar]
- 2.Labeit S., Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 3.Rief M., Gautel M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 4.Carrion-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 5.Lu H., Schulten K. The key event in force-induced unfolding of Titin's immunoglobulin domains. Biophys. J. 2000;79:51–65. doi: 10.1016/S0006-3495(00)76273-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H., Krammer A., Schulten K. Computer modeling of force-induced titin domain unfolding. Adv. Exp. Med. Biol. 2000;481:143–160. doi: 10.1007/978-1-4615-4267-4_9. [DOI] [PubMed] [Google Scholar]
- 7.Forman J.R., Clarke J. Mechanical unfolding of proteins: insights into biology, structure and folding. Curr. Opin. Struct. Biol. 2007;17:58–66. doi: 10.1016/j.sbi.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Chyan C.L., Lin F.C., Yang G. Reversible mechanical unfolding of single ubiquitin molecules. Biophys. J. 2004;87:3995–4006. doi: 10.1529/biophysj.104.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Craig D., Gao M., Vogel V. Tuning the mechanical stability of fibronectin type III modules through sequence variations. Structure. 2004;12:21–30. doi: 10.1016/j.str.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 10.Matthew J.B. Electrostatic effects in proteins. Annu. Rev. Biophys. Biophys. Chem. 1985;14:387–417. doi: 10.1146/annurev.bb.14.060185.002131. [DOI] [PubMed] [Google Scholar]
- 11.Perl D., Mueller U., Schmid F.X. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 2000;7:380–383. doi: 10.1038/75151. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez-Ruiz J.M., Makhatadze G.I. To charge or not to charge? Trends Biotechnol. 2001;19:132–135. doi: 10.1016/s0167-7799(00)01548-1. [DOI] [PubMed] [Google Scholar]
- 13.Perl D., Schmid F.X. Some like it hot: the molecular determinants of protein thermostability. ChemBioChem. 2002;3:39–44. doi: 10.1002/1439-7633(20020104)3:1<39::AID-CBIC39>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 14.Alber T. Mutational effects on protein stability. Annu. Rev. Biochem. 1989;58:765–798. doi: 10.1146/annurev.bi.58.070189.004001. [DOI] [PubMed] [Google Scholar]
- 15.Kumar S., Nussinov R. Close-range electrostatic interactions in proteins. ChemBioChem. 2002;3:604–617. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 16.Koide A., Jordan M.R., Koide S. Stabilization of a fibronectin type III domain by the removal of unfavorable electrostatic interactions on the protein surface. Biochemistry. 2001;40:10326–10333. doi: 10.1021/bi010916y. [DOI] [PubMed] [Google Scholar]
- 17.Ibarra-Molero B., Loladze V.V., Sanchez-Ruiz J.M. Thermal versus guanidine-induced unfolding of ubiquitin. An analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 1999;38:8138–8149. doi: 10.1021/bi9905819. [DOI] [PubMed] [Google Scholar]
- 18.Lindman S., Xue W.F., Linse S. Salting the charged surface: pH and salt dependence of protein G B1 stability. Biophys. J. 2006;90:2911–2921. doi: 10.1529/biophysj.105.071050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindman S., Linse S., André I. pK(a) values for side-chain carboxyl groups of a PGB1 variant explain salt and pH-dependent stability. Biophys. J. 2007;92:257–266. doi: 10.1529/biophysj.106.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans E., Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans E., Ritchie K. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 1999;76:2439–2447. doi: 10.1016/S0006-3495(99)77399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng S.P., Clarke J. Experiments suggest that simulations may overestimate electrostatic contributions to the mechanical stability of a fibronectin type III domain. J. Mol. Biol. 2007;371:851–854. doi: 10.1016/j.jmb.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexander P., Fahnestock S., Bryan P. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: why small proteins tend to have high denaturation temperatures. Biochemistry. 1992;31:3597–3603. doi: 10.1021/bi00129a007. [DOI] [PubMed] [Google Scholar]
- 24.Cao Y., Yoo T., Li H. Single molecule force spectroscopy reveals engineered metal chelation is a general approach to enhance mechanical stability of proteins. Proc. Natl. Acad. Sci. USA. 2008;105:11152–11157. doi: 10.1073/pnas.0803446105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrion-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 1999;96:3694–3699. doi: 10.1073/pnas.96.7.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gronenborn A.M., Filpula D.R., Clore G.M. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]
- 27.Cao Y., Li H. Polyprotein of GB1 is an ideal artificial elastomeric protein. Nat. Mater. 2007;6:109–114. doi: 10.1038/nmat1825. [DOI] [PubMed] [Google Scholar]
- 28.Cao Y., Lam C., Li H. Nonmechanical protein can have significant mechanical stability. Angew. Chem. Int. Ed. Engl. 2006;45:642–645. doi: 10.1002/anie.200502623. [DOI] [PubMed] [Google Scholar]
- 29.Li P.C., Makarov D.E. Ubiquitin-like protein domains show high resistance to mechanical unfolding similar to that of the 127 domain in titin: evidence from simulations. J. Phys. Chem. B. 2004;108:745–749. [Google Scholar]
- 30.Glyakina A.V., Balabaev N.K., Galzitskaya O.V. Mechanical unfolding of proteins L and G with constant force: similarities and differences. J. Chem. Phys. 2009;131:045102. doi: 10.1063/1.3183974. [DOI] [PubMed] [Google Scholar]
- 31.Edgcomb S.P., Murphy K.P. Variability in the pKa of histidine side-chains correlates with burial within proteins. Proteins. 2002;49:1–6. doi: 10.1002/prot.10177. [DOI] [PubMed] [Google Scholar]
- 32.Poulsen K.R., Snabe T., Petersen S.B. Quantization of pH: evidence for acidic activity of triglyceride lipases. Biochemistry. 2005;44:11574–11580. doi: 10.1021/bi050634o. [DOI] [PubMed] [Google Scholar]
- 33.Haynes W.M. 91st ed. CRC Press/Taylor and Francis; Boca Raton, FL: 2011. CRC Handbook of Chemistry and Physics (Internet Version 2011) [Google Scholar]
- 34.Marko J.F., Siggia E.D. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 35.Cao Y., Er K.S., Li H. A force-spectroscopy-based single-molecule metal-binding assay. ChemPhysChem. 2009;10:1450–1454. doi: 10.1002/cphc.200900333. [DOI] [PubMed] [Google Scholar]
- 36.Hass M.A., Vlasie M.D., Led J.J. Conformational exchange in pseudoazurin: different kinds of microsecond to millisecond dynamics characterized by their pH and buffer dependence using 15N NMR relaxation. Biochemistry. 2009;48:50–58. doi: 10.1021/bi801858f. [DOI] [PubMed] [Google Scholar]
- 37.Finkelstein A.V., Ptitsyn O. Academic Press; London: 2002. Protein Physics: A Course of Lectures. [Google Scholar]
- 38.Tanford C. Protein denaturation. Adv. Protein Chem. 1968;23:121–282. doi: 10.1016/s0065-3233(08)60401-5. [DOI] [PubMed] [Google Scholar]
- 39.Tanford C. Protein denaturation. C. Theoretical models for the mechanism of denaturation. Adv. Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 40.Tan Y.J., Oliveberg M., Fersht A.R. Titration properties and thermodynamics of the transition state for folding: comparison of two-state and multi-state folding pathways. J. Mol. Biol. 1996;264:377–389. doi: 10.1006/jmbi.1996.0647. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.