

Abstract
The lipid matrix present in the uppermost layer of the skin, the stratum corneum, plays a crucial role in the skin barrier function. The lipids are organized into two lamellar phases. To gain more insight into the molecular organization of one of these lamellar phases, we performed neutron diffraction studies. In the diffraction pattern, five diffraction orders were observed attributed to a lamellar phase with a repeat distance of 5.4 nm. Using contrast variation, the scattering length density profile could be calculated showing a typical bilayer arrangement. To obtain information on the arrangement of ceramides in the unit cell, a mixture that included a partly deuterated ceramide was also examined. The scattering length density profile of the 5.4-nm phase containing this deuterated ceramide demonstrated a symmetric arrangement of the ceramides with interdigitating acyl chains in the center of the unit cell.
Introduction
The skin forms the interface between the human body and the environment. It protects our body against various biological and chemical hazards and from desiccation in a dry environment. The outermost layer of the skin, the stratum corneum (SC), forms the main barrier against diffusion of substances across the skin (1). This layer consists of overlapping flattened dead skin cells. Each cell is surrounded by lipids, which serve as the mortar between the cells. The lipids form multiple sheets of lamellae and are mainly composed of ceramides (CERs), cholesterol (CHOL), and free fatty acids (FFAs). These lipid classes are present in an approximately equimolar ratio (2). The lipids are organized in two lamellar phases with repeat distances of ∼6 and 13 nm, also referred to as the short periodicity phase (SPP) and long periodicity phase (LPP), respectively (3–7). Despite the high level of CHOL, the lipids form predominantly a crystalline lateral packing (4,8). This is different from phospholipid membranes, in which high levels of CHOL induce the formation of a liquid-ordered phase (9–11). In the SC, the CERs are a crucial component in the formation of the lamellar phases that constitute the main barrier function. In previous studies, we mainly focused on the formation and molecular organization of the LPP (3,4,12–14). In this study, we focus on the molecular organization of the SPP. Not only in SC, but also in membranes of living cells the CERs play an important role, especially in the formation of lipid rafts (15,16).
Previous studies showed that mixtures prepared with either synthetic CER or native CER mixed with CHOL and with FFA mimic the SC lipid organization very closely (17–19). Although CER and CHOL play a prominent role in the formation of the two lamellar phases, the addition of FFAs is crucial for the formation of the densely packed orthorhombic crystalline structure (17,20). A more-detailed analysis of the lipid composition revealed that the FFAs are predominantly saturated and have a wide distribution of chain lengths, in which the chain lengths of 22 and 24 C atoms are most abundantly present (21).
In addition, there are 11 subclasses of CER identified in human SC (22–24). However, in the synthetic CER mixture that we used in our previous studies, five subclasses of CER are present that closely mimic the composition of pig SC. These subclasses consist of either a sphingosine (S) or phytosphingosine (P) base, whereas the acyl chain is a nonhydroxy (n), α-hydroxy (A), or ω-hydroxy chain (25). The corresponding nonhydroxy and α-hydroxy CERs that are present in this synthetic CER mixture are denoted as CER NP, CER NS, CER AS, and CER AP. The molecular structure of these CERs is displayed in Fig. 1. In the synthetic CER mixture used in previous studies, an ω-hydroxy CER is present (26). This CER possesses a longer acyl chain length (C30) and has a linoleic acid chemically bound to its ω-hydroxy group (indicated with EO). It is denoted as CER EOS. Using these CER subclasses, mixed with CHOL and FFA, it was demonstrated that EOS is very important for the formation of the LPP and that the mixtures closely mimicked the lamellar phase behavior of mixtures prepared from either isolated human or pig CER (13,26).
Figure 1.
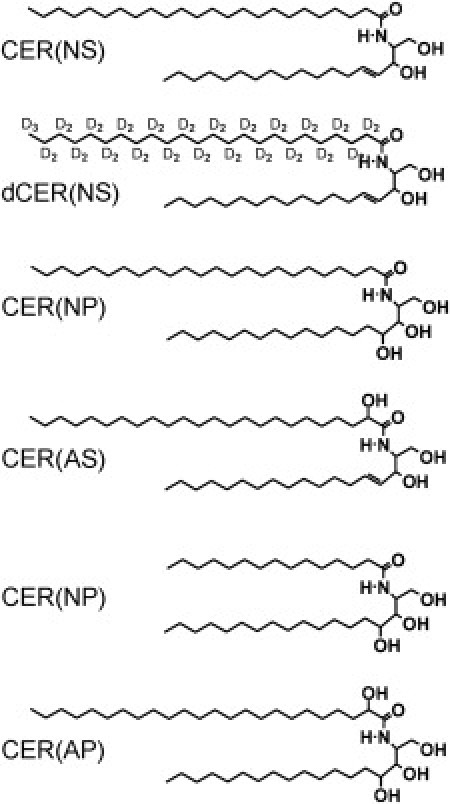
Molecular structure of the synthetic CER used in the mixtures.
Although the above studies were very relevant for providing information on the role the various lipid classes play in the lipid organization, no detailed information was obtained about the localization and molecular arrangement of the molecules within the unit cell. In this respect in several other studies, progress has been made using simplified ternary or quaternary lipid mixtures. The mixtures included mainly CER AP with a short acyl-chain length of 18 C atoms, CHOL, and cholesterol sulfate. In these studies, the neutron scattering length density profile was determined of CER-rich phases with a short periodicity (27–29). In some studies, CER EOS and a single (deuterated) fatty acid were also included, which allowed the localization of the deuterated fatty acid in the unit cell of the phases formed by these simplified mixtures (29).
In this article, we investigate the SPP of SC in more detail, using a complex lipid mixture of synthetic CERs, CHOL, and FFAs that closely mimics the SPP observed in stratum corneum (18). The aim is to obtain information on the localization and conformation of CER NS in the SPP. We chose CER NS because it is the most abundant CER subclass in the model mixture. As CER EOS is crucial for the formation of the LPP (31), it is excluded from these mixtures. In the studies focused on the conformation and localization of CER NS in the SPP, part of the CER NS is replaced by CER NS with a perdeuterated acyl chain (dCER NS). First, the neutron scattering length density profile of the SPP is determined by using contrast variation. In the second part of our studies, information on the arrangement of CER NS in the unit cell of the SPP is obtained by using its partially deuterated counterpart.
Materials and Methods
Materials
Synthetic CER NS (C24), dCER NS (C24), Cer NP (C24), CER AS (C24), CER NP (C16), and CER AP (C24) were generously provided by Cosmoferm (Delft, The Netherlands). The number given in parentheses denotes the number of carbon (C) atoms in the acyl chain of the ceramides. All the lipids studied were acyl-chain saturated. The palmitic acid (C16:0), stearic acid (C18:0), arachidic acid (C20:0), behenic acid (C22:0), tricosanoic acid (C23:0), lignoceric acid (C24:0), cerotic acid (C26:0), cholesterol, and acetate buffer salts were purchased from Sigma-Aldrich Chemie (Schnelldorf, Germany). The silicon substrates were cut from a wafer (P/Boron (110), thickness 380 ± 10 μm) obtained from Okmetic (Vantaa, Finland). All organic solvents used were of analytical grade and manufactured by Labscan (Dublin, Ireland). The water used was of Millipore quality.
Preparation of the lipid models
For preparation of the model with protonated lipids, synthetic CER, CHOL, and FFA were used. The following synthetic CER composition was selected (see also Fig. 1): CER NS C24, CER NP C24, CER AS C24, CER NP C16, and CER AP C24 in a 60:19:5:11:6 molar ratio which, apart from the absence of CER EOS, closely resembles the CER composition in pig SC (32). For the free fatty acids mixture (FFA), the following composition was selected: C16:0, C18:0, C20:0, C22:0, C23:0, C24:0, and C26:0 at molar ratios of 1.8, 4.0, 7.7, 42.6, 5.2, 34.7, and 4.1, respectively. This chain length distribution is based on the FFA composition in human SC (21). To achieve the desired 1:1:1 ratio of CER/CHOL/FFA, for each model the appropriate amounts of individual lipids were dissolved in chloroform/methanol (2:1) and combined into one solution with a final lipid concentration of ∼10 mg/mL. This solution was sprayed over an area of 1 × 3.8 cm2 on a silicon substrate using a Camag Linomat IV sample applicator (Muttenz, Switzerland). The spraying rate was 5 μL/min and the solvent was evaporated by a stream of nitrogen gas. The silicon substrate with applied lipid film was then equilibrated twice for 10 min at a temperature of ∼80°C. After each heating step the sample was cooled to room temperature over ∼30 min. After equilibration, the lipid layer was immersed in acetate buffer (50 mM, pH 5) and kept at 37°C for 24 h to achieve maximum hydration. After the hydration step, the sample was kept at 100% RH until measured in neutron diffraction. This sample preparation leads to oriented multilayers with a low mosaicity, as is demonstrated in Fig. 2.
Figure 2.
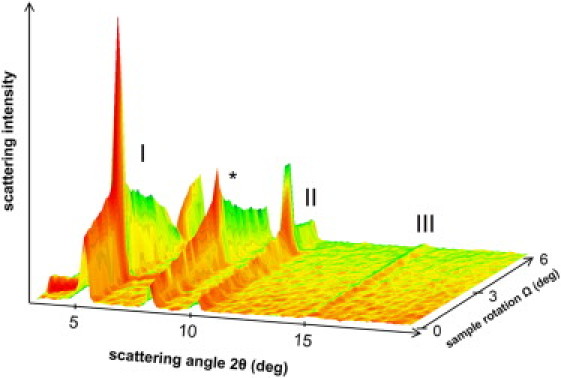
Neutron diffraction profile of the deuterated sample at 100% RH with a buffer of 100% D2O. The reflections of the SPP are indicated by Roman numbers I–III and a reflection of crystalline CHOL is indicated by an asterisk.
The same preparation method was used for the model in which 30 mol % of the CER NS was replaced by dCER NS. This deuterated sample contains, in total, 2.5 mol % of deuterated lipids. For the determination of the signs of the structure factors, acetate buffers containing 0, 33, 67, and 100% D2O were used. When a neutron diffraction measurement of a sample at a selected H2O/D2O ratio was completed, the sample was exposed to a buffer with another H2O/D2O ratio for a period of 24 h at 37°C and subsequently mounted in the chamber for the next neutron diffraction measurement. This procedure was repeated until the sample was measured with all four H2O/D2O buffer concentrations.
Weighing of the samples to determine the hydration level
To determine the degree of hydration of the mixtures, all samples were weighed in a dry state (24 h dehydration over P2O5) and then in a fully hydrated state (see procedure above) using a Sartorius SE 2 microbalance (Nieuwegein, The Netherlands). This was performed for both the protonated (in duplicate) and deuterated samples (in triplicate) and the weighing procedure of each sample was repeated three times.
X-ray diffraction experiments
Before the neutron diffraction measurements were performed, the equilibration temperature and equilibration period of the lipid mixtures during sample preparation were optimized to form only one lamellar phase with maximum orientation parallel to the silicon support. The samples were measured by x-ray diffraction. An X'pert Pro-Alpha Diffractometer from PANalytical (Almelo, The Netherlands) was used equipped with a sealed Cu x-ray tube, 0.01 rad primary and secondary Soller slits, and an elliptical mirror that produces a convergent beam of Cu Kα1 and Kα2 radiation with wavelengths of, respectively, 1.5406 and 1.5444 Å. The analysis of the one-dimensional diffraction patterns was the same as described previously (19).
Neutron diffraction experiments
Experiments were performed on the D16 diffractometer at the Institute Laue-Langevin (ILL, Grenoble, France) operating at λ = 4.75 Å in the reflection mode. Samples were mounted on a goniometer placed in a sealed temperature-controlled aluminum humidity chamber in the presence of an acetate buffer bath to maintain constant maximum humidity. The temperature in the chamber was kept at 25°C during the measurements. The sample to detector distance was 1.0 m. The intensity of the diffracted beam was recorded by a position-sensitive two-dimensional 3He detector with 128 × 128 channels and 2 mm resolution between channels. The two-dimensional detector readout was integrated in the vertical direction, which results in a one-dimensional intensity projection as a function of the detector channel position (2θ).
Intensities on the detector surface were corrected by normalization to a water calibration and by subtraction of the empty chamber background. The measurement time per sample varied between 7 and 12 h, depending on the signal/noise ratio during the measurements. A longer measurement time was used for the higher diffraction orders (i.e., 3–5). The lamellar spacing (d) was obtained by fitting the peak positions (in 2θ) of all diffraction orders (h) according to Bragg's law: 2dsinθ = hλ. Data analysis was performed using the ILL in-house software LAMP (33). Since it was not possible to integrate over the complete range in ω (due to cut off by the substrate and due to the ω-range used in the measurements), we chose to integrate only the peak intensities originating from the lamellae-oriented parallel to the silicon support. This was achieved by selecting only the small angle high intensity part of the diffraction peaks at sample rotations of Ω = −0.1, 0.0, and +0.1° around the Bragg angle (see also Fig. 2). A summing of these pixel intensities resulted in the total peak intensity value (Ih). Using this integration method, we obtain an excellent signal/noise ratio. Then, the structure factor amplitudes |Fh| were calculated from the total peak intensity (Ih) by
| (1) |
where Ah is the correction factor for sample absorption, which can be calculated via (34)
| (2) |
In this equation, θ is the Bragg angle, μ the linear attenuation coefficient, and L the thickness of the lipid film. With a lipid density of ∼0.87 g/cm3 and weight of 10 mg in an area of 3.8 cm2, the thickness of the lipid film (L) was calculated to be ∼30 μm. The linear attenuation coefficient was calculated from the wavelength used in combination with the density and the chemical composition of the lipid film (35). For the protonated sample, μ ranges from 5.48 to 5.09 cm−1 for 0–100% D2O, respectively, and for the deuterated sample, μ = 5.03 to 4.65 cm−1. The error in |Fh| was determined from the standard deviation of the summed pixel intensities (σI) by
| (3) |
If the unit cell of the SPP bilayer is centrosymmetric, the phases of the structure factors are either 0 or π. For this situation, the structure factors display a linear correlation as function of H2O:D2O ratio (34). For a hydrated bilayer, we may assume that water is distributed near the hydrophilic headgroups, at the boundaries of the unit cell. Mathematically, a Gaussian water distribution at the boundaries of the unit cell (±d/2) results in structure factor signs for the water layer being − + − + −. Because the water structure factors are defined as the structure factors at 100% D2O minus that at 100% H2O, the signs of the structure factors can be determined from the plot of the H2O/D2O contrast versus the obtained structure factor amplitudes |Fh|. The procedure is as follows: The sign of Fh must be chosen such that Fh at 100% D2O minus Fh at 0% D2O results in the correct sign for the hth order of the water layer structure factor.
When the structure factor phase signs are determined, the neutron scattering length density profile for the SPP bilayer, ρ(x), can be calculated via a Fourier transform of the structure factors (36),
| (4) |
where x is the direction normal to the bilayer surface. The significance of features in the density profile can be visualized by the error in the scattering length density, depicted in the equation (37)
| (5) |
where t is Student's t-statistic. The error in the structure factors (ΔFh) was determined with Eq. 3. A 99.7% confidence limit for the scattering length density was obtained by using a Student's t-statistic of t = 2.97.
Furthermore, after a normalization of the patterns by setting the sum of the structure factor amplitudes (F1–F5) equal for the protonated and deuterated sample, the difference density profile (resulting from the deuterated CER NS tails) can be calculated by subtracting the density of the protonated sample from that of the deuterated sample. To obtain an absolute scaling for the density patterns the total scattering length density (F0) must be determined and additional information on the features in the difference density pattern is needed. F0 was calculated using the chemical composition and the mass density of the sample (38,39).
Results
Optimal sample preparation
Before neutron measurements were performed, the samples were assessed by x-ray diffraction measurements to ensure a proper lamellar organization and orientation. Our original procedure of equilibrating the samples twice for 1 h at a temperature slightly above the onset of melting, followed by cooling to room temperature, resulted not only in reflections attributed to the SPP, but also in reflections attributed to an additional phase with a repeat distance of 4.4 nm (see Fig. S1 A in the Supporting Material). To examine the formation of this additional phase in more detail, the sample was heated during which diffraction curves were recorded. At 80°C the diffraction peaks attributed to this additional phase disappeared. A subsequent cooling to room temperature resulted in a diffraction pattern with reflections attributed to the SPP with a repeat distance of 5.4 nm and crystalline CHOL only (see Fig. S1 B). Based on these results, the lipid mixtures for the neutron diffraction studies were equilibrated for 10 min at a temperature of ∼80°C.
Number of water molecules per lipid molecule
Increasing the D2O concentration in the hydration buffer resulted in a moderate increase in neutron scattering signal, indicating that a relatively low number of water molecules are present in the sample. To determine the amount of water in the samples, the samples are weighed with a microbalance in dry and hydrated state. As expected, between the protonated and deuterated samples no difference in the hydration level was observed. The resulting water/lipid molar ratio determined by weighing is 1.91 ± 0.42, which indicates that in the lipid mixtures ∼2 water molecules are present per lipid molecule.
Neutron diffraction pattern
In Fig. 2, a typical example of a diffraction pattern is shown of the mixture containing dCER NS hydrated at 100% D2O. Three reflections that can be attributed to the SPP are visible at scattering angles of 2θ = 5.13, 10.16, and 15.29°, corresponding to a repeat distance of d = 5.36 ± 0.04 nm. In addition, one reflection attributed to crystalline CHOL is visible at 2θ = 8.0°. The maximum intensity of the SPP third diffraction order is not visible in Fig. 2, as it is located at the sample rotation of Ω = 7.65°, which is recorded at the second detector position (not shown). The diffraction pattern of the SPP reveals very sharp peaks indicating that a higher number of lipid lamellae is oriented parallel to the substrate surface as compared to other orientations. The mosaicity of the parallel-ordered lamellae was calculated (taking the full width at half-maximum of a Gaussian fit to the sharp peak in Ω for orders 1–5) to be 0.275 ± 0.014°. A very similar pattern is observed for the protonated sample. The second reflection of CHOL is too low in intensity to be visible in Fig. 2 and its maximum intensity is located at a higher sample rotation of Ω = 8.0°, also recorded at the second detector position. After integration of the peak at this second detector position, the second order of CHOL was determined at 2θ = 16.0°. This reflection did not overlap with the third-order reflection of the SPP.
Determination of the phase signs
The absolute structure factor amplitudes with corresponding errors and absorption correction factors are calculated from the diffraction patterns using Eqs. 1–3. They are provided in Table 1. As shown in the table, the errors in the structure factor amplitudes are small. The linear fits of the structure factor amplitudes with the H2O/D2O ratio are displayed in Fig. 3, A and B, for the protonated and deuterated sample, respectively. From these fits, it is clear that the structure factors correlate linearly with the H2O/D2O ratio, demonstrating that the lipids in the SPP form a centrosymmetric structure—and this is also true when CER NS is partially substituted by dCER NS. Furthermore, clear differences are observed between the structure factors of the protonated sample and those of the deuterated sample, which indicates a difference between their scattering length density profiles.
Table 1.
Structure factor amplitudes with errors and absorption correction factors
| [D2O] | F(1) | A1 | F(2) | A2 | F(3) | A3 | F(4) | A4 | F(5) | A5 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protonatedsample | 0% | 41.23 ± 0.08 | 1.191 | 19.67 ± 0.13 | 1.094 | 13.99 ± 0.09 | 1.063 | 14.17 ± 0.11 | 1.047 | 5.19 ± 0.25 | 1.038 |
| 33% | 62.83 ± 0.11 | 1.186 | 33.72 ± 0.08 | 1.092 | 22.65 ± 0.08 | 1.061 | 24.52 ± 0.08 | 1.046 | 12.65 ± 0.11 | 1.037 | |
| 67% | 80.82 ± 0.13 | 1.182 | 46.56 ± 0.10 | 1.090 | 33.91 ± 0.09 | 1.060 | 33.27 ± 0.08 | 1.045 | 18.37 ± 0.10 | 1.036 | |
| 100% | 97.22 ± 0.15 | 1.177 | 58.39 ± 0.11 | 1.088 | 42.64 ± 0.08 | 1.058 | 40.96 ± 0.07 | 1.044 | 24.93 ± 0.06 | 1.035 | |
| 0% | 43.68 ± 0.11 | 1.175 | 26.04 ± 0.09 | 1.087 | 13.99 ± 0.09 | 1.058 | 18.03 ± 0.11 | 1.043 | 6.10 ± 0.22 | 1.035 | |
| Deuteratedsample | 33% | 57.52 ± 0.11 | 1.170 | 39.33 ± 0.11 | 1.084 | 22.45 ± 0.08 | 1.056 | 25.52 ± 0.11 | 1.042 | 9.80 ± 0.24 | 1.034 |
| 67% | 64.57 ± 0.13 | 1.166 | 44.72 ± 0.10 | 1.082 | 25.97 ± 0.08 | 1.055 | 30.60 ± 0.10 | 1.041 | 14.21 ± 0.09 | 1.033 | |
| 100% | 81.48 ± 0.15 | 1.161 | 59.71 ± 0.14 | 1.080 | 35.96 ± 0.08 | 1.053 | 38.48 ± 0.10 | 1.040 | 19.56 ± 0.11 | 1.032 |
Figure 3.
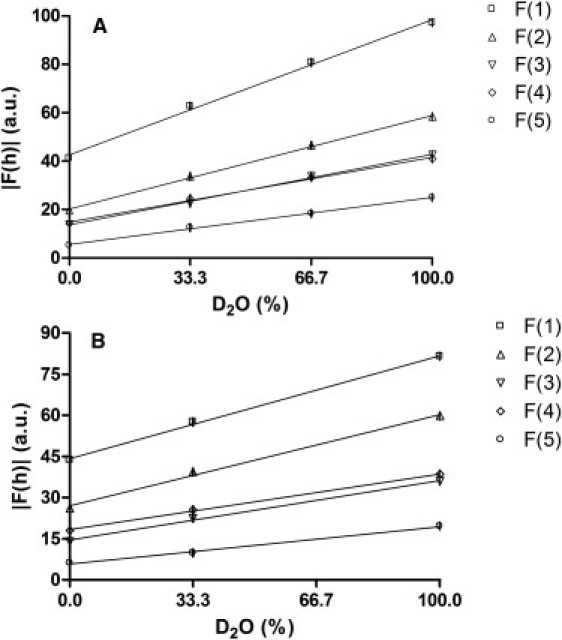
(A) Linear fits of the structure factor amplitudes of the protonated sample with the ratio of H2O/D2O in the hydration buffer. (B) Linear correlation of the structure factor amplitudes of the deuterated sample with the H2O/D2O ratio. The error bars represent the standard deviations.
The phase signs are obtained from the H2O/D2O plots as follows: A Gaussian water distribution is assumed near the lipid headgroups at the unit cell boundaries (±d/2). The phase signs for the structure factors of this water layer are, according to its distribution, − + − + −. Subsequently, because the water layer structure factors are defined as the total structure factors at 100% D2O minus those at 0% D2O, the structure factor phase signs for the protonated and deuterated sample can be deduced (see Fig. 3, A and B). Because the structure factor amplitudes at 100% D2O in Fig. 3, A and B, are all higher than those at 0% D2O, the structure factor signs of both the protonated and deuterated sample are also − + − + −, for diffraction orders 1–5, respectively.
Scattering length density profiles
The neutron scattering length density profiles of the SPP at 0% D2O concentration in the protonated and deuterated sample were calculated using Eq. 4. The profiles are displayed in Fig. 4, A and B, for the protonated and deuterated sample, respectively, with a 99.7% confidence interval calculated with Eq. 5. The profile of the protonated sample displays a high density at the boundaries of the unit cell, a low density in the center, and a submaximum at −1.5 and +1.5 nm from the center. Minima in the profile are located at −0.75 and +0.75 nm from the center. The profile of the deuterated sample is very similar to that of the protonated sample, except for an elevated density in the center of the unit cell.
Figure 4.
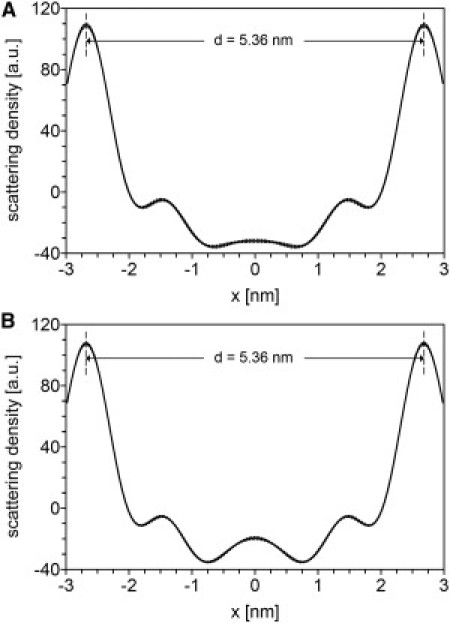
(A) Scattering length density profile of the SPP in the protonated sample (solid line). (B) Scattering length density profile of the SPP in the deuterated sample (solid line). (Dotted lines) The 99.7% confidence interval.
Subsequently, both density patterns are normalized and a difference density pattern is calculated by subtracting the protonated from the deuterated pattern. This difference profile represents the density of the deuterated acyl chains. The resulting difference pattern displays an elevated density in the center of the unit cell. To put the density patterns on an absolute scale as described in the Materials and Methods, we use information on the molecular structure given by the shape of the difference density pattern as follows: The dCER NS is deuterated along the total length of the acyl chain (D47). Because the acyl chain is also located in the headgroup region (see Fig. 1) and the intermembrane space is very small (∼0.1 nm), the neutron scattering length density is expected to increase throughout the entire length of the unit cell. Because the shape of the difference density pattern only displays an elevation in the center of the unit cell, this indicates an interdigitation of deuterated acyl chains in the center of the unit cell. We use this information to scale the difference pattern so that the density (D-H) in the center of the unit cell (resulting from two overlapping acyl chains) is exactly twice the density in the remaining part of the unit cell (resulting from a single acyl chain). In Fig. 5 the protonated, deuterated, and difference density patterns are displayed on an absolute density scale. It must be emphasized that the scaling procedure has no influence on the shape of the difference density pattern, only on the range of the scattering length density on the y axis.
Figure 5.
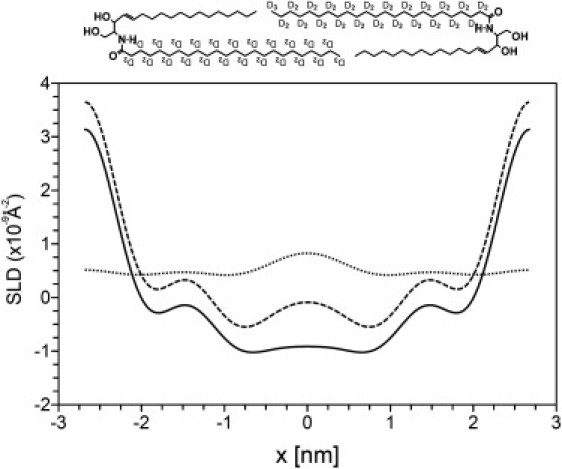
Scattering length density profiles of the SPP in the protonated sample (solid line) and deuterated sample (dashed line) and their difference profile (dotted line) plotted on an absolute density scale. A possible arrangement of the dCER NS molecules in the SPP bilayer is also presented.
Discussion
In human SC the lipids form two lamellar phases referred to as the LPP and SPP (3–7,40). Very recently, the electron density profile of the unit cell in the LPP was calculated by means of x-ray diffraction studies (14). These studies indicated that in the unit cell of the LPP the lipids are organized in three layers that are almost equal in width. This lipid arrangement, which is very characteristic for the lipid organization in SC, is dictated by the molecular architecture of CER EOS. These studies also demonstrated that the lipid organization of mixtures prepared from either synthetic CER mixtures or isolated CER mixtures is very similar to that in SC, not only with respect to the repeat distances of the lipid phases, but also with respect to the electron density profile of the unit cell of the LPP. The aim of this study was to gain more insight into the molecular structure of the SPP.
It is well known that the repeat distances of the lamellar phases in mixtures prepared from CERs, CHOL, and FFAs are very insensitive to hydration and that especially for the SPP only a limited number of diffraction orders are obtained with x-ray diffraction. Therefore it is difficult to obtain an electron density profile by x-ray diffraction analysis. In addition, by using a deuterated lipid in neutron experiments, information can be obtained about the arrangement of this lipid in the unit cell. For this reason it was decided to perform neutron diffraction studies, as this permits contrast variation by changing the H2O/D2O ratio to obtain the phase signs of the structure factors of the various diffraction orders. Using this method we were able to calculate the scattering length density profile of the SPP. However, our final goal is to obtain information on the arrangement of the CER in the unit cell. Therefore we partly replaced the (most abundant) CER NS in the lipid mixture by dCER NS to determine the position of the acyl chain in the unit cell. To the authors' knowledge, these are the first neutron diffraction measurements reported for a lipid mixture that closely mimics the SPP in SC.
In our previous studies, using x-ray diffraction on mixtures with CER, CHOL, and FFA prepared in the absence of CER EOS, we were able to form the SPP and partly phase-separated crystalline CHOL without the presence of additional phases. However, during the sample preparation for the neutron diffraction experiments, in which we used silicon as the supporting substrate, the equilibration method had to be adjusted to obtain only the SPP and phase-separated crystalline CHOL. Our studies demonstrate that the formation of an additional 4.4-nm phase can be avoided by equilibration of the sample at ∼10°C above the melting region of the lipid mixture. The presence of the CHOL is not avoided, as phase-separated crystalline CHOL is also present in SC (3,4,41) and the diffraction peaks of CHOL do not interfere with those of the SPP. In previous studies it was shown that at a ratio of CER/CHOL/FFA of 1:0.4:1, CHOL is incorporated in the lamellar phases (unpublished data). From this we may conclude that CHOL is partly dissolved in the lamellar phases. In studies using mixtures with only a few CERs, CHOL, and a single fatty acid, phase separation of the fatty acid fraction has been reported (29,42,43). However, the mixtures in the latter studies contained a lower number of CER subclasses and only a single fatty acid. Either a mismatch between the CER and fatty acid chain lengths or a different equilibration procedure most probably causes the phase separation in these mixtures. We not only measured the lamellar phases of our mixtures, but we also examined the lateral packing with infrared spectroscopy (unpublished results). There was no indication of phase separation between FFA and CER within the lateral packing of the SPP. This suggests that rather homogenous mixtures are formed—not only with respect to the lamellar phase behavior, but also with respect to the lateral packing.
Hydration level of the lipid mixtures
In previous studies on SC or SC lipid models it has been observed that, even at 100% humidity, the repeat distance of the lipid lamellae is almost insensitive to the level of hydration, suggesting that very little water is present in the headgroup region (12,18,44). Our studies show that based on weight measurements, ∼2 water molecules are present per lipid molecule. This is a very low hydration level as compared to phospholipid bilayers, which can contain up to 12 (in the gel phase) or even 35 (in the fluid phase) water molecules per lipid molecule (36). In a related study on a SC model mixture with CER EOS, CER AP, CHOL, and palmitic acid, the number of water molecules per lipid molecule was estimated to be only 1, based on the available intermembrane space and the molecular volume of H2O (29). Because of the limited amount of water in the headgroup region, ceramide-containing mixtures are less sensitive to the H2O/D2O contrast variation. However, in this study the change in the structure factor values was significant when increasing the D2O level from 0 to 100%. This allowed us to determine the phase signs and to calculate a neutron scattering length density profile for the SPP with a high precision. Due to the low water levels in the lipid mixtures, it was decided to hydrate the samples at 100% humidity.
The low hydration level of CER-containing mixtures implies that the hydration level of the SC lipid matrix is also very low, as compared to phospholipid layers in a cell membrane. Regarding the barrier function of the lipid matrix in SC, because the amount of water molecules in the headgroup region is very low, the penetration for hydrophilic molecules is expected to be significantly reduced as compared to crystalline phospholipid membranes. Thus, the very low hydration level of the SC lipid matrix may play an important role in the barrier function of the skin.
Bilayer structure of the SPP and the arrangement of CER NS in the unit cell
When considering the scattering length density profiles for the unit cell of the SPP, a high density is located at the boundaries of the unit cell and a low density in the center. This suggests that the headgroups of the lipids are located at the boundaries of the unit cell, whereas the hydrocarbon chains are located in the center. This is similar to the formation of a typical lipid bilayer driven by hydrophobic-hydrophilic interactions, as often observed for phospholipid membranes (45–47). Furthermore, the experimentally obtained repeat distance of 5.36 nm is in excellent agreement with the bilayer arrangement of two CER molecules: the total length of the extended sphingosine C18 base and C24 acyl chain, when assuming a 0.127 nm length per C-C bond, is 5.33 nm (48). Therefore it is likely that the CER subclasses with an acyl chain length of C24 (90% of the CER) dictates the repeat distance of the SPP unit cell in our systems. This corresponds with our previous findings that the CER dictate the formation of the lamellar phases in SC (14,31,49). In a study by McIntosh et al. (44) on a mixture with isolated CER from pig SC, consisting of CER/CHOL/palmitic acid in a 2:1:1 molar ratio, a SPP with a repeat distance of 5.4 nm was also observed. In a study by our group using similar mixtures with isolated pig CER, CHOL, and FFA a SPP was observed with a repeat distance of 5.2–5.4 nm depending on the CER/CHOL ratio and the presence of FFAs (31). Because the mixtures in both studies were prepared in the absence of pig CER EOS, but with the remaining CER isolated from pig SC, the observation of a 5.4 nm repeat distance equals that in our synthetic mixtures, suggesting that the SPP in our mixtures is very similar to the SPP present in mixtures prepared with isolated CER.
In a related neutron diffraction study on a mixture with CER AP, CHOL, cholesterol sulfate, and palmitic acid, a density profile for a bilayer structure with a smaller periodicity phase of 4.56 nm was presented (27). In that profile the lowest density is located exactly in the center of the unit cell, whereas in our profile the lowest density is located outside the center at a distance of ±0.75 nm. This difference may be explained by the different CER used: The acyl and sphingosine chains in the CER AP mixture are equal in length (both C18) and thus no interdigitation is expected to occur. Therefore in the CER AP mixture the terminal methyl groups are located in the center of the bilayer, resulting in a low density region at this location. In contrast, in our mixtures the acyl and sphingosine chains are not equal in length, being C24 and C18, respectively. This difference in chain length is likely to result in an interdigitation of the acyl chains, which could explain the slightly elevated density observed in the center of the unit cell. This is demonstrated in Fig. 5, in which an arrangement of dCER NS molecules is displayed (discussed into more detail below). The interdigitation is in agreement with the lower density regions on both sides of the elevated density in the center of the unit cell (at ±0.75 nm), as these minima correlate with the positions of the terminal CH3 groups of the acyl and sphingosine chains.
The submaxima in the scattering length density profile located at −1.5 and +1.5 nm from the center of the unit cell may be correlated to a superimposition of CHOL and the methylene chains of CER and FFA. This is similar to the submaxima observed in the profile of the CER AP mixture at ±1.3 nm from the center of the unit cell, which were also correlated to a superimposition of methylene chains and CHOL (32). Previously, for phosphatidylcholine molecules with unequal chain lengths of C10 and C18, a bilayer arrangement with interdigitating chains was also suggested (45), supporting our arrangement for the CER.
As far as the arrangement of the CER in the bilayer is concerned, in previous studies an asymmetric fully extended arrangement is suggested for CER NP (50,51). Whether an asymmetric arrangement also occurs in mixtures prepared with CER, CHOL, and FFA can be deduced from our data.
First of all, the magnitude of the structure factors correlates linearly with the D2O concentration, which is only observed in systems with a centrosymmetric unit cell. When dealing with only protonated mixtures this observation does not provide conclusive information, as also in the asymmetric fully extended configuration of the CER a symmetric unit cell is expected. However, when the mixture is prepared with dCER NS, an asymmetric fully extended configuration of the CER will result in an asymmetric density distribution for the deuterated tails (see Fig. 6 A). But because a linear relationship between structure factors and D2O concentration is observed using this mixture, a symmetric unit cell must be present.
Figure 6.
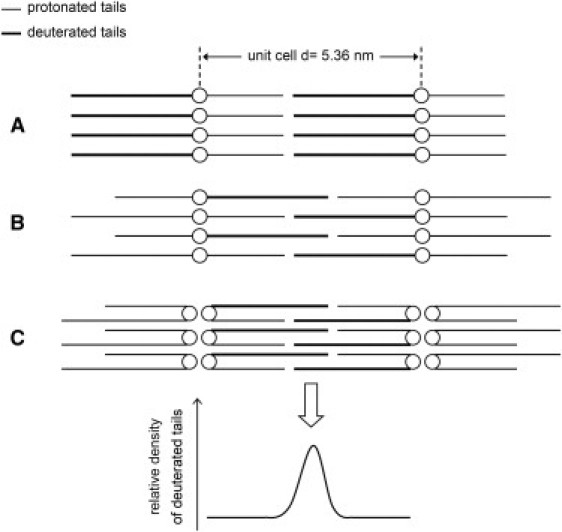
(A) Schematic of asymmetrically arranged fully extended dCER NS in the unit cell of the SPP. (B) Symmetric arrangement of fully extended dCER NS molecules in the unit cell of the SPP. (C) Symmetric arrangement in the unit cell with dCER NS in the hairpin configuration. Only in the arrangements presented in panels B and C are the deuterated acyl chains interdigitating, resulting in the difference profile that is also displayed in the figure.
Secondly, the elevated density observed in the center of the dCER NS profile can only result from an interdigitation of dCER NS tails in the center of the unit cell, which is only possible when assuming a symmetric arrangement of the CER NS. Therefore, the CER must be arranged symmetrically, either in the hairpin or in the fully extended configuration.
These two arrangements are schematically depicted in Fig. 6, B and C. However, with the current data it is not possible to determine whether the CER NS is in the fully extended configuration or in the hairpin configuration, for the following reasons: In the CER-containing mixtures, the water level between the headgroups is ∼2 water molecules per lipid, resulting in a very small intermembrane space. In addition, the expected width of a single unfolded headgroup (fully extended conformation) is almost equal to the expected width of two headgroups in the hairpin conformation. Therefore, considering that the maximum resolution in the scattering length density profile is 0.54 nm (d/2hmax) and the full width at half-maximum of the high density region in the unit cell is 1 nm, it is impossible to distinguish between the two configurations. Thus, the CER molecules in the SPP bilayer structure may be either in the hairpin, or in the fully extended configuration.
Our studies show that a 5.4-nm lamellar phase is formed that consists of homogeneously mixed CER, CHOL, and FFA. This is important for the skin barrier function, because different lipid domains may lead to an increase in the diffusivity in these lipid membranes. In addition, the low hydration level of the lamellar phase suggests a poor hydrophilic pathway, which minimizes the permeation of hydrophilic compounds across these lamellar phases. Furthermore, the symmetric arrangement observed for the bilayer arrangement of the SPP, excluding the fully extended asymmetric arrangement, may also exist in the LPP because this phase is prepared using the same CER subclasses, with the exception of CER EOS being present only in the LPP.
We may compare our results obtained for the CER in SC lipid models to the role of CER in cell membranes: Previously, monolayers of CER/CHOL in varying ratios were examined to gain insight into the formation of lipid rafts (52). In that study it was concluded that the CER/CHOL mixtures form a crystalline phase with the CER configured in a hairpin structure. This is in agreement with our findings for the CER/CHOL/FFA mixture and it may suggest that a configuration in the hairpin structure is preferred over a fully extended configuration of the CER.
Conclusions
A mixture that, apart from the absence of CER EOS, closely mimics the lipid composition in SC was examined by neutron diffraction. The high-resolution density distribution for the 5.36-nm lamellar phase in this mixture demonstrated a bilayer arrangement. The symmetry in the unit cell and the shape of the density profile of deuterated CER NS excluded an asymmetric fully extended arrangement of CER NS in the bilayer. To our knowledge, it is the first time that neutron diffraction studies were performed using deuterated CER providing insights into the arrangement of the SPP.
Acknowledgments
We thank Cosmoferm (Evonik Industries) for the provision of the ceramides, and we thank the ILL for the allocation of beamtime for neutron diffraction measurements.
This work was supported by a grant from the Dutch Technology Foundation (STW grant No. LGP7503).
Supporting Material
References
- 1.Simonetti O., Hoogstraate A.J., Ponec M. Visualization of diffusion pathways across the stratum corneum of native and in-vitro-reconstructed epidermis by confocal laser scanning microscopy. Arch. Dermatol. Res. 1995;287:465–473. doi: 10.1007/BF00373430. [DOI] [PubMed] [Google Scholar]
- 2.Weerheim A., Ponec M. Determination of stratum corneum lipid profile by tape stripping in combination with high-performance thin-layer chromatography. Arch. Dermatol. Res. 2001;293:191–199. doi: 10.1007/s004030100212. [DOI] [PubMed] [Google Scholar]
- 3.White S.H., Mirejovsky D., King G.I. Structure of lamellar lipid domains and corneocyte envelopes of murine stratum corneum. An x-ray diffraction study. Biochemistry. 1988;27:3725–3732. doi: 10.1021/bi00410a031. [DOI] [PubMed] [Google Scholar]
- 4.Bouwstra J.A., Gooris G.S., Bras W. Structural investigations of human stratum corneum by small-angle x-ray scattering. J. Invest. Dermatol. 1991;97:1005–1012. doi: 10.1111/1523-1747.ep12492217. [DOI] [PubMed] [Google Scholar]
- 5.Cornwell P.A., Barry B.W., Bouwstra J.A. Wide-angle x-ray diffraction of human stratum corneum: effects of hydration and terpene enhancer treatment. J. Pharm. Pharmacol. 1994;46:938–950. doi: 10.1111/j.2042-7158.1994.tb03248.x. [DOI] [PubMed] [Google Scholar]
- 6.Ohta N., Ban S., Hatta I. Swelling of intercellular lipid lamellar structure with short repeat distance in hairless mouse stratum corneum as studied by x-ray diffraction. Chem. Phys. Lipids. 2003;123:1–8. doi: 10.1016/s0009-3084(02)00126-3. [DOI] [PubMed] [Google Scholar]
- 7.Hatta I., Ohta N., Yagi N. Coexistence of two domains in intercellular lipid matrix of stratum corneum. Biochim. Biophys. Acta. 2006;1758:1830–1836. doi: 10.1016/j.bbamem.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 8.Gooris G.S., Bouwstra J.A. Infrared spectroscopic study of stratum corneum model membranes prepared from human ceramides, cholesterol, and fatty acids. Biophys. J. 2007;92:2785–2795. doi: 10.1529/biophysj.106.094292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chachaty C., Rainteau D., Wolf C. Building up of the liquid-ordered phase formed by sphingomyelin and cholesterol. Biophys. J. 2005;88:4032–4044. doi: 10.1529/biophysj.104.054155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clarke J.A., Heron A.J., Law R.V. The diversity of the liquid ordered (Lo) phase of phosphatidylcholine/cholesterol membranes: a variable temperature multinuclear solid-state NMR and x-ray diffraction study. Biophys. J. 2006;90:2383–2393. doi: 10.1529/biophysj.104.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marsh D. Liquid-ordered phases induced by cholesterol: a compendium of binary phase diagrams. Biochim. Biophys. Acta. 2010;1798:688–699. doi: 10.1016/j.bbamem.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 12.Bouwstra J.A., Gooris G.S., Bras W. The lipid and protein structure of mouse stratum corneum: a wide and small angle diffraction study. Biochim. Biophys. Acta. 1994;1212:183–192. doi: 10.1016/0005-2760(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 13.McIntosh T.J. Organization of skin stratum corneum extracellular lamellae: diffraction evidence for asymmetric distribution of cholesterol. Biophys. J. 2003;85:1675–1681. doi: 10.1016/S0006-3495(03)74597-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groen D., Gooris G.S., Bouwstra J.A. New insights into the stratum corneum lipid organization by x-ray diffraction analysis. Biophys. J. 2009;97:2242–2249. doi: 10.1016/j.bpj.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Megha E.L. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J. Biol. Chem. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 16.Johnston I., Johnston L.J. Ceramide promotes restructuring of model raft membranes. Langmuir. 2006;22:11284–11289. doi: 10.1021/la061636s. [DOI] [PubMed] [Google Scholar]
- 17.Bouwstra J.A., Gooris G.S., Ponec M. Phase behavior of lipid mixtures based on human ceramides: coexistence of crystalline and liquid phases. J. Lipid Res. 2001;42:1759–1770. [PubMed] [Google Scholar]
- 18.de Jager M., Groenink W., Bouwstra J. Preparation and characterization of a stratum corneum substitute for in vitro percutaneous penetration studies. Biochim. Biophys. Acta. 2006;1758:636–644. doi: 10.1016/j.bbamem.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Groen D., Gooris G.S., Bouwstra J.A. Two new methods for preparing a unique stratum corneum substitute. Biochim. Biophys. Acta. 2008;1778:2421–2429. doi: 10.1016/j.bbamem.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 20.Chen X., Kwak S., Thewalt J. Fatty acids influence “solid” phase formation in models of stratum corneum intercellular membranes. Langmuir. 2007;23:5548–5556. doi: 10.1021/la063640+. [DOI] [PubMed] [Google Scholar]
- 21.Wertz P. Epidermal lipids. In: Goldsmith L.A., editor. Physiology, Biochemistry and Molecular Biology of the Skin. Oxford University Press; Oxford, UK: 1991. pp. 205–235. [Google Scholar]
- 22.Wertz P.W., Miethke M.C., Downing D.T. The composition of the ceramides from human stratum corneum and from comedones. J. Invest. Dermatol. 1985;84:410–412. doi: 10.1111/1523-1747.ep12265510. [DOI] [PubMed] [Google Scholar]
- 23.Ponec M., Weerheim A., Wertz P. New acylceramide in native and reconstructed epidermis. J. Invest. Dermatol. 2003;120:581–588. doi: 10.1046/j.1523-1747.2003.12103.x. [DOI] [PubMed] [Google Scholar]
- 24.Masukawa Y., Narita H., Kita K. Characterization of overall ceramide species in human stratum corneum. J. Lipid Res. 2008;49:1466–1476. doi: 10.1194/jlr.M800014-JLR200. [DOI] [PubMed] [Google Scholar]
- 25.Motta S., Monti M., Ghidoni R. Ceramide composition of the psoriatic scale. Biochim. Biophys. Acta. 1993;1182:147–151. doi: 10.1016/0925-4439(93)90135-n. [DOI] [PubMed] [Google Scholar]
- 26.de Jager M.W., Gooris G.S., Bouwstra J.A. Lipid mixtures prepared with well-defined synthetic ceramides closely mimic the unique stratum corneum lipid phase behavior. J. Lipid Res. 2005;46:2649–2656. doi: 10.1194/jlr.M500221-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Kiselev M.A., Ryabova N.Y., Neubert R.H. New insights into the structure and hydration of a stratum corneum lipid model membrane by neutron diffraction. Eur. Biophys. J. 2005;34:1030–1040. doi: 10.1007/s00249-005-0488-6. [DOI] [PubMed] [Google Scholar]
- 28.Ruettinger A., Kiselev M.A., Neubert R.H. Fatty acid interdigitation in stratum corneum model membranes: a neutron diffraction study. Eur. Biophys. J. 2008;37:759–771. doi: 10.1007/s00249-008-0258-3. [DOI] [PubMed] [Google Scholar]
- 29.Schröter A., Kessner D., Neubert R.H. Basic nanostructure of stratum corneum lipid matrices based on ceramides [EOS] and [AP]: a neutron diffraction study. Biophys. J. 2009;97:1104–1114. doi: 10.1016/j.bpj.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reference deleted in proof.
- 31.Bouwstra J.A., Gooris G.S., Ponec M. Role of ceramide 1 in the molecular organization of the stratum corneum lipids. J. Lipid Res. 1998;39:186–196. [PubMed] [Google Scholar]
- 32.Bouwstra J.A., Gooris G.S., Ponec M. Phase behavior of isolated skin lipids. J. Lipid Res. 1996;37:999–1011. [PubMed] [Google Scholar]
- 33.Richard, D. 2008. http://www.ill.eu/instruments-support/computing-for-science/data-analysis/. Accessed May 2010.
- 34.Franks N.P., Lieb W.R. The structure of lipid bilayers and the effects of general anesthetics. An x-ray and neutron diffraction study. J. Mol. Biol. 1979;133:469–500. doi: 10.1016/0022-2836(79)90403-0. [DOI] [PubMed] [Google Scholar]
- 35.NCNR. 2005. www.ncnr.nist.gov/instruments/bt1/neutron.html. Accessed: May 2010.
- 36.Nagle J.F., Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dante S., Hauss T., Dencher N.A. Beta-amyloid 25 to 35 is intercalated in anionic and zwitterionic lipid membranes to different extents. Biophys. J. 2002;83:2610–2616. doi: 10.1016/S0006-3495(02)75271-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harroun T.A., Katsaras J., Wassall S.R. Cholesterol hydroxyl group is found to reside in the center of a polyunsaturated lipid membrane. Biochemistry. 2006;45:1227–1233. doi: 10.1021/bi0520840. [DOI] [PubMed] [Google Scholar]
- 39.NCNR. 2010. http://www.ncnr.nist.gov/resources/sldcalc.html. Accessed: January 2011.
- 40.Fujii M., Takeda Y., Watanabe Y. Comparison of skin permeation enhancement by 3-l-menthoxypropane-1,2-diol and l-menthol: the permeation of indomethacin and antipyrine through Yucatan micropig skin and changes in infrared spectra and x-ray diffraction patterns of stratum corneum. Int. J. Pharm. 2003;258:217–223. doi: 10.1016/s0378-5173(03)00205-9. [DOI] [PubMed] [Google Scholar]
- 41.Bouwstra J.A., Gooris G.S., Downing D.T. Lipid organization in pig stratum corneum. J. Lipid Res. 1995;36:685–695. [PubMed] [Google Scholar]
- 42.Percot A., Lafleur M. Direct observation of domains in model stratum corneum lipid mixtures by Raman microspectroscopy. Biophys. J. 2001;81:2144–2153. doi: 10.1016/S0006-3495(01)75862-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velkova V., Lafleur M. Influence of the lipid composition on the organization of skin lipid model mixtures: an infrared spectroscopy investigation. Chem. Phys. Lipids. 2002;117:63–74. doi: 10.1016/s0009-3084(02)00042-7. [DOI] [PubMed] [Google Scholar]
- 44.McIntosh T.J., Stewart M.E., Downing D.T. X-ray diffraction analysis of isolated skin lipids: reconstitution of intercellular lipid domains. Biochemistry. 1996;35:3649–3653. doi: 10.1021/bi952762q. [DOI] [PubMed] [Google Scholar]
- 45.McIntosh T.J., Simon S.A., Porter N.A. New structural model for mixed-chain phosphatidylcholine bilayers. Biochemistry. 1984;23:4038–4044. doi: 10.1021/bi00313a005. [DOI] [PubMed] [Google Scholar]
- 46.Tero R., Watanabe H., Urisu T. Supported phospholipid bilayer formation on hydrophilicity-controlled silicon dioxide surfaces. Phys. Chem. Chem. Phys. 2006;8:3885–3894. doi: 10.1039/b606052h. [DOI] [PubMed] [Google Scholar]
- 47.Creczynski-Pasa T.B., Millone M.A., Vela M.E. Self-assembled dithiothreitol on Au surfaces for biological applications: phospholipid bilayer formation. Phys. Chem. Chem. Phys. 2009;11:1077–1084. doi: 10.1039/b811964c. [DOI] [PubMed] [Google Scholar]
- 48.Small D.M. The physical chemistry of lipids. In: Hanahan D.J., editor. Handbook of Lipid Research. Plenum Press; New York: 1986. [Google Scholar]
- 49.de Jager M.W., Gooris G.S., Bouwstra J.A. Modeling the stratum corneum lipid organization with synthetic lipid mixtures: the importance of synthetic ceramide composition. Biochim. Biophys. Acta. 2004;1664:132–140. doi: 10.1016/j.bbamem.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 50.Dahlén B., Pascher I. Molecular arrangements in sphingolipids. Crystal structure of N-tetracosanoylphytosphingosine. Acta Crystallogr. B. 1972;28:2396–2404. [Google Scholar]
- 51.Rerek M.E., Chen H., Moore D.J. Phytosphyngosine and sphingosine ceramide headgroup hydrogen bonding: structural insights through thermotropic hydrogen/deuterium exchange. J. Phys. Chem. B. 2001;105:9355–9362. [Google Scholar]
- 52.Scheffer L., Solomonov I., Addadi L. Structure of cholesterol/ceramide monolayer mixtures: implications to the molecular organization of lipid rafts. Biophys. J. 2005;88:3381–3391. doi: 10.1529/biophysj.104.051870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.