

Abstract
The abnormal self-assembly of a number of proteins or peptides is a hallmark of >20 amyloidogenic diseases. Recent studies suggest that the pathology of amyloidogenesis can be attributed primarily to cytotoxic, soluble, intermediate oligomeric species rather than to mature amyloid fibrils. Despite the lack of available structural information regarding these transient species, many therapeutic efforts have focused on inhibiting the formation of these aggregates. One of the most successful approaches has been to use small molecules, many of which have been found to inhibit toxic species with high efficacy. A significant issue that remains to be resolved is the mechanism underlying the inhibitory effects of these molecules. In this article, we present extensive replica-exchange molecular dynamics simulations to study the early aggregation of the human islet amyloid polypeptide segment 22–27 in the presence and absence of the small-molecule inhibitor resveratrol. The simulations indicate that aggregation of these peptides was hindered by resveratrol via a mechanism of blocking the lateral growth of a single-layered β-sheet oligomer (rather than preventing growth by elongation along the fibril axis). Intersheet side-chain stacking, especially stacking of the aromatic rings, was blocked by the presence of resveratrol molecules, and the overall aggregation level was reduced.
Introduction
Amyloidogenic diseases belong to a range of neurodegenerative or metabolic disorders characterized by abnormal self-assembly of locally expressed proteins or peptides (1) whose end-products of self-assembly exhibit a similar pleated β-sheet fibrillar morphology (2). It is interesting to note that recent studies indicate that the main cytotoxicity of these amyloidogenic peptides is not linked to mature fibrils but rather to soluble, intermediate oligomeric species (2–5). To interrupt the oligomer formation by chemical substances could be one promising way to alleviate the toxicity.
The most common antiamyloid small-molecule inhibitors belong to the family of polyphenol compounds and contain two common functional moieties: aromatic rings and polar groups on the rings (2). Novel inhibitor molecules were designed based on the unique scaffold obtained from these compounds (2,6). The resulting small-molecule inhibitors can be very active even in the presence of membranes (7,8). Different molecules show distinct inhibition behaviors: some inhibitors block the formation of oligomers but promote fibril formation, some block the formation of fibrils but not oligomers, and others block both (9–11). However, the underlying inhibition mechanism is not yet completely clear.
In this study, we attempted to characterize the interactions between a small molecule, resveratrol, and human islet amyloid polypeptide segment 22–27 (hIAPP22–27), an aggregating peptide derived from the human amyloid islet peptide implicated in Type II diabetes. Resveratrol is a well-known constituent of red wine and a small polyphenol found to be capable of inhibiting hIAPP fibril formation efficiently (7,8,12). The hIAPP22–27 segment alone was found to be amyloidogenic and capable of forming typical amyloid fibrils (13). The sequence NFGAIL in humans differs by three amino acids from the rodent IAPP (NLGPVL), which together with three other mutations in full-length IAPPs defines the divergent aggregation ability of rodent species (13,14). The hIAPP22–27 sequence is hydrophobic, and the aromatic residue F23 is believed to play a key role in aggregation through the formation of aromatic interactions. Given the aromatic nature of resveratrol, similar aromatic interactions could be important in inhibition processes (2,15). Therefore, hIAPP22–27 is a suitable model peptide to study amyloid aggregation. Our goal was to investigate the effect of the inhibitor on the early steps of aggregation rather than the interaction of the inhibitor with a preformed fibril. We used extensive replica-exchange molecular-dynamics (REMD) simulations, with seven copies of the short peptide model. We considered a preformed β-sheet tetramer as the aggregation nucleus together with three free peptides (15). The self-assembly of the short peptide was studied with and without resveratrol (7,8,12). The simulation results revealed that lateral association occurs at a very early phase of aggregation and that the impact of resveratrol on early oligomers lies in its blockage of the lateral association. Competing with free peptides, resveratrol molecules bind to the hydrophobic side chains, which play key roles in mediating β-sheet stacking.
Methods
Parameterization and validation of the resveratrol model
The structure of the small molecule resveratrol is shown in Fig. 1 A. The initial topology for trans-resveratrol was generated in Spartan and energy-minimized (16). After geometry optimization at the HF/6-31G∗ level using Gaussian09 (17), atomic partial charges were derived using the R.E.D. III package (18). Other bonded and nonbonded parameters of resveratrol were taken from the optimized potentials for liquid simulations all-atom (OPLS-AA) force field (19). The details of validation of the parameters can be found in the Supporting Material.
Figure 1.
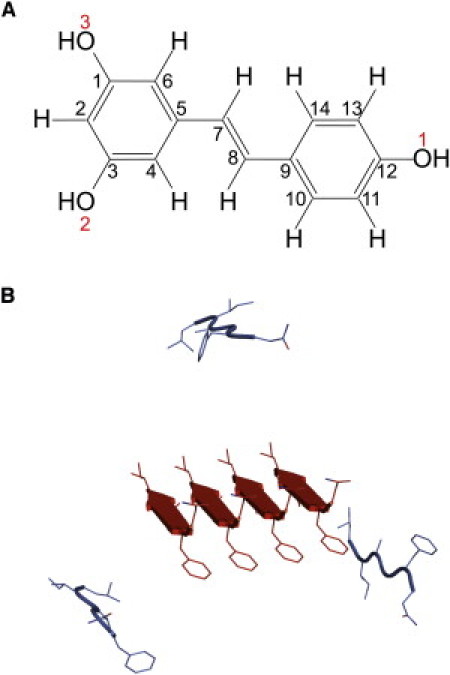
(A) Molecular geometry of trans-resveratrol. All carbon atoms are distinguished by sequential numbers (black), and oxygen atoms on three hydroxyl groups are labeled with red numbers. (B) Cartoon representation of seven strands of hIAPP22–27. A preformed parallel-β-sheet tetramer (red online) at center is surrounded by three APMs (blue online) placed at least 10 Å away from the tetramer.
Replica-exchange molecular-dynamics simulations
We chose a short segment of hIAPP22–27, known to be amyloidogenic, as a peptide model to study amyloid aggregation (15). Recent studies have shown that other segments from hIAPP can also form an amyloid structure (20–22). The initial conformation for our simulations consists of a parallel β-sheet (23) consisting of four strands centered in a dodecahedron box with a size of 40 Å and surrounded by three monomers, each rotated 120° from the others and placed at least 10 Å from the β-sheet (Fig. 1 B). We will refer to the four strands of peptide segment in the preformed β-sheet as preformed β-peptides (PBPs). The three peptide monomers that were initially positioned away from the PBPs are referred to as added peptide monomers (APMs). To cover more sampling space, the APMs were rotated around the PBPs 30° each time to generate a total of four different initial configurations (after four rotations, APM peptides adopt the same conformational configuration as one with no rotation, so we do not consider further rotations). The N- and C-termini of hIAPP22–27 were capped by acetyl (ACE) and N-methyl (NME) groups. We expect that the PBP tetramer can act as an aggregation seed to accelerate the overall aggregation process. However, the PBP was not subject to any type of restraint during simulations, because the tetrameric β-sheet for such a short peptide is neither stable nor competent to work as an aggregation nucleus. In fact, the PBP was found to be partially stabilized and undergo large structural fluctuation and reorganization, as we expected. A temperature-swapping REMD simulation scheme was performed using the Gromacs simulation package with the OPLS-AA force field for the peptides (20,26), with 32 replicas. If not explicitly marked, the data used were obtained from the lowest temperature, 315 K. All the error bars were obtained through block averages: the last 300 ns of data were divided into three 100-ns segments. The details of the REMD method can be found in the Supporting Material.
Binding energy calculation
The binding energy of resveratrol on hIAPP22–27 peptides can be estimated using the molecular mechanics generalized Born/surface area (MM-GBSA) method (24) in the equation
where , , and are the total potential energies of the resveratrol-peptide complex, peptide alone, and resveratrol alone, respectively. Each of the three energy values in the equation can be decomposed into three terms: the intraprotein potential energy (the MM term), calculated by the Gromacs package; the polar part of the solvation energy (the GB term), estimated by using the generalized Born model; and the nonpolar part of the solvation energy (the SA term). The last two solvation parts were calculated in the sander module of the AMBER 9 package (25,26). The source code of the Amber leap program was modified in-house to import nonbonded parameters from the OPLS-AA force field.
Results
Evolution of β secondary structure in the hIAPP22–27 peptide in the absence of resveratrol
The assembly progress of the hIAPP22–27 peptide was monitored by examining the β secondary structure evolution in the presence and absence of the inhibitor (Fig. 2). All the data are obtained from the lowest temperature ensemble. The β secondary structure formation was quantified by β-content, the ratio of the number of residues in β secondary structure to the total number of residues (42 in this study). As an initial condition for our study, a parallel β-sheet composed of four peptide strands (PBPs) was formed, with three APMs, as described in the Methods section. During the first 50 ns of the simulation, the β-content in PBPs sharply decreases and loses nearly half the β-structure (black solid line). Afterward, the β-content of the PBP fluctuates around 0.25 until the end of the 500-ns simulation. The β-content of the APM grows constantly during the first 400 ns. After that, the β-content arrives at the plateau of 0.25 (gray solid line). It is interesting to find that regardless of the initial configuration of the peptides, either in preformed β-sheet or in random monomers, the β-content is approaching 0.25 at the end of the 500-ns simulation.
Figure 2.
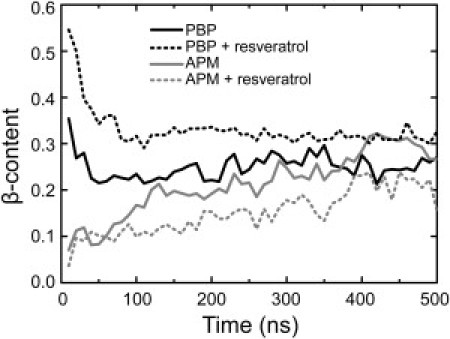
Time evolution of the β-content of PBPs and APMs in the absence and presence of resveratrol. β-content is the ratio of the number of residues in β secondary structure to the total residue number for each species, 24 for PBP and 18 for APM. Each data point on the curves is an average over 10,000 time frames (10 ns). The secondary structures of peptides were assessed by the DSSP algorithm (45). All data are from the ensemble of T = 315 K.
Aggregation of the hIAPP22–27 peptide in the absence of resveratrol
We classified the hIAPP22–27 peptide oligomers formed in our simulations based on two separate criteria. The first is that at least two interstrand backbone hydrogen bonds are formed. Under this criterion, peptide aggregates can be grouped into different β-sheet oligomers (BSOs). The second criterion is based only on interresidue distances: at least four pairs of residues must form interstrand atomic contacts. An atomic contact between two residues is considered to be formed when the minimum interresidue distance between any pair of heavy atoms is <5 Å. Based on this criterion, peptide aggregates can be divided into different collapsed oligomers (CLO). In our classification, the criteria of two hydrogen bonds in BSOs and four atomic contacts in CLOs are somewhat arbitrarily chosen. We found that changing these thresholds changed the population of different aggregates; however, the results regarding the behavior in the absence or presence of resveratrol were tolerant to the changes.
We observed in our simulations that the seven peptide strands form many types of oligomers. The system configuration can be described by the number of oligomers and the related oligomer size, which is the number of strands in one oligomer. We used the following conventions, based on size, to label the configurations. For example, 7 or 7∗ means the seven peptides form a heptameric BSO or CLO. A label such as 6.1 indicates the presence of two BSO oligomers in which one is a hexamer and one is a monomer. The 1 is omitted for simplicity, so that 6.1 is simplified to 6. A system label of 4 is in fact 4.1.1.1, which means four different oligomers of which one is a tetramer and three are monomers. The reason we used two criteria is that they can provide complementary information. For example, for a configuration with two BSOs, the information about whether the two BSOs are close to each other in space is lost. If one can check that the two BSOs belong to one CLO, then it is clear that the two BSOs are close in space, whereas if the two BSOs belong to two CLOs, then the two BSOs are far away from each other. The distributions of aggregate patterns are shown in Fig. 3.
Figure 3.
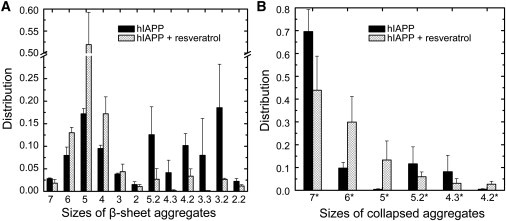
(A) Species distribution of β-sheet aggregates. (B) Species distribution of collapsed aggregates. See text for definition of β-sheet aggregates and collapsed aggregates. Aggregates with very small populations are excluded from the figures. All data are from the ensemble of T = 315 K.
The distribution of BSOs (Fig. 3 A) indicates that the peptides have an equal possibility of forming single-sheet (or single-BSO, including sizes 7, 6, 5, 4, 3, and 2) aggregates or double-sheet aggregates (sizes 5.2, 4.3, 4.2, 3.3, 3.2, and 2.2). The evidence that 70% of aggregates are heptameric CLOs (size 7∗ (Fig. 3 B)) implies that 70% of aggregates in all BSO configurations have their component strands packed into condensed aggregates. This result shows that single-layer β-sheets, i.e., BSOs of 7 and 6, have very small populations, which indicates that elongation along a single β-sheet may not be the primary mode of nucleation formation. The formation of double-layered β-sheets is an important event that is also unexpected in our simulations.
To characterize the structure of the double-sheet aggregates, an root-mean-squared deviation (RMSD) clustering (27) was performed. For different aggregate species, the structures of the cluster center of the top-ranked clusters were taken as representative structures (Fig. 4). The percentage of each top-ranked cluster in each species is also shown in parentheses. The RMSD calculation was based on heptamers, and unstructured monomers can cause large RMSD fluctuations that result in classification without an especially dominant top-ranked cluster, such as for size 2.2 (only 8.0%).
Figure 4.
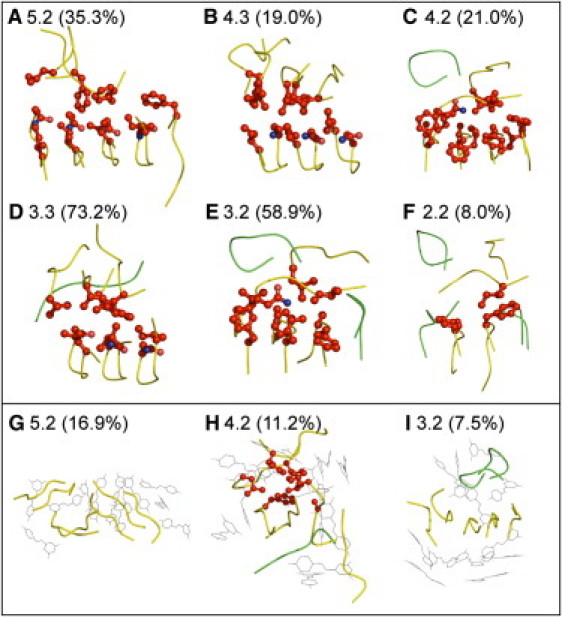
Representative structures of aggregates with double β-sheets. Percentages in parentheses show the size of the first-ranked cluster in the overall aggregates with certain size. The stacking side chains are indicated by red balls and sticks. Peptide backbones are shown as ribbons, yellow for oligomers and green for monomers. Resveratrol molecules are shown in black, line representation. All six aggregates in the absence of resveratrol (A–F) show a multilayer arrangement. (G–I) Three aggregates in the presence of resveratrol. Except in the case of 4.2 (H), there are no side-chain contacts between the two β sheets.
In these double-sheet aggregates, we found that one layer of β-sheets usually evolves from the PBPs, and we refer to this layer as the primary β-sheet; the other layer is formed by APM self-assembly and referred to as the secondary β-sheet. The secondary β-sheet (upper β-sheet layer in Fig. 4, A–F) tends to lie parallel to the primary β-sheet (the lower layer), involving extensive side-chain stacking interactions (explicitly shown as red balls and sticks), in a face-to-face configuration. Such an arrangement prevents exposure of hydrophobic residues to solvents and thereby stabilizes stacking β-sheets. Even in the small aggregates, such as double dimers (Fig. 4 F), the face-to-face intersheet interaction is already formed, suggesting a natural pathway in which lateral extension occurs simultaneously with elongation at the very early stage of amyloid aggregation. In fact, the structure of single-layer aggregates of larger sizes, such as heptamers (size 7), is not a normal planar β-sheet. Instead, it takes the form of a β-barrel by wrapping inward several side chains (see Fig. S1 in the Supporting Material). The planar β-sheet structures of short peptides resolved by microcrystal also demonstrate intimately steric zippering of side chains (28,29). Thus, side-chain interactions, as well as backbone hydrogen bonds are the main driving forces during the early aggregation process.
Distinct evolution of β-content in the presence of resveratrol
When resveratrol molecules are added into the system, the peptides show distinct self-assembly behaviors. After 100 ns, the β-content of the PBP is stabilized around 0.3 (Fig. 2, black dotted line), which is higher than the value of its counterpart in the absence of resveratrol (∼0.25). The evolution of β-structures in APMs is much slower, taking nearly 400 ns to reach a steady level (Fig. 2, gray dotted line) of ∼0.20, which is smaller than the value of its counterpart in the absence of resveratrol (∼0.25). It seems that resveratrol acts to maintain the preformed β-structure but prevent the monomers from forming new β-structure (with the PBPs). Ideally, the β-content of PBPs and APMs should be identical. Our simulations indicate that such an equilibration process is far beyond the timescale of 500 ns. Of more importance, we found that even though the β-content does not show significant differences between the two ensembles (with and without resveratrol), it does not mean that the two ensembles are the same. The different inhibitory effects of resveratrol on PBPs and APMs are further analyzed from a thermodynamic perspective.
Thermodynamic evidence of the distinct effects of resveratrol
We compared the population of monomeric peptides and β-content as a function of temperature for the system with and without resveratrol (Fig. 5, A and B). Data were analyzed based on the last 300 ns of simulations at all temperatures. The amount of monomeric peptides remaining in solution is a normal measure in experiments to detect the extent of amyloid aggregation (30). In the presence of resveratrol, the abundance of monomers unambiguously exceeds that in the absence of resveratrol at all temperatures, indicating the inhibitory effect of the compound on peptide aggregation (Fig. 5 A). The β-content results illustrate the same effect (Fig. 5 B). Although at low temperatures the amount of β-structure is equal in the two systems, at higher temperatures, the β-content is evidently lower in the presence of resveratrol than in its absence.
Figure 5.
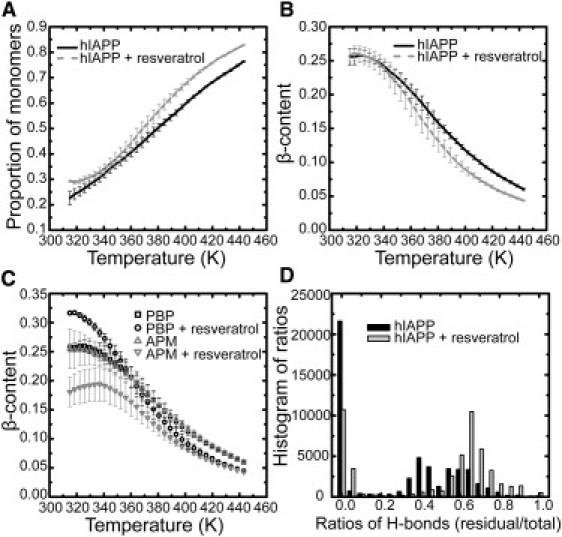
Temperature-dependent features of peptide aggregation. (A) Proportion of peptide monomers, the ratio of the number of monomers to the total number of peptide strands. (B) β-content. (C) β-content contributions from PBPs and APMs. (D) Histogram statistics of survival ratios of the preformed hydrogen bonds. Calculations of A–C are based on data from the last 300 ns of simulations. Error bars are obtained from the standard deviation of three average values from nonoverlapping time blocks: 200–300 ns, 300–400 ns, and 400–500 ns. The survival ratio is the ratio of the number of hydrogen bonds maintained in the PBP to the total number of 15 in the intact PBP. The histogram in D is based on a subset of data from the last 300 ns of simulations.
In Fig. 5 C, the calculation of β-content is broken down into PBP and APM peptides. The plot clearly shows that resveratrol acts differently on aggregates than on free peptides in solution. PBP peptides have ∼17% more β-structure than APM peptides at 315 K in the presence of resveratrol, and the difference eventually vanishes when the temperature is >400 K. In contrast, without resveratrol, β-structure populations are not dependent on the initial structures at any temperature. Thus, the effects of resveratrol are dependent on conformational state: resveratrol inhibits the β-sheet transition of monomeric peptides while holding the existing β-structure.
In the initial PBP configuration, 15 backbone hydrogen bonds were formed that can be regarded as preformed hydrogen bonds. To trace their survival during the simulation, in each snapshot, a ratio between the number of preformed hydrogen bonds and the total number of hydrogen bonds was calculated. The evolution of this ratio is plotted in Fig. S2. The related population of the ratio is shown in Fig. 5 D. Data were obtained from the last 100 ns simulation at the lowest temperature. At a ratio of 0, where all preformed hydrogen bonds are lost, the population nearly doubles in the case without resveratrol compared to the case with resveratrol. The loss of preformed hydrogen bonds can happen in the process of dissociation and reassociation of peptides, as well as during reorientation from parallel to antiparallel β-sheets. On the other hand, the high peak at a ratio of 0.65 for the system in the presence of resveratrol indicates that resveratrol stabilizes the preformed hydrogen bonds. The population analysis clarifies our aforementioned speculation about the higher β-content found in PBPs, that is, that resveratrol molecules protect the existing β-sheet oligomers against reorganization.
Destabilization of double-layered β-sheet aggregates by resveratrol
The distributions of aggregate patterns in the presence of resveratrol are clearly different from those in the absence of resveratrol (see Fig. 3, gray bars). Compared with peptides without inhibitors, it is clear that those with resveratrol have a greatly increased population of monomers such as species 6, 5, and 4, as well as 6∗, 5∗, and 4∗. This result agrees well with our previous observation suggesting the induction of a higher population of monomers by resveratrol molecules (Fig. 5 A). On the other hand, the population of configurations with double-β-sheet oligomers is clearly reduced (see also Fig. S6). The double-sheet aggregates are shown in Fig. 4, G–I, and Fig. S3. In the presence of resveratrol, the hydrophobic residues are buried more favorably by the amphiphilic compounds than by side chains of other strands. The double-sheet aggregates are more disordered than those in the absence of resveratrol. The double β-sheets do not have the feature of parallel stacking seen in the inhibitor-free simulations (Fig. 4, A–F).
Inhibitory activities of resveratrol caused by blocking of intersheet side-chain contacts
A β-sheet pentamer is the dominant configuration (>50%) in the heptameric aggregates in the presence of resveratrol (Fig. 3). The formation of pentamers is via elongation of the PBP tetramer by adding one APM peptide. Because of the abundance of this kind of aggregate, we focused on studying the binding preference of inhibitors on the pentamer. As a simple model, the β-sheet pentamer can be viewed as a truncated single-layer protofibril. The spatial distribution of resveratrol around the pentamer is illustrated in Fig. 6. The six-residue peptide is short and forms a turnlike, twisted parallel β-sheet instead of a flat β-sheet. The centers of mass of resveratrol molecules are marked as colored dots. Different colors differentiate the six binding sites that are the largest populated clusters in the rank of all 12 clustering resveratrol molecules in the aggregate species of BSO labeled as 5. Structures belong to the six binding sites cover 90.1% of all structures we examined. A generous RMSD cutoff of heavy atoms of 1.25 nm was used to consider only the positions of small molecules surrounding the pentamer and neglecting their internal conformation and overall rotation. The preferences of the six binding sites are illustrated by the population of bound resveratrol therein, as well as by binding energies calculated by the MM-GBSA model (Table 1).
Figure 6.
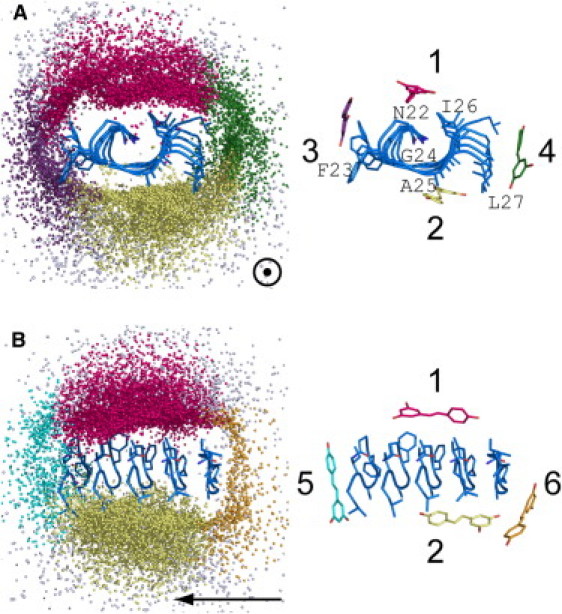
Six binding sites of resveratrol molecules on β-sheet hIAPP22–27 pentamers (left), with the representative structures at each binding site (right). Oligomers with size 5 in Fig. 4 were chosen and distribution of small molecules (colored dots) surrounding the β-sheet pentamers (blue) was drawn by superimposing the peptides in the middle. Roughly six binding sites are identified and represented by different colors (hot pink, Site 1; yellow, Site 2; violet, Site 3; green, Site 4; cyan, Site 5; orange, Site 6; white, all other sites). The corresponding representative structures are shown on the right. Arrows point to the longitude direction of the fibril. Binding sites are shown along the direction of fibril growth (A) and perpendicular to the direction of growth (B). For the sake of clarity, sites on the front and back sides of the structures are not shown, corresponding to sites 5 and 6 in A and sites 3 and 4 in B.
Table 1.
Percentages and binding energies of resveratrol at six binding sites of β-sheet pentamers
| Binding site | Percentage (%)∗ | Binding energy (kJ/mol) |
||
|---|---|---|---|---|
| ΔEtotal† | ΔEnp‡ | ΔEp§ | ||
| 1 | 33.6 | −51.0 ± 8.7 | −77.7 ± 8.6 | 26.7 ± 8.1 |
| 2 | 30.1 | −36.6 ± 7.2 | −65.6 ± 8.8 | 29.1 ± 4.5 |
| 3 | 9.8 | −21.7 ± 5.6 | −44.1 ± 7.8 | 22.4 ± 4.3 |
| 4 | 8.9 | −35.7 ± 8.2 | −57.4 ± 9.6 | 21.7 ± 4.3 |
| 5 | 4.0 | −34.1 ± 6.4 | −57.2 ± 8.5 | 23.2 ± 4.2 |
| 6 | 3.7 | −18.6 ± 6.4 | −34.7 ± 8.9 | 16.1 ± 3.9 |
Values are represented as the mean ± SD of 100 structures from each class with resveratrol binding at different binding sites.
Percentage of binding at each binding site in relation to the total binding on the β-sheet pentamers. Only those aggregates with a well-formed β-sheet pentamer were taken into consideration, and the percentage of these aggregates is ∼50%, as shown in Fig. 4.
ΔEtotal is the sum of ΔEnp and ΔEp.
ΔEnp is the sum of binding energies induced by van der Waals interactions between resveratrol and peptides as well as the surface-area term in solvation energy.
ΔEp is the sum of binding energies induced by electrostatic interactions between resveratrol and peptides, as well as the generalized Born term in solvation energy.
The high binding preference of resveratrol for the two surfaces of the protofibril (Sites 1 and 2 in Fig. 6) is clear, as indicated by highest percentages (33.6% and 30.1% in Table 1) and lowest binding energies (−51.0 kJ/mol and −36.6 kJ/mol in Table 1). Note that polar components, ΔEp, are even higher than those binding at other sites. However, the unfavorable polar interaction is compensated by an extremely favorable nonpolar component ΔEnp. The favorable van der Waals interactions benefit from the hydrophobicity of both side chains at the sites and the compound. In contrast, the compound is less likely to bind at the growth edges (Sites 5 and 6), as shown by the sparser dot clouds along the longitudinal direction (Fig. 6 B). Percentages are lowest for these two sites (4.0% and 3.7%), and binding energies are also higher (−34.1 kJ/mol and −18.6 kJ/mol). Although the energy penalty originating from polar contacts is not as high as Sites 1 and 2 (23.2 kJ/mol and 16.1 kJ/mol at Sites 5 and 6 vs. 26.7 kJ/mol and 29.1 kJ/mol at Site 1 and 2), the hydrophilic nature of Sites 5 and 6, where backbone hydrogen-bond donors and acceptors are located, impedes the nonpolar contacts and makes them less preferential binding sites. The binding preference for Sites 3 and 4 ranks between the aforementioned two groups of binding sites in terms of percentages, ΔEtotal, ΔEnp, and ΔEp.
Lateral association of β-sheets is a prerequisite for further growth of amyloid fibrils, and intersheet side-chain interactions mainly drive and stabilize this kind of association. However, the extensive binding of resveratrol to the surface of sheets can efficiently block the intersheet side-chain interactions. We counted the atomic contacts between side chains of the primary BSO and the other peptides, and between side chains of the primary BSO and resveratrol molecules (Fig. S4). The two quantities have a strong anticorrelation, which unambiguously indicates that resveratrol binding reduces the possibility of β-sheet formation in a double-layer arrangement.
To characterize the details of the atomic interaction between resveratrol and peptides, contact maps of four types of peptide-inhibitor interaction were drawn (Fig. 7). Four types of interaction between resveratrol and side chains or main chains of BSOs or monomers were considered separately. A contact is believed to form when the distance separating a pair of heavy atoms between two partners is <5 Å. We calculated the inhibitor-peptide contact numbers/frame/strand: 12.9 for BSO side chains; a similar value, 12.7, for monomer side chains; 3.7 for BSO backbones; and 6.1 for monomer backbones. The transition from unstructured monomers to BSOs limits exposure of the backbones to inhibitors by reducing the contact numbers by nearly half (from 6.1 to 3.7). Despite the similar contact numbers on side chains, when the peptide is in a monomeric state, resveratrol has an overwhelming preference for the aromatic F23 over the other two hydrophobic residues, I26 and L27 (Fig. 7 D). This preference is attenuated when the peptide is part of a protofibril. The result is consistent with the less frequent binding at Site 3, where F23 locates, as shown in Fig. 6. When inhibitors bind to specific positions on ordered oligomers, stereospecific features, such as the existence of hydrophobic grooves on the surface, are recognized by compounds. However, when inhibitors interact with unstructured monomers, there is no particular structural motif on the unfolded peptides. Therefore, physicochemical properties of the compound and single residue, such as aromaticity or hydrophobicity, rather than tertiary structures, determine the interactions. In addition, the middle region of resveratrol shows the largest contact intensities with side chains of peptides (Fig. 7, B and D). In contrast, three hydroxyl groups on the resveratrol make polar contacts with peptides that are mainly located on the terminal residues (F23 and L27) and exclusively involved with backbones (Fig. 7 A). The information derived from contact maps confirms the previous results of favorable nonpolar binding energies on Sites 1–4 and less favorable polar binding energies on Sites 5 and 6 (Table 1). Also, the differences in binding energy at different sites are mainly caused by physiochemical properties of the binding site.
Figure 7.
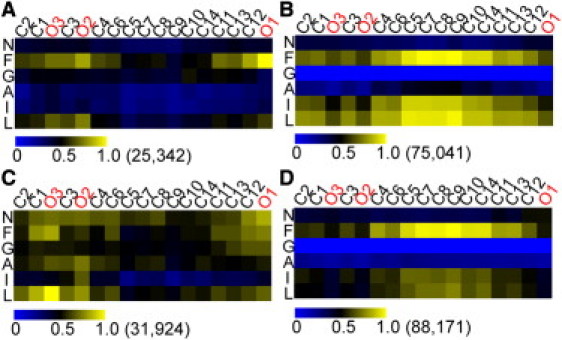
Contact maps of heavy atoms on resveratrol (abscissa) and on the residues of peptides (ordinate). The contact intensities have been normalized by dividing the individual largest contact occurrence number (in parentheses next to the color bar). Contacts on residues are divided into four groups: backbones of BSOs (A), side chains of BSOs (B), backbones of monomers (C), and side chains of monomers (D). The atoms of resveratrol are ordered according to spatial proximity in chemical structure rather than sequence atomic numbers (see Fig. 1 for the chemical structure).
Discussions
The main focus of this study is to elucidate the inhibition mechanism of an antiaggregation compound, resveratrol. A short segment of hIAPP22–27 (NFGAIL) was studied as a model system. To better observe the inhibitory effects of the compound, simulations of peptides with and without resveratrol were performed. Our results suggest that the inhibitory mechanism of resveratrol is to block lateral association that occurs at the very early stage of aggregation. Such a mechanism does not contradict an inhibitory mechanism that comes into play later in the fibril elongation process, in which inhibitor molecules occupy the surface grooves on the well-formed fibril edge (31–33).
There are quite a few interesting experimental studies on the inhibitory mechanism of fibril formation of IAPP and Aβ proteins by resveratrol (7,8,12,34,35). IAPP and Aβ amyloid show a similar secondary structure in the fibrillar state and share amino acid sequence in the ordered region (36). When resveratrol is added, the usual fibrillation pathway is diverted to an off-pathway product consisting of spherical amorphous oligomers with dominant secondary structure of random coil. It is important to note that these spherical structures are not cytotoxic (7,34,35). The link between cytotoxicity and IAPP fibril formation could be multifactorial, e.g., involving the incorporation of lipid upon IAPP fibril formation (12,37), or hydrophobic exposure of the fibrillar intermediates (38).
In our simulations, the presence of resveratrol causes the redistribution of oligomer species. One of the prominent features is that the ordered hydrophobic matching patterns demonstrated in the double-layered oligomer species (Fig. 5) are greatly diminished. Even though the molecules we studied are peptide segments, we believe that the mechanism identified in this study will apply to the full-length protein as well. We propose the following putative inhibitory mechanism based on the results of our simulations (Fig. 8). In the path leading to the native end-product (path 1), aggregates grow both laterally and longitudinally, driven by hydrophobic matching by the side chains as well as hydrogen bonds on the backbone. When resveratrol interferes with this path, aggregates can only elongate in the longitudinal direction (path 2). Based on this study, there is no hint about whether these single-layer β-sheets can continue to grow into a single-layer fibril or the fibril intermediate species is just too fragile to grow further. We found no indication that typical fibrils formed when full-length hIAPP was incubated with resveratrol in vitro (7,12). The observations seem consistent with available protofibril structure models (29,39–44) in which the steric zippering of side chains is one major factor in stabilization of the fibril structures. Thus, if resveratrol acts on the full-length hIAPP with the same mechanism observed here for hIAPP22–27, it appears that inhibition of lateral growth is active enough to fully abolish fibril formation.
Figure 8.
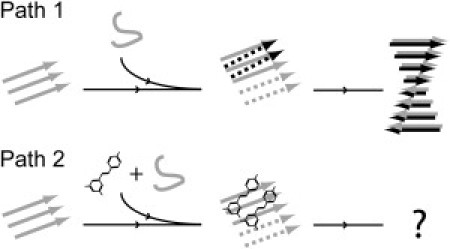
Self-assembly of amyloidogenic peptides and suggested inhibitory mechanism of resveratrol. When monomers attempt to integrate into a fibril nucleus, aggregates grow in two directions, by lateral association and longitudinal elongation (path 1). The end product is a multiple-layer fibril. When peptides are incubated with resveratrol, resveratrol is capable of blocking the lateral growth, whereas longitudinal elongation remains nearly intact (path 2). Black and gray are used to distinguish β-strands belonging to different layers. Dashed arrows represent the nascent β-strands in oligomers.
Acknowledgments
This work was supported by grants from Nanyang Technological University (RG65/06) and the Ministry of Education academic research fund AcRF Tier 2 (T206B3210RS). The authors gratefully acknowledge the generous provision of computation facility time by the Nanyang Technological University High-Performance Computing Center. J.E.S. acknowledges support from the David and Lucile Packard Foundation and the National Science Foundation (MCB 0642086). Parts of simulations were performed on the Ranger cluster at the Texas Advanced Computing Center (LRAC MCA 05S027).
Supporting Material
References
- 1.Kopito R.R., Ron D. Conformational disease. Nat. Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 2.Porat Y., Abramowitz A., Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 3.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 5.Kayed R., Head E., Glabe C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 6.Mishra R., Bulic B., Winter R. Small-molecule inhibitors of islet amyloid polypeptide fibril formation. Angew. Chem. Int. Ed. Engl. 2008;47:4679–4682. doi: 10.1002/anie.200705372. [DOI] [PubMed] [Google Scholar]
- 7.Mishra R., Sellin D., Winter R. Inhibiting islet amyloid polypeptide fibril formation by the red wine compound resveratrol. ChemBioChem. 2009;10:445–449. doi: 10.1002/cbic.200800762. [DOI] [PubMed] [Google Scholar]
- 8.Evers F., Jeworrek C., Winter R. Elucidating the mechanism of lipid membrane-induced IAPP fibrillogenesis and its inhibition by the red wine compound resveratrol: a synchrotron x-ray reflectivity study. J. Am. Chem. Soc. 2009;131:9516–9521. doi: 10.1021/ja8097417. [DOI] [PubMed] [Google Scholar]
- 9.Taniguchi S., Suzuki N., Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005;280:7614–7623. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 10.Necula M., Kayed R., Glabe C.G. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 11.Berhanu W.M., Masunov A.E. Natural polyphenols as inhibitors of amyloid aggregation. Molecular dynamics study of GNNQQNY heptapeptide decamer. Biophys. Chem. 2010;149:12–21. doi: 10.1016/j.bpc.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Radovan D., Opitz N., Winter R. Fluorescence microscopy studies on islet amyloid polypeptide fibrillation at heterogeneous and cellular membrane interfaces and its inhibition by resveratrol. FEBS Lett. 2009;583:1439–1445. doi: 10.1016/j.febslet.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 13.Tenidis K., Waldner M., Kapurniotu A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties. J. Mol. Biol. 2000;295:1055–1071. doi: 10.1006/jmbi.1999.3422. [DOI] [PubMed] [Google Scholar]
- 14.Westermark P., Engström U., Betsholtz C. Islet amyloid polypeptide: pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA. 1990;87:5036–5040. doi: 10.1073/pnas.87.13.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gazit E. Mechanisms of amyloid fibril self-assembly and inhibition. Model short peptides as a key research tool. FEBS J. 2005;272:5971–5978. doi: 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- 16.Shao Y., Molnar L.F., Head-Gordon M. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- 17.Frisch M.J., Trucks G.W., Fox D.J. Gaussian; Wallingford, CT: 2009. Gaussian 09 User's Manual. [Google Scholar]
- 18.Dupradeau F.Y., Pigache A., Cieplak P. The R.E.D. tools: advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010;12:7821–7839. doi: 10.1039/c0cp00111b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaminski G.A., Friesner R.A., Jorgensen W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- 20.Abedini A., Raleigh D.P. Destabilization of human IAPP amyloid fibrils by proline mutations outside of the putative amyloidogenic domain: is there a critical amyloidogenic domain in human IAPP? J. Mol. Biol. 2006;355:274–281. doi: 10.1016/j.jmb.2005.10.052. [DOI] [PubMed] [Google Scholar]
- 21.Wiltzius J.J.W., Sievers S.A., Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wiltzius J.J.W., Landau M., Eisenberg D. Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 2009;16:973–978. doi: 10.1038/nsmb.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiltzius J.J.W., Sievers S.A., Eisenberg D. Atomic structure of the cross-β spine of islet amyloid polypeptide (amylin) Protein Sci. 2008;17:1467–1474. doi: 10.1110/ps.036509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Srinivasan J., Cheatham T.E., Case D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 1998;120:9401–9409. [Google Scholar]
- 25.Onufriev A., Bashford D., Case D.A. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 26.Case D.A., Darden T.A., Kollman P.A. University of California, San Francisco; San Francisco, CA: 2006. AMBER 9. [Google Scholar]
- 27.Daura X., van Gunsteren W.F., Mark A.E. Folding-unfolding thermodynamics of a β-heptapeptide from equilibrium simulations. Proteins. 1999;34:269–280. doi: 10.1002/(sici)1097-0134(19990215)34:3<269::aid-prot1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 28.Nelson R., Eisenberg D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006;16:260–265. doi: 10.1016/j.sbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Nelson R., Sawaya M.R., Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanning J.D., Hawk A.J., Meredith S.C. Chaperone-like N-methyl peptide inhibitors of polyglutamine aggregation. Biochemistry. 2010;49:7108–7118. doi: 10.1021/bi1006095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raman E.P., Takeda T., Klimov D.K. Molecular dynamics simulations of Ibuprofen binding to Aβ peptides. Biophys. J. 2009;97:2070–2079. doi: 10.1016/j.bpj.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takeda T., Chang W.L.E., Klimov D.K. Binding of nonsteroidal anti-inflammatory drugs to Aβ fibril. Proteins. 2010;78:2849–2860. doi: 10.1002/prot.22804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Convertino M., Pellarin R., Caflisch A. 9,10-Anthraquinone hinders β-aggregation: how does a small molecule interfere with Aβ-peptide amyloid fibrillation? Protein Sci. 2009;18:792–800. doi: 10.1002/pro.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ladiwala A.R.A., Lin J.C., Tessier P.M. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Aβ into off-pathway conformers. J. Biol. Chem. 2010;285:24228–24237. doi: 10.1074/jbc.M110.133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ladiwala A.R.A., Dordick J.S., Tessier P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid β through three independent pathways. J. Biol. Chem. 2011;286:3209–3218. doi: 10.1074/jbc.M110.173856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jayasinghe S.A., Langen R. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 2004;279:48420–48425. doi: 10.1074/jbc.M406853200. [DOI] [PubMed] [Google Scholar]
- 37.Jiang P., Xu W.X., Mu Y.G. Amyloidogenesis abolished by proline substitutions but enhanced by lipid binding. PLOS Comput. Biol. 2009;5:e1000357. doi: 10.1371/journal.pcbi.1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campioni S., Mannini B., Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 39.Paravastu A.K., Leapman R.D., Tycko R. Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lührs T., Ritter C., Riek R. 3D structure of Alzheimer's amyloid-β (1–42) fibrils. Proc. Natl. Acad. Sci. USA. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ritter C., Maddelein M.L., Riek R. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawaya M.R., Sambashivan S., Eisenberg D. Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 43.Luca S., Yau W.M., Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505–13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jahn T.R., Makin O.S., Serpell L.C. The common architecture of cross-β amyloid. J. Mol. Biol. 2010;395:717–727. doi: 10.1016/j.jmb.2009.09.039. [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.