

Abstract
By using a Raman microscope, we show that it is possible to probe the conformational states in protein crystals and crystal fragments under growth conditions (in hanging drops). The flavin cofactor in the enzyme para-hydroxybenzoate hydroxylase can assume two conformations: buried in the protein matrix (“in”) or essentially solvent-exposed (“out”). By using Raman difference spectroscopy, we previously have identified characteristic flavin marker bands for the in and out conformers in the solution phase. Now we show that the flavin Raman bands can be used to probe these conformational states in crystals, permitting a comparison between solution and crystal environments. The in or out marker bands are similar for the respective conformers in the crystal and in solution; however, significant differences do exist, showing that the environments for the flavin's isoalloxazine ring are not identical in the two phases. Moreover, the Raman-band widths of the flavin modes are narrower for both in and out conformers in the crystals, indicating that the flavin exists in a more limited range of closely related conformational states in the crystal than in solution. In general, the ability to compare detailed Raman data for complexes in crystals and solution provides a means of bridging crystallographic and solution studies.
The flavoprotein para-hydroxybenzoate hydroxylase (PHBH; EC 1.14.13.2) catalyzes the reaction of para-hydroxybenzoic acid (p-OHB), O2, and NADPH to form 3,4-dihydroxybenzoate, water, and NADP+ (1, 2). In the absence of NADPH, the enzyme and substrate form a nonreacting 1:1 complex. Many crystal structures of PHBH have been solved, with a variety of substituted benzoic acid ligands bound to the wild-type or mutant enzymes (3–8). In the majority of enzyme–ligand complexes, the reactive isoalloxazine moiety of the flavin is buried essentially in the protein and is adjacent to the aromatic substrate. In this “in” configuration, only the dimethyl edge and some of the re face are exposed to the solvent. However, certain substrate analogs yield an alternative “out” flavin conformation in which the isoalloxazine ring rotates about the C1′—C2′ ribityl bond by ≈33° into a largely solvent-exposed position. The in and out conformations are shown in Fig. 1. These two conformational options may explain how PHBH stabilizes the diversity of transition states required in its catalytic cycle. It has been proposed that the hydroxylation reaction occurs when the flavin is in the in position, and that flavin reduction by NADPH occurs in the out conformation (9, 10). This property of in and out flavin conformations has been observed also in phenol hydroxylase (11), which demonstrates that flavin conformational mobility occurs for some members of the flavoenzyme family.
Figure 1.

Structures of the active sites of the PHBH complexes. Water molecules are shown as red spheres, and putative hydrogen bonds are depicted with dotted lines. (A) Holo-PHBH + p-OHB (in conformation). (B) Holo-PHBH + 2,4-di-OHB (out conformation). Structures were obtained from the Protein Data Bank [PDB ID 1PBE, holo-PHBH + p-OHB (in); PDB ID 1DOD, holo-PHBH + 2,4-di-OHB (out)].
By using 752-nm excitation and a high-throughput Raman spectrometer (12, 13), we previously have obtained Raman difference spectra of the flavin ring of FAD bound to PHBH in solution (14). This experiment was done by subtracting the spectrum of the apoenzyme (without flavin) from that of the holoenzyme (flavin bound) for either the in or out conformational states in solution. The spectra consist of a rich assortment of isoalloxazine ring modes, the normal mode origins of which have been assigned by using density functional theory and ab initio calculations. There are several regions in which characteristic differences in peak positions were shown for flavin in the in or out conformation. These differences occur near 1,700, 1,430, 1,400, 1,360, 1,340, 1,240, and 1,150 cm−1 and can be used as empirical marker bands to identify the conformational state of the flavin in a complex of PHBH.
This study can be extended to explore the protein–ligand interaction in single crystals of PHBH. Studies comparing the Raman spectra of proteins in solution and in single crystals (without using a microscope) were undertaken first by Nai-Teng Yu in the early 1970s (15, 16). Later, resonance Raman studies of heme protein crystals were recorded by using a microprobe (17) or by mounting the crystals in a standard Raman instrument (18). Although these studies did find small differences between the solution and crystal phases, the work was limited severely by low sensitivity and the requirement for the resonance condition when working with heme proteins.
We have used a Raman microscope to acquire Raman spectra of protein crystals. The system is calibrated to match the Raman spectrometer we use to study samples in solution. This method allows us to accurately compare the Raman spectra of bound flavin in both the solution and crystalline phases.
Materials and Methods
Enzyme and Substrate Samples.
p-OHB and 2,4-dihydroxybenzoic acid were purchased from Aldrich and used without further purification. PHBH cloned from Pseudomonas aeruginosa was expressed in Escherichia coli (19) and purified as described (20). Apo-PHBH (lacking the flavin cofactor) was prepared according to the protocol of Muller and van Berkel (21). To stabilize the protein (both apo- and holo-), p-OHB is included in all steps of its preparation. p-OHB was removed by precipitating the apoenzyme with 70% ammonium sulfate, centrifuging, and resuspending the apoenzyme precipitate in fresh p-OHB-free 70% saturated ammonium sulfate; this process was repeated at least four times, and the apoenzyme was stored as a precipitate in the presence of 70% saturated ammonium sulfate. For spectroscopy, apoenzyme was centrifuged, the pellet was dissolved in 100 mM potassium phosphate buffer (pH 7.5), and the solution was filtered quickly (0.22 μm) before Raman spectra were collected.
Raman Spectroscopy of Aqueous Samples.
For the enzyme complexes in solution, Raman spectra were acquired by using 1,000 mW 647-nm laser excitation from an Innova 400 krypton laser (Coherent Radiation, Palo Alto, CA) and a modified Holospec f/1.4 axial transmission spectrometer (12). The x (frequency shift) and y (intensity response) axes of the instrument were calibrated by using a HoloLab Calibration Accessory (Kaiser Optical Systems, Ann Arbor, MI), which uses standardized neon and tungsten lamps and permits the transfer of calibration models between various Raman instrument configurations. Reproducibility of peak positions was better than ±1 cm−1. Typically, sample volumes were ≈50 μl, and the spectra were recorded at 22°C with the total data collection time of 5 min. Spectra of the holoenzyme and apoenzyme samples at protein concentrations of ≈15 mg⋅ml−1 were recorded, and the subtraction of holoenzyme minus apoenzyme gave the spectrum of bound FAD. The computer subtraction was done with GRAMS/32 software (Galactic Industries Company, Salem, NH). For some experiments, a spectrum of free ligand (e.g., p-OHB) was recorded also.
Crystal Preparation.
Crystals of holo-PHBH in complex with substrate (p-OHB) or substrate analog [2,4-dihydroxybenzoic acid (2,4-di-OHB)] were grown by using the hanging-drop method (8). The protein solution contained ≈10 mg⋅ml−1 enzyme in 100 mM potassium phosphate buffer (pH 7.0). The reservoir solution contained 50% saturated ammonium sulfate, 0.15 mM EDTA, 0.1 mM reduced glutathione, and 100 mM potassium phosphate buffer (pH 7.0). Hanging drops were prepared as follows: for PHBH + p-OHB, 3 μl protein + 1.5 μl well solution; for PHBH + 2,4-di-OHB, 1 μl protein + 3 μl well solution. Hanging drops were allowed to equilibrate at 4°C against 1 ml of well solution. Crystals with dimensions of approximately 50 × 50 × 30 μm grew within 1–2 weeks.
Raman Spectroscopy of Single Crystals.
Raman spectra of single crystals in situ (i.e., in hanging
drops) were acquired by using a HoloLab Series 5000 Raman microscope
(Kaiser Optical Systems) interfaced with fiber optics to the Holospec
spectrometer previously described (Fig.
2). The 647-nm laser light was injected
into the microscope's HoloProbe by using a fiber mounted on an XYZ
stage. A single crystal in a hanging drop, within the growth tray, was
placed on the microscope stage and viewed by using the charge-coupled
device video camera incorporated into the microscope. The sample was
excited with ≈100 mW 647-nm laser light, and 180° back-scattered
Raman light was collected from the protein crystal by using a
long-focal-length ×20 lens. The Raman microscope also was calibrated
by using standardized neon and tungsten lamps as described for the
Holospec f/1.4 spectrometer. Comparison of neon line frequencies as
well as the Raman spectra of sulfate (SO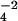 ) show that
the peak positions measured with the microscope are within 1
cm−1 of those measured with the standard
Holospec spectrometer. Ambient temperature, data collection time, and
data analysis were the same as described above.
) show that
the peak positions measured with the microscope are within 1
cm−1 of those measured with the standard
Holospec spectrometer. Ambient temperature, data collection time, and
data analysis were the same as described above.
Figure 2.

Schematic diagram (not to scale) of a Raman microscope. (Inset) Depiction of collection of Raman data from a single protein crystal in a hanging drop. Both video (①) and spectrographic data (②) can be displayed on the computer screen.
Results and Discussion
In and Out Conformations of Flavin Bound to PHBH in Solution.
The Raman difference spectra of flavin bound to PHBH in solution were obtained by using 647-nm laser excitation instead of the 752-nm excitation used previously (14) to compare these data more precisely with Raman spectra of crystals taken with the Raman microscope (also at 647 nm). Raman spectra of the flavin molecule, acquired with 647-nm excitation, use the nonresonance or “prepre-resonance” condition, in which the excitation wavelength is more than 150 nm from the flavin's absorption maxima. We calculated the difference spectra by taking a spectrum of the holoenzyme (PHBH + FAD) and subtracting from it a spectrum of apoenzyme (PHBH alone) that was prepared separately. This process differs slightly from previous work in which we created holoenzyme by adding FAD directly to the apoenzyme sample (14), and as a result our subtractions are somewhat less precise. This difference is most pronounced in the 1,600–1,700 cm−1 region, in which water and protein both have relatively large contributions that must be subtracted away. Fig. 3 shows the Raman difference spectra of FAD bound to wild-type PHBH in aqueous solution. The spectrum in Fig. 3B is caused by flavin assuming the in conformation in the presence of p-OHB, whereas the spectrum in Fig. 3C is caused by flavin adopting the out conformation in the presence of 2,4-di-OHB. Several Raman peaks shift to a higher value when the flavin moves from the in to the out conformation. These “marker bands” include the peaks near 1,540, 1,400, 1,360, 1,340, 1,240, and 1,150 cm−1. We can eliminate the possible contribution of a systematic error to these “up shifts,” because several other Raman peaks remain unperturbed, such as the peaks near 1,575, 1,500, 1,460, 1,225, and 1,175 cm−1 as well as the sulfate peak at 980 cm−1. (Ammonium sulfate is used in protein preparation and storage as described above and is detected by Raman spectroscopy even at low concentrations.) Moreover, these results essentially reproduce the earlier 752-nm-excited data (14), with minor variations caused by improved intensity and frequency calibration, narrower slit width, and different excitation wavelength.
Figure 3.

Raman difference spectra for FAD bound to wild-type PHBH. (A) Holo-PHBH + p-OHB (in), single crystal. (B) Holo-PHBH + p-OHB (in), in solution. (C) Holo-PHBH + 2,4-di-OHB (out), in solution. (D) Holo-PHBH + 2,4-di-OHB (out), single crystal.
The differences between the in and out spectra can be attributed generally to the movement of the isoalloxazine ring from its ring-binding pocket to a solvent-exposed conformation, the environment of which is less hydrophobic. Our present model for this effect is that the high-dielectric water environment that surrounds the flavin in the out conformation induces an increased electron density in the center of the ring because of transient interactions between the positive dipoles of the water molecules and the partial negative charges associated with the core of the flavin-ring system (22). In turn, this electron density could increase the bond strengths between ring atoms, and the increase in associated force constants would lead to an up shift of several normal modes. This shift of key flavin peaks to higher wavenumbers has been observed also in a model system by using lumiflavin dissolved in acetonitrile-d3 (having a small propensity for dipole-ring electron interactions) or water (corresponding to the out conformer). Here, too, several key flavin peaks remain unchanged while others move to a higher frequency shift after going to aqueous solution (data not shown). The Raman markers for in and out flavins and the spectral differences between the two conformations are summarized in Table 1.
Table 1.
Comparisons of in vs. out flavin conformations and solution vs. crystalline phases
| Approx. peak location | Difference between in → out
(cm−1)
|
Difference between solution → crystal (cm−1) for out conformation | Peak assignment | |
|---|---|---|---|---|
| Solution | Crystals | |||
| 1,575 cm−1 | N.S. | N.S. | N.S. | νN5—C4a, νC10a—N1 |
| 1,540 cm−1 | +5 | +8 | +3 | Ring I stretch and νC10a—N1 |
| 1,500 cm−1 | N.S. | +3 | N.S. | νN5—C4a, νC10a—N1, and Ring I stretch |
| 1,460 cm−1 | N.S. | N.S. | N.S. | Rings I and II and methyl deformation |
| 1,400 cm−1 | +6 | +8 | N.S. | Rings I and II, and methyl deformation |
| 1,360 cm−1 | +4 | +6 | +3 | a. Rings I, II, and III stretch and N10—C1′ |
| 1,340 cm−1 | +11 | +14 | N.S. | b. N3—H bend |
| c. Rings I, II, and III stretch and N3—H bend | ||||
| 1,270 cm−1 | * | +11 | +5 | C6—H bend + C9—H bend + N3—H bend |
| 1,240 cm−1 | +17 | +20 | +3 | + νN5—C5a + νN10—C1′ |
| 1,225 cm−1 | N.S. | +8 | +10 | C6—H bend and C9—H bend |
| 1,150 cm−1 | +7 | +8 | N.S. | C6—H bend, N3—H bend + Ring II |
N.S., not significant (<3 cm−1).
Broad shoulder.
In and Out Conformations of Flavin Bound to PHBH in Single Crystals.
In single protein crystals, Raman difference spectra of FAD bound to PHBH in both the in and out conformations were obtained by using the Raman microscope as depicted in Fig. 2. High-quality spectra were obtained from well formed crystals in hanging drops, as well as cracked crystals, crystal fragments, rod clusters, and very small crystals (smaller than 20 μm on a side). Flat crystals (<10-μm thick) yielded poor Raman data, suggesting that a limiting factor is the depth of the sample being studied. The diameter of the focused laser beam on the crystal was approximately 25 μm—somewhat smaller than the size of the crystal face. The orientation of the crystal relative to the laser beam appeared to be important but was difficult to assess systematically because the crystals are fragile and difficult to manipulate. This point will be discussed in more detail below.
Because the isoalloxazine ring system is a strong Raman scatterer even under nonresonance Raman conditions, its peaks can be detected even before subtraction is performed. An unsubtracted Raman spectrum of a holo-PHBH crystal in the presence of 2,4-di-OHB (out conformation) is shown in Fig. 4, with asterisks indicating the prominent Raman peaks that are caused by vibrational modes of the flavin moiety. We could obtain a Raman difference spectrum of FAD bound to PHBH in crystals by using the same technique as described above for the solution phase: acquiring a Raman spectrum of apo-PHBH and subtracting it from the holo-PHBH spectrum. This Raman difference spectrum would contain very little contribution from protein or buffer components, because FAD is a much stronger scatterer. However, apo-PHBH is very difficult to crystallize (B.A.P., unpublished work), and we were forced to explore two other methods of obtaining an apo-PHBH spectrum from crystals.
Figure 4.

Unsubtracted Raman spectra of a single crystal of holo-PHBH + 2,4-di-OHB (out). Raman peaks identified as prominent flavin-marker bands are indicated (*). Crystal dimensions are approximately 50 × 50 × 30 μm.
First, sodium dithionite (Na2S2O4), a strong reducing agent, can be used to reduce the conjugated diimine involving N1 and N5 (see Fig. 5 for numbering), destroying the three-ring delocalized π-electron system of the isoalloxazine moiety. Thus, adding sodium dithionite to the holo-PHBH crystal greatly reduces the strong Raman scattering of the flavin and renders it “invisible” to Raman spectroscopy, effectively yielding an apo-PHBH spectrum. By using this reduced holo-PHBH as the subtrahend, a Raman difference spectrum can be obtained.
Figure 5.

Isoalloxazine ring system of FAD, in which —X represents adenosine diphosphate.
Second, the intensity of the flavin peaks in the Raman spectrum of the in crystal (holo-PHBH + p-OHB) depends on the orientation of the crystal in the laser beam. In certain orientations, the peaks caused by the flavin modes were completely absent. However, when the crystal was turned or when a cluster of crystals, each in a different orientation, was analyzed, the flavin signal was clearly evident. Because the observed flavin bands are essentially in-plane ring modes, they will give rise only to strong Raman scattering when the E vector of the incident laser source is in the plane of the isoalloxazine ring system. In the Raman microscope, the path of the laser beam is normal to the face of the crystal, and the E vector is parallel to the crystal face. Thus, when Raman peaks caused by the flavin are not observed, the flavin-ring system is perpendicular to the top face of the crystal, the electric field of the laser beam is oriented at right angles to the plane of the ring, and no flavin signal is detected. The x-ray crystallographic data (3) confirm that the isoalloxazine ring is almost perpendicular to four of the six faces of the in PHBH crystal such that any spectra obtained with the laser beam normal to one of these faces would contain no contribution from the flavin and thus effectively yield an apo-PHBH spectrum. Such spectra are virtually identical to those of dithionite-reduced crystals, so properly oriented in crystals can be used as subtrahends in place of apo-PHBH crystals to obtain a Raman difference spectrum of FAD bound to holo-PHBH.
Raman difference spectra of the out crystals (holo-PHBH + 2,4-di-OHB) consistently showed a strong flavin signal, indicating that the isoalloxazine ring does not lie perpendicular to any of the crystal faces. This result is consistent with the published x-ray crystallographic data (3) that demonstrate that the isoalloxazine ring system is not perpendicular to any of the faces of the protein crystal.
Fig. 3 shows the single-crystal Raman spectra of FAD bound to wild-type PHBH + p-OHB, where the flavin is in the in conformation (Fig. 3A), and bound to wild-type PHBH + 2,4-di-OHB, where the flavin is in the out conformation (Fig. 3D). The Raman spectra of flavin in the protein crystal share many key features with the spectra in solution, and almost all the flavin markers can be identified. This finding demonstrates that Raman difference spectra can be obtained from samples in the crystalline state, and the key Raman markers of flavin conformation can be identified reproducibly in both the solution and solid phases.
It is important to note that the Raman modes associated with protein features do not depend on the orientation of the crystal in the laser beam because of their complex three-dimensional (i.e., nonplanar) structure. As a result, the protein features subtract to undetectable intensities in the Raman difference spectrum, and the flavin signal dominates.
Although it is valid to discuss intensity changes in isoalloxazine ring modes for the in and out conformers in solution, it is difficult to make the same comparison for the crystal forms. The difficulty lies in the tensorial nature of Raman scattering and the dependence of band intensities on the relative orientations of the isoalloxazine ring and the E vector of the laser beam. In future studies, controlled orientation of the protein crystals could provide information on ring orientation within the crystals. However, at this time we will focus the discussion on factors affecting band positions only.
Comparison of Flavin Conformations Between the Crystalline and Solution States.
When we compare the Raman difference spectra of the in and out conformations of flavin in single crystals, we see many of the same differences as previously observed in the solution state; the same Raman marker bands shift to a higher value when the flavin moves from the in to the out conformation, but other nonmarker flavin bands remain unperturbed. The most apparent distinction between the solution and crystalline phases is that the frequency shifts between Raman markers in the in and out conformations in solution are smaller than the corresponding shifts in single crystals. Furthermore, the in spectra show very few differences between the solution and crystalline states. In other words, the out spectrum exhibits significant changes when the system is crystallized, but the in conformer does not change at all within the error limit of our experiments. These data are summarized in Table 1.
To analyze these changes, we must have a more detailed understanding of the position of the flavin ring in the crystal and the groups of atoms that make up its environment. In the enzyme–substrate complex (PHBH + p-OHB), crystallographic studies have shown that the flavin is in the in conformation with ring III of the isoalloxazine group forming numerous hydrogen bonds with surrounding protein residues (Fig. 1). The data show that the C4⩵O carbonyl likely forms two hydrogen bonds with the peptide NHs of Gly-46 and Val-47 (3, 5). Other putative hydrogen bonds for ring III exist between N1 and the peptide NH of Gly-298 between the C2⩵O carbonyl and the peptide NHs of Leu-299 and Asn-300 and between N3—H and the backbone carbonyl of Val-47. The atoms in ring II are surrounded by peptide backbone atoms (not shown in figure) and two waters, whereas ring I is exposed to solvent and is surrounded almost completely by water molecules. When the substrate analog 2,4-di-OHB is complexed with PHBH, the steric overlap between its 2-OH group and the flavin pushes the flavin into the out position (7). Solvent molecules occupy the space vacated by the isoalloxazine ring such that almost all of the ring III atoms are now hydrogen-bonded to water instead of peptide NH and carbonyl groups. Ring I still is surrounded by water molecules, and the atoms in ring II are surrounded now by several additional water molecules as well. A summary of the putative ring III hydrogen bonds is given in Table 2.
Table 2.
Putative hydrogen bonds between flavin ring and PHBH for in and out flavin conformers
| Flavin atom | In conformer | Distance, Å | Out conformer | Distance, Å |
|---|---|---|---|---|
| N1: | Gly-298 NH | 3.10 | HOH 430 | 3.17 |
| HOH 503 | 3.30 | |||
| Leu-299 NH | 2.95 | HOH 430 | 2.82 | |
| C2⩵O | Asn-300 NH | 3.12 | HOH 428 | 3.04 |
| Asn-300 Nδ | 3.15 | |||
| N3—H | Val-47 O | 2.91 | DOB* 396 O | 2.75 |
| C4⩵O | Gly-46 NH | 3.10 | Trp-222 OH | 3.00 |
| Val-47 NH | 3.13 | HOH 492 | 3.01 | |
| N5: | — | — | HOH 426 | 2.94 |
2,4-di-OHB.
With this model of the interactions between the flavin ring and its environment, we can begin to understand why the out conformer is so much more sensitive to the change in phase (from solution to crystal) than the in conformer. When the flavin assumes the in position, it is held in place almost exclusively by hydrogen-bond interactions with nearby protein residues. These residues are in structurally well defined regions of the protein as shown by the low B-factor (temperature factor) values of the atoms in these amino acids (<20 Å2). We expect that the structural integrity of these residues will be preserved regardless of whether the protein is in solution or in a crystal. Thus, the effect of the phase change on the geometry of the flavin ring bound to these residues will be minimal. In contrast, the flavin in the out conformation is surrounded almost entirely by water molecules. In the solution phase, this is a rapidly changing fluid environment, and the ability of the water molecules to induce an increased electron density in the center of the isoalloxazine ring (as discussed above) is limited by the average residence time the molecule can interact with the flavin before it moves away. However, crystallization causes an increase in order, and as a result the mobility of the water environment decreases as it conforms to the constraints of the crystal lattice. Consequently, the water molecules have a higher average residence time to interact with the isoalloxazine and support the increased partial negative charge in the center of the ring. This charge causes the peaks in the out conformation to shift even further to higher wavenumbers (cm−1) in crystals than they do in solution.
In the crystalline phase, both the solvent and the protein are
subjected to a more restricted, well ordered environment compared with
solution. We expect this situation to limit the flavin's ability to
undergo small structural fluctuations, and this effect should be
reflected as a narrowing of the widths of Raman peaks compared with
solution. Our Raman spectrophotometer has an instrument-imposed minimum
peak width of 8 cm−1 at half height measured by
using a standard neon emission lamp. The
SO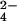 Raman signal in every spectrum (caused by
the presence of ammonium sulfate buffer) has a peak width at half
height of 10 cm−1. Thus, any further broadening
of Raman peaks is due to a property of the system being studied. The
spectra depicted in Fig. 3 show that the Raman band widths for the
flavin are, on average, 30% wider at half height in the solution state
than in crystals for both in and out conformers. The band widths at
half height for key Raman peaks are shown in Table
3.
Raman signal in every spectrum (caused by
the presence of ammonium sulfate buffer) has a peak width at half
height of 10 cm−1. Thus, any further broadening
of Raman peaks is due to a property of the system being studied. The
spectra depicted in Fig. 3 show that the Raman band widths for the
flavin are, on average, 30% wider at half height in the solution state
than in crystals for both in and out conformers. The band widths at
half height for key Raman peaks are shown in Table
3.
Table 3.
Band widths of Raman peaks (cm−1)
| Sample | 1,575 | 1,545 | 1,500 | 1,400 | 1,340 | 1,220 | 1,150 | 980* |
|---|---|---|---|---|---|---|---|---|
| Crystal in | 9 | 14 | 12 | 14 | 21 | 11 | 9 | 10 |
| Solution in | 12 | 15 | 16 | 18 | 28 | 16 | 12 | 10 |
| Solution out | 16 | 20 | 15 | 16 | 22 | 21 | 12 | 10 |
| Crystal out | 13 | 19 | 11 | 12 | 13 | 17 | 10 | 10 |
SO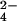 buffer peak.
buffer peak.
It is likely that changes in the band widths of the flavin's Raman peaks represent changes in the protein and solvent environments that surround the flavin molecule. Flavin bands broaden as the flavin-ring system experiences a number of slightly different environments. This is the so-called inhomogeneous broadening mechanism. Thus, the broadening of the isoalloxazine Raman peaks in PHBH illustrates increased “fluidity” of the protein in the solution phase, which allows the flavin molecule to experience a wider range of conformational excursions of the protein from its mean conformation. This situation is in contrast to the crystalline state, in which the more ordered environment impedes the protein's ability to make thermally induced conformational excursions from its optimal structural configuration. Decreased solvent mobility in the crystal also may help reduce flavin band width, because part or all of the isoalloxazine ring is in contact with water molecules. In this way, the flavin is being used to probe protein dynamics.
Study of Mixed Conformational Populations.
Certain PHBH mutants contain flavin molecules in both the in and out conformations. X-ray crystallographic data have demonstrated that the Y222F mutant bound to p-OHB contains a mixed population of the in and out forms in the ratio of approximately 30:70, respectively (7). Previously we have shown that a similar situation occurs in the solution phase, in which the Raman difference spectrum of flavin in the Y222F mutant contains both in and out flavin markers, suggesting a mixed population (14). Band-shape analysis and deconvolution in the marker regions could provide a quantitative estimate of the relative amounts of the two forms. Furthermore, this analysis can be carried out over a range of temperatures to determine enthalpic and entropic differences between the two conformers. If the in and out forms do indeed catalyze chemical steps at different points along the reaction pathway, these thermodynamic values may be necessary for a complete understanding of the PHBH mechanism. The same experiment can be done also on single crystals, and the thermodynamic parameters in the crystalline phase can be compared with the data obtained from complexes in solution to provide a unique and novel insight into the thermodynamics of the same conformational change within two different environments.
The ability of Raman spectroscopy to identify mixed-population conformers may be useful for interpreting existing x-ray crystallographic structures. Although x-ray crystal data do include parameters that indicate their reliability (R-value, temperature factor) and the presence of alternative positions for certain atoms (occupancy), the interpretation of the initial electron-density map requires a substantial human contribution. A region that contains several alternative structural conformations or has substantial dynamic disorder may be interpreted instead as a single poorly defined region. Under those circumstances, the Raman features associated with this region would have wide and poorly resolved peaks. However, if there were two distinct conformations, each might have its own distinct Raman signature, and a multiplicity of structural features would be apparent. Each conformer would have its own set of well defined peaks with their relative intensities indicating their relative populations.
Characterization of Unknown Samples in Crystalline and Solution Phases.
We have used Raman spectroscopy and Raman microscopy to demonstrate that the Raman difference spectrum of flavin bound to PHBH in single crystals can be compared closely with the Raman difference spectrum in solution. Although differences do exist between the crystalline and solution phases, key Raman marker bands are identified easily, and the conformation of the flavin can be determined under either set of conditions. This method can be used to identify the flavin conformation of PHBH mutants, the crystal structures of which have not been solved yet and whose flavin conformations are unknown. In general, Raman microscopy can be used to screen the properties of protein crystals in hanging drops.
Acknowledgments
This work was supported by National Institutes of Health Grant GM-54072 (to P.R.C.). B.A.P. was supported by Public Health Service Grants GM-20877 to David P. Ballou and GM-11106 to Vincent Massey.
Abbreviations
- PHBH
para-hydroxybenzoate hydroxylase
- p-OHB
para-hydroxybenzoic acid
- 2,4-di-OHB
2,4-dihydroxybenzoic acid
References
- 1.Palfey B A, Massey V. In: Comprehensive Biological Catalysis: A Mechanistic Reference. Sinnott M, editor. Vol. 4. San Diego: Academic; 1998. [Google Scholar]
- 2.Palfey B A, Ballou D P, Massey V. In: Active Oxygen in Biochemistry. Valentine J S, Foote C S, Greenburg A, Lieberman J F, editors. Vol. 3. London: Blackie; 1995. [Google Scholar]
- 3.Schreuder H A, Prick P A, Wierenga R K, Vriend G, Wilson K S, Hol W G, Drenth J. J Mol Biol. 1989;208:679–696. doi: 10.1016/0022-2836(89)90158-7. [DOI] [PubMed] [Google Scholar]
- 4.Schreuder H A, van der Laan J M, Swarte M B, Kalk K H, Hol W G, Drenth J. Proteins. 1992;14:178–190. doi: 10.1002/prot.340140205. [DOI] [PubMed] [Google Scholar]
- 5.Schreuder H A, Mattevi A, Obmolova G, Kalk K H, Hol W G, van der Bolt F J, van Berkel W J. Biochemistry. 1994;33:10161–10170. doi: 10.1021/bi00199a044. [DOI] [PubMed] [Google Scholar]
- 6.Lah M S, Palfey B A, Schreuder H A, Ludwig M L. Biochemistry. 1994;33:1555–1564. doi: 10.1021/bi00172a036. [DOI] [PubMed] [Google Scholar]
- 7.Gatti D L, Palfey B A, Lah M S, Entsch B, Massey V, Ballou D P, Ludwig M L. Science. 1994;266:110–114. doi: 10.1126/science.7939628. [DOI] [PubMed] [Google Scholar]
- 8.van Berkel W J, Eppink M H, Schreuder H A. Protein Sci. 1994;3:2245–2253. doi: 10.1002/pro.5560031210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palfey B A, Ballou D P, Massey V. Biochemistry. 1997;36:15713–15723. doi: 10.1021/bi971427u. [DOI] [PubMed] [Google Scholar]
- 10.Palfey B A, Moran G R, Entsch B, Ballou D P, Massey V. Biochemistry. 1999;38:1153–1158. doi: 10.1021/bi9826613. [DOI] [PubMed] [Google Scholar]
- 11.Enroth C, Neujahr H, Schneider G, Lindqvist Y. Structure (London) 1998;6:605–617. doi: 10.1016/s0969-2126(98)00062-8. [DOI] [PubMed] [Google Scholar]
- 12.Dong J, Dinakarpandian D, Carey P R. Appl Spectrosc. 1998;52:1117–1122. [Google Scholar]
- 13.Kim M, Owen H, Carey P R. Appl Spectrosc. 1993;47:1780–1783. [Google Scholar]
- 14.Zheng Y, Dong J, Palfey B A, Carey P R. Biochemistry. 1999;38:16727–16732. doi: 10.1021/bi9918893. [DOI] [PubMed] [Google Scholar]
- 15.Yu N-T, Jo B H. J Am Chem Soc. 1973;95:5033–5037. doi: 10.1021/ja00796a041. [DOI] [PubMed] [Google Scholar]
- 16.Yu N-T. J Am Chem Soc. 1974;96:4664–4668. doi: 10.1021/ja00821a049. [DOI] [PubMed] [Google Scholar]
- 17.Smulevich G, Spiro T G. Methods Enzymol. 1993;226:397–408. doi: 10.1016/0076-6879(93)26018-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhu L, Sage J T, Champion P M. Biochemistry. 1993;32:11181–11185. doi: 10.1021/bi00092a030. [DOI] [PubMed] [Google Scholar]
- 19.Moran G R, Entsch B. Protein Expression Purif. 1995;6:164–168. doi: 10.1006/prep.1995.1020. [DOI] [PubMed] [Google Scholar]
- 20.Palfey B A, Entsch B, Ballou D P, Massey V. Biochemistry. 1994;33:1545–1554. doi: 10.1021/bi00172a035. [DOI] [PubMed] [Google Scholar]
- 21.Muller F, van Berkel W J. Eur J Biochem. 1982;128:21–27. [PubMed] [Google Scholar]
- 22.Creighton T E. Proteins: Structures and Molecular Properties. New York: Freeman; 1993. pp. 139–169. [Google Scholar]