

Summary
Little is known about metabolic regulation in stem cells and how this modulates tissue regeneration or tumour suppression. We studied the Lkb1 tumour suppressor, and its substrate AMPK, kinases that coordinate metabolism with cell growth. Lkb1 deletion caused increased haematopoietic stem cell (HSC) division, rapid HSC depletion, and pancytopenia. HSCs depended more acutely on Lkb1 for cell cycle regulation and survival than many other haematopoietic cells. HSC depletion did not depend on mTOR activation or oxidative stress. Lkb1-deficient HSCs, but not myeloid progenitors, had reduced mitochondrial membrane potential and ATP. AMPK-deficient HSCs showed similar changes in mitochondrial function but remained able to reconstitute irradiated mice. Lkb1-deficient HSCs, but not AMPK-deficient HSCs, exhibited defects in centrosomes and mitotic spindles in culture, and became aneuploid. Lkb1 is therefore required for HSC maintenance through AMPK-dependent and AMPK-independent mechanisms, revealing differences in metabolic and cell cycle regulation between HSCs and some other haematopoietic progenitors.
Lkb1 coordinates cell growth with energy metabolism. Energy stress prompts Lkb1 to activate catabolic processes and mitochondrial biogenesis and to inactivate anabolic processes including mammalian target of rapamycin (mTOR)-mediated protein synthesis1. Lkb1 exerts these effects by activating AMP-activated protein kinase (AMPK) and AMPK-related kinases2. AMPK activates the Tuberous sclerosis complex (TSC), which inhibits mTOR complex 1 (mTORC1), reducing protein translation and cell growth3,4. AMPK also inactivates mTORC1 by phosphorylating Raptor5. AMPK can promote the function of Foxo family transcription factors6,7, which regulate energy metabolism, cell cycle, apoptosis, and oxidative stress8.
Lkb1 regulates embryogenesis and the metabolism and polarity of differentiated adult cells. The Lkb1 homolog in C. elegans regulates embryo asymmetry9. Drosophila Lkb1 and AMPK regulate cell polarity, asymmetric division, and mitotic spindle formation in embryos10,11,12. Mice deficient for Lkb1 (encoded by the gene Stk11; henceforth called Lkb1 for clarity) die at midgestation with vascular and neural tube defects13,14. In adult tissues, Lkb1 regulates the metabolism of muscle15,16, liver17, pancreas18,19,20 and T cells21,22. Deletion of Lkb1 in mammalian neurons23, epithelial cells24 and pancreatic ß cells18,19,20 disrupts their polarity or differentiation; however, Lkb1 is not known to regulate stem cell maintenance or adult tissue regeneration.
Lkb1-deficiency increases the proliferation of many tissues20,25,26,27 and immortalizes mouse embryonic fibroblasts28. Lkb1 is mutated in Peutz-Jeghers syndrome patients 29,30, who have a high incidence of epithelial cancers1. These data suggest that the primary function of Lkb1 in adult tissues is to negatively regulate cell division, preventing tissue overgrowth. To test whether Lkb1 positively or negatively regulates stem cell function we conditionally deleted Lkb1 from haematopoietic cells.
Deletion of Lkb1 depletes HSCs
Lkb1 mRNA was expressed at approximately two fold higher levels in HSCs (CD150+CD48−CD41−lineage−Sca-1+c-kit+), transiently reconstituting multipotent progenitors (MPPs) (CD150−CD48−CD41−lineage−Sca-1+c-kit+) 31,32, and granulocyte-macrophage progenitors (GMPs; lineage−Sca-1−c-kit+CD34+CD16/32+ 33) as compared to whole bone marrow (WBM) cells by quantitative real-time PCR (qPCR) (Suppl. Fig. 1d).
We generated a floxed allele of Lkb1 (Lkb1fl) by gene-targeting in Bruce4 ES cells 34 (Suppl. Fig.1) then conditionally deleted Lkb1 from haematopoietic cells in adult Mx1-Cre; Lkb1fl/fl mice by injecting polyinosine-polycytidine (pIpC)35,36 (Suppl. Fig. 1e, f). All control (Lkb1fl/fl mice) and mutant (Mx1-Cre; Lkb1fl/fl) mice were treated with 3 injections of pIpC over 6 days and the time of analysis is indicated in days after the first pIpC injection. We used a low dose of pIpC (0.5 μg/gram body mass) that was titrated to completely delete Lkb1 (Fig. 3a) without significantly altering HSC surface marker phenotype or cell cycle kinetics.
Figure 3. AMPK signaling requires Lkb1 in HSCs/MPPs but HSC depletion could not be rescued with rapamycin.
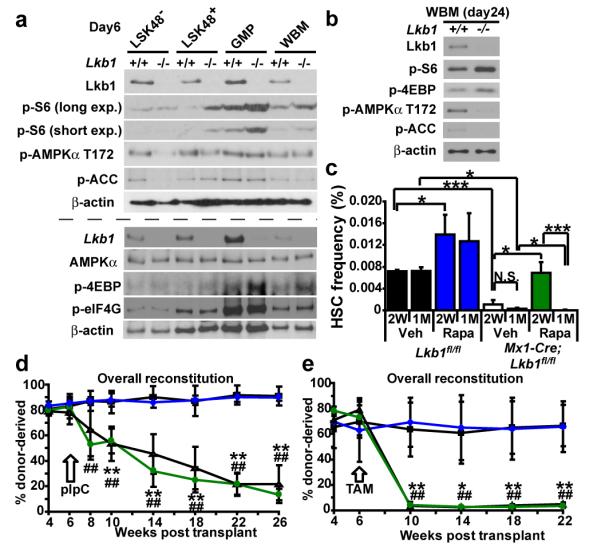
a, Six days after pIpC treatment, Lkb1 deletion increased mTORC1 activation (phospho-S6 and phospho-4EBP levels) in restricted progenitors (LSK48+ cells, GMPs, and WBM cells) but not in LSK48- cells (HSCs/MPPs). Decreased phospho-AMPKa T172 was noted in Lkb1-deficient LSK48− and to a lesser extent in LSK48+ cells but not in GMPs or WBM cells. phospho-ACC was decreased in Lkb1-deficient LSK48− cells but not in other populations. We did not observe a consistent change in phospho-eIF4G levels after Lkb1-deletion in any population. Each lane contained protein from 30,000 sorted cells. +/+ indicates Lkb1fl/fl cells and −/− indicates Mx1-Cre; Lkb1fl/fl cells after pIpC treatment. This panel reflects two independent experiments (upper and lower panels separated by the dashed line). b, 24 days after pIpC treatment, phospho-AMPKα T172 and phospho-ACC were decreased and phospho-S6 and phospho-4EBP levels were increased in Lkb1-deficient WBM cells. c-e, Rapamycin failed to rescue the depletion of Lkb1-deficient HSCs. c, Mice were treated with rapamycin after pIpC treatment for two weeks (2W) or one month (1M). Data are from 3-4 independent experiments. d-e, Rapamycin failed to rescue the reconstituting capacity of Lkb1-deficient HSCs, irrespective of whether Lkb1 was deleted using pIpC in Mx1-Cre; Lkb1fl/fl mice (d) or tamoxifen in Ubc-CreERT2 mice (e). In each case, 1×106 donor WBM cells from untreated mutant (Mx1-Cre; Lkb1fl/fl in d; Ubc-CreERT2; Lkb1fl/fl in e) or control (Lkb1fl/fl) mice were transplanted into irradiated mice along with 500,000 recipient WBM cells. Six weeks after transplantation, all recipients were treated with pIpC (d) or tamoxifen (e), then treated with rapamycin or vehicle. One representative experiment is shown out of 2-3 independent experiments for each mode of Lkb1 deletion (**, p<0.005 for Lkb1fl/fl versus Mx1-Cre/Ubc-CreERT2; Lkb1fl/fl recipients treated with vehicle; ##, p<0.005 for Lkb1fl/fl versus Mx1-Cre/Ubc-CreERT2; Lkb1fl/fl recipients treated with rapamycin).
Deletion of Lkb1 had little acute effect on the cellularity or composition of haematopoietic tissues 6 to 18 days after starting pIpC treatment (Fig. 1a-d; Suppl. Fig. 2a-c). However, pancytopenia was observed by 24 to 34 days after pIpC treatment (Fig. 1a, e; Suppl. Fig. 2d-l). Two and six days after starting pIpC treatment, HSC frequency significantly increased (p<0.0005) in pIpC-treated Mx1-Cre; Lkb1fl/fl mice compared to littermate controls (Fig. 1f). HSC frequency declined to one seventh of normal levels in pIpC-treated Mx1-Cre; Lkb1fl/fl mice by 18 days after pIpC treatment (p< 0.0005; Fig. 1f). MPPs transiently expanded and then were depleted in parallel with HSCs (Suppl. Fig. 4c, d). The absolute number of HSCs and MPPs followed similar trends (Suppl. Fig. 4). HSCs were therefore profoundly depleted by 18 days after pIpC treatment, before pancytopenia was evident.
Figure 1. Lkb1 deletion causes HSCs to go into cycle before being depleted.

a, Lkb1 deletion had a limited effect on the cellularity of whole bone marrow (WBM), spleen (SPL) or thymus (THY) 6 to 18 days after starting pIpC but WBM and thymus cellularity declined significantly by 24 to 34 days (all panels show mean±standard deviation from at least 3 independent experiments; *, p<0.05; **, p<0.005; and ***, p<0.0005 by Student’s t-test in all figures). +/+ indicates Lkb1fl/fl mice and −/− indicates Mx1-Cre; Lkb1fl/fl mice. b-d, Lkb1 deletion had little effect on T (b), myeloid or erythroid (c), or B (d) lineage cells 18 days after pIpC treatment. e, White blood cells (WBC), red blood cells (RBC) and platelets (PLT) were significantly depleted in the blood of Lkb1-deficient mice by 24 to 34 days after pIpC treatment. f, HSC (CD150+CD48−CD41−lineage−Sca-1+c-kit+) frequency significantly increased 2-6 days and significantly reduced 18 days following pIpC treatment in Lkb1-deficient mice. g, Lkb1-deficient HSCs and MPPs, but not WBM cells, incorporated significantly more BrdU (24 hour pulse) 6 days after pIpC treatment. h, Lkb1 deletion drove HSCs and MPPs into cycle, increasing the frequency of these cells in G1 (Ki-67+ cells with 2N DNA content, 2.5-fold, p<0.05) and S/G2/M phases of the cell cycle (Ki67+ cells with >2N DNA content, 2.4-fold, p<0.05) at day 6, but did not affect the cell cycle distribution of GMPs or WBM cells. Lkb1-deficient HSCs had significantly increased caspase activity (i, 2.6-fold) at day 11, but other haematopoietic progenitors did not significantly increase caspase activity until day 24 (j).
Lkb1 deletion acutely increased the division of HSCs and MPPs but not most WBM cells. Five days after starting pIpC, Mx1-Cre; Lkb1fl/fl mice and Lkb1fl/fl controls were administered BrdU for 24 hours. We observed a significant increase in BrdU incorporation in HSCs (p<0.005) and MPPs (p<0.0005) from Mx1-Cre; Lkb1fl/fl mice (Fig. 1g, Suppl. Fig. 5a). This increase in BrdU incorporation within Lkb1-deficient HSCs continued 18 days after pIpC treatment, when HSC depletion was already evident (Suppl. Fig. 5b). Lkb1-deficiency also significantly (p<0.05) increased the frequencies of HSCs and MPPs in G1 and S/G2/M phases of the cell cycle (Fig. 1h; Suppl. Fig. 5c). In contrast, we observed no effect of Lkb1 deletion on the rate of BrdU incorporation or the frequency of cycling GMPs or WBM cells (Fig. 1g, h; Suppl. Fig. 5a, c).
Lkb1 deletion induced cell death in HSCs. Eleven days after starting pIpC, we observed significant (p<0.05) increases in caspase activity (Fig. 1i) and the frequency of Annexin-V+DAPI+ dead cells (Suppl. Fig. 5d) in Lkb1-deficient HSCs but not in MPPs, GMPs or WBM cells. LSK cells, GMPs, and WBM cells from Lkb1-deficient mice eventually underwent cell death, with significantly (p<0.05) increased caspase activity 24 days after pIpC (Fig. 1j). HSCs therefore depend more acutely upon Lkb1 for survival than many other haematopoietic progenitors.
Lkb1-deficient HSCs fail to long-term reconstitute
Lkb1-deficient HSCs failed to long-term multilineage reconstitute irradiated mice. One million donor (CD45.2+) WBM cells from Mx1-Cre; Lkb1fl/fl or Lkb1fl/fl mice 6 days after starting pIpC were transplanted into irradiated recipient (CD45.1+) mice along with 500,000 recipient cells. Lkb1-deficient cells gave significantly lower levels of overall (Fig. 2a), myeloid, B, and T (Suppl. Fig. 6a-c) lineage reconstitution.
Figure 2. Lkb1-deficient HSCs have a cell autonomous defect in their ability to reconstitute irradiated mice and to form colonies in culture.
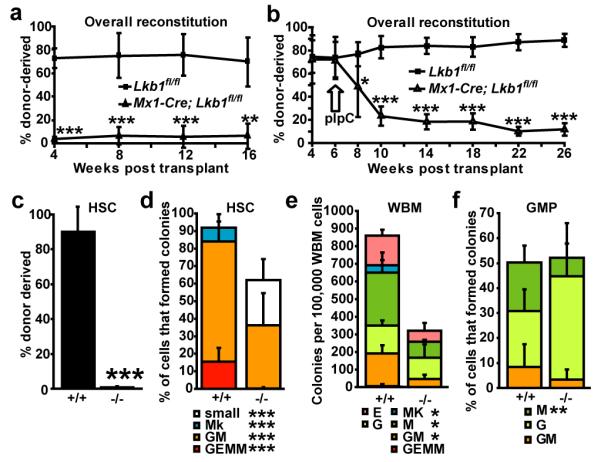
a-d, 1×106 donor WBM cells from Lkb1fl/fl or Mx1-Cre; Lkb1fl/fl mice were transplanted into irradiated recipient mice along with 500,000 recipient WBM cells. The transplant was performed 6 days after (a) or 6 weeks before (b) pIpC treatment. Reconstitution levels were monitored for 16-20 weeks after transplantation (a) or after pIpC treatment (b). Data are from one representative experiment of each type out of 3 independent experiments of each type. c, Donor HSCs (CD150+CD48−CD41−lineage−Sca-1+c-kit+) were depleted in recipients of Mx1-Cre; Lkb1fl/fl (Lkb1-deficient) cells two month after pIpC treatment. Data are from 4 independent experiments. 6 days after pIpC treatment, the frequencies of HSCs (d), WBM cells (e), and GMPs (f) that formed granulocyte, erythroid, macrophage, megakaryocyte (GEMM), granulocyte, macrophage (GM), megakaryocyte (Mk), “small” colonies with fewer than 100 cells, or single lineage (G, or M, or E) colonies in culture. Data (mean±standard deviation) are from 3-16 independent experiments per cell type (*, significantly different between Lkb1-deficient and control by Student’s t-test).
This reflected an autonomous requirement for Lkb1 in HSCs. We transplanted one million donor WBM cells from Mx1-Cre; Lkb1fl/fl mice or Lkb1fl/fl controls, without pIpC treatment, along with 500,000 wild-type recipient cells into irradiated recipient mice. Six weeks after transplantation, when donor cells had stably engrafted, we treated all the recipients with pIpC. Reconstitution by Mx1-Cre; Lkb1fl/fl cells, but not Lkb1fl/fl cells, dropped precipitously (Fig. 2b; Suppl. Fig. 6d-f). The low level of residual reconstitution by Mx1-Cre; Lkb1fl/fl cells was from cells that had not fully deleted Lkb1 (Suppl. Fig. 7b). Two months after pIpC treatment we recovered donor HSCs from recipients of Lkb1fl/fl cells, but not from recipients of Mx1-Cre; Lkb1fl/fl cells (Fig. 2c).
Lkb1-deficient HSCs were also unable to form normal colonies in culture. Significantly (p<0.0005) fewer Lkb1-deficient HSCs formed colonies as compared to control HSCs (Fig. 2d). The Lkb1-deficient colonies that did form were often much smaller than control colonies (Suppl. Fig. 7a) and failed to form any secondary colonies upon subcloning (Suppl. Fig. 5e). Lkb1-deficient WBM cells also formed significantly fewer colonies (p<0.005, Figure 2e); however, not all colony-forming progenitors required Lkb1. Approximately 50% of sorted GMP cells formed colonies irrespective of whether they were wild-type or Lkb1-deficient (Fig. 2f; Suppl. Fig. 7a). Complete deletion of Lkb1 was confirmed by western blotting of freshly isolated cells (Fig. 3a).
mTORC1-independent depletion of Lkb1-deficient HSCs
To test if AMPK was inactivated by Lkb1 deletion, we isolated 30,000 Lin−Sca-1+c-kit+CD48− (LSK48−) cells (highly enriched for HSCs31), LSK48+ cells (a mixed population of restricted progenitors), GMPs, or WBM cells from Lkb1-deficient and littermate control mice 6 days after pIpC treatment and analyzed protein extracts by western blotting. Lkb1 was expressed by each cell population from control mice but not by cells from mutant mice (Fig. 3a). AMPKα T172 phosphorylation (the site phosphorylated by Lkb11,2,37) was reduced in Lkb1-deficient LSK48− HSCs and to a lesser extent in LSK48+ progenitors, but not in GMPs or WBM cells (Fig. 3a). Phosphorylation of acetyl-CoA carboxylase (ACC), a known substrate of AMPK37, was substantially reduced in Lkb1-deficient LSK48− HSCs but not in other cell populations 6 days after pIpC treatment (Fig. 3a). AMPKα T172 and ACC phosphorylation levels were ultimately reduced in Lkb1-deficient WBM cells by 24 days after pIpC treatment (Fig. 3b). Lkb1 therefore regulates AMPK activation in many haematopoietic cells but HSCs depend more acutely upon Lkb1 for AMPK regulation.
AMPK negatively regulates mTOR activation1,2,37 and increased mTOR activation leads to HSC depletion36. We assessed mTORC1 activation based on S6, 4EBP, and eIF4G phosphorylation. Phosphorylation of eIF4G did not change significantly after Lkb1 deletion in any population (Fig. 3a). Phospho-S6 and phospho-4EBP levels increased in Lkb1-deficient LSK48+ restricted progenitors, GMPs and WBM cells but not in Lkb1-deficient LSK48− HSCs (Fig. 3a). Lkb1/AMPK signaling is either not required to regulate mTORC1 in HSCs or mTORC1 activity in Lkb1-deficient HSCs reflects a complex balance of effects on AMPK/TSC activation versus mitochondrial function/ATP levels38. Either way, Lkb1 appears to regulate PI-3kinase/mTORC1 pathway signaling differently in HSCs as compared to many other haematopoietic progenitors.
To test whether mTOR activation contributes to HSC depletion, we tested whether rapamycin could rescue the depletion of Lkb1-deficient HSCs. Mx1-Cre; Lkb1fl/fl mice and Lkb1fl/fl controls were treated with pIpC, then injected daily with rapamycin for two weeks or one month. Rapamycin treatment increased HSC frequency in both wild-type and Lkb1-deficient mice after 2 weeks but rapamycin did not rescue the depletion of HSCs one month after Lkb1 deletion (Fig. 3c). We also transplanted one million Mx1-Cre; Lkb1fl/fl or Lkb1fl/fl WBM cells into irradiated mice along with 500,000 recipient WBM cells. We treated the recipient mice with pIpC 6 weeks later then administered rapamycin daily to half of the recipient mice. In contrast to what we observed after Pten deletion36, rapamycin did not significantly affect reconstitution levels by Lkb1-deficient cells (Fig. 3d; Suppl. Fig. 6g-i). Our data suggest that increased mTOR activation is not a major mediator of HSC depletion after Lkb1 deletion.
These results were confirmed using tamoxifen inducible Ubc-Cre-ERT2; Lkb1fl/fl mice 39. Tamoxifen-induced deletion of Lkb1 led to a rapid loss of donor cell reconstitution that was not attenuated by rapamycin treatment (Fig. 3e; Suppl. Fig. 6j-l). The depletion of Lkb1-deficient HSCs therefore does not require pIpC.
Lkb1 regulates HSC mitochondrial function
Prkaa1 and Prkaa2, which encode the two catalytic α-subunits of AMPK, were more highly expressed in various haematopoietic stem/progenitor cell populations than in unfractionated WBM cells (Suppl. Fig. 8a, b). To test whether AMPK regulates HSC function, we generated floxed alleles of both catalytic subunits of AMPK (Prkaa1fl and Prkaa2fl; Suppl. Fig. 9). pIpC-treated Mx1-Cre; Prkaa1fl/fl; Prkaa2fl/fl mice are hereafter described as AMPK-deficient mice (Mx1-Cre; AMPKα1/α2fl/fl or AMPKα−/−) for simplicity. After pIpC treatment, AMPKα expression, AMPK T172 phosphorylation, and ACC phosphorylation were significantly reduced in all cell populations analyzed from Mx1-Cre; AMPKα1/α2fl/fl mice (Fig. 4a). In contrast to Lkb1, AMPK deletion increased phospho-S6 levels in all populations including LKS48− HSCs, LSK48+ restricted progenitors, GMPs, and WBM cells. These results suggest that AMPK negatively regulates mTORC1 signaling in many haematopoietic stem and progenitor cells.
Figure 4. AMPK deficiency partially phenocopies the mitochondrial defects but not the HSC depletion observed after Lkb1 deletion.

a, AMPKα1/α2 deficiency reduced phospho-AMPKα T172 and phospho-ACC levels and increased phospho-S6 levels, as expected. Each lane contained protein from 30,000 sorted cells. +/+ indicates AMPKα1/α2fl/fl cells and −/− indicates Mx1-Cre; AMPKα1/α2fl/fl cells 6 days after pIpC treatment. b, Lkb1 or AMPKα deletion did not significantly affect DCFDA staining (ROS levels) in HSCs (b, c), MPPs or WBM (c) cells 11 days after pIpC treatment. d, NAC treatment for two weeks did not rescue the depletion of Lkb1-deficient HSCs (*, p<0.05 by Student’s t-test). e, Mitochondrial DNA copy number was significantly reduced 6 days after Lkb1 or AMPKα deletion (*, p<0.05; **, p<0.005 in all panels). f, g, Mitochondrial mass significantly increased 11 days after Lkb1 deletion in HSCs, and MPPs, but not in WBM cells. AMPKα deletion significantly increased mitochondrial mass in all populations 11 days after pIpC treatment. A representative histogram shows Mitotracker staining in HSCs after Lkb1 or AMPKα deletion (f). h, ATP levels were significantly reduced in HSCs after Lkb1 or AMPKα deletion, 6 or 11 days after pIpC treatment. i, j, Mitochondrial membrane potential (Δψ) was significantly reduced after Lkb1 deletion in HSCs (i, j) and MPPs but not in WBM cells (j) or GMPs (k) 11 days after pIpC treatment. AMPKα deletion did not reduce Δψ in any cell population (i, j). l, AMPKα deletion did not cause transient expansion or rapid depletion of HSCs, but did modestly reduce HSC frequency 70 days after pIpC treatment (p<0.05). m, AMPKα-deficient HSCs were capable of long-term multilineage reconstitution 6 days after pIpC treatment, in contrast to Lkb1-deficient HSCs (Fig. 2a, b). All data (mean±standard deviation) are from 3 to 7 independent experiments.
Elevated reactive oxygen species (ROS) contribute to HSC depletion in Foxo-deficient mice 40,41. However, neither Lkb1 deletion nor AMPK deletion significantly affected ROS levels, measured by 2′-7′-dichlorofluorescein diacetate (DCFDA) staining 11 days after pIpC (Fig. 4b, c). Oxidative stress contributed little to the depletion of Lkb1-deficient HSCs as N-Acetyl-cysteine (NAC) treatment of Mx1-Cre; Lkb1fl/fl mice did not rescue HSC depletion (Fig. 4d).
Mitochondria were misregulated in Lkb1-deficient and AMPK-deficient haematopoietic cells. Eleven days after pIpC treatment, mitochondrial mass was significantly (p<0.05) increased in both Lkb1-deficient and AMPK-deficient HSCs (Fig. 4f, g). This could reflect negative regulation of mitochondrial mass by Lkb1-AMPK or a compensatory expansion of mitochondria in response to mitochondrial dysfunction and ATP depletion, as observed with Tfam deficiency42. Consistent with the latter possibility, we observed a significant reduction in mitochondrial DNA copy number in both Lkb1-deficient and AMPK-deficient HSCs 6 days after pIpC treatment (Fig. 4e), as observed in Tfam-deficient cells42. We also observed a significant (p<0.05) reduction in mitochondrial membrane potential (Δψ) within Lkb1-deficient HSCs but not in AMPK-deficient HSCs (Fig. 4i, j). This reduction in Δψ was not observed in Lkb1-deficient GMPs or WBM cells (Fig. 4j, k). It is formally possible that the reduction in Δψ in Lkb1-deficient HSCs was caused by the induction of apoptosis. However, we did not observe reduced Δψ in early apoptotic cells (Annexin-V+DAPI−) compared to cells that showed no sign of initiating cell death (Annexin-V−DAPI−) (Suppl. Fig. 11a). These results suggest that Lkb1 regulates mitochondrial function by AMPK-dependent and AMPK-independent mechanisms.
To test ATP levels we sorted live cells from each population to ensure that ATP levels were not confounded by the presence of dead cells or debris, and to normalize ATP levels on a per cell basis. At 6 and 11 days after starting pIpC treatment, we observed a 10 to 15% reduction in cellular ATP levels in Lkb1-deficient HSCs (p<0.05; Fig. 4h) but not in Lkb1-deficient MPPs and GMPs (Suppl. Fig. 11b). AMPK-deficient HSCs also had significantly reduced levels of ATP 11 days after pIpC treatment (p<0.005) (Fig. 4h). The Lkb1-AMPK pathway is thus required for mitochondrial function and energy homeostasis in HSCs.
Although AMPK deficiency phenocopied some of the effects of Lkb1 deficiency in HSCs, AMPK was not required for HSC maintenance. We did not observe a transient increase (day 6) or a rapid depletion (day 18) of HSCs after pIpC treatment of Mx1-Cre; AMPKα1/α2fl/fl mice (Fig. 4l). We did, however, observe a 2-fold reduction in HSC frequency in AMPK-deficient mice 70 days after pIpC treatment (p<0.05, Fig. 4l). AMPK-deficient HSCs also gave long-term multilineage reconstitution (Fig. 4m, Suppl. Fig. 10). We confirmed that the reconstituting cells in these experiments were AMPK-deficient (data not shown). This suggests Lkb1 promotes HSC maintenance through mechanisms that are largely AMPK-independent.
Lkb1-deficient HSCs became aneuploid
To carefully examine HSC division we sorted HSCs from Mx1-Cre; Lkb1fl/fl mice and Lkb1fl/fl controls into culture after pIpC treatment. Almost all HSCs, regardless of Lkb1, divided during the first three days of culture (Fig. 5a). However, wild-type HSCs subsequently expanded geometrically, while Lkb1-deficient HSCs exhibited little further division (Fig. 5b). The limited size of Lkb1-deficient HSC colonies was not due to reduced cell cycle entry as wild-type and Lkb1-deficient colonies contained similar frequencies of BrdU+ cells after a one hour pulse on the third day of culture (Fig. 5c). Instead, Lkb1-deficient colonies contained significantly (p<0.05) fewer cells that stained positively for the mitosis marker phospho-Histone H3 (Fig. 5d). This suggested Lkb1-deficient HSCs were often unable to enter mitosis or they failed to complete mitosis due to cell death.
Figure 5. Lkb1-deficient HSCs exhibit defects in mitotic spindles, aneuploidy, and cell death.

Lkb1-deficient HSCs (6 days after pIpC) only underwent a few divisions in culture. (a, the fraction of cells that divided and (b), the total number of cells/HSC colony). Lkb1-deficient HSCs entered S-phase normally in culture (c) but failed to enter or complete mitosis, perhaps due to cell death (d). e, Lkb1-deficient HSCs, but not GMPs, exhibited supernumerary centrosomes (red arrowheads) and defective mitotic spindles: α-tubulin (green) marks mitotic spindles, γ-tubulin (red) marks centrosomes, and phospho-H3 Ser10 (blue) marks M phase cells. f, g, Increased cell death within Lkb1-deficient HSC colonies but not GMP colonies based on Annexin-V (f) or wright-giemsa (g, see the cell fragments, arrowheads) staining. h-j, Cells within Lkb1-deficient LSK (Lineage-Sca-1+c-kit+) colonies, but not within GMP colonies, became aneuploid within 2 days in culture. Representative chromosome spreads of wild-type and Lkb1-deficient LSKs with 40 and 39 chromosomes, respectively (h). k, AMPKα-deficient LSK cells did not become aneuploid. All data (mean±standard deviation) are from 3-8 independent experiments, with the indicated numbers of cells scored for chromosome numbers.
Strikingly, many (32±9%) of the mitotic cells within Lkb1-deficient HSC colonies had supernumerary centrosomes and aberrant mitotic spindles, phenotypes not observed in control HSC colonies (Fig. 5e). We did not observe supernumerary centrosomes or aberrant mitotic spindles in GMPs (data not shown). This raised the possibility that many Lkb1-deficient HSCs may die or produce aneuploid progeny. Indeed, Lkb1 deficient HSC colonies, but not GMP colonies, contained significantly more Annexin-V+ cells and dead cells (Figure 5f, g). While cells in colonies formed by wild-type (LSK) stem/progenitor cells rarely (6.3±6.1%) had abnormal chromosome numbers, cells in Lkb1-deficient colonies were often (40.5±19.9%) aneuploid (p<0.0005, Fig. 5h, i). Lkb1-deficient GMPs from the same mice did not show a significant increase in aneuploidy (Fig. 5j, p=0.75), consistent with their ability to form normal colonies in culture (Fig. 2f). AMPK-deficient LSK cells also did not show increased aneuploidy (Fig. 5k), indicating that Lkb1 regulates chromosome stability through AMPK-independent pathways.
Discussion
Consistent with our results, Gan et al.43 and Gurumurthy et al.44 also concluded that Lkb1 is autonomously required for cell cycle regulation, survival, mitochondrial function, and energy homeostasis in HSCs and that HSCs depend more acutely upon Lkb1 than many other haematopoietic cells. The earlier onset of pancytopenia observed by Gan et al.43 and Gurumurthy et al.44 after Lkb1 deletion could be explained by the different allele of Lkb1 28 or different genetic background used in those studies, by the ubiquitous deletion of Lkb1 with Rosa26-CreER in the study by Gan et al.43, or by the use of a higher dose of pIpC in the study by Gurumurthy et al.44 (Suppl. Fig. 3).
HSCs were more rapidly depleted after Lkb1 deletion (Fig. 1f) than after Pten 36 or Foxo 41 deletion. Whereas the mTOR inhibitor rapamycin and/or the anti-oxidant NAC rescued the depletion of Pten-deficient HSCs 36 and Foxo-deficient HSCs 41, they failed to rescue the depletion of Lkb1-deficient HSCs (Fig. 3c-e; Fig. 4d). Lkb1-deficient HSCs are therefore depleted by mechanisms that do not depend upon increased mTOR activation or ROS levels. Lkb1 also regulated energy metabolism and mitochondrial function in HSCs through AMPK-dependent and AMPK-independent mechanisms. However, while Lkb1 deficiency and AMPK deficiency both reduced ATP levels in HSCs, AMPK deficiency had much less effect on HSC frequency or function. Lkb1 also maintained chromosome stability n HSCs through AMPK-independent mechanisms as AMPK-deficient primitive progenitors did not show increased aneuploidy (Fig. 5k). It is unclear whether Lkb1 prevents aneuploidy by regulating mitosis or whether the mitotic defects were secondary to other defects, such as in mitotic entry45. Our results indicate that in adult mammals Lkb1 promotes stem cell maintenance and tissue regeneration by regulating energy metabolism and by preventing aneuploidy.
Methods
Flow-cytometry and isolation of HSCs
Bone marrow cells were either flushed from the long bones (tibias and femurs) or isolated by crushing the long bones (tibias and femurs), pelvic bones, and vertebrae with mortar and pestle in Hank’s buffered salt solution (HBSS) without calcium and magnesium, supplemented with 2% heat-inactivated bovine serum (GIBCO, Grand Island, NY). Cells were triturated and filtered through nylon screen (45 μm, Sefar America, Kansas City, MO) or a 40μm cell strainer (Fisher Scientific, Pittsburg, PA) to obtain a single-cell suspension. For isolation of CD150+CD48−CD41−lineage−Sca-1+c-kit+ HSCs, bone marrow cells were incubated with PE-conjugated anti-CD150 (TC15-12F12.2; BioLegend), FITC-conjugated anti-CD48 (HM48-1; BioLegend), FITC-conjugated anti-CD41 (MWReg30; BD Biosciences), biotin- or APC- or PerCP-Cy5.5- conjugated anti-Sca-1 (Ly6A/E; E13-6.7), and biotin- or APC-conjugated anti-c-kit (2B8) antibody, in addition to antibodies against the following FITC-conjugated lineage markers: Ter119, B220 (6B2), Gr-1 (8C5), and CD2 (RM2-5), CD3 (KT31.1) and CD8 (53-6.7). Unless otherwise mentioned, antibodies were obtained from BioLegend, BD Biosciences, or eBioscience (San Diego, CA). Biotin-conjugated antibodies were visualized using streptavidin-conjugated APC-Cy7. HSCs were sometimes pre-enriched by selecting Sca-1+ or c-kit+ cells using paramagnetic microbeads and autoMACS (Miltenyi Biotec, Auburn, CA). Nonviable cells were excluded from sorts and analyses using the viability dye 4′,6-diamidino-2-phenylindole (DAPI) (1 μg/ml). Apoptotic cells were identified using APC Annexin V (BD biosciences). Flow cytometry was performed with FACSAria II or FACSCanto II flow-cytometers (BD Biosciences).
For isolation of lineage−Sca-1+c-kit+ cells (LSKs) and lineage−Sca-1−c-kit+CD34+CD16/CD32+ GMPs, whole bone marrow cells were incubated with FITC conjugated anti-CD34 (RAM34) for 90 minutes on ice followed by addition of PE-conjugated monoclonal antibodies to lineage markers including B220 (6B2), CD3 (KT31.1), CD4 (GK1.5), CD8 (53-6.7), Gr-1 (8C5), Mac-1 (M1/70), and Ter119 in addition to APC-conjugated anti-Sca-1 (Ly6A/E; E13-6.7), biotin-conjugated anti-c-kit (2B8) and Alexa Fluor 700 conjugated anti-CD16/32 (93) antibodies. Biotin-conjugated c-kit staining was visualized using streptavidin APC-Cy7.
B-cells were analyzed using FITC-conjugated anti-B220, PE-conjugated anti-CD43 (S7) and APC-conjugated anti-sIgM. T-cells were analyzed using FITC-conjugated anti-CD4, PE-conjugated anti-CD8 and APC-conjugated anti-CD3. Myeloid cells were analyzed using FITC-conjugated anti-Ter119, PE-conjugated anti-Gr-1 and APC-conjugated anti-Mac-1.
To measure mitochondrial mass the HSC stain was modified to make the APC channel available for Mitotracker Deep Red staining (Molecular Probes, Eugene, OR). After antibody staining cells were incubated with 1 nM Mitotracker Deep Red and 50 μM verapamil (Sigma, St. Louis, MO) for 15 min at 37 °C followed by flow-cytometry.
To measure ROS levels the HSC stain was modified to make the FITC channel available for DCFDA staining (2′-7′-dichlorofluorescein diacetate, Molecular Probes, Eugene, OR). To do this, antibodies for HSC isolation were PE/Cy5-conjugated anti-CD150 (TC15-12F12.2; BioLegend), PE-conjugated anti-CD48 (HM48-1; BioLegend), PE-conjugated anti-CD41 (MWReg30; BD PharMingen), APC-conjugated anti-Sca-1 (Ly6A/E; E13-6.7), biotin-conjugated anti-c-kit (2B8) antibody, and PE-conjugated antibodies against lineage markers. Biotin-conjugated c-kit staining was visualized using streptavidin APC-Cy7. After antibody staining cells were incubated with 5μM DCFDA for 15 min at 37 °C followed by flow-cytometry.
To measure mitochondrial membrane potential, the HSC stain was modified to make the PE channel available for tetramethyl rhodamine methyl ester (TMRM; Molecular Probes, Eugene, OR)39. Antibodies for HSC isolation were PE/Cy5-conjugated anti-CD150 (TC15-12F12.2; BioLegend), FITC-conjugated anti-CD48 (HM48-1; BioLegend), FITC-conjugated anti-CD41 (MWReg30; BD BioSciences), APC-conjugated anti-Sca-1 (Ly6A/E; E13-6.7), and biotin-conjugated anti-c-kit (2B8) antibody and FITC-conjugated antibodies against lineage markers. Biotin-conjugated c-kit staining was visualized using streptavidin APC-Cy7. After antibody staining cells were incubated with 25 nM TMRM for 15 min at 37 °C followed by flow-cytometry.
Cell cycle analysis
BrdU incorporation in vivo was measured by flow-cytometry using the APC BrdU Flow Kit (BD Biosciences, San Jose, CA). Mice were given an intraperitoneal injection of 1 mg of BrdU (Sigma, St. Louis, MO, St. Louis, MO) per 6 g of body mass in PBS and maintained on 1 mg/ml of BrdU in the drinking water for 24 hours. Cell cycle analysis in vitro was performed as follows. 500 HSCs were sorted into SF-O3 medium containing SCF and TPO (see below) and cultured for 3 days. BrdU (10 μM final concentration) was added for an hour before cells were cytospun to a slide. Slides were fixed with cold methanol for 5 minutes at −20 °C, then washed with PBS containing 0.01 % NP-40 and treated with 2N HCl for 20 minutes. Slides were blocked in PBS containing 4 % goat serum, 4 mg/ml BSA and 0.1% NP-40 followed by staining overnight at 4 °C with antibodies against BrdU (BU1/75, 1:100, Abcam, Cambridge, MA) and phospho-Histone H3 Serine10 (3H10, 1:2500, Millipore, Temecula, CA) diluted in blocking buffer. Primary antibody staining was developed with secondary antibodies conjugated to Alexa fluor 488 or 594 (Invitrogen, Eugene, OR) together with DAPI (2 μg/ml). Slides were analyzed on an Olympus microscope equipped with 40× objective lens.
For Ki-67/propidium iodide staining, HSCs were sorted into 70% ethanol and kept at - 20°C for at least 24 hours. Ki-67 staining was performed using a FITC Ki-67 kit (BD Biosciences), followed by staining with 50μg/ml propidium iodide (Molecular Probes, Eugene, OR) and analyzed by flow-cytometry.
Long-Term Competitive Repopulation Assay
Adult recipient mice (CD45.1) were irradiated with an Orthovoltage X-ray source delivering approximately 300 rad/min in two equal doses of 540 rad, delivered at least 2 hr apart. Cells were injected into the retro-orbital venous sinus of anesthetized recipients. Beginning 4 weeks after transplantation and continuing for at least 16 weeks, blood was obtained from the tail veins of recipient mice, subjected to ammonium-chloride potassium red cell lysis, and stained with directly conjugated antibodies to CD45.2 (104), CD45.1 (A20), B220 (6B2), Mac-1 (M1/70), CD3 (KT31.1), and Gr-1 (8C5) to monitor engraftment.
Western blotting
The same number of cells (25,000-35,000) from each population to be analyzed were sorted into Trichloroacetic acid (TCA) and adjusted to a final concentration of 10% TCA. Extracts were incubated on ice for 15 minutes and spun down for 10 minutes at 16.1 rcf at 4°C. The supernatant was removed and the pellets were washed with acetone twice then dried. The protein pellets were solubilized with Solubilization buffer (9 M Urea, 2% Triton X-100, 1% DTT) before adding LDS loading buffer (Invitrogen, Carlsbad, CA). Proteins were separated on a Bis-Tris polyacrylamide gel (Invitrogen) and transferred to a PVDF membrane (Millipore, Billerica, MA). Antibodies were anti-Lkb1 (#3047), anti-phospho-AMPKα (Thr172) (#2535), anti-AMPKα (#2532), anti-phospho-Acetyl-CoA Carboxylase (Ser79) (#3661), anti-phospho-S6 (#2215), anti-phospho-4EBP1 (#2855), anti-phospho-eIF4G (#2441) (all from Cell Signaling Technology, Danvers, MA) and anti-ß-actin (A1978, Sigma).
Caspase activity and ATP measurement
The same numbers of cells (approximately 5000 cells, depending on the experiment) were sorted into microcentrifuge tubes containing HBSS with 2% calf serum and pelleted. Cell pellets were lysed using Caspase-Glo2 reagent (Promega, Madison, WI) and luminescence was measured using a luminometer. Background luminescence from HBSS plus 2% calf serum was measured and the value was subtracted from sample values, then the values were divided by the cell number used to calculate the caspase activity/cell. Cellular ATP levels were measured using the ATP Bioluminescence Assay Kit CLS II (Roche Applied Science, Indianapolis, IN). Cells were sorted in PBS and boiled in the presence of 100 mM Tris, 4 mM EDTA then luciferase reagents were added. Background was measured using buffer without cells and subtracted from the values of each cell sample. ATP level/cell was calculated by dividing the measured value with the cell number used in the assay.
Methylcellulose Culture
Methylcellulose culture of bone marrow cells, HSCs and GMPs were performed as described36. Primary colonies were resuspended and replated in secondary cultures and counted 14 days later.
Cell culture for analysis of mitotic spindles and chromosome numbers
HSCs were sorted into SF-O3 media (Sankyo Junyaku, Japan) supplemented with 1% Heat-inactivated fetal bovine serum, 1% Penicillin-Streptomycin-Glutamine (GIBCO, Grand Island, NY), 50 μM 2-Mercaptoethanol, 50 ng/ml SCF and 50 ng/ml TPO (both from Peprotech, Rocky Hill, NJ). Similar result were obtained by culturing in STIF medium consisting of StemSpan serum-free medium (StemCell Technologies) supplemented with 10 μg/mL heparin (Sigma, St. Louis, MO, St Louis, MO), 10 ng/mL mouse SCF, 20 ng/mL mouse TPO, 20 ng/mL mouse IGF-2 (R&D Systems, Minneapolis, MN), and 10 ng/mL human FGF-1 (Peprotech). LSKs and GMPs were cultured in STIF supplemented with 10 ng/ml IL-3 and IL-6 (both from Peprotech). Single HSCs were sorted into each well and their cell numbers were monitored as indicated. To prepare cytospins for immunostaining, 500 to 1000 cells were sorted into each well and cultured for 3 days. Annexin-V staining was performed after 3 days of culture.
For chromosome counts, LSK or GMP cells were cultured in STIF medium supplemented with 10 ng/ml IL-3 and IL-6 for 2 days then arrested in metaphase by a 2 h incubation with 100 ng/ml colcemid (KaryoMAX solution, GIBCO). Cells were treated with hypotonic solution (0.56 % KCl) for 15 minutes at 37 °C, then fixed with 3:1 methanol:glacial acetic acid and spread on a slide to prepare metaphase spreads.
Immunostaining
Cytospin slides prepared as above without acid treatment were stained overnight at 4°C with antibodies against α-tubulin (clone YL1/2, 1:100), γ-tubulin (clone C-11, 1:100, both from Santa Cruz Biotechnology, Santa Cruz, CA) and phospho-Histone H3 Serine10 (3H10) diluted in blocking buffer. Primary antibody staining was developed with secondary antibodies conjugated to Alexa fluor 488, 555 and 647 together with DAPI (2 μg/ml). Slides were analyzed on a Leica confocal microscope.
Quantitative real-time (reverse transcription) PCR
HSCs and WBM cells were sorted into Trizol (Invitrogen) and RNA was isolated according to manufacturer’s instructions. cDNA was made with random primers and SuperScript III reverse transcriptase (Invitrogen). Quantitative PCR was performed using a SYBR Green Kit and a LightCycler 480 (Roche Applied Science). Each sample was normalized to β-actin. Primers to quantify Lkb1 cDNA levels were Lkb1 F, 5′-CACACTTTACAACATCACCA-3′, Lkb1 R, 5′-CTCATACTCCAACATCCCTC-3′, Prkaa1 F, 5′- CACCCTCACATCATCAAACTG-3′, Prkaa1 R, 5′- CTCCTCCAGAGACATATTCCA-3′, Prkaa2 F, 5′- CTTAAACTCTTTCGTCATCCTC-3′, Prkaa2 R, 5′- AACAATTCACCTCCAGACAC-3′, β-actin F, 5′-CGTCGACAACGGCTCCGGCATG-3′ and β-actin R, 5′- GGGCCTCGTCACCCACATAGGAG-3′. To quantify mitochondrial DNA copy number, cells were sorted into Trizol and DNA was isolated according to manufacturer’s instructions. Quantitative PCR was performed with primers for mtND4 (mtND4 F, 5′-ggaaccaaactgaacgccta-3′ and mtND4 R, 5′- atgagggcaattagcagtgg-3′) and β2 microglobulin intron (B2m F, 5′-tcattagggaggagccaatg-3′ and B2m R, 5′- atcccctttcgtttttgctt-3′).
PCR of genomic DNA for genotyping
To assess the degree of Lkb1 excision in genomic DNA from donor cells after transplantation, approximately 1000 donor Gr-1+ cells were sorted into alkaline lysis buffer (25 mM NaOH, 0.2 mM EDTA) and boiled, then neutralized by addition of an equal volume of neutralizing buffer (40 mM Tris-HCl). The neutralized extract was used for PCR with the following primers; R1 5′-CTGTGCTGCCTAATCTGTCG-3′, F2 5′-TTCACCATCCCTTGTGACTG-3′ and F4 5′-ATCGGAATGTGATCCAGCTT-3′. To genotype tail DNA from mice for the presence of the Lkb1fl allele primers R1 and F2 were used.
Supplementary Material
Acknowledgements
This work was supported by the Howard Hughes Medical Institute. Flow-cytometry was partially supported by the University of Michigan (UM) Comprehensive Cancer National Institutes of Health (NIH) CA46592. D.N. was supported by a postdoctoral fellowship from the Japan Society for the Promotion of Science. Thanks to Andrew Prendergast and Chris Sifuentes for technical assistance. Thanks to Elizabeth Hughes and the UM Transgenic Animal Model Core for help generating Lkb1fl mice. Thanks to David Adams and Martin White for flow-cytometry. Thanks to Elizabeth Smith (UM Hybridoma Core) for antibody production. Thanks to Chris Mountford, Sara Grove and Rose Coolon for mouse colony management. Thanks to Lewis Cantley and Craig Thompson for helpful discussions.
Footnotes
Author Information The authors declare no competing financial interests.
References
- 1.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessi DR, Sakamoto K, Bayascas JR. LKB1-dependent signaling pathways. Annual review of biochemistry. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 3.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 4.Corradetti MN, Inoki K, Bardeesy N, DePinho RA, Guan KL. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greer EL, et al. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282:30107–30119. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 7.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watts JL, Morton DG, Bestman J, Kemphues KJ. The C. elegans par-4 gene encodes a putative serine-threonine kinase required for establishing embryonic asymmetry. Development. 2000;127:1467–1475. doi: 10.1242/dev.127.7.1467. [DOI] [PubMed] [Google Scholar]
- 10.Lee JH, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447:1017–1020. doi: 10.1038/nature05828. [DOI] [PubMed] [Google Scholar]
- 11.Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature. 2003;421:379–384. doi: 10.1038/nature01296. [DOI] [PubMed] [Google Scholar]
- 12.Bonaccorsi S, et al. The Drosophila Lkb1 kinase is required for spindle formation and asymmetric neuroblast division. Development. 2007;134:2183–2193. doi: 10.1242/dev.02848. [DOI] [PubMed] [Google Scholar]
- 13.Ylikorkala A, et al. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293:1323–1326. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]
- 14.Jishage K, et al. Role of Lkb1, the causative gene of Peutz-Jegher’s syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci U S A. 2002;99:8903–8908. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakamoto K, et al. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakamoto K, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab. 2006;290:E780–788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw RJ, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu A, et al. Loss of Lkb1 in adult beta cells increases beta cell mass and enhances glucose tolerance in mice. Cell Metab. 2009;10:285–295. doi: 10.1016/j.cmet.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Granot Z, et al. LKB1 regulates pancreatic beta cell size, polarity, and function. Cell Metab. 2009;10:296–308. doi: 10.1016/j.cmet.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hezel AF, et al. Pancreatic LKB1 deletion leads to acinar polarity defects and cystic neoplasms. Mol Cell Biol. 2008;28:2414–2425. doi: 10.1128/MCB.01621-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamas P, et al. LKB1 is essential for the proliferation of T cell progenitors and mature peripheral T cells. Eur J Immunol. 2010;40:242–253. doi: 10.1002/eji.200939677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Y, et al. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Res. 2010;20:99–108. doi: 10.1038/cr.2009.141. [DOI] [PubMed] [Google Scholar]
- 23.Barnes AP, et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 24.Shorning BY, et al. Lkb1 deficiency alters goblet and paneth cell differentiation in the small intestine. PLoS One. 2009;4:e4264. doi: 10.1371/journal.pone.0004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Contreras CM, et al. Loss of Lkb1 provokes highly invasive endometrial adenocarcinomas. Cancer Res. 2008;68:759–766. doi: 10.1158/0008-5472.CAN-07-5014. [DOI] [PubMed] [Google Scholar]
- 26.Pearson HB, McCarthy A, Collins CM, Ashworth A, Clarke AR. Lkb1 deficiency causes prostate neoplasia in the mouse. Cancer Res. 2008;68:2223–2232. doi: 10.1158/0008-5472.CAN-07-5169. [DOI] [PubMed] [Google Scholar]
- 27.Gurumurthy S, et al. LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis. Cancer Res. 2008;68:55–63. doi: 10.1158/0008-5472.CAN-07-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bardeesy N, et al. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 29.Jenne DE, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 30.Hemminki A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 31.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 32.Kiel MJ, Yilmaz OH, Morrison SJ. CD150-cells are transiently reconstituting multipotent progenitors with little or no stem cell activity. Blood. 2008;111:4413–4414. doi: 10.1182/blood-2007-12-129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–197. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 34.Kontgen F, Suss G, Stewart C, Steinmetz M, Bluethmann H. Targeted disruption of the MHC Class-II AA gene in C57BL/6 mice. International Immunology. 1993;5:957–964. doi: 10.1093/intimm/5.8.957. [DOI] [PubMed] [Google Scholar]
- 35.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 36.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 37.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 38.Schieke SM, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- 39.Ruzankina Y, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyamoto K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell stem cell. 2007;1:101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Larsson NG, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nature genetics. 1998;18:231–236. doi: 10.1038/ng0398-231. [DOI] [PubMed] [Google Scholar]
- 43.Gan B, et al. Lkb1-mediated energy sensing maintains hematopoietic stem cell reserves. Submitted.
- 44.Gurumurthy S, Scadden D, Bardeesy N. Lkb1 is a critical regulator of quiescence, energy metabolism, and survival in hematopoietic stem cells. Submitted.
- 45.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodriguez CI, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nature genetics. 2000;25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.