

Abstract
Human phosphate homeostasis is regulated at the level of intestinal absorption of phosphate from the diet, release of phosphate through bone resorption, and renal phosphate excretion and involves the actions of parathyroid hormone (PTH), 1,25-dihydroxy-vitamin D (1,25-(OH)2-D), and fibroblast growth factor 23 (FGF23) to maintain circulating phosphate levels within a narrow normal range, which is essential for numerous cellular functions, for the growth of tissues and for bone mineralization. Prokaryotic and single cellular eukaryotic organisms such as bacteria and yeast “sense” ambient phosphate with a multi-protein complex located in their plasma membrane, which modulates the expression of genes important for phosphate uptake and metabolism (pho pathway). Database searches based on amino acid sequence conservation alone have been unable to identify metazoan orthologs of the bacterial and yeast phosphate sensors. Thus little is known about how human and other metazoan cells sense inorganic phosphate to regulate the effects of phosphate on cell metabolism (“metabolic” sensing) or to regulate the levels of extracellular phosphate via feedback system(s) (“endocrine” sensing). Whether the “metabolic” and the “endocrine” sensor use the same or different signal transduction cascades is unknown. This chapter will review the bacterial and yeast phosphate sensors, and then discuss what is currently known about the metabolic and endocrine effects of phosphate in multicellular organisms and humans.
Introduction
Inorganic phosphate, the variably charged anion of phosphoric acid, i.e. [H2PO41−] and [HPO42−] (for the purpose of this review referred to as “phosphate” or Pi) is required for cellular functions such as DNA/RNA and membrane phospho-lipid synthesis, generation of high-energy phosphate esters, and intracellular signaling (1). The intracellular inorganic phosphate concentration can be measured using 31P-NMR, which is a non-destructive method with little artifactual hydrolysis of labile organophosphates such as phosphocreatine; phosphocreatine is typically present intracellularly at 20 times the apparent free intracellular phosphate concentration of 0.5–5 mM (2). In contrast, the methods used to measure serum phosphate are usually based on a photometric approach using ammonium molybdate, which forms a chromogenic complex with inorganic phosphate (3).
The intracellular concentration of inorganic phosphate is maintained by membrane transporters, which accumulate phosphate at concentrations larger than would be predicted, if phosphate were distributed passively across the membrane by coupling with plasma membrane H+ (4) or Na+ gradients (5). Concentrations of intracellular phosphate are influenced by pH, hormones, and subcellular compartmentalization; levels in these compartments may be regulated by separate transporters in mitochondria (6, 7), lysosomes(8, 9), and the endoplasmic or sarcoplasmic reticulum (10). Many enzymes of key metabolic pathways are regulated by phosphate; these pathways include those for anaerobic glycolysis, gluconeogenesis, mitochondrial metabolism, glutamine, purine and nucleic acid metabolism. Although most studies were performed with purified enzymes in vitro, some enzymes respond to phosphate concentrations that could reasonably be expected intracellularly in the organs/cells from which they were isolated (Kd=1–10 mM), and thus these enzymes may also be regulated by intracellular phosphate in vivo (1). Intracellular phosphate levels are elevated in pathological conditions such as ischemia, hypoxia, and skeletal muscle fatigue, as well as in some inherited disorders such as mitochondrial myopathies. Conversely, decreased intracellular levels of phosphate are observed in disorders with severe hypophosphatemia, such as X-linked hypophosphatemia. In addition to the metabolic changes phosphate appears to activate distinct nutrient sensing pathways. These are best understood in unicellular organisms like bacteria and yeast, which will be described in detail below. In mammals circulating phosphate, in addition to serving to maintain intracellular phosphate levels for cell metabolism and growth, serves to regulate extracellular mineralization (complexes of phosphate with calcium). To control mineralization and cellular delivery, extracellular phosphate levels and total body phosphate content are tightly regulated by a number of hormones, including parathyroid hormone (PTH), 1,25-dihydroxy vitamin D (1,25(OH)2D), and fibroblast growth factor 23 (FGF23) and serum phosphate feeds back to regulate these factors in an endocrine fashion (11): high phosphate reduces secretion of PTH and increases secretion FGF23, while low phosphate stimulates the synthesis of 1,25(OH)2D, the active form of vitamin D.
Misregulation of phosphate homeostasis can cause serious human disorders (11): the clinical consequences of severe hypophosphatemia for example in tumor-induced osteomalacia or familial forms of rickets, such as X-linked hypophosphatemia, involve disruption of both the metabolic and mineralization functions of phosphate and result in hemolysis, skeletal muscle myopathy, cardiomyopathy, neuropathy, and osteomalacia; in some cases it can contribute to death. Hyperphosphatemia on the other hand leads to tissue calcifications and metabolic changes, which are to date poorly understood. Hyperphosphatemia is encountered most frequently in patients with chronic kidney disease (CKD)(12 –14), which affects 20 Million Americans today and the serum phosphate level is an important predictor of mortality in this population. Furthermore, mouse models with hyperphosphatemia due to loss-of-function mutations in Klotho or FGF23 die prematurely. This early lethality can be rescued and their life-spans extended when fed a phosphate-restricted diet (15, 16). A similar benefit is seen with dietary phosphate-restriction in humans with CKD and it is possible that similar mechanisms are at work in mice and men. An understanding of the molecular basis underlying the metabolic and endocrine phosphate effects is therefore of great significance for human disease.
The ability of organisms to detect presence and levels of various metabolites (“metabolic sensing”) is important for their normal physiology. G protein-coupled receptors have been shown to play an important role in metabolic sensing of metabolites like glucose in yeast (17), or for calcium in multicellular organisms (18). Amino acids activate metabotropic G protein-coupled receptors, but also rely on import by specialized proton assisted transporters to activate the intracellular TSC (tuberous sclerosis complex)/TOR (target of rapamycin) sensing system (19). While the TSC/TOR pathway is highly conserved between yeast and multicellular organisms, glucose sensing has evolved from use of an extracellular membrane sensor, to an intracellular sensing mechanism, which requires import of glucose by Glut2 transporters in pancreatic beta cells of multicellular organisms (20). Based on these considerations it is possible that the molecular components of the human phosphate sensor are conserved, although new mechanisms could likewise have evolved. Phosphate sensing in bacteria and yeast appears to involve uptake of extracellular phosphate, activation of an intracellular sensor composed of a kinase/kinase inhibitor complex, which leads to transcriptional events mediated by phosphate-specific transcription factors (Fig. 1A). Emerging evidence suggests that similar events are at work to mediate the cellular effects of phosphate in metazoan species including humans. However, the exact molecular nature of these mechanisms is still unclear. This chapter will review the bacterial and yeast phosphate sensors and then discuss what is currently known about the metabolic and endocrine effects of phosphate in multicellular organisms and humans.
Fig. 1.
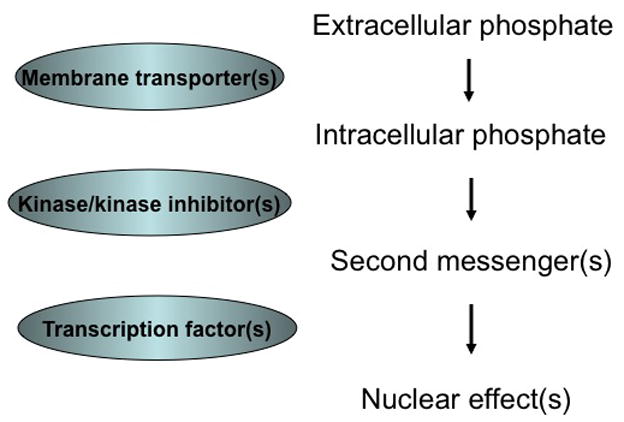


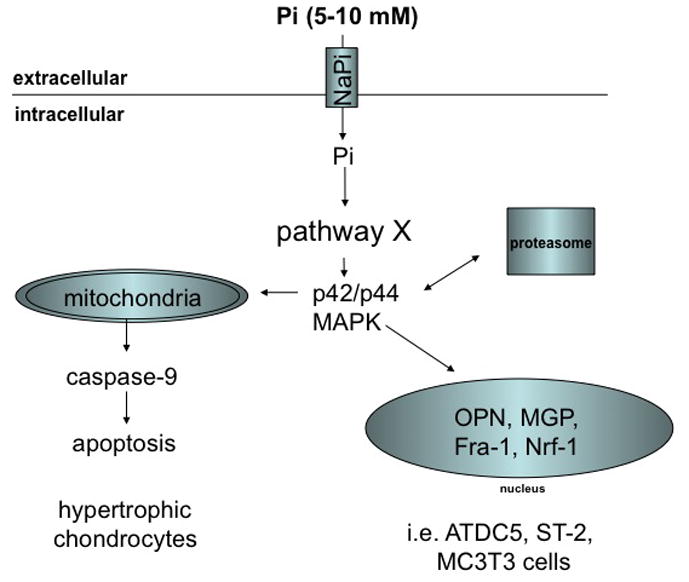
Fig. 1A: An evolutionarily conserved mechanism of phosphate sensing
Fig. 1B: Bacterial phosphate sensing
Modified from: (22), see table 1 and text for an explanation of the abbreviations
Fig. 1C: Yeast phosphate sensing
Modified from: (29), see table 1 and text for an explanation of the abbreviations
Fig. 1D: Regulation of MAPK and gene expression by phosphate in murine and human cell lines
See text for an explanation of the abbreviations
Phosphate sensing in Bacteria
In bacteria Pi is sensed by binding to the four-component Pst transporter (PstS, PstA, PstB, PstC). Phosphate thereby activates a two component signaling system composed of the sensory histidine kinase PhoR (an integral membrane protein) and the transcription factor, PhoB. This interaction is modified by a chaperone/Hsp70-like PhoR/PhoB inhibitory protein called PhoU. Excess phosphate turns the system off, while the default state is active. Thus, phosphate sensing in bacteria appears to involve a negative feed-back mechanism, which permits the pho operon to get up-regulated under conditions of phosphate limitation (21, 22).
PhoR is required for the activation (phosphorylation) of PhoB under conditions of Pi limitation. It is comprised of five domains, a domain important for anchoring in the membrane, a positively charged linker region, a Per-Arnt-Sim (PAS) domain, a dimerization and histidine phosphoacceptor domain, and a catalytic domain. The transmembrane domain anchors PhoR in the membrane, which is necessary for interaction with the Pst transporter. The PAS domain of PhoR interacts directly with the PstB component of the Pst transporter and binds PhoU. Binding of phosphate to the PstS portion of the transporter in the periplasmic space leads to inhibition of PhoR, resulting in deactivation, i.e. dephosphorylation, of phospho-PhoB (Fig. 1B, Table 1). Cellular phosphate-uptake per se is not required for phosphate-dependent signaling by the Pst system, since mutations that abolish Pi-uptake still allow for inhibition of PhoR by phosphate (21).
Table 1.
Molecular components of the bacterial, and yeast phosphate sensor
| Name | Accession | Species | Function |
|---|---|---|---|
| phoB | ECK0393 | E. coli K-12 | DNA-binding response regulator |
| phoR | ECK0394 | E. coli K-12 | Sensory histidine kinase |
| phoU | ECK3717 | E. coli K-12 | Chaperone-like PhoR/PhoB inhibitory protein |
| pstA | ECK3719 | E. coli K-12 | Phosphate transporter subunit, membrane component |
| pstB | ECK3718 | E. coli K-12 | Phosphate transporter subunit, ATP-binding component |
| pstC | ECK3720 | E. coli K-12 | Phosphate subunit, membrane component |
| pstS | ECK3721 | E. coli K-12 | Phosphate periplasmic-binding component |
| PHO80 | YOL001 W | S. cerevisiae | Cyclin: inhibitor of Pho4p |
| PHO81 | YGR233C | S. cerevisiae | Cyclin-dependent protein kinase inhibitor |
| PHO85 | YPL031C | S. cerevisiae | Cyclin-dependent protein kinase regulator |
| PHO4 | YFR034C | S. cerevisiae | DNA-binding transcriptional activator |
| PHO84 | YML123C | S. cerevisiae | MFS Pi transporter (H+-coupled) |
| PHO89 | YBR296C | S. cerevisiae | Pi transporter (Na+-coupled) |
| PHO90 | YJL198 W | S. cerevisiae | Putative Pi transporter |
| PHO91 | YNR013C | S. cerevisiae | Putative Pi transporter |
| PHO87 | YCR037C | S. cerevisiae | Pi transporter-associated |
PhoB belongs to the OmpR/PhoB subfamily of response regulators in E. coli, and, like transcription factors in higher species, it is comprised of a N-terminal regulatory receiver domain and a C-terminal DNA-binding domain. Other (non-PhoR) histidine kinases or small molecule phosphoryl donor(s) such as acetyl phosphate can activate (phosphorylate) PhoB (23), which may be important for integrating different steps of Pi metabolism and other metabolic pathways. Estimates for the number of Pi-regulated genes vary but may involve as many as 400 proteins (almost 10% of the E. coli proteome) based on proteomic data (24) and computational predictions (25). These genes encode the above phosphate sensing complex, and other genes important for phosphate scavenging and metabolism such as aldolases, phosphonate transporter subunits, carbon-phosphorus lyase complex subunits, bacterial alkaline phosphatase, glycerol-3-phosphate transporter subunits, and glycosyl transferases (as reviewed in (21)).
In addition to the Pst system, bacteria express a second family of phosphate transporters, which are related to eukaryotic type III sodium-phosphate co-transporters (Pit). However, the high-affinity Pst system appears to be the predominant system for bacterial phosphate-uptake both in conditions of phosphate limitation and abundance, while the low affinity phosphate transporters of the Pit system (i.e. PitA) serve primarily as a transporter of divalent metal cations (Zn2+) that are transported in complex with Pi (26).
The bacterial Pst transporter shows similarity to the large group of mammalian ABC-transporters, however, no specific mammalian ortholog has been identified. Furthermore, an apolipoprotein highly similar to the phosphate binding protein PstS was purified from human plasma (27), but it was later realized that this likely represents a bacterial contaminant (28). Likewise, no orthologs were identified for PhoR or PhoB. Interestingly, PhoU contains a domain similar to eukaryotic type II sodium-phosphate co-transporters, the significance of which is unclear.
Phosphate sensing in Yeast
As in bacteria, phosphate controls a specialized transcription factor, called Pho4, in yeast (29, 30). However, unlike the bacterial transporters, the phosphate transporters in yeast belong to the major facilitator superfamily (MFS) or Pit-family and the activity and subcellular localization of Pho4 is regulated by the cyclin-dependent kinase (CDK) complex, Pho80–Pho85 (31). When cells are starved of phosphate, the CDK inhibitor Pho81 inactivates Pho80–Pho85, thereby allowing unphosphorylated Pho4 to associate with the nuclear import receptor Pse1 and to enter the nucleus and bind to a phosphate response element (PRE)(32) in genes belonging to the yeast Pho-regulon (Fig. 1C, Table 1) (33) The yeast Pho-regulon includes genes coding for high affinity phosphate transporters (PHO84, PHO89) and secreted acid phosphatases (PHO5, PHO11, PHO12), which permit the cell to better assimilate phosphate from the surroundings (34).
High-affinity uptake of extracellular phosphate under phosphate-limited growth conditions is mediated by Pho84 (4). Pho84 belongs to the major facilitator superfamily of transporters, and studies of intact yeast cells (4) and inverted plasma membrane vesicles (35) have shown that the Pho84 transporter catalyses a bi-directional proton-coupled Pi uptake, where the direction of transport is determined by the directionality of the driving force rather than by the orientation of the protein. Expression of Pho84 is de-repressed when the external phosphate concentration of the growth medium decreases to about 100 uM (30). Low intracellular phosphate may serve as an activation signal for the Pho-pathway, since the disruption of Pho84 results in the constitutive expression of Pho5 in Δpho84 yeast strains (4, 36).
Following the addition of repressive amounts of phosphate (10mM) to low-Pi-grown cells, Pho84 is rapidly internalized from the plasma membrane and degraded within 60 minutes, a process that involves PKA signaling (30, 37, 38). Endocytosis and vacuolar processing of Pho84, like many other plasma membrane proteins in yeast, may require phosphorylation and/or ubiquitination (37). Down-regulation and degradation of Pho84 is also triggered by the non-metabolized phosphate analogue methylphosphonate (MP) at a concentration of 10 mM (38). MP appears to be recognized as a Pho84 substrate and to mimic phosphate’s inhibition of the phosphate-response at the level of Pho84 protein expression and degradation within 60 minutes and causes repression of Pho5 activity by 120 minutes Lack of Pho84 (Δpho84) can be rescued by over-expression of the high-affinity Pho89 and low-affinity Pho87, Pho90, and Pho91 inorganic phosphate transporters, or the glycerophosphoinositol transporter (Git1). This supports the idea that intracellular phosphate is the regulatory signal. However, de-repression of secreted acid phosphatase Pho5 does not require activation of the PKA signaling pathway (39) and is not rescued by over-expression of the Pho89, Pho87, Pho90 and Pho91, and thus binding of phosphate to Pho84 alone may be sufficient to cause some down-stream effects of phosphate.
Under conditions of high phosphate (above 100 uM), the Pho80–Pho85 cyclin/CDK complex is gradually activated to phosphorylate the transcription factor Pho4. Phospho-Pho4 associates with the nuclear export receptor Msn5, leading to the rapid export of Pho4 from the nucleus to the cytoplasm and thus to repression of the Pho-regulon (Fig. 1C). The multiple ankyrin-repeat containing CDK inhibitor Pho81 has been proposed as the intracellular phosphate sensor because of its regulatory activity in the Pho-pathway (40, 41), and because a minimal domain of Pho81 containing 80 amino acids is both necessary and sufficient to inhibit Pho80–Pho85 CDK activity (41). Inhibition of Pho81 in conditions of phosphate excess requires accumulation of myo-d-inositol heptakisphosphate (IP7) (42), which appears to involve binding of IP7 to the Pho80/81/85 complex (43). Pho81 is also required to control trehalose metabolism, the postdiauxic shift stress-response (44), and the activity of the protein kinase Rim15, which plays a central role in the integration of phosphate, glucose, nitrogen and amino acid availability during the cell cycle (45). Thus, Pho81 may integrate several nutrient-sensing pathways to control the cell cycle in yeast. Further indication for a cross-talk between several nutrient sensing pathways is that limitation in carbon sources results in rapid degradation of Pho84 despite the presence of de-repressing levels of Pi (38, 46).
Pho84 belongs to the major facilitator family, which comprises more than 200 members in humans and includes the type I sodium-phosphate transporters (SLC17A1-9). Pho87, 90, 91 are related to metazoan sodium-sulfate transporters (SLC13A1-4), and Pho89 is related to the type III sodium-phosphate transporters SLC20A1 (Pit-1) and SLC20A2 (Pit-2) (47, 48). Using DNA footprint analysis, Kido et al. identified a phosphate responsive element (PRE) in the mouse NaPi-IIa promotor, which has similarity to the yeast PRE and isolated a transcription factor mE3 (TFE3), which is similar to yeast Pho4, and may mediate the up-regulation of NaPi-IIa gene expression in response to low-phosphate diet (49).
Regulation of phosphate sensing by intracellular phosphate stores
Inorganic polyphosphate (Poly P) is a linear polymer comprising ten to many hundred phosphate residues; these are linked by the same phosphoanhydride bonds found in ATP. It is present in intracellular vacuoles in all cell types, where it functions as a phosphate store.
In bacteria, Poly P can serve both as a phosphate reservoir and as an energy source in different biological processes, where it substitutes for ATP in phosphorylation reactions by kinases to trigger signal transduction by in inhibiting or activating effector molecules (50, 51). The poly P kinase gene PPK, which is involved in poly P synthesis, appears to be under the control of the PhoR/PhoB two component signaling system (52, 53). Conversely, poly P is able to regulate PitA expression, and furthermore, PPK, Pst and PitA are able to compensate for each other to regulate cellular phosphate uptake (54).
Similarly in yeast, Poly P is synthesized and accumulated in the vacuole and represents a phosphate reserve used during periods of phosphate starvation (55–57). This is illustrated in cell culture experiments when transfer of cells to a phosphate-deficient medium results in a rapid decrease of poly P content (58). In addition to serving as a buffer that can be mobilized during periods of Pi limitation (59), the capacity to synthesize Poly P, furthermore, exerts control on the rate at which Pi is taken up, possibly via a transient increase in the level of phosphate in the cytosol (38). Finally, Poly P strongly influences the expression of Pho5, the main secreted acid phosphatase (36) by which yeast mobilizes phosphate from extracellular sources.
Little is known about Poly P in animal cells beyond its widespread abundance in tissues and subcellular compartments (60). Poly P, at the concentrations of 0.15–1.5 μM found in mammalian cells (61) and with Poly P chain lengths ranging from 15 to 750 phosphate residues, appears to stimulate mTOR (mammalian target of rapamycin), a ser-thr protein kinase that is at the center of signaling pathways in cell growth and proliferation and is activated by autophosphorylation (62). mTOR function is controlled by nutrient availability to ensure that protein synthesis is repressed when the supply of precursor amino acids is insufficient. The key downstream targets of mTOR are PHAS-I (also called eukaryotic initiation factor 4E binding protein (eIF4E-BP)), and the kinase for the 40S ribosomal S6 protein (p70 S6 kinase) (63). PHAS-I is inhibited by mTOR, while S6K is activated. PHAS-I in its unphosphorylated state sequesters eIF4E, a mRNA cap-binding protein in the eIF4G elongation complex (64, 65). Via activation of mTOR polyP thus stimulates global protein translation. Poly P also appears to enhance the mitogenic activities of acidic fibroblast growth factor (FGF-1) and basic fibroblast growth factor (FGF-2) in human fibroblasts by functioning as co-activator similar to heparin (66). Finally, addition of Poly P to human plasma cells (PCs) inhibited secretion of immunoglobulin and stimulated apoptosis by activating caspase-3 and arresting the cell cycle (67), suggesting that poly P plays a role in immunity.
Phosphate sensing in multicellular organism
Metabolic effects of activation of MAPK by phosphate
It has long been recognized that differentiation of primary or immortalized osteogenic cells requires the addition of inorganic phosphate in the form of hydroxyapatite (68–70) or beta-glycerophosphate (71) along with ascorbic acid, which is believed to support formation of collagen matrix (72). Expression of several bone-cell specific genes, such as dentin matrix protein 1 (DMP1), osteopontin (OPN), and matrix-gla-protein (MGP), is stimulated by phosphate at the transcriptional level, while expression of alkaline phosphatase (TNSALP) is suppressed (73–75). Cellular uptake of phosphate by specialized transporters appears to be required since addition of phosphonoformic acid (PFA, forscarnet), an inhibitor of sodium-phosphate co-transporters, blocks these effects of Pi (75–77). The downstream effects that result in suppression of TNSALP may require BMP-signaling in ST2 murine bone marrow stromal cells (78). In contrast, dependence of phosphate signaling on the p42/p44-MAPK/ERK pathway was shown in ATDC5 cells (79). Likewise, induction of OPN and MGP requires activation of the p42/p44-MAPK/ERK pathway and can be blocked by UO126, a specific inhibitor of MAPK-kinase, MEK (80, 81).
Activation of MAPK by calcium-phosphate crystals in primary fibroblasts was initially described by Nair and colleagues (82). Beck and colleagues subsequently showed that phosphate at concentrations between 5–10 mM alone is sufficient to activate MAPK in MC3T3 mouse fibroblast cells (80), and subsequently activation of MAPK by inorganic phosphate was demonstrated in multiple other cell lines, including chondrogenic ATDC5 cells, MC3T3-E1 osteoblasts and ST2 murine bone marrow stromal cells (81), HEK293 human proximal tubular cells (83), and lung alveolar cells (77) (Fig. 1D). Although some cell lines, for example C2C12 or L929 cells, were less responsive than others (81), activation of MAPK by phosphate appears to be quite universal in metazoans. This mechanism is furthermore conserved evolutionarily, since similar observations were made in Drosophila S2R+ hemocyte cells (84). Addition of PFA (76, 85) or siRNA-mediated knockdown of Pit1 sodium-phosphate co-transporters blocks activation of MAPK by phosphate (83) indicating that cellular uptake of phosphate is required for the activation of MAPK. Furthermore, using cell lines expressing a Pi-transport-deficient Pit1 transporter, Beck and colleagues recently reported that Pit1 may have transport-independent effects on cell proliferation and tumor growth in vitro and in vivo, although it remains to be shown whether these effects depend on phosphate-binding to Pit1 (86). Pit1 together with its mammalian paralogue Pit2, the bacterial pitA, and yeast pho89 transporters belongs to the NaPi-III class of sodium-phosphate co-transporters (47, 48).
Phosphate appears to induce phosphorylation of FRS2alpha, a mediator of FGF receptor action (79, 83). In addition siRNA-mediated knockdown of FGFR1 is able to interfere with Pi-induced MAPK (79, 83). Furthermore, phosphorylation of Akt and c-Raf by phosphate was shown in human bronchial epithelial cells (77) and proliferating chondrocytes (79), respectively, suggesting that phosphate interacts with the MAPK pathway relatively close to the cell membrane. Activation of p42/p44-MAPK by phosphate is rapid and occurs within 5 min., but biphasic up-regulation with a second peak after 8–12 hrs. has been reported in some cell lines, while the related kinases JNK and p38-MAPK appear not to be affected by phosphate at least in those cell lines reported to date (80, 81).
It is possible that binding of extracellular phosphate to Pit1 activates the MAPK pathway. However, since knockdown of members of the MFS family (i.e. NaPi-I) blocks phosphate-induced MAPK at least in Drosophila S2R+ cells (84), intracellular phosphate after uptake via multiple transporter classes may be the signal for MAPK and the activation of possibly other pathways. Furthermore, blocking phosphate-induced MAPK by knockdown of FGFR1 or FRS2alpha either suggests a cooperative interaction between Pit1 and FGFR1 to sense extracellular phosphate after binding to Pit1 or that intracellular phosphate acts to modulate flow through the MAPK pathway at one or multiple levels downstream of the FGFR1/FRS2alpha.
Egr-1 was shown to be a downstream effector of phosphate-induced MAPK (87, 88). Pi also induces the expression of Fra-1 (87) and Nrf2 (89, 90), although dependence of these factors on MAPK as the intermediary has only thus far been demonstrated for Fra-1 (87). Phosphate-induced MAPK has been implicated in proliferation of the chondrogenic cell line ATDC5, possibly by activating cyclin D1 (79). Finally, activation of MAPK by phosphate stimulates apoptosis of hypertrophic chondrocytes (91), which also requires changes of the mitochondrial membrane potential and activation of caspase-9 (92). Conversely, inhibition of phosphate uptake or phosphate restriction prevent apoptosis of hypertrophic chondrocytes in vitro (85, 93, 94), and lead to a 2.5-fold increase in parathyroid hormone-related protein mRNA expression pointing to an important role for phosphate in regulating growth plate maturation (92). Both effects may explain expansion of the growth plates in the growing skeleton in vivo, for example in children with hypophosphatemic rickets (95).
Endocrine effects of phosphate
1. Human phosphate homeostasis
Over the last two decades, information derived from genetic analyses of familial disorders has led to the discovery of novel genes involved in the regulation of phosphate homeostasis, which in contrast to the regulation of calcium homeostasis (96, 97), remains less well understood to date. Among these novel genes is fibroblast growth factor 23 (FGF23), named after its similarity to members of the fibroblast growth factor family. Different from most other members of this family, it is a circulating hormone and acts at the level of the kidney to regulate phosphate reabsorption (98). FGF23 is produced by osteocytes and osteoblasts in bone and up-regulated by an increase in dietary phosphate intake and the active form of vitamin D, 1,25(OH)2D, while it is down-regulated, through yet unknown mechanisms, by PHEX, DMP1, ENPP1 and probably several additional proteins (Fig. 2) (99). FGF23 acts through one or more FGF receptors, with Klotho as co-receptor (100), to reduce renal phosphate re-absorption (5, 101, 102), to decrease circulating 1,25(OH)2D levels, and possibly to inhibit PTH secretion by the parathyroid glands. Its net effect is to lower the serum phosphate concentration. PTH on the other hand acts, like FGF23, to decrease renal phosphate re-absorption and thus also reduces the serum phosphate concentration. In contrast to FGF23, however, PTH stimulates renal 1-alpha hydroxylase and thus increases serum 1,25(OH)2D levels. Serum phosphate feeds back to stimulate FGF23 and PTH secretion (see below). It also appears to be sensed in the intestinal lumen to regulate renal retention of phosphate independent of FGF23 and PTH, which may involve novel, yet to be discovered factors (103). Through its effect on the regulation of FGF23, it also reduces 1,25(OH)2D levels (104), although serum phosphorus may also have direct effects on 1-alpha-hydroxylase and/or 24-hydroxylase (105). 1,25(OH)2D acts through VDR/RXR heterodimers to enhance the intestinal absorption of phosphate (106) and to stimulate FGF23 synthesis and secretion by osteocytes; it furthermore inhibits PTH synthesis and secretion by the parathyroid glands. The net 1,25(OH)2D effect is an increase in the serum phosphate concentration. For detailed discussion of the receptors and signal transduction cascades for PTH, 1,25(OH)2D, and FGF23 the reader is referred to several recent reviews (98, 99, 107–115).
Fig. 2.

Endocrine regulation of mammalian phosphate homeostasis
Modified from: (115), see text for an explanation of the abbreviations
2. Regulation of Parathyroid hormone secretion by phosphate
The major physiological role of the parathyroid glands is to function as a “calciostat” (116–119). Consequently, parathyroid hormone (PTH) secretion by the parathyroid glands is tightly regulated by extracellular calcium on a transcriptional and post-transcriptional level. Similarly, 1,25(OH)2D suppresses PTH gene expression via vitamin D responsive elements in the PTH promotor (120). Acute infusion of phosphate has little effect on PTH secretion in dogs (121) and cows (118). However, chronic elevation of serum phosphate stimulates PTH secretion by direct and indirect mechanisms: persistent hyperphosphatemia stimulates PTH-secretion presumably by lowering extracellular calcium (122, 123), while hypophosphatemia suppresses PTH-secretion indirectly by up-regulation of 1,25(OH)2D. Up-regulation of 1,25(OH)2D production also induces of FGF23 synthesis, which acts at the parathyroids to suppress PTH mRNA synthesis (as already mentioned above) and secretion in vitro and in vivo in an alpha klotho (KL)-dependent fashion (124–127). Finally, KL may also have an FGF23-independent role by facilitating PTH-secretion through maintenance of membrane Na/K-ATPase activity in the setting of hypocalcemia and KL null mice are thus desensitized to hypocalcemic stress (128).
In addition to these indirect effects, phosphate directly acts on the parathyroids to stimulate PTH synthesis through posttranscriptional mechanisms, which have been elucidated in recent years. Using Northwestern blot analysis, RNA electrophoretic mobility shift and affinity cross-linking studies of parathyroid extracts, cytosolic transacting factors were identified that bind to a 63 bp cis-acting instability element in the PTH mRNA 3′-untranslated region (UTR) (122, 129) (Fig. 3). It appears that there is a balanced interaction of the PTH mRNA with the stabilizing proteins AUF1 (AU-rich element binding protein, isoforms p37, p40, p42 and p45) and Unr (upstream of N-ras) (130) and the destabilizing protein KSRP (K-homology splicing regulator protein)in vivo (131). In the setting of hypocalcemia or chronic kidney disease the peptidyl-prolyl isomerase Pin1 is inactive (132), resulting in KSRP phosphorylation and hence its inactivation. This presumably allows AUF1 and Unr to bind the PTH mRNA 3′ UTR ARE with a greater affinity, leading to increased PTH mRNA stability. Conversely, hypercalcemia and hypophosphatemia lead to destabilization of the PTH mRNA, which then is targeted for degradation by the exosome. Because parathyroid cells that sense phosphate, as they do in vivo, have not been established as stable cell lines, the mechanistic links between the regulation of PTH mRNA stability by phosphate in vivo and the Pin1/KSRP pathway remain to be established.
Fig. 3.
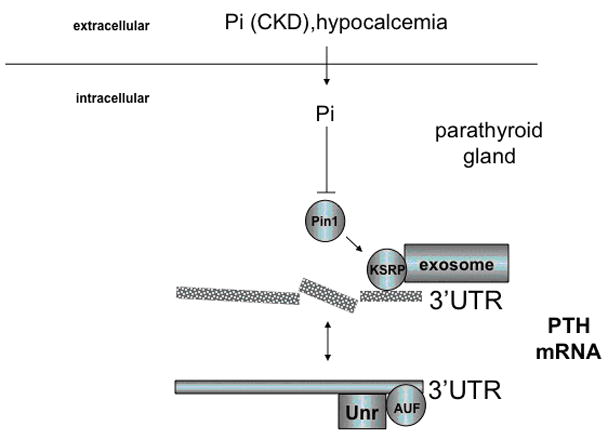
Regulation of PTH mRNA stability by phosphate
Modified from: (132), see text for an explanation of the abbreviations
3. 1-alpha hydroxylase regulation by phosphate
Cholecalciferol is generated from its precursors 7-dehydrocholesterol and ergosterol in the skin, subjected to 25-hydroxylation in the liver and converted to the active 1,25-dihydroxycholecalciferol (1,25(OH)2D) in the kidney (133, 134). 1-alpha-hydroxylation in the kidney is tightly regulated. It is induced by PTH, hypocalcemia and hypophosphatemia, which all appear to induce expression of CYP27B1, the gene encoding 1-alpha hydroxylase (135) (Fig. 4). FGF23, hypercalcemia and hyperphosphatemia, on the other hand, reduce CYP27B1 expression (135, 136). Also FGF7 and sFRP-4 appear to inhibit synthesis of 1,25(OH)2D (137, 138). FGF23 and 1,25(OH)2D also increase renal CYP24 activity (136, 139, 140), which converts 25-OH-vitamin D and 1,25(OH)2D into inactive metabolites. It will be of interest to elucidate the upstream sensing mechanism and whether phosphate regulates CYP27B1 at a transcriptional or posttranscriptional level, as these two events are not properly understood.
Fig. 4.

Regulation of 1-alpha hydroxylase by phosphate
Modified from: (133), see text for an explanation of the abbreviations
4. Regulation of FGF23 secretion by phosphate
The main sources of FGF23 are osteocytes and osteoblasts in the skeleton (141). The phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX) and dentin matrix protein 1 (DMP1), bone-specific proteins which were discovered in genome-wide linkage studies of hypophosphatemic disorders (108, 142), suppress expression of FGF23 in bone most likely through indirect mechanisms (99). Intact DMP1 is cleaved into a 35 and a 57 kDa fragment, possibly by bone morphogenic protein 1 (BMP1) (143), which, in turn, is activated by a complex consisting of the subtilisin propeptide convertase SPC2 and the co-activator 7B2 (144). Transgenic over-expression of the C-terminal 57 kDa DMP1 fragment is both necessary and sufficient to rescue the bone phenotype (and probably the hypophosphatemia) resulting from increased FGF23 secretion in Dmp1-null mice (145). This 57 kDa DMP1 fragment appears to have nuclear effects, which may be required for suppression and/or feedback regulation of FGF23 gene transcription and/or FGF23 secretion (145, 146). Recently, ecto-nucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) was found to be another negative regulator of FGF23 secretion. ENPP1 is a membrane-bound ecto-enzyme responsible for the generation of the mineralization inhibitor pyrophosphate (PPi) (147). Loss-of function mutations in this enzyme can cause generalized arterial calcifications of infancy (GACI) (148, 149) and/or hypophosphatemic rickets due to FGF23-dependent renal phosphate-wasting (150, 151). FGF23 excess may be directly related to lack of PPi production, or may be due to accumulation of precursors of PPi, such as ATP in the extracellular space. However, presence of mild hyperphosphatemia in individuals and carriers suffering from hypophosphatasia, which is caused by loss-of-function mutations in the PPi-degrading enzyme, namely tissue non-specific alkaline phosphatase (TNALP) (152), suggests that PPi is the intermediary suppressing FGF23 production.
In contrast, dietary phosphate and serum 1,25(OH)2D stimulate FGF23 synthesis in human (153) (154, 155) or animal studies (156); both factors appear to regulate FGF23 expression independently from each other. For example, injection of 1,25-dihydroxyvitamin D3 increases serum FGF23 levels within hours in rodents without significant increase in serum phosphate levels, indicating that vitamin D can increase FGF23 independently of phosphate (156). The effect of 1,25-dihydroxyvitamin D3 is via its nuclear vitamin D receptor (VDR), which heterodimerizes with RXR, another nuclear receptor. The VDR-RXR heterodimer in turn binds to the promoter region of the FGF23 gene and transactivates its expression (109). Similarly, phosphate positively regulates FGF23 expression, although the exact mechanisms need to be defined. VDR-deficient mice have low serum levels of phosphate and FGF23, but when placed on a calcium and phosphate-rich “rescue” diet serum FGF23 levels are restored to normal, indicating that phosphate can increase FGF23 independent of the actions of vitamin D (157).
Due to the lack of suitable in vitro cell culture models, which show regulation of FGF23 secretion by phosphate, it remains unclear, whether the in vivo effects of phosphate on FGF23 transcription and/or secretion are cell autonomous or mediated via intermediary factors. Insights from cell lines transfected with plasmids encoding the full-length human hormone indicate that FGF23 is subjected to post-translational modification, and that this modification may be regulated by phosphate (Fig. 5): After the cleavage and removal of the signal sequence comprising 24 amino acids, FGF23(25–251) is O-glycosylated by UDP-N-acetyl-alpha-D-galactosamine: polypeptide N-acetylgalactosaminyl-transferase 3 (GALNT3). O-glycosylation is essential for secretion of FGF23 by CHO cells (158). Similar observations are made for the secretion of FGF7 by human embryonic kidney cells (HEK293) (159). Mutations that impair O-glycosylation of FGF23 lead to hypophosphatemic familial tumoral calcinosis in humans (160, 161) and similar observations are made in mice (162) due to the lack of bioactive FGF23. Expression of GALNT3 appears to be stimulated by extracellular phosphate and is suppressed by extracellular calcium and 1,25(OH)2D in HEK293 cells (159). It also appears to be stimulated by cAMP-mediated mechanisms in vitro (163) and by fibrous dysplastic lesions that are caused by gain-of-function mutations of Gs-alpha in vivo (164). This suggests that GALNT3 may be an important component of the regulatory mechanism of FGF23 secretion by phosphate and cAMP.
Fig. 5.

Regulation of FGF23 secretion by phosphate
Modified from: (159), (163), see text for an explanation of the abbreviations
Summary and outlook
Phosphate sensing in bacteria and yeast requires binding and cellular uptake of phosphate by transporters of the ABC and MFS superfamilies, respectively. Intracellular phosphate acts to negatively regulate histidine or cyclin-dependent kinases in these species. These kinases, in turn, control specialized transcription factors that are required for the expression of phosphate-scavenging genes, which are collectively referred to as Pho-regulon, and encode secreted phosphatases, phosphate transporters, along with members of the phosphate signal transduction cascade. Database searches based on sequence homology have so far been unsuccessful in identifying metazoan orthologs of the bacterial and yeast phosphate sensors and the Pho-regulon. However, there is growing evidence indicating that binding and cellular uptake of phosphate by specialized sodium-phosphate transporters is required also for phosphate effects in mammalian cells. This suggests that similar processes govern phosphate sensing in higher species. Elevated intracellular phosphate leads to the activation of MAPK, which is required for the regulation of gene expression and apoptosis by phosphate in mammalian cells. Furthermore, phosphate appears to have post-transcriptional and post-translational effects on PTH mRNA stability and O-glycosylation of FGF23 protein, respectively, two important endocrine regulators of phosphate homeostasis. We currently perform functional screens using genome-wide siRNA technology to identify components of the mammalian phosphate-sensing pathway. An important unanswered question is whether the regulators of the metabolic functions of intracellular phosphate and the regulators of the extracellular functions of phosphate are similar. For this purpose, generation of PTH-, FGF23- and CYP27B1 producing cell lines will be helpful to further characterize the transcriptional, post-transcriptional and post-translational control of these hormones by phosphate.
Acknowledgments
We would like to thank Norbert Perrimon, Ph.D., Dept. of Genetics, Harvard Medical School/Howard Hughes Medical Institute, as well as John T. Potts, Henry M. Kronenberg and Lawrence Mensah, Endocrine Unit, Massachusetts General Hospital, Boston for helpful comments to the manuscript.
Footnotes
Disclosures: The authors have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bevington A, Kemp GJ, Graham R, Russell G. Phosphate-sensitive enzymes: possible molecular basis for cellular disorders of phosphate metabolism. Clin Chem Enzym Comms. 1992;4:235–257. [Google Scholar]
- 2.Bevington A, Mundy KI, Yates AJ, et al. A study of intracellular orthophosphate concentration in human muscle and erythrocytes by 31P nuclear magnetic resonance spectroscopy and selective chemical assay. Clin Sci (Lond) 1986;71:729–735. doi: 10.1042/cs0710729. [DOI] [PubMed] [Google Scholar]
- 3.Gaddum JH. The Estimation of Phosphorus in Blood. Biochem J. 1926;20:1204–1207. doi: 10.1042/bj0201204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bun-Ya M, Nishimura M, Harashima S, Oshima Y. The PHO84 gene of Saccharomyces cerevisiae encodes an inorganic phosphate transporter. Mol Cell Biol. 1991;11:3229–3238. doi: 10.1128/mcb.11.6.3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch. 2004;447:763–767. doi: 10.1007/s00424-003-1072-5. [DOI] [PubMed] [Google Scholar]
- 6.Wohlrab H. Molecular aspects of inorganic phosphate transport in mitochondria. Biochim Biophys Acta. 1986;853:115–134. doi: 10.1016/0304-4173(86)90007-8. [DOI] [PubMed] [Google Scholar]
- 7.Ferreira GC, Pedersen PL. Phosphate transport in mitochondria: past accomplishments, present problems, and future challenges. J Bioenerg Biomembr. 1993;25:483–492. doi: 10.1007/BF01108405. [DOI] [PubMed] [Google Scholar]
- 8.Pisoni RL, Lindley ER. Incorporation of [32P]orthophosphate into long chains of inorganic polyphosphate within lysosomes of human fibroblasts. J Biol Chem. 1992;267:3626–3631. [PubMed] [Google Scholar]
- 9.Pisoni RL. Characterization of a phosphate transport system in human fibroblast lysosomes. J Biol Chem. 1991;266:979–985. [PubMed] [Google Scholar]
- 10.Burchell A. Endoplasmic reticulum phosphate transport. Kidney Int. 1996;49:953–958. doi: 10.1038/ki.1996.134. [DOI] [PubMed] [Google Scholar]
- 11.Bringhurst FR, Leder BZ. Regulation of calcium and phosphate homeostasis. In: DeGroot LJ, Jameson JL, editors. Endocrinology. 5. Philadelphia: W.B. Saunders Co; 2006. pp. 805–843. [Google Scholar]
- 12.Razzaque MS. Does FGF23 toxicity influence the outcome of chronic kidney disease? Nephrol Dial Transplant. 2009;24:4–7. doi: 10.1093/ndt/gfn620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. J Am Soc Nephrol. 2009;20:1453–1464. doi: 10.1681/ASN.2008070692. [DOI] [PubMed] [Google Scholar]
- 15.Stubbs JR, Liu S, Tang W, et al. Role of hyperphosphatemia and 1,25-dihydroxyvitamin d in vascular calcification and mortality in fibroblastic growth factor 23 null mice. J Am Soc Nephrol. 2007;18:2116–2124. doi: 10.1681/ASN.2006121385. [DOI] [PubMed] [Google Scholar]
- 16.Morishita K, Shirai A, Kubota M, et al. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr. 2001;131:3182–3188. doi: 10.1093/jn/131.12.3182. [DOI] [PubMed] [Google Scholar]
- 17.Thevelein JM, Gelade R, Holsbeeks I, et al. Nutrient sensing systems for rapid activation of the protein kinase A pathway in yeast. Biochem Soc Trans. 2005;33:253–256. doi: 10.1042/BST0330253. [DOI] [PubMed] [Google Scholar]
- 18.Brown EM, Gamba G, Riccardi D, et al. Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 19.Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 20.MacDonald PE, Joseph JW, Rorsman P. Glucose-sensing mechanisms in pancreatic beta-cells. Philos Trans R Soc Lond B Biol Sci. 2005;360:2211–2225. doi: 10.1098/rstb.2005.1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh YJ, Wanner BL. Global regulation by the seven-component Pi signaling system. Curr Opin Microbiol. 2010;13:198–203. doi: 10.1016/j.mib.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamarche MG, Wanner BL, Crepin S, Harel J. The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev. 2008;32:461–473. doi: 10.1111/j.1574-6976.2008.00101.x. [DOI] [PubMed] [Google Scholar]
- 23.Zundel CJ, Capener DC, McCleary WR. Analysis of the conserved acidic residues in the regulatory domain of PhoB. FEBS Lett. 1998;441:242–246. doi: 10.1016/s0014-5793(98)01556-7. [DOI] [PubMed] [Google Scholar]
- 24.VanBogelen RA, Olson ER, Wanner BL, Neidhardt FC. Global analysis of proteins synthesized during phosphorus restriction in Escherichia coli. J Bacteriol. 1996;178:4344–4366. doi: 10.1128/jb.178.15.4344-4366.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan ZC, Zaheer R, Morton R, Finan TM. Genome prediction of PhoB regulated promoters in Sinorhizobium meliloti and twelve proteobacteria. Nucleic Acids Res. 2006;34:2686–2697. doi: 10.1093/nar/gkl365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beard SJ, Hashim R, Wu G, Binet MR, Hughes MN, Poole RK. Evidence for the transport of zinc(II) ions via the pit inorganic phosphate transport system in Escherichia coli. FEMS Microbiol Lett. 2000;184:231–235. doi: 10.1111/j.1574-6968.2000.tb09019.x. [DOI] [PubMed] [Google Scholar]
- 27.Morales R, Berna A, Carpentier P, et al. Serendipitous discovery and X-ray structure of a human phosphate binding apolipoprotein. Structure. 2006;14:601–609. doi: 10.1016/j.str.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 28.Berna A, Scott K, Chabriere E, Bernier F. The DING family of proteins: ubiquitous in eukaryotes, but where are the genes? Bioessays. 2009;31:570–580. doi: 10.1002/bies.200800174. [DOI] [PubMed] [Google Scholar]
- 29.Mouillon JM, Persson BL. New aspects on phosphate sensing and signalling in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:171–176. doi: 10.1111/j.1567-1364.2006.00036.x. [DOI] [PubMed] [Google Scholar]
- 30.Persson BL, Lagerstedt JO, Pratt JR, et al. Regulation of phosphate acquisition in Saccharomyces cerevisiae. Curr Genet. 2003;43:225–244. doi: 10.1007/s00294-003-0400-9. [DOI] [PubMed] [Google Scholar]
- 31.Toh-e A, Tanaka K, Uesono Y, Wickner RB. PHO85, a negative regulator of the PHO system, is a homolog of the protein kinase gene, CDC28, of Saccharomyces cerevisiae. Mol Gen Genet. 1988;214:162–164. doi: 10.1007/BF00340196. [DOI] [PubMed] [Google Scholar]
- 32.Oshima Y. The phosphatase system in Saccharomyces cerevisiae. Genes Genet Syst. 1997;72:323–334. doi: 10.1266/ggs.72.323. [DOI] [PubMed] [Google Scholar]
- 33.Kaffman A, Herskowitz I, Tjian R, O’Shea EK. Phosphorylation of the transcription factor PHO4 by a cyclin-CDK complex, PHO80-PHO85. Science. 1994;263:1153–1156. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 34.Lenburg ME, O’Shea EK. Signaling phosphate starvation. Trends Biochem Sci. 1996;21:383–387. [PubMed] [Google Scholar]
- 35.Fristedt U, Berhe A, Ensler K, Norling, Persson BL. Isolation and characterization of membrane vesicles of Saccharomyces cerevisiae harboring the high-affinity phosphate transporter. Arch Biochem Biophys. 1996;330:133–141. doi: 10.1006/abbi.1996.0235. [DOI] [PubMed] [Google Scholar]
- 36.Auesukaree C, Homma T, Tochio H, Shirakawa M, Kaneko Y, Harashima S. Intracellular phosphate serves as a signal for the regulation of the PHO pathway in Saccharomyces cerevisiae. J Biol Chem. 2004;279:17289–17294. doi: 10.1074/jbc.M312202200. [DOI] [PubMed] [Google Scholar]
- 37.Lagerstedt JO, Voss JC, Wieslander A, Persson BL. Structural modeling of dual-affinity purified Pho84 phosphate transporter. FEBS Lett. 2004;578:262–268. doi: 10.1016/j.febslet.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Pratt JR, Mouillon JM, Lagerstedt JO, Pattison-Granberg J, Lundh KI, Persson BL. Effects of methylphosphonate, a phosphate analogue, on the expression and degradation of the high-affinity phosphate transporter Pho84, in Saccharomyces cerevisiae. Biochemistry. 2004;43:14444–14453. doi: 10.1021/bi049327t. [DOI] [PubMed] [Google Scholar]
- 39.Mouillon JM, Persson BL. Inhibition of the protein kinase A alters the degradation of the high-affinity phosphate transporter Pho84 in Saccharomyces cerevisiae. Curr Genet. 2005;48:226–234. doi: 10.1007/s00294-005-0019-0. [DOI] [PubMed] [Google Scholar]
- 40.Huang K, Ferrin-O’Connell I, Zhang W, Leonard GA, O’Shea EK, Quiocho FA. Structure of the Pho85-Pho80 CDK-cyclin complex of the phosphate-responsive signal transduction pathway. Mol Cell. 2007;28:614–623. doi: 10.1016/j.molcel.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang S, Jeffery DA, Anthony MD, O’Shea EK. Functional analysis of the cyclin-dependent kinase inhibitor Pho81 identifies a novel inhibitory domain. Mol Cell Biol. 2001;21:6695–6705. doi: 10.1128/MCB.21.19.6695-6705.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee YS, Mulugu S, York JD, O’Shea EK. Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science. 2007;316:109–112. doi: 10.1126/science.1139080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YS, Huang K, Quiocho FA, O’Shea EK. Molecular basis of cyclin-CDK-CKI regulation by reversible binding of an inositol pyrophosphate. Nat Chem Biol. 2008;4:25–32. doi: 10.1038/nchembio.2007.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swinnen E, Rosseels J, Winderickx J. The minimum domain of Pho81 is not sufficient to control the Pho85-Rim15 effector branch involved in phosphate starvation-induced stress responses. Curr Genet. 2005;48:18–33. doi: 10.1007/s00294-005-0583-3. [DOI] [PubMed] [Google Scholar]
- 45.Swinnen E, Wanke V, Roosen J, et al. Rim15 and the crossroads of nutrient signalling pathways in Saccharomyces cerevisiae. Cell Div. 2006;1:3. doi: 10.1186/1747-1028-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petersson J, Pattison J, Kruckeberg AL, Berden JA, Persson BL. Intracellular localization of an active green fluorescent protein-tagged Pho84 phosphate permease in Saccharomyces cerevisiae. FEBS Lett. 1999;462:37–42. doi: 10.1016/s0014-5793(99)01471-4. [DOI] [PubMed] [Google Scholar]
- 47.Werner A, Kinne RK. Evolution of the Na-P(i) cotransport systems. Am J Physiol Regul Integr Comp Physiol. 2001;280:R301–312. doi: 10.1152/ajpregu.2001.280.2.R301. [DOI] [PubMed] [Google Scholar]
- 48.Hubbard TJ, Aken BL, Beal K, et al. Ensembl 2007. Nucleic Acids Res. 2007;35:D610–617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kido S, Miyamoto K, Mizobuchi H, et al. Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. 1999;274:28256–28263. doi: 10.1074/jbc.274.40.28256. [DOI] [PubMed] [Google Scholar]
- 50.Kornberg A, Rao NN, Ault-Riche D. Inorganic polyphosphate: a molecule of many functions. Annu Rev Biochem. 1999;68:89–125. doi: 10.1146/annurev.biochem.68.1.89. [DOI] [PubMed] [Google Scholar]
- 51.Brown MR, Kornberg A. Inorganic polyphosphate in the origin and survival of species. Proc Natl Acad Sci U S A. 2004;101:16085–16087. doi: 10.1073/pnas.0406909101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato J, Yamamoto T, Yamada K, Ohtake H. Cloning, sequence and characterization of the polyphosphate kinase-encoding gene (ppk) of Klebsiella aerogenes. Gene. 1993;137:237–242. doi: 10.1016/0378-1119(93)90013-s. [DOI] [PubMed] [Google Scholar]
- 53.Ghorbel S, Kormanec J, Artus A, Virolle MJ. Transcriptional studies and regulatory interactions between the phoR-phoP operon and the phoU, mtpA, and ppk genes of Streptomyces lividans TK24. J Bacteriol. 2006;188:677–686. doi: 10.1128/JB.188.2.677-686.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rao NN, Kornberg A. Inorganic polyphosphate regulates responses of Escherichia coli to nutritional stringencies, environmental stresses and survival in the stationary phase. Prog Mol Subcell Biol. 1999;23:183–195. doi: 10.1007/978-3-642-58444-2_9. [DOI] [PubMed] [Google Scholar]
- 55.Kulaev I, editor. The biochemistry of inorganic polyphosphates. New York: Wiley; 1979. [DOI] [PubMed] [Google Scholar]
- 56.Kulaev IS, Vagabov VM. Polyphosphate metabolism in micro-organisms. Adv Microb Physiol. 1983;24:83–171. doi: 10.1016/s0065-2911(08)60385-9. [DOI] [PubMed] [Google Scholar]
- 57.Kulaev I, Kulakovskaya T. Polyphosphate and phosphate pump. Annu Rev Microbiol. 2000;54:709–734. doi: 10.1146/annurev.micro.54.1.709. [DOI] [PubMed] [Google Scholar]
- 58.Kulaev I, Vagabov V, Kulakovskaya T. New aspects of inorganic polyphosphate metabolism and function. J Biosci Bioeng. 1999;88:111–129. doi: 10.1016/s1389-1723(99)80189-3. [DOI] [PubMed] [Google Scholar]
- 59.Thomas MR, O’Shea EK. An intracellular phosphate buffer filters transient fluctuations in extracellular phosphate levels. Proc Natl Acad Sci U S A. 2005;102:9565–9570. doi: 10.1073/pnas.0501122102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Docampo R, de Souza W, Miranda K, Rohloff P, Moreno SN. Acidocalcisomes - conserved from bacteria to man. Nat Rev Microbiol. 2005;3:251–261. doi: 10.1038/nrmicro1097. [DOI] [PubMed] [Google Scholar]
- 61.Kumble KD, Kornberg A. Inorganic polyphosphate in mammalian cells and tissues. J Biol Chem. 1995;270:5818–5822. doi: 10.1074/jbc.270.11.5818. [DOI] [PubMed] [Google Scholar]
- 62.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–262. doi: 10.1016/s0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 63.Tang H, Hornstein E, Stolovich M, et al. Amino acid-induced translation of TOP mRNAs is fully dependent on phosphatidylinositol 3-kinase-mediated signaling, is partially inhibited by rapamycin, and is independent of S6K1 and rpS6 phosphorylation. Mol Cell Biol. 2001;21:8671–8683. doi: 10.1128/MCB.21.24.8671-8683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jacinto E, Hall MN. Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol. 2003;4:117–126. doi: 10.1038/nrm1018. [DOI] [PubMed] [Google Scholar]
- 65.Martin KA, Blenis J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv Cancer Res. 2002;86:1–39. doi: 10.1016/s0065-230x(02)86001-8. [DOI] [PubMed] [Google Scholar]
- 66.Shiba T, Nishimura D, Kawazoe Y, et al. Modulation of mitogenic activity of fibroblast growth factors by inorganic polyphosphate. J Biol Chem. 2003;278:26788–26792. doi: 10.1074/jbc.M303468200. [DOI] [PubMed] [Google Scholar]
- 67.Hernandez-Ruiz L, Gonzalez-Garcia I, Castro C, Brieva JA, Ruiz FA. Inorganic polyphosphate and specific induction of apoptosis in human plasma cells. Haematologica. 2006;91:1180–1186. [PubMed] [Google Scholar]
- 68.Beck GR., Jr Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem. 2003;90:234–243. doi: 10.1002/jcb.10622. [DOI] [PubMed] [Google Scholar]
- 69.Schoeters GE, de Saint-Georges L, Van den Heuvel R, Vanderborght O. Mineralization of adult mouse bone marrow in vitro. Cell Tissue Kinet. 1988;21:363–374. doi: 10.1111/j.1365-2184.1988.tb00794.x. [DOI] [PubMed] [Google Scholar]
- 70.Xie J, Baumann MJ, McCabe LR. Osteoblasts respond to hydroxyapatite surfaces with immediate changes in gene expression. J Biomed Mater Res A. 2004;71:108–117. doi: 10.1002/jbm.a.30140. [DOI] [PubMed] [Google Scholar]
- 71.Quarles LD, Yohay DA, Lever LW, Caton R, Wenstrup RJ. Distinct proliferative and differentiated stages of murine MC3T3-E1 cells in culture: an in vitro model of osteoblast development. J Bone Miner Res. 1992;7:683–692. doi: 10.1002/jbmr.5650070613. [DOI] [PubMed] [Google Scholar]
- 72.Otsuka E, Yamaguchi A, Hirose S, Hagiwara H. Characterization of osteoblastic differentiation of stromal cell line ST2 that is induced by ascorbic acid. Am J Physiol. 1999;277:C132–138. doi: 10.1152/ajpcell.1999.277.1.C132. [DOI] [PubMed] [Google Scholar]
- 73.Berendsen AD, Smit TH, Schoenmaker T, et al. Inorganic phosphate stimulates DMP1 expression in human periodontal ligament fibroblasts embedded in three-dimensional collagen gels. Cells Tissues Organs. 2010;192:116–124. doi: 10.1159/000289585. [DOI] [PubMed] [Google Scholar]
- 74.Beck GR, Jr, Sullivan EC, Moran E, Zerler B. Relationship between alkaline phosphatase levels, osteopontin expression, and mineralization in differentiating MC3T3-E1 osteoblasts. J Cell Biochem. 1998;68:269–280. doi: 10.1002/(sici)1097-4644(19980201)68:2<269::aid-jcb13>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 75.Beck GR, Jr, Zerler B, Moran E. Phosphate is a specific signal for induction of osteopontin gene expression. Proc Natl Acad Sci U S A. 2000;97:8352–8357. doi: 10.1073/pnas.140021997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoshiko Y, Candeliere GA, Maeda N, Aubin JE. Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol. 2007;27:4465–4474. doi: 10.1128/MCB.00104-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang SH, Yu KN, Lee YS, et al. Elevated inorganic phosphate stimulates Akt-ERK1/2-Mnk1 signaling in human lung cells. Am J Respir Cell Mol Biol. 2006;35:528–539. doi: 10.1165/rcmb.2005-0477OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goseki-Sone M, Yamada A, Hamatani R, Mizoi L, Iimura T, Ezawa I. Phosphate depletion enhances bone morphogenetic protein-4 gene expression in a cultured mouse marrow stromal cell line ST2. Biochem Biophys Res Commun. 2002;299:395–399. doi: 10.1016/s0006-291x(02)02646-3. [DOI] [PubMed] [Google Scholar]
- 79.M Kimata, T Michigami, K Tachikawa, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na(+)/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone. 2010 doi: 10.1016/j.bone.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 80.Beck GR, Jr, Knecht N. Osteopontin regulation by inorganic phosphate is ERK1/2-, protein kinase C-, and proteasome-dependent. J Biol Chem. 2003;278:41921–41929. doi: 10.1074/jbc.M304470200. [DOI] [PubMed] [Google Scholar]
- 81.Julien M, Magne D, Masson M, et al. Phosphate stimulates matrix Gla protein expression in chondrocytes through the extracellular signal regulated kinase signaling pathway. Endocrinology. 2007;148:530–537. doi: 10.1210/en.2006-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nair D, Misra RP, Sallis JD, Cheung HS. Phosphocitrate inhibits a basic calcium phosphate and calcium pyrophosphate dihydrate crystal-induced mitogen-activated protein kinase cascade signal transduction pathway. J Biol Chem. 1997;272:18920–18925. doi: 10.1074/jbc.272.30.18920. [DOI] [PubMed] [Google Scholar]
- 83.Yamazaki M, Ozonoa K, Okada T, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem. 2010 doi: 10.1002/jcb.22842. [DOI] [PubMed] [Google Scholar]
- 84.Bergwitz C, DeRobertis C, Chen H, Friedman A, Jüppner H, Perrimon N. Role of Sodium-Phosphate Co-Transporters in the Activation of the MAPK Pathway by Phosphate. JASN. 2009 abstract F-FC258. [Google Scholar]
- 85.Mansfield K, Teixeira CC, Adams CS, Shapiro IM. Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone. 2001;28:1–8. doi: 10.1016/s8756-3282(00)00409-9. [DOI] [PubMed] [Google Scholar]
- 86.Beck L, Leroy C, Salaun C, Margall-Ducos G, Desdouets C, Friedlander G. Identification of a novel function of PiT1 critical for cell proliferation and independent of its phosphate transport activity. J Biol Chem. 2009;284:31363–31374. doi: 10.1074/jbc.M109.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Julien M, Khoshniat S, Lacreusette A, et al. Phosphate-dependent regulation of MGP in osteoblasts: role of ERK1/2 and Fra-1. J Bone Miner Res. 2009;24:1856–1868. doi: 10.1359/jbmr.090508. [DOI] [PubMed] [Google Scholar]
- 88.Meng Z, Camalier CE, Lucas DA, Veenstra TD, Beck GR, Jr, Conrads TP. Probing early growth response 1 interacting proteins at the active promoter in osteoblast cells using oligoprecipitation and mass spectrometry. J Proteome Res. 2006;5:1931–1939. doi: 10.1021/pr060009l. [DOI] [PubMed] [Google Scholar]
- 89.Beck GR, Jr, Moran E, Knecht N. Inorganic phosphate regulates multiple genes during osteoblast differentiation, including Nrf2. Exp Cell Res. 2003;288:288–300. doi: 10.1016/s0014-4827(03)00213-1. [DOI] [PubMed] [Google Scholar]
- 90.Conrads KA, Yu LR, Lucas DA, et al. Quantitative proteomic analysis of inorganic phosphate-induced murine MC3T3-E1 osteoblast cells. Electrophoresis. 2004;25:1342–1352. doi: 10.1002/elps.200405892. [DOI] [PubMed] [Google Scholar]
- 91.Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A. 2005;102:9637–9642. doi: 10.1073/pnas.0502249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miedlich SU, Zalutskaya A, Zhu ED, Demay MB. Phosphate-induced apoptosis of hypertrophic chondrocytes is associated with a decrease in mitochondrial membrane potential and is dependent upon Erk1/2 phosphorylation. J Biol Chem. 2010;285:18270–18275. doi: 10.1074/jbc.M109.098616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oshima Y, Akiyama T, Hikita A, et al. Pivotal role of Bcl-2 family proteins in the regulation of chondrocyte apoptosis. J Biol Chem. 2008;283:26499–26508. doi: 10.1074/jbc.M800933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Magne D, Bluteau G, Faucheux C, et al. Phosphate is a specific signal for ATDC5 chondrocyte maturation and apoptosis-associated mineralization: possible implication of apoptosis in the regulation of endochondral ossification. J Bone Miner Res. 2003;18:1430–1442. doi: 10.1359/jbmr.2003.18.8.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. 2002;143:3691–3694. doi: 10.1210/en.2002-220454. [DOI] [PubMed] [Google Scholar]
- 96.Jüppner H, Gardella TJ, Brown EM, Kronenberg HM, Potts JT., Jr . Parathyroid Hormone and Parathyroid Hormone-Related Peptide in the Regulation of Calcium Homeostasis and Bone Development. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Philadelphia: Elsevier Saunders; 2006. pp. 1377–1417. [Google Scholar]
- 97.Potts JT, Jr, Jüppner H. Parathyroid Hormone and parathyroid hormone-related peptide in calcium homeostasis, bone metabolism, and bone development: the proteins, their genes, and receptors. In: Avioli L, Krane S, editors. Metabolic Bone Disease. New York: Academic Press; 1997. pp. 51–94. [Google Scholar]
- 98.White KE, Larsson TE, Econs MJ. The roles of specific genes implicated as circulating factors involved in normal and disordered phosphate homeostasis: frizzled related protein-4, matrix extracellular phosphoglycoprotein, and fibroblast growth factor 23. Endocr Rev. 2006;27:221–241. doi: 10.1210/er.2005-0019. [DOI] [PubMed] [Google Scholar]
- 99.Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–3828. doi: 10.1172/JCI36479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 101.Miyamoto KI, Ito M, Tatsumi S, Kuwahata M, Segawa H. New Aspect of Renal Phosphate Reabsorption: The Type IIc Sodium-Dependent Phosphate Transporter. Am J Nephrol. 2007;27:503–515. doi: 10.1159/000107069. [DOI] [PubMed] [Google Scholar]
- 102.Forster IC, Hernando N, Biber J, Murer H. Proximal tubular handling of phosphate: A molecular perspective. Kidney Int. 2006;70:1548–1559. doi: 10.1038/sj.ki.5001813. [DOI] [PubMed] [Google Scholar]
- 103.Berndt T, Thomas LF, Craig TA, et al. Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A. 2007;104:11085–11090. doi: 10.1073/pnas.0704446104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stubbs J, Liu S, Quarles LD. Role of fibroblast growth factor 23 in phosphate homeostasis and pathogenesis of disordered mineral metabolism in chronic kidney disease. Semin Dial. 2007;20:302–308. doi: 10.1111/j.1525-139X.2007.00308.x. [DOI] [PubMed] [Google Scholar]
- 105.Segawa H, Onitsuka A, Kuwahata M, et al. Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol. 2009;20:104–113. doi: 10.1681/ASN.2008020177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilz DR, Gray RW, Dominguez JH, Lemann J., Jr Plasma 1,25-(OH)2-vitamin D concentrations and net intestinal calcium, phosphate, and magnesium absorption in humans. Am J Clin Nutr. 1979;32:2052–2060. doi: 10.1093/ajcn/32.10.2052. [DOI] [PubMed] [Google Scholar]
- 107.Nabeshima Y. Discovery of alpha-Klotho unveiled new insights into calcium and phosphate homeostasis. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:125–141. doi: 10.2183/pjab/85.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Strom TM, Jüppner H. PHEX, FGF23, DMP1 and beyond. Curr Opin Nephrol Hypertens. 2008;17:357–362. doi: 10.1097/MNH.0b013e3282fd6e5b. [DOI] [PubMed] [Google Scholar]
- 109.Kuro-o M. Endocrine FGFs and Klothos: emerging concepts. Trends Endocrinol Metab. 2008;19:239–245. doi: 10.1016/j.tem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 110.Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94:26–34. doi: 10.1210/jc.2008-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Silver J, Levi R. Regulation of PTH synthesis and secretion relevant to the management of secondary hyperparathyroidism in chronic kidney disease. Kidney Int Suppl. 2005:S8–12. doi: 10.1111/j.1523-1755.2005.09501.x. [DOI] [PubMed] [Google Scholar]
- 112.Gardella TJ. Mimetic Ligands for the PTHR1: Approaches Developments, and Considerations. IBMS BoneKey; 2009. pp. 71–85. [Google Scholar]
- 113.Kronenberg HM. PTH regulates the hematopoietic stem cell niche in bone. Adv Exp Med Biol. 2007;602:57–60. doi: 10.1007/978-0-387-72009-8_7. [DOI] [PubMed] [Google Scholar]
- 114.Berndt T, Kumar R. Novel mechanisms in the regulation of phosphorus homeostasis. Physiology (Bethesda) 2009;24:17–25. doi: 10.1152/physiol.00034.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu Rev Med. 2010;61:91–104. doi: 10.1146/annurev.med.051308.111339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kemper B, Habener JF, Rich A, Potts JT., Jr Parathyroid secretion: discovery of a major calcium-dependent protein. Science. 1974;184:167–169. doi: 10.1126/science.184.4133.167. [DOI] [PubMed] [Google Scholar]
- 117.Slatopolsky E, Finch J, Denda M, et al. Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion in vitro. J Clin Invest. 1996;97:2534–2540. doi: 10.1172/JCI118701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sherwood LM, Mayer GP, Ramberg CF, Jr, Kronfeld DS, Aurbach GD, Potts JT., Jr Regulation of parathyroid hormone secretion: proportional control by calcium, lack of effect of phosphate. Endocrinology. 1968;83:1043–1051. doi: 10.1210/endo-83-5-1043. [DOI] [PubMed] [Google Scholar]
- 119.Almaden Y, Canalejo A, Hernandez A, et al. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J Bone Miner Res. 1996:970–976. doi: 10.1002/jbmr.5650110714. [DOI] [PubMed] [Google Scholar]
- 120.Silver J, Russell J, Sherwood LM. Regulation by vitamin D metabolites of messenger ribronucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci USA. 1985;82:4270–4273. doi: 10.1073/pnas.82.12.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Estepa J, Aguilera-Tejero E, Lopez I, Almaden Y, Rodriguez M, Felsenfeld A. Effect of Phosphate on Parathyroid Hormone Secretion In Vivo. J Bone Miner Res. 1999;14:1848–1854. doi: 10.1359/jbmr.1999.14.11.1848. [DOI] [PubMed] [Google Scholar]
- 122.Moallem E, Kilav R, Silver J, Naveh-Many T. RNA-Protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J Biol Chem. 1998;273:5253–5259. doi: 10.1074/jbc.273.9.5253. [DOI] [PubMed] [Google Scholar]
- 123.Naveh-Many T, Rahaminov R, Livini N, Silver J. Parathyroid cell proliferation in normal and chronic renal failure in rats. The effects of calcium, phosphate, and vitamin D. J Clin Invest. 1995;96:1786–1793. doi: 10.1172/JCI118224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Galitzer H, Ben-Dov I, Lavi-Moshayoff V, Naveh-Many T, Silver J. Fibroblast growth factor 23 acts on the parathyroid to decrease parathyroid hormone secretion. Curr Opin Nephrol Hypertens. 2008;17:363–367. doi: 10.1097/MNH.0b013e328303e172. [DOI] [PubMed] [Google Scholar]
- 125.Lavi-Moshayoff V, Silver J, Naveh-Many T. Human PTH gene regulation in vivo using transgenic mice. Am J Physiol Renal Physiol. 2009;297:F713–719. doi: 10.1152/ajprenal.00161.2009. [DOI] [PubMed] [Google Scholar]
- 126.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Krajisnik T, Bjorklund P, Marsell R, et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J Endocrinol. 2007;195:125–131. doi: 10.1677/JOE-07-0267. [DOI] [PubMed] [Google Scholar]
- 128.Imura A, Tsuji Y, Murata M, et al. alpha-Klotho as a regulator of calcium homeostasis. Science. 2007;316:1615–1618. doi: 10.1126/science.1135901. [DOI] [PubMed] [Google Scholar]
- 129.Kilav R, Silver J, Naveh-Many T. Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest. 1995;96:327–333. doi: 10.1172/JCI118038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dinur M, Kilav R, Sela-Brown A, Jacquemin-Sablon H, Naveh-Many T. In vitro evidence that upstream of N-ras participates in the regulation of parathyroid hormone messenger ribonucleic acid stability. Mol Endocrinol. 2006;20:1652–1660. doi: 10.1210/me.2005-0333. [DOI] [PubMed] [Google Scholar]
- 131.Nechama M, Ben-Dov IZ, Briata P, Gherzi R, Naveh-Many T. The mRNA decay promoting factor K-homology splicing regulator protein post-transcriptionally determines parathyroid hormone mRNA levels. Faseb J. 2008;22:3458–3468. doi: 10.1096/fj.08-107250. [DOI] [PubMed] [Google Scholar]
- 132.Nechama M, Uchida T, Mor Yosef-Levi I, Silver J, Naveh-Many T. The peptidyl-prolyl isomerase Pin1 determines parathyroid hormone mRNA levels and stability in rat models of secondary hyperparathyroidism. J Clin Invest. 2009;119:3102–3114. doi: 10.1172/JCI39522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bikle D, Adams J, Christakos C. Vitamin D: Production, Metabolism, Mechanism of Action, and Clinical Requirements: Primer on Metabolic Bone Disease and Disorders of Mineral Metabolism. Philadelphia, PA: Lippincott-Raven; 2008. pp. 141–149. [Google Scholar]
- 134.Boullion R. Vitamin D: From photosynthesis, metabolism, and action to clinical application. In: DeGroot LJ, Jameson JL, editors. Endocrinology. Philadelphia: Elsevier Saunders; 2006. pp. 1435–1463. [Google Scholar]
- 135.Sakaki T, Kagawa N, Yamamoto K, Inouye K. Metabolism of vitamin D3 by cytochromes P450. Front Biosci. 2005;10:119–134. doi: 10.2741/1514. [DOI] [PubMed] [Google Scholar]
- 136.Bai XY, Miao D, Goltzman D, Karaplis AC. The autosomal dominant hypophosphatemic rickets R176Q mutation in fibroblast growth factor 23 resists proteolytic cleavage and enhances in vivo biological potency. J Biol Chem. 2003;278:9843–9849. doi: 10.1074/jbc.M210490200. [DOI] [PubMed] [Google Scholar]
- 137.Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. Fibroblast growth factor 7: an inhibitor of phosphate transport derived from oncogenic osteomalacia-causing tumors. J Clin Endocrinol Metab. 2005;90:1012–1020. doi: 10.1210/jc.2004-0357. [DOI] [PubMed] [Google Scholar]
- 138.Berndt T, Craig T, Bowe A, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest. 2003;112:785–794. doi: 10.1172/JCI18563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.DeLuca HF. The metabolism and functions of vitamin D. Adv Exp Med Biol. 1986;196:361–375. doi: 10.1007/978-1-4684-5101-6_24. [DOI] [PubMed] [Google Scholar]
- 140.Inoue Y, Segawa H, Kaneko I, et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem J. 2005;390:325–331. doi: 10.1042/BJ20041799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun. 2000;277:494–498. doi: 10.1006/bbrc.2000.3696. [DOI] [PubMed] [Google Scholar]
- 142.Bastepe M, Jüppner H. Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord. 2008;9:171–180. doi: 10.1007/s11154-008-9075-3. [DOI] [PubMed] [Google Scholar]
- 143.von Marschall Z, Fisher LW. Dentin matrix protein-1 isoforms promote differential cell attachment and migration. J Biol Chem. 2008;283:32730–32740. doi: 10.1074/jbc.M804283200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yuan B, Meudt J, Feng JO, Drezner MK. 7B2 protein mediated inhibition of DMP1 cleavage in osteoblasts enhances FGF-23 production in hyp-mice. JBMR. 2008;23:s16. abstract 1053. [Google Scholar]
- 145.Lu Y, Qin C, Xie Y, Bonewald LF, Feng JQ. Studies of the DMP1 57-kDa functional domain both in vivo and in vitro. Cells Tissues Organs. 2009;189:175–185. doi: 10.1159/000151727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lorenz-Depiereux B, Bastepe M, Benet-Pages A, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250. doi: 10.1038/ng1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Terkeltaub R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2006;2:371–377. doi: 10.1007/s11302-005-5304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rutsch F, Vaingankar S, Johnson K, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158:543–554. doi: 10.1016/S0002-9440(10)63996-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 150.Levy-Litan V, Hershkovitz E, Avizov L, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86:273–278. doi: 10.1016/j.ajhg.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86:267–272. doi: 10.1016/j.ajhg.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Chodirker BN, Evans JA, Seargeant LE, Cheang MS, Greenberg CR. Hyperphosphatemia in infantile hypophosphatasia: implications for carrier diagnosis and screening. Am J Hum Genet. 1990;46:280–285. [PMC free article] [PubMed] [Google Scholar]
- 153.Burnett SM, Gunawardene SC, Bringhurst FR, Jüppner H, Lee H, Finkelstein JS. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–1196. doi: 10.1359/jbmr.060507. [DOI] [PubMed] [Google Scholar]
- 154.Nishi H, Nii-Kono T, Nakanishi S, et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism. Nephron Clin Pract. 2005;101:c94–99. doi: 10.1159/000086347. [DOI] [PubMed] [Google Scholar]
- 155.Kolek OI, Hines ER, Jones MD, et al. 1alpha, 25-Dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1036–1042. doi: 10.1152/ajpgi.00243.2005. [DOI] [PubMed] [Google Scholar]
- 156.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 157.Yu X, Sabbagh Y, Davis SI, Demay MB, White KE. Genetic dissection of phosphate- and vitamin D-mediated regulation of circulating Fgf23 concentrations. Bone. 2005;36:971–977. doi: 10.1016/j.bone.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 158.Frishberg Y, Ito N, Rinat C, et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22:235–242. doi: 10.1359/jbmr.061105. [DOI] [PubMed] [Google Scholar]
- 159.Chefetz I, Kohno K, Izumi H, Uitto J, Richard G, Sprecher E. GALNT3, a gene associated with hyperphosphatemic familial tumoral calcinosis, is transcriptionally regulated by extracellular phosphate and modulates matrix metalloproteinase activity. Biochim Biophys Acta. 2009;1792:61–67. doi: 10.1016/j.bbadis.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bergwitz C, Banerjee S, Abu-Zahra H, et al. Defective O-glycosylation due to a novel homozygous S129P mutation is associated with lack of fibroblast growth factor 23 secretion and tumoral calcinosis. J Clin Endocrinol Metab. 2009;94:4267–4274. doi: 10.1210/jc.2009-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Larsson T, Davis SI, Garringer HJ, et al. Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology. 2005;146:3883–3891. doi: 10.1210/en.2005-0431. [DOI] [PubMed] [Google Scholar]
- 162.Ichikawa S, Sorenson AH, Austin AM, et al. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology. 2009;150:2543–2550. doi: 10.1210/en.2008-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bhattacharyya N, Andreopoulou P, Dumitrescu C, Miwa H, Gafni R, Collins M. Cyclic AMP-mediated processing of FGF23: Regulation by O-glycosylation. JBMR. 2009 abstract A09003028. [Google Scholar]
- 164.Riminucci M, Collins MT, Fedarko NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]