

Abstract
Genomic integrity is maintained via the concerted action of proteins that coordinate and control DNA replication and those that respond to DNA damage. The Mre11 complex is a mediator of the DNA damage response through its functions in DNA double strand break (DSB) sensing, checkpoint activation and recombinational DNA repair. The complex responds to mitotic and meiotic DSBs, and is also activated in cells experiencing DNA replication stress. The Mre11 complex’s role in recombinational repair primarily concerns the promotion of homologous recombination (HR), but it is also implicated in non-homologous end joining (NHEJ)—a DSB repair mechanism prevalent in non-dividing cells. We recently characterized deletion of the Mre11 complex member, Rad50, in a number of postmitotic and proliferative tissues of the mouse. These studies indicated that the complex is dispensable in postmitotic tissues, but loss of Rad50 in proliferating cells resulted in accumulation of unrepaired, DNA replication-dependent lesions. The data suggest that the Mre11 complex is not a major contributor to NHEJ and support the interpretation that its role in recombinational DNA repair is largely restricted to dividing cells, in which repair involving sister chromatids predominates. An exception to this concept is manifest in previous work from our laboratory revealing that the mammalian Mre11 complex promotes meiotic DSB repair, an event involving recombination between sister chromatids of homologous chromosomes and taking place in cells not undergoing replication. Together these studies highlight the importance of cell cycle and cell type specific modulation of the Mre11 complex’s repair activities in vivo.
Keywords: Mre11, Rad50, Nbs1, DSB repair, postmitotic, meiosis, cell cycle, telomere
Introduction
DNA double strand breaks are potentially genotoxic lesion that can result in mutation or translocation if left unchecked or repaired improperly. The ability to sense and respond to DNA DSBs is therefore a principal means of preserving genomic integrity. The Mre11 complex, composed of Mre11, Rad50 and Nbs1, is a key component of the cellular response to DNA double strand breaks. Mutations in these genes result in impaired cell cycle checkpoint activation, DNA repair and apoptosis in response to DSB inducing agents (reviewed in ref. 1).
DNA double strand breaks can be repaired via homologous recombination or non-homologous end joining. Defects in these repair pathways result in significant pathology including immunodeficiency, infertility, impaired motor coordination, tumor predisposition, premature aging and inviability2–4 (reviewed in refs. 1 and 5). The choice of HR versus NHEJ in DSB repair appears to be primarily determined by cell cycle status and developmental cues; nevertheless whereas HR is essential for viability of proliferating cells and embryonic development, it has been unclear whether HR is required in postmitotic cells.
The Mre11 complex is implicated in both HR and NHEJ. Efforts to understand the Mre11 complex’s role in these processes in mammalian systems have been hampered by the fact that it is required for embryonic viability.6,7 To address this, we have analyzed a number of proliferative and postmitotic tissues of the mouse in which a conditional allele of Rad50 was deleted post-embryonically.8 Additionally, meiotic DSB repair was previously characterized in mice carrying hypomorphic alleles of Mre11 and Nbs1.9 These studies have illuminated the Mre11 complex’s functions in response to endogenous damage and have implications with respect to its roles in HR and NHEJ.
The Mre11 Complex and Somatic Cells: Compartmentalization of Repair
The Mre11 complex is implicated in both HR and NHEJ in S. cerevisiae (reviewed in ref. 10). While the complex functions in HR in vertebrates, studies suggest that it is not required for NHEJ in these organisms. For example, deletion of Mre11 or Nbs1 in chicken DT40 cells results in reduced HR and sister chromatid exchange but does not affect end joining frequencies in plasmid based assays; and unlike G1 irradiated Ku70 deficient cells (which lack a vital component of the end joining machinery), viability of G1 irradiated Mre11 deficient cells is not compromised.11,12 Additionally, data from Nbs1 deficient mouse cells suggests that the Mre11 complex may play an inhibitory role in NHEJ in mammals.13
To study the effects of Mre11 complex deficiency in vivo, we generated a conditionally-null allele of Rad50 that can be deleted via introduction of a tissue-specific Cre transgene.8 We used these mice to examine the role of the Mre11 complex in repair in postmitotic cells, thus addressing its potential involvement in NHEJ. To circumvent the confounding issue of whether Mre11 complex deficiency in proliferating precursor cells account for phenotypic outcomes manifest later in development in postmitotic compartments, we chose the Purkinje cell-specific Pcp2-Cre and the interferon-inducible Mx-Cre lines. These lines allow for deletion in postmitotic Purkinje cells of the cerebellum14 or in quiescent hepatocytes of adult mice.15
In contrast to loss of Rad50 from proliferating cells (see Fig. 1), deletion of Rad50 in nonproliferative settings had no apparent effect at either the cellular or organismal level. Since loss of bona fide NHEJ components results in growth defects, elevated levels of apoptosis, and inviability in some cases, with particularly profound effects observed in neuronal compartments,2–4 our data support the interpretation that the Mre11 complex does not play an important role in NHEJ in vertebrates.
Figure 1.
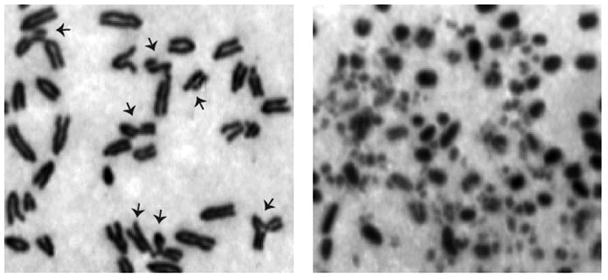
Genomic instability in Rad50 deficient cells. Deletion of Rad50 from proliferating tissue culture cells leads to the appearance of spontaneous chromosomal aberrations (arrows, left) visible in metaphase spread preparations. Aberrations include chromatid breaks, gaps, chromosome breaks, fusions and exchanges. In extreme cases, several cell division cycles in the absence of Rad50 results in severe chromosome fragmentation (right).
The fact that Rad50 is dispensable for maintenance of post-mitotic cells also suggests that HR is not essential in postmitotic settings. Conversely, the strikingly high level of DNA damage observed in Rad50-deficient hepatocytes upon S-phase entry brightly underscores the critical importance of the Mre11 complex, and by extension HR to successful DNA replication. The re-entry of quiescent hepatocytes into the cell cycle upon stimulation via partial hepatectomy (PH) has been shown to be a precisely timed event. In mice, cell proliferation is relatively synchronized for the first wave of replication following PH; this wave is initiated between 16 and 24 hours, and peaks at approximately 36 hours post-PH.16 Coincident with this time frame, DNA damage foci were observed in Rad50 deficient livers beginning at 24 hours post-PH and rose to peak levels (between 70 and 90% γ-H2AX positive hepatocytes) at 72 hours. This result is reflective of the proportion of hepatocytes in adult mice that proliferate following two-thirds PH (77–93%).17 Since the frequency of spontaneous γ-H2AX foci in quiescent Rad50 null hepatocytes was comparable to controls (<5% in both cases), these data demonstrate that damage arising in Mre11 complex deficient cells is dependent on passage through S-phase.
Our data indicating that Rad50 is required for genome maintenance during replication are in agreement with additional observations linking the Mre11 complex with events in S-phase. Localization of the complex to sites of replication fork stalling, its role in activation of the intra-S phase checkpoint in response to DNA replication stress, and engagement of the complex with chromatin during normal S-phase indicate Mre11 complex functions are intimately related to molecular transactions attendant to DNA replication.18–21 The Mre11 complex’s role in promotion of sister chromatid mediated HR in S. cerevisiae22,23 may account for the observed dynamics of the complex during S-phase and the phenotypic outcomes in its absence. These data, together with the observation that proliferating cells appear to require the Mre11 complex for viability6–8,24,25 support the conclusion that its activities are a vital component of the DNA damage response to spontaneous lesions arising during DNA replication.
In light of the replication specific repair functions of the Mre11 complex, the complex’s presence throughout the cell cycle raises the question of how its activities are restricted to particular phases. This may be explained in part by the cell cycle stage specificity of HR and NHEJ. Studies in S. cerevisiae indicate that HR mediated repair of DSBs and concomitant dampening of NHEJ correlate with increased cyclin dependent kinase (CDK) activity in S and G2 phase cells. The switch between NHEJ and HR appears to be controlled by the ability of cell cycle specific CDK activity to promote DSB end resection,26,27 one of the first steps required for HR mediated repair. These data are corroborated by studies in mouse cells demonstrating NHEJ mediated fusion of dysfunctional telomeres is restricted to G1 and is dependent on low levels of CDK activity.28
The mammalian CtIP protein and its yeast orthologs, Sae2 (S. cerevisiae) and Ctp1 (S. pombe), are directly implicated in mediating the cell cycle specific HR activities of the Mre11 complex during DSB repair.29–31 Like the Mre11 complex, DSB end resection and HR are disrupted in CtIP/Ctp1/Sae2 deficient cells. Expression of Ctp1 oscillates during the cell cycle whereas Sae2 activity is controlled by CDK mediated phosphorylation, indicating two potential mechanisms of Mre11 complex modulation during the cell cycle.
Although the Mre11 complex exhibits exonuclease activity, and both Sae2 and Mre11 exhibit endonuclease activity,31,32 their role in DNA double strand break end resection remained ambiguous until recently. Two separate studies revealed that Sae2 and the Mre11 complex cooperate during initial processing of DSBs to produce short 3′ overhangs.33,34 These overhangs are then resected processively via apparently redundant pathways involving Sgs1 and Exo1. In the absence of Sgs1 and Exo1, resection is severely impaired indicating that the Mre11 complex does not function efficiently in long-range processing of breaks.
The structural characteristics of the Mre11 complex also appear to be a key determinant of its function in HR mediated repair of DSBs. The complex consists of a globular domain, comprising its DNA binding and nucleolytic domains, and an extended coiled-coil region that facilitates intermolecular dimerization of the complex.35,36 This structure allows for bridging of double stranded DNA ends.37,38 Disruption of intermolecular dimerization results in a phenotype that is essentially indistinguishable from null mutants,39 suggesting that the Mre11 complex’s function in repair is largely related to its ability to tether DSB ends. Whether this structural arrangement may also potentiate the DNA end processing activity of the complex remains an intriguing question.
The Mre11 Complex in Meiotic DSB Repair: Life after S-Phase
Homologous recombinational DSB repair is utilized exclusively during the specialized prophase preceding meiotic cell division. Repair of meiotic breaks is carried out by a combination of meiosis-specific damage response factors and accessory proteins and canonical DSB repair proteins that also function in mitotic cells (reviewed in refs. 40 and 41). In contrast to mitotic cells where sister chromatids are the preferred substrate for HR, meiotic DSBs are repaired via HR between maternal and paternal homologs. This process facilitates homolog alignment and crossover formation, and ultimately allows for segregation of homologs during the first meiotic division.
The Mre11 complex is implicated in meiotic recombination in all species studied to date. The complex is required for double strand break formation, processing and checkpoint signaling in S. cerevisiae, whereas in A. thaliana and S. pombe the complex is dispensable for break formation but required for DSB processing and/or repair (reviewed in ref. 42). In mammals, the complex is associated with meiotic chromatin and it relocalizes to the XY body during late prophase in males43—a specialized structure required for XY silencing that is enriched in DSB response factors.
Further studies of the complex in mammalian meiosis were once again hindered by the fact that the Mre11 complex is required for embryonic viability. However, viable mouse models of hypomorphic mutations of Mre11 complex members found in human disease had been previously developed in our laboratory,44,45 allowing us to examine meiocytes in which Mre11 complex function was perturbed. These studies revealed that the complex is required for proper synapsis between homologous chromosomes, timely resolution of repair intermediates, and formation of appropriate crossover numbers and position.9 These results indicate that the Mre11 complex is dispensable for break formation in mice but like its counterparts in A. thaliana and S. pombe, plays an early role in meiotic repair in mammals. Moreover, perturbation of crossover numbers and position suggest that deviations in repair efficiency can influence crossover control.
A role for the Mre11 complex in meiotic repair is not surprising given its function in HR in mitotic cells. Nevertheless, in light of data indicating that the complex largely promotes HR between sister chromatids in mitotic cells, an important question that remains to be addressed is how these functions are restricted or redirected during meiotic repair. As in mitotic cells, it appears that regulation of meiotic HR and the Mre11 complex may be achieved in part by the activity of CDK or meiosis-specific kinases. For example, induction of meiotic DSBs in S. cerevisiae is controlled by CDK activity.46,47 Additionally, the meiosis specific Mek1 kinase is required to promote interhomolog mediated repair of DSBs, presumably via inhibition of sister chromatid repair mediated by the Mek1 target, Red1.48 Whether the Mre11 complex is similarly modulated is still unclear. The Mre11 complex interacting partner, Sae2 however, also functions in meiotic DSB formation and processing.49,50 Given that Sae2 is a target of CDK activity in mitotic cells,51 it is plausible that phosphorylation events mediate its activity, and in turn the Mre11 complex’s activity, in meiotic cells.
In contrast to the situation in postmitotic and quiescent cells, the implication of the Mre11 complex in meiotic DSB repair highlights an important repair function for the complex that is not associated with replication. Aside from lesions generated during the process of replication, genetically programmed DSBs are one of the most prevalent sources of breaks naturally encountered in vivo. The requirement for HR mediated repair of these breaks surely accounts for the Mre11 complex’s involvement in this repair process. This may also be generally reflective of post-replication functions for the complex in G2 cells. Indeed, the complex operates in G2/M checkpoint activation in mitotic cells in response to ionizing radiation.45,52,53 Although spontaneous DSBs would be a rare occurrence in G2 cells, the Mre11 complex’s roles in HR during S-phase and in meiotic cells suggest that its activities may extend beyond checkpoint activation, encompassing repair of breaks during this phase as well.
The End of the Story: the Mre11 Complex and Telomeres
Double strand breaks are not the only DNA ends that must be managed by the cell: the terminal ends of chromosomes also engage the DSB repair machinery. In contrast to a true break however, repair of telomeric ends is normally suppressed. The paradoxical relationship between telomeres and DSB response proteins is not fully understood, but it is clear that the specialized telomere binding proteins recruited to telomeres are essential to suppression of the DNA damage response (reviewed in ref. 54). Erosion or deprotection of telomeres results in activation of the DNA damage response, triggering senescence, apoptosis or induction of fusions—all of which effectively lead to removal of the cell from the gene pool.
The Mre11 complex is implicated in telomere maintenance. In S. cerevisiae the complex is required for telomere length homeostasis;55,56 its ability to recruit telomerase (the enzyme responsible for telomeric repeat synthesis) to telomere ends, likely accounts for this.57 In mammalian cells however, the Mre11 complex associates with the telomere binding protein Trf2.58 Trf2 deletion results in telomere deprotection followed by end fusions,59 raising the possibility that the Mre11 complex plays a similar role in telomere protection.
To address the question of whether the Mre11 complex functions in telomere protection, telomeres were examined in cultured Rad50 deficient cells.8 We assessed telomere fusions, erosion and telomere dysfunction induced foci formation, all possible endpoints of telomere deprotection. In contrast to results in Trf2 deficient cells, we observed no evidence of telomere dysfunction upon acute loss of Rad50. These findings are consistent with the lack of telomere-related phenotypes in human and mouse cells carrying hypomorphic mutations of Mre11 complex members. Ongoing studies in telomerase deficient mice will address the outcome of Mre11 complex hypomorphism in the context of telomere erosion.
Concluding Remarks
The results of our studies with regard to the roles of the Mre11 complex in the response to meiotic DSBs and spontaneous damage in postmitotic and replicating cells in vivo have illustrated the advantages of examining the DNA damage response in the organismal context. These studies allowed us to access compartments that are typically not amenable to ex vivo analyses, including meiotic and postmitotic cells. In conjunction with previously published work, the data helped define the Mre11 complex’s role in mammalian systems to those functions relevant to HR mediated DSB repair, a role that appears to be limited to replicating and possibly G2 cells. This role also extends to the specialized post-replication repair carried out in meiotic cells. Defining the precise functions of the Mre11 complex in repair and telomere maintenance remains an important challenge facing the field in the future.
References
- 1.Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair. 2004;3:845–54. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 2.Orii KE, Lee Y, Kondo N, McKinnon PJ. Selective utilization of nonhomologous end-joining and homologous recombination DNA repair pathways during nervous system development. Proc Natl Acad Sci USA. 2006;103:10017–22. doi: 10.1073/pnas.0602436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993–1002. doi: 10.1016/s1097-2765(00)80264-6. [DOI] [PubMed] [Google Scholar]
- 4.Gu Y, Sekiguchi J, Gao Y, Dikkes P, Frank K, Ferguson D, et al. Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proc Natl Acad Sci USA. 2000;97:2668–73. doi: 10.1073/pnas.97.6.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinnon PJ. ATM and ataxia telangiectasia. EMBO Rep. 2004;5:772–6. doi: 10.1038/sj.embor.7400210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo G, Yao MS, Bender CF, Mills M, Bladl AR, Bradley A, et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development and sensitivity to ionizing radiation. Proc Natl Acad Sci USA. 1999;96:7376–81. doi: 10.1073/pnas.96.13.7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Petersen S, Tessarollo L, Nussenzweig A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Current Biology. 2001;11:105–9. doi: 10.1016/s0960-9822(01)00019-7. [DOI] [PubMed] [Google Scholar]
- 8.Adelman CA, De S, Petrini JH. Rad50 is dispensable for the maintenance and viability of postmitotic tissues. Mol Cell Biol. 2009;29:483–92. doi: 10.1128/MCB.01525-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cherry SM, Adelman CA, Theunissen JW, Hassold TJ, Hunt PA, Petrini JH. The Mre11 complex influences DNA repair, synapsis and crossing over in murine meiosis. Curr Biol. 2007;17:373–8. doi: 10.1016/j.cub.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haber JE. The many interfaces of Mre11. Cell. 1998;95:583–6. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi-Iwai Y, Sonoda E, Sasaki MS, Morrison C, Haraguchi T, Hiraoka Y, et al. Mre11 is essential for the maintenance of chromosomal DNA in vertebrate cells. EMBO J. 1999;18:6619–29. doi: 10.1093/emboj/18.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature. 2002;420:93–8. doi: 10.1038/nature01125. [DOI] [PubMed] [Google Scholar]
- 13.Yang YG, Saidi A, Frappart PO, Min W, Barrucand C, Dumon-Jones V, et al. Conditional deletion of Nbs1 in murine cells reveals its role in branching repair pathways of DNA double-strand breaks. EMBO J. 2006;25:5527–38. doi: 10.1038/sj.emboj.7601411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barski JJ, Dethleffsen K, Meyer M. Cre recombinase expression in cerebellar Purkinje cells. Genesis. 2000;28:93–8. [PubMed] [Google Scholar]
- 15.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–9. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 16.Matsuo T, Yamaguchi S, Mitsui S, Emi A, Shimoda F, Okamura H. Control mechanism of the circadian clock for timing of cell division in vivo. Science. 2003;302:255–9. doi: 10.1126/science.1086271. [DOI] [PubMed] [Google Scholar]
- 17.Fausto N. Liver regeneration: from laboratory to clinic. Liver Transpl. 2001;7:835–44. doi: 10.1053/jlts.2001.27865. [DOI] [PubMed] [Google Scholar]
- 18.Taalman RD, Jaspers NG, Scheres JM, de Wit J, Hustinx TW. Hypersensitivity to ionizing radiation, in vitro, in a new chromosomal breakage disorder, the Nijmegen Breakage Syndrome. Mutat Res. 1983;112:23–32. doi: 10.1016/0167-8817(83)90021-4. [DOI] [PubMed] [Google Scholar]
- 19.Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 20.Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel R, et al. The MRE11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol. 2001;21:6006–16. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirzoeva OK, Petrini JH. DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res. 2003;1:207–18. [PubMed] [Google Scholar]
- 22.Bressan DA, Baxter BK, Petrini JH. The Mre11-Rad50-Xrs2 protein complex facilitates homologous recombination-based double-strand break repair in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:7681–7. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ivanov EL, Korolev VG, Fabre F. XRS2, a DNA repair gene of Saccharomyces cerevisiae, is needed for meiotic recombination. Genetics. 1992;132:651–64. doi: 10.1093/genetics/132.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demuth I, Frappart PO, Hildebrand G, Melchers A, Lobitz S, Stockl L, et al. An inducible null mutant murine model of Nijmegen breakage syndrome proves the essential function of NBS1 in chromosomal stability and cell viability. Hum Mol Genet. 2004;13:2385–97. doi: 10.1093/hmg/ddh278. [DOI] [PubMed] [Google Scholar]
- 25.Reina-San-Martin B, Nussenzweig MC, Nussenzweig A, Difilippantonio S. Genomic instability, endoreduplication and diminished Ig class-switch recombination in B cells lacking Nbs1. Proc Natl Acad Sci USA. 2005;102:1590–5. doi: 10.1073/pnas.0406289102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–7. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004;23:4868–75. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konishi A, de Lange T. Cell cycle control of telomere protection and NHEJ revealed by a ts mutation in the DNA-binding domain of TRF2. Genes Dev. 2008;22:1221–30. doi: 10.1101/gad.1634008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–46. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007;28:638–51. doi: 10.1016/j.molcel.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trujillo KM, Yuan SSF, Lee E, Sung P. Nuclease activities in a complex of human recombination and DNA repair factors rad50, mre11 and p95. J Biol Chem. 1998;273:21447–50. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 33.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–4. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell. 2001;105:473–85. doi: 10.1016/s0092-8674(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 36.Hopfner KP, Craig L, Moncalian G, Zinkel RA, Usui T, Owen BA, et al. The Rad50 zinc-hook is a structure joining Mre11 complexes in DNA recombination and repair. Nature. 2002;418:562–6. doi: 10.1038/nature00922. [DOI] [PubMed] [Google Scholar]
- 37.de Jager M, van Noort J, van Gent DC, Dekker C, Kanaar R, Wyman C. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell. 2001;8:1129–35. doi: 10.1016/s1097-2765(01)00381-1. [DOI] [PubMed] [Google Scholar]
- 38.Moreno-Herrero F, de Jager M, Dekker NH, Kanaar R, Wyman C, Dekker C. Mesoscale conformational changes in the DNA-repair complex Rad50/Mre11/Nbs1 upon binding DNA. Nature. 2005;437:440–3. doi: 10.1038/nature03927. [DOI] [PubMed] [Google Scholar]
- 39.Wiltzius JJ, Hohl M, Fleming JC, Petrini JH. The Rad50 hook domain is a critical determinant of Mre11 complex functions. Nat Struct Mol Biol. 2005;12:403–7. doi: 10.1038/nsmb928. [DOI] [PubMed] [Google Scholar]
- 40.Richardson C, Horikoshi N, Pandita TK. The role of the DNA double-strand break response network in meiosis. DNA Repair (Amst) 2004;3:1149–64. doi: 10.1016/j.dnarep.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Zickler D, Kleckner N. Meiotic chromosomes: integrating structure and function. Annu Rev Genet. 1999;33:603–754. doi: 10.1146/annurev.genet.33.1.603. [DOI] [PubMed] [Google Scholar]
- 42.Borde V. The multiple roles of the Mre11 complex for meiotic recombination. Chromosome Res. 2007;15:551–63. doi: 10.1007/s10577-007-1147-9. [DOI] [PubMed] [Google Scholar]
- 43.Goedecke W, Eijpe M, Offenberg HH, van Aalderen M, Heyting C. Mre11 and Ku70 interact in somatic cells, but are differentially expressed in early meiosis. Nat Genet. 1999;23:194–8. doi: 10.1038/13821. [DOI] [PubMed] [Google Scholar]
- 44.Williams BR, Mirzoeva OK, Morgan WF, Lin J, Dunnick W, Petrini JH. A murine model of nijmegen breakage syndrome. Curr Biol. 2002;12:648–53. doi: 10.1016/s0960-9822(02)00763-7. [DOI] [PubMed] [Google Scholar]
- 45.Theunissen JW, Kaplan MI, Hunt PA, Williams BR, Ferguson DO, Alt FW, et al. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol Cell. 2003;12:1511–23. doi: 10.1016/s1097-2765(03)00455-6. [DOI] [PubMed] [Google Scholar]
- 46.Henderson KA, Kee K, Maleki S, Santini PA, Keeney S. Cyclin-dependent kinase directly regulates initiation of meiotic recombination. Cell. 2006;125:1321–32. doi: 10.1016/j.cell.2006.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wan L, Niu H, Futcher B, Zhang C, Shokat KM, Boulton SJ, et al. Cdc28-Clb5 (CDK-S) and Cdc7-Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev. 2008;22:386–97. doi: 10.1101/gad.1626408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wan L, de los Santos T, Zhang C, Shokat K, Hollingsworth NM. Mek1 kinase activity functions downstream of RED1 in the regulation of meiotic double strand break repair in budding yeast. Mol Biol Cell. 2004;15:11–23. doi: 10.1091/mbc.E03-07-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keeney S, Kleckner N. Covalent protein-DNA complexes at the 5′ strand termini of meiosis-specific double-strand breaks in yeast. Proc Natl Acad Sci USA. 1995;92:11274–8. doi: 10.1073/pnas.92.24.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huertas P, Cortes-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature. 2008;455:689–92. doi: 10.1038/nature07215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buscemi G, Savio C, Zannini L, Micciche F, Masnada D, Nakanishi M, et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol Cell Biol. 2001;21:5214–22. doi: 10.1128/MCB.21.15.5214-5222.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. The Mre11 complex is required for ATM activation and the G(2)/M checkpoint. EMBO J. 2003;22:6610–20. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 55.Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–28. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nugent CI, Bosco G, Ross LO, Evans SK, Salinger AP, Moore JK, et al. Telomere maintenance is dependent on activities required for end repair of double-strand breaks. Curr Biol. 1998;8:657–60. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- 57.Goudsouzian LK, Tuzon CT, Zakian VA. S. cerevisiae Tel1p and Mre11p are required for normal levels of Est1p and Est2p telomere association. Mol Cell. 2006;24:603–10. doi: 10.1016/j.molcel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 58.Zhu XD, Kuster B, Mann M, Petrini JH, Lange T. Cell cycle-regulated association of RAD50/MRE11/NBS1 with TRF2 and human telomeres. Nat Genet. 2000;25:347–52. doi: 10.1038/77139. [DOI] [PubMed] [Google Scholar]
- 59.Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–8. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]