

Abstract
Using adoptive transfer techniques, we explored the immune cells implicated in a model of anti-RNP autoimmunity that presents either with pneumonitis or glomerulonephritis. Unfractionated splenocytes from donors without renal disease induced predominantly lung disease (8/14 (57%) lung versus 2/14 (14%) renal, p = 0.046). However, the CD4+ cells taken from these splenocytes induced renal disease more frequently than lung disease (7/10 (70%) renal versus 2/10 (20%) lung, p = 0.01). Adoptive transfer of RNP+ CD4+ T cells from short-term culture gave results similar to those with donor splenic CD4+ cells (8/11 (73%) of recipients with renal disease versus 3/11 (27%) with lung disease). Co-transfer of myeloid dendritic cells (MDCs) from the spleens of immunized mice along with CD4+ cells from immunized donors prevented the induction of renal disease (0/5 mice, p = 0.026 versus recipients of fresh CD4+ cells), though lung disease was still seen in 1/5 mice. Transfer of MDCs alone from immunized donors induced lung disease in 3/5 (60%) of recipients, with no nephritis. Co-transfer of splenocytes from mice with nephritis along with splenocytes from mice without nephritis led to renal disease in 4/5 recipients, with lung disease in 0/5 recipients. These findings indicate that RNP+ CD4+ T cells are sufficient to induce anti-RNP autoimmunity, that the tissue targeting of anti-RNP autoimmunity can be deviated to either a renal or pulmonary phenotype depending upon the presence of accessory cells including MDCs, and that dendritic cell subsets can play roles in both propagation of autoimmunity and end organ targeting.
Keywords: Lupus Erythematosus, Systemic, Mixed Connective Tissue Disease, Lung Diseases, Interstitial, Ribonucleoproteins, small nuclear, Models, animal
INTRODUCTION
Autoimmunity to ribonucleoprotein (RNP) autoantigens is frequently seen in systemic autoimmune diseases including lupus and mixed connective tissue disease (MCTD) (1). The induction of anti-RNP autoantibodies are closely linked in time with the initial presentation of autoimmune disease clinical manifestations in patients, suggesting that anti-RNP responses may have direct pathogenic roles in autoimmune diseases (2,3). We recently demonstrated that a single direct immunization with an RNP peptide and adjuvant into mice that were not otherwise disease-prone can induce a persistent autoimmune response including autoantibodies and end organ injury that is consistent with human anti-RNP syndromes (4). This model also suggested that the pattern of tissue injury that emerged could be influenced to either be MCTD-like lung disease or lupus-like nephritis depending upon the immune context present. We have since found that serum transfer from immunized mice could exacerbate lung injury in the setting of acute inflammation, but caused minimal tissue injury alone (5). Prior studies have indicated that T cells may be sufficient to convey anti-RNP autoimmunity (6). This has led us to investigate the role of cellular immunity in mediating anti-RNP autoimmunity.
In this report, we examine the ability of immune cells from anti-RNP immunized mice to induce disease after adoptive transfer into naïve syngeneic animals. We find that whole splenocytes transfer anti-RNP autoimmunity and lung disease from donors to recipients, while transfer of only CD4+ splenocytes transfers anti-RNP autoimmunity but changes the clinical phenotype from lung disease to nephritis in the recipients. We identify a population of non-plasmacytoid splenic dendritic cells, referred to here as myeloid dendritic cells (MDCs), that can prevent the induction of nephritis when co-transferred with CD4+ cells, or can induce anti-RNP immunity with lung disease when transferred alone. However, splenocytes from mice without nephritis fail to prevent nephritis when transferred along with splenocytes from mice with established nephritis, and a small number of plasmacytoid dendritic cells from immunized mice with renal disease are sufficient to convey nephritis to naïve recipients. These results suggest that adaptive and innate immune cells collaboratively shape the tissue expression of systemic autoimmune disease, and identify distinct steps in the development of autoimmune responses and the differentiation of autoimmune tissue targeting.
METHODS
Mice
Experiments were performed using C57BL/6Ntac-[KO]Abb-[Tg]DR-4 mice (Taconic, Germantown, NY), C57BL/6 mice transgenic for the expression of a chimeric human/mouse class II MHC in which the extracellular antigen presentation domains of HLA-DR4 have replaced the native murine class II regions, with the remainder of the native murine molecule intact, as we have previously reported (4), that we will refer to as DR4 mice. Earlier studies have used the same mouse strain used in this report successfully in adoptive transfers after immunization with type II collagen (CII), with successful transfer of anti-CII immunity and induction of arthritis in recipients, but without indications of lupus or MCTD-like immunity (7). All experiments were performed according to IACUC-approved protocols in AAALAC-certified facilities. DR4 mice were immunized subcutaneously in the flank with 50 micrograms of U1-70kD small nuclear ribonucleoprotein fusion protein (70k) and 50 micrograms of U1-RNA adjuvant once at 8–10 weeks of age. As previously reported, the directly immunized mice developed a high frequency of anti-RNP autoantibodies (4), RNP-specific T cells (8), and interstitial pneumonitis that persisted for months after a single immunization, and was associated with anti-70k T cell epitope spreading (4). In order to be used as an “immunized” mouse for the current study, anti-RNP antibodies were required to be demonstrated by ELISA and/or immunoblot.
Adoptive transfer
To assess the contribution of immune cells to this process, we harvested splenocytes 2 months after immunization from anti-RNP immunized mice or similar control mice. Whole spleens were mechanically disrupted and passed through a fine wire mesh in cold PBS to create a single cell suspension. The cells were treated with RBC lysis buffer, washed, and resuspended in sterile PBS for infusion or further cell separation. We infused cells (10 × 106 whole splenocytes, 2 × 106 CD4+ cells, 0.1 × 106 myeloid dendritic cells, or 0.1 × 106 plasmacytoid dendritic cells) in a final volume of 50 microliters of sterile PBS into 8–10 week old naïve syngeneic (non-irradiated) recipients via the tail vein, and followed recipient mice for 2 months. As we have reported, whole splenocyte recipient mice developed anti-RNP antibodies that were typically of lower titer than donor mice (4,8). Anti-RNP T cells could be isolated from recipients that had T cell fine specificity and TCR V gene use similar to anti-RNP-specific T cells derived from donor mice (8).
Cell separation
CD4+ cells were obtained from RBC-depleted splenocytes by positive selection with anti-CD4 microbeads by AutoMACS (Miltenyi, Auburn, CA). Myeloid dendritic cells were obtained from RBC-depleted splenocytes by Optiprep (Sigma-Aldrich, St. Louis, MO) density centrifugation to isolate the dendritic cell layer following the manufacturer’s instructions, followed by negative selection with anti-mPDCA-1 microbeads (Miltenyi) by AutoMACS to deplete plasmacytoid dendritic cells, and positive selection with anti-CD11c microbeads (Miltenyi) by AutoMACS. Plasmacytoid dendritic cells were obtained by Optiprep desity centrifugation as above followed by Miltenyi murine PDC negative selection kit by AutoMACS, and mPDCA-1 positive selection by AutoMACS.
Cell Culture
Murine T cell lines were generated and characterized using an approach similar to that which we have used extensively for generating human autoantigen-specific T cell lines and more recently used to generate murine autoantigen-specific T cell lines (1–8). In brief, spleen and lymph node cells were obtained sterilely at necropsy, mechanically disrupted, filtered through a sterile 100 μm nylon mesh filter and then subjected to density gradient centrifugation using Histopaque (Sigma). Cells obtained from immunized mice were used immediately for the generation of T cell lines. Cells obtained from naïve mice were irradiated with 30 Grey (Gy) and used as APC to stimulate T cells and in proliferation assays. Approximately 5 × 106 cells were cultured in DMEM with 2 mM L-glutamine (complete medium), supplemented with 20 μg/ml gentamicin, 15% fetal calf serum and containing fusion protein at a final concentration of 50 μg/ml. As antigen, 70kDa fusion protein was used, as described (8–12). Cells in a final volume of 5 ml were placed in a 25-cm2 flask and incubated in 5% carbon dioxide at 37°C. Cells were restimulated with 5 × 106 murine APC that had been irradiated with 30 Gy plus antigen in fresh medium on days 7–10. Following one or two cycles of stimulation, cells were harvested for adoptive transfer, in vitro proliferation assays, and/or TCR analysis. RNA was extracted from the cells which had been stimulated with plate-bound anti-CD3 in the absence of APC at 48 h for TCR analyses using RNeasy (InVitrogen, Carlsbad, CA).
Disease assessment
The presence of active urinary sediment was determined by urinalysis on urine specimen taken at the time of sacrifice. The presence of histologic abnormalities was determined by blinded reading of UM-Fix prepared tissues stained with Hematoxylin and Eosin, as previously reported (4,13). Additional sections of kidney at the time of sacrifice were embedded in OCT and snap-frozen for immunofluorescence assays with FITC-conjugated anti-mouse C3 and anti-mouse IgG antibodies. Serum complement C3 levels were measured in terminal bleeds using a commercial ELISA assay (Immunology Consultants Laboratory, Newberg, OR).
Flow Cytometry
Cells were quantitated on a BD LSRII instrument (Franklin Lakes, NJ), and data analyzed using BD FACSDiva Software. Fluorochrome-conjugated antibodies to CD3, CD4, CD8, CD11c, and HLA-DR4 were from BD. CFSE was from Molecular Probes (Eugene, OR). Fluorochrome-conjugated anti-mPDCA-1 was from Miltenyi. Purified MDCs were labeled with CFSE by 10 minute incubation in complete medium containing 2.5μM CSFE final concentration. Cells were washed and resuspended in cold sterile PBS prior to infusion via tail vein.
Anti-RNP Assays
Anti-RNP autoantibodies were detected by immunoblot against intact and apoptotic Jurkat lysate and by ELISA against purified 70k fusion protein antigen, both as previously reported (14). Anti-RNP T cells were detected by the presence of elevated stimulation indices in response to known DR4-restricted 70k T cell epitopes presented by syngeneic naïve irradiated APCs, as previously described (8,12). T cell lines proliferating in response to RNP peptides were generated as described above. Using primers for the TCR heavy chain Vbeta region to isolate the recombined area of T cell receptors from RNP-responsive cells, the Vbeta regions were sequenced, as previously described (8).
Statistics
Categorical variables were compared using Fisher’s Exact test, calculated using Prism 3.0 (GraphPad, San Diego, CA).
RESULTS
Disease Induction with anti-RNP Splenocyte Adoptive Transfer
Direct immunization with one subcutaneous dose of 50 micrograms of 70k peptide and 50 micrograms of U1-RNA leading to anti-70k immune responses by ELISA and/or immunoblot were confirmed in 80 DR4 mice. Of these, 47 (59%) had no protienuria on urinalysis while 33 (41%) had at least trace proteinuria. The mice without proteinuria developed histologic manifestations of interstitial lung disease in 18 mice (38%), with histologic manifestations of renal disease in 0 mice (0%). In contrast, the mice with proteinuria developed histologic manifestations of lung disease in 5 mice (15%, Fisher’s Exact p = 0.027 versus nonproteinuric mice), and histologic manifestations of renal disease in 9 mice (27%, Fisher’s Exact p = 0.0002). We therefore hypothesized that the presence or absence or proteinuria may be a marker for immunologic conditions favorable for induction of nephritis versus immunologic conditions favorable for induction of lung disease.
We performed adoptive transfer studies from immunized DR4 mice into naïve syngeneic recipients. Two months after transfer of 10 million RBC-depleted whole splenocytes from mice without proteinuria, 8/14 recipients (57%) developed interstitial lung disease while only 2/14 mice (14%) showed evidence of renal disease (without lung disease) (Figure 1, Table 1). Examination of the joints, heart, liver, spleen, skin, esophagus, salivary glands, and intestines revealed no gross or histologic differences between directly immunized mice and splenocyte adoptive transfer recipients. In contrast, two months after transfer of 10 million splenocytes from proteinuric mice to naïve syngeneic recipients, 0/5 (0%) developed lung disease and 3/5 (60%) developed renal disease. Similar transfer of 10 million splenocytes from naïve syngeneic donors led to no identifiable lung or renal disease in any of 5 recipients. We could identify anti-RNP antibodies and anti-RNP T cells in the recipients of immunized mouse cells but not the recipients of naïve cells.
Figure 1.
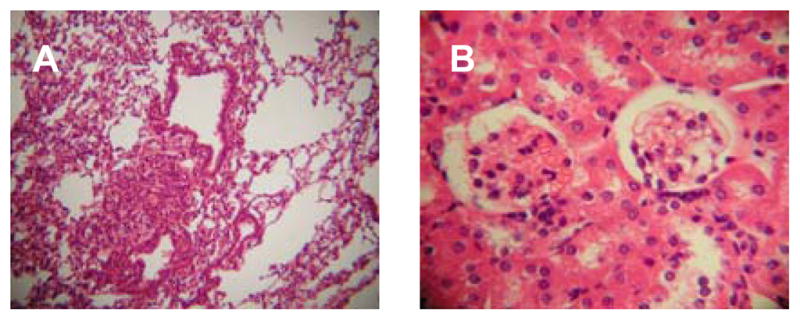
Adoptive transfer of splenocytes from immunized DR4 mice transfers lung disease. Representative H&E sections from a naïve syngeneic recipient of whole splenocytes from a 70k + U1-RNA immunized HLA-DR4+ donor are shown. A: 10x image of lung with MCTD-like lung disease. B: 40x image of kidney with no evidence of renal pathology (urinalysis, not shown, was also normal).
Table 1.
Protective Effect of MDCs on CD4 Cell Transfer-Induced Nephritis
| Whole Splenocyte Transfer | N | Number (%)lung disease | Number (%)renal disease | |
|---|---|---|---|---|
| Yes | 14 | 8 (57%) | 2 (14%) | |
| CD4 Cell Transfer | Myeloid DC Transfer | |||
| Yes, Fresh | No | 10 | 2 (20%) | 7 (70%)* |
| Yes, Cultured | No | 11 | 3 (27%) | 8 (73%)# |
| Yes, Fresh | Yes | 5 | 1 (20%) | 0 (0%)† |
| No | Yes | 5 | 3 (60%) | 0 (0%) |
Lung disease and renal disease defined as in methods section, in naïve syngeneic female mice 2 months after adoptive transfer of 10 × 106 RBC-depleted whole splenocytes, 2 × 106 purified splenic CD4+ cells (CD4 Cell Transfer) that were either immediately transferred after purification (Fresh) or cultured for two weeks in vitro with naïve syngeneic irradiated APCs and 70k protein (Cultured), and/or 1 × 105 myeloid dendritic cells (Myeloid DC Transfer). All donor mice were syngeneic animals sacrificed two months after anti-70k immunization, and were confirmed to have anti-70k antibodies by ELISA, and to have consistently normal urinalyses. N = number of adoptive transfer recipient mice receiving the indicated cell transfers.
Fisher’s Exact p = 0.01 versus transfer of whole splenocytes.
Fisher’s Exact p = 0.005 versus transfer of whole splenocytes.
Fisher’s Exact p = 0.026 versus transfer of fresh CD4+ cells alone.
Renal Disease after Adoptive Transfer of RNP+ CD4+ Splenocytes from Donors without Proteinuria
Using immunized RNP+ DR4 mice without proteinuria, we separated CD4+ cells from spleens by magnetic bead positive selection. We adoptively transferred 2 million CD4+ cells, approximating the number of CD4+ cells recoverable from a single immunized donor mouse spleen, to naïve syngeneic recipients as above. In contrast to our findings in recipients of whole splenocytes, renal disease was common in recipients of CD4 cells from non-proteinuric donors, manifesting in 7/10 (70%), of recipient mice (Fisher’s Exact p = 0.01 versus whole splenocyte transfer recipients from similar donors) (Figure 2, Table 1). Lung disease was less common, present in only 2/10 (20%) of recipients of the immunized CD4+ cells. Transfer of 2 million CD4+ cells from naïve mice to syngeneic donors under the same protocol led to no lung or kidney disease in 5/5 recipients.
Figure 2.
Adoptive transfer of CD4+ splenocytes from non-nephritic donors induces nephritis rather than lung disease. Representative images are shown of H&E-stained sections of lung and kidney from a naïve syngeneic recipient of CD4+ cells directly isolated from a 70k-immunized mouse (A & B), and from a naïve syngeneic recipient of CD4+ cells harvested from a syngeneic 70k-immunized donor and grown in vitro for 2 weeks with 70k-loaded irradiated APCs from a naïve syngeneic APC donor (C & D). In both cases, minimal interstitial pulmonary infiltrates are noted, but renal lesions consistent with nephritis are present. In both cases, urinalysis also showed active urinary sediment (not shown).
To assure that the nephritis induction was mediated by anti-RNP-specific T cells, we isolated CD4+ splenocytes as above, then cultured the CD4+ cells with 70k antigen and irradiated syngeneic APCs from naïve APC donors for two weeks, and separated the T cells by Histopaque density centrifugation of the nonadherent cells. T cells thus isolated from the recipients remained homologous to those from the donors with regard to RNP epitope responses and TCR VB region sequence (data not shown). Using 2 million of these T cells from short-term culture to adoptively transfer into recipient mice, we found the same manifestations of nephritis without lung disease as with direct CD4+ cell transfers (Figure 2, Table 1). The results shown are representative of those from two separate experiments, in which CD4+ cells from 70k+ donors without renal disease expanded significantly in vitro, and a total of 11 recipients (6 from one experiment and 5 from another) received 2 million T cells each. Lung disease was observed in 3/11 (27%) of recipients, while renal disease was seen in 8/11 (73%) of recipients (p = 0.005 versus whole splenocyte transfers).
The renal lesions seen after CD4+ cell transfers were typified by histologic findings of glomerular proliferation, mesangial hypertrophy, and hematoxylin body formation, characteristic of lupus nephritis, although immune complex deposition and complement depletion were generally not observed (data not shown). In both the fresh CD4+ cell recipients and the recipients of short-term cultured cells, the infrequent mice with lung disease were equally divided between those without renal disease (1/10 fresh CD4 and 1/11 cultured CD4 recipients) and those that also had renal disease (1/10 fresh CD4 and 2/11 cultured CD4 recipients). Recipients of immunized CD4 cells showed anti-RNP T cell reactivity, as previously described (8).
These results suggest that anti-RNP-reactive T cells are sufficient to transfer anti-RNP autoimmunity from immunized donor to otherwise naïve recipients, and that in the absence of other signals RNP-reactive T cells can direct the induction of glomerulonephritis.
Myeloid Dendritic Cell Adoptive Transfers from RNP+ Donors
The divergence in tissue targeting between recipients of whole splenocyte transfers (frequent lung disease, less renal disease) and CD4+ cell transfers (frequent renal disease, less lung disease) from non-proteinuric donors suggests that non-CD4+ splenocytes participate in the development of the lung targeting pattern seen with whole splenocyte transfers. Based on our previous observations that TLR3-null mice were more susceptible to renal disease and less susceptible to lung disease than TLR3-intact mice after direct immunization of 70k + U1-RNA (4,13), we considered whether TLR3 expressing splenocytes could similarly protect against nephritis induction. We therefore investigated myeloid dendritic cells (MDCs), a major known TLR3-expressing immune cell type in the spleen (15).
We separated MDCs from RBC-depleted spleens by a three step process of Optiprep density centrifugation, negative selection for plasmacytoid dendritic cells with mPDCA-1 magnetic beads, then positive selection for CD11c surface expression with magnetic beads. We found that this yielded approximately 200,000 cells per immunized mice spleen, with an over 95% pure population of MDCs by flow cytometry for the CD11c and mPDCA-1 selection markers (data not shown).
To establish that MDC adoptive transfers from immunized mice would lead to engraftment in syngeneic naïve recipients, we adoptively transferred naïve mice with 100,000 MDCs from immunized non-proteinuric mice that were labeled with CFSE prior to infusion. Recipient mice were then sacrificed 48 hours and 7 days after cell transfer. Substantial and stable numbers of CFSE-expressing CD11c+ cells could be detected in the lungs (but not the spleens) of recipient mice at 48 hours and 7 days after cell transfer (Figure 3A). However, the accumulation of lung MDCs was insufficient to manifest as histologically significant lung inflammation at either the 48 hour or 7 day time points (data not shown). Using CFSE-labeled MDCs from identically immunized donors with proteinuria (and no lung disease), we found dramatically less trafficking of donor MDCs to the lungs of naïve recipient mice, with similar trafficking to spleen (Figure 3A).
Figure 3.

Adoptively transferred myeloid dendritic cells persist after transfer, with variable tissue specificity for the lungs. MDCs were purified from spleens of immunized mice, labeled with CFSE, and transferred to 10 week old naïve syngeneic mice by tail vein injection. At 48 hours and 1 week after cell transfer, single cell suspensions of the spleens and lungs were prepared, treated with Optiprep centrifugation to enhance for identification of dendritic cells, stained with CD11c-APC, and subjected to flow cytometry. Histograms show CFSE fluorescence on the x axis and CD11c staining on the y axis. Identical gating conditions were used for all experiments. A. With MDCs from immunized non-proteinuric donors, over 30% of the gated cells from lung (versus 1% from spleen) have CD11c+/CFSE+ staining consistent with donor-derived MDCs at both 48 hours and 1 week after transfer, with approximately equal intensity of CFSE staining suggesting minimal proliferation or other dilution of these CD11c+ cells between 48 hours and 7 days. B. With MDCs from immunized nephritic donors, fewer than 4% CD11c+/CFSE+ cells appear in the lungs at 48 hours or at one week, while the migration of labeled MDCs to spleen is similar to that seen in A.
In mice receiving 100,000 MDCs from RNP+ mice with lung disease that were followed for 2 months, 3/5 (60%) developed lung disease, while 0/5 developed renal disease (Figure 4, Table 1). These results were similar to the rate of end organ manifestations in recipients of whole splenocytes. To assess the ability of MDCs to inhibit CD4+ cell-induced nephritis, we performed co-transfers. When the same 100,000 MDCs from RNP+ mice were transferred along with 2 million CD4+ cells from RNP+ mice without renal disease, a dramatic drop in the incidence of nephritis compared to mice receiving CD4+ cells alone was seen, from 7/10 (70%) to 0/5 (0%) (Fisher’s Exact p = 0.026) (Figure 4, Table 1). These MDC + CD4 co-transferred mice, additionally, showed lung disease in only 1/5 (20%) of recipients. These results support the hypothesis that MDCs from immunized mice can mediate a nephritis-protective effect in anti-RNP autoimmunity. On the other hand, when we co-transferred CD4+ cells from immunized mice without renal disease and MDCs from unimmunized syngeneic mice, we observed nephritis in 5/5 recipients (100%), and lung disease in only 1/5 recipients (20%). The protective effect of MDCs on the CD4 cell-mediated kidney disease was thus not present with unimmunized MDCs subjected to the same separation protocol.
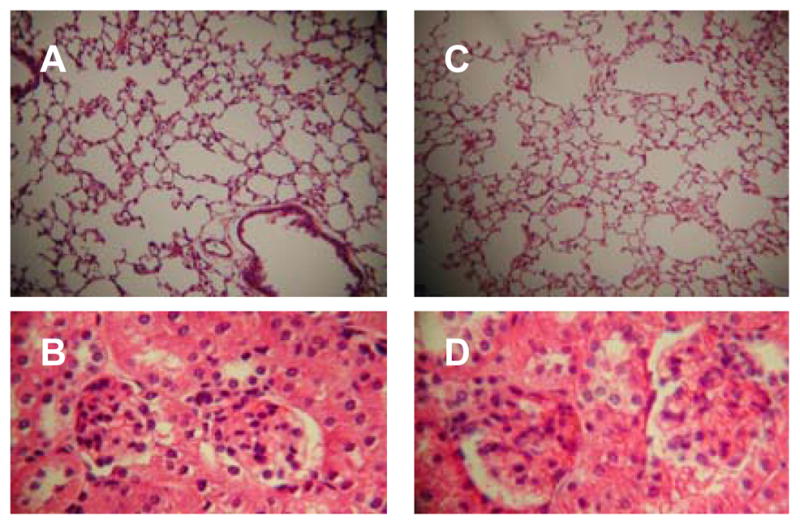
Figure 4.

Effect of myeloid dendritic cell (MDC) adoptive transfers. Syngeneic naïve recipient mice received 100,000 myeloid dendritic cells from RNP-immunized mice without renal disease (A, B), or the same cells along with 2 million CD4+ cells from immunized mice with renal disease (C,D). The H&E stained images are representative findings of lungs at 10x magnification and kidneys at 40x magnification taken from the same mice. MDC-only adoptive transfer was associated with pulmonary infiltrates but not renal disease, whereas MDC + CD4+ adoptive transfer typically led to neither lung nor renal disease.
Adoptive Transfer from Nephritic Donors
Directly immunized RNP+ DR4 mice that had active urinary sediment were also studied. Histologic lesions identified, as in the T cell recipient mice described above, consisted of glomerular proliferation, mesangial hypertrophy, and hematoxylin body formation, without prominent immune complex deposition or complement depletion (data not shown). Lung disease was observed in 14/43 (33%) of these mice.
For adoptive transfer studies, mice with renal disease and no lung disease (on the basis of active urinary sediment and subsequent confirmatory histologic findings) were sacrificed 2 months after immunization, and RBC-depleted splenocytes were purified, as above. We performed mixing studies, in which naïve syngeneic DR4 recipient mice received 5 million splenocytes each from an RNP+ donor with lung disease and no kidney disease, and from an RNP+ donor with renal disease but no lung disease. When these recipients were sacrificed 2 months after adoptive transfer, renal disease was present in 4/5 (80%) of mice and lung disease in 0/5 (0%) of mice (Figure 5).
Figure 5.

Co-transfer of splenocytes from nephritic and non-nephritic immunized mice induces nephritis. Syngeneic HLA-DR4 transgenic B6 mice were immunized with 70k + U1-RNA. CD4+ and CD4- splenocytes from a typical mouse with MCTD lung disease and no renal lesions (A & B) were mixed with CD4+ and CD4- splenocytes from an unusual mouse with active urinary sediment and histologic manifestations of nephritis but no lung disease (C & D). Representative sections are taken from an adoptive transfer recipient of the mixed cells, demonstrating nephritis in the absence of lung lesions typical for the recipient mice in this experiment (E & F).
Using dendritic cell subsets derived from immunized donors with active urinary sediment, 4/5 (80%) of recipients of plasmacytoid dendritic cells developed renal disease, and 0/5 of these PDC recipients developed lung disease. In contrast, 0/4 recipients of myeloid dendritic cells from nephritic donors developed renal disease. Notably, lung disease was also seen in none (0/4) of the recipients of MDCs from immunized donors with proteinuria and no lung disease. We were able to detect anti-RNP antibody responses in recipient mice by ELISA and/or immunoblot after transfer of immunized MDCs from non-proteinuric donors either alone or when co-transferred with CD4+ cells, and similarly detect anti-RNP antibodies after transfer of PDCs alone from nephritic mice (data not shown). Anti-RNP antibodies were not detected, however, in recipients of MDCs from immunized nephritic donors.
DISCUSSION
These studies demonstrate that whole splenocytes, CD4+ T cells, or purified dendritic cells subsets from RNP-immunized mice can be sufficient to convey anti-RNP autoimmunity to naïve syngeneic mice. Furthermore, anti-RNP-reactive T cells can either lead to SLE-like nephritis or to MCTD-like lung disease depending upon the cell types that are transferred or co-transferred. Effects mediated by TLR3-expressing myeloid dendritic cells on tissue targeting of anti-RNP responses appear to potentially account for the previously reported divergence in pulmonary and renal manifestations in directly immunized anti-RNP mice (4). Consistent with their reported roles in lupus pathogenesis (16), adoptive transfer of a relatively small number of purified immunized plasmacytoid dendritic cells were also able to induce the development of glomerulonephritis.
The lung disease observed in our mice appears to be indistinguishable from cases of human MCTD lung disease by conventional hematoxylin and eosin staining. We do not observe substantial fibrosis in the mouse lungs, as in the majority of cases of human MCTD lung disease (17). Further studies using immunohhistochemistry to characterize the murine and human infiltrates are warranted. The renal lesions in our study mice differ from SLE nephritis in rarely having immunoglobulin or complement component deposits at the glomerular basement membrane, but do demonstrate diffuse or focal proliferative patterns, mesangial involvement, plus the frequent development of hematoxylin bodies as in lupus nephritis.
Reports have linked TLR3 signals to exacerbated lung inflammation, and TLR7 signals to reduced lung inflammation in models of asthma, viral infection, and direct TLR agonist challenge (18–21). We hypothesize that in our system, lung-predominant disease similarly develops when TLR3-activated MDCs direct anti-RNP T cells to pulmonary rather than renal targets. The mechanism of action of MDCs in mediating lung-specific tissue targeting has not yet been defined and further study will be needed to establish the role, if any, of TLR3 in this process. Conversely, we hypothesize that renal disease develops when anti-RNP autoimmunity is augmented at an early stage by a TLR7 signal rather than being modulated by a strong TLR3 signal. The inability of 70k-immunized splenocytes from non-nephritic mice to inhibit established nephritis in co-transfer experiments may reflect the involvement of immune mediators downstream of the point where MDCs are able to inhibit nephritis induction, or the elaboration of factors that inhibit (putatively) TLR3-induced MDC activity. Since anti-RNP CD4+ T cell induced nephritis was able to be inhibited by MDCs in our studies, this suggests that: i. development of anti-RNP immunity can occur prior to a commitment of an autoimmune process to renal injury, and ii. cells other than CD4+ T cells emerge after the induction of anti-RNP immunity that drive the renal targeting in a manner that is resistant to reversal by activated MDCs. Given the associations identified between lupus nephritis and TLR7 (22), TLR7-expressing cells such as plasmacytoid dendritic cells or B cells appear to be logical candidates for playing roles in renal disease downstream of anti-RNP T cells that MDCs cannot reverse.
Since MCTD was first proposed as a diagnostic entity clinically, debate has occurred regarding whether MCTD is truly distinct from other established rheumatic diseases, including lupus (23–25). Consistent with this experience, the results of our study indicate that subtle shadings of difference exist in a mouse model of anti-RNP autoimmunity between MCTD-like (lung disease) and lupus-like (renal disease) manifestations of tissue injury. In mice, the same T cells appear to be relevant to either lung disease or renal disease induction, with the tissue targeting determined by accessory cell types, including MDCs and PDCs. Our finding that in adoptive transfer mixing studies, cells from mice with established renal disease are dominant over cells from mice with established lung disease may inform the clinical observation that MCTD can evolve into SLE, but that SLE seldom evolves into MCTD.
While our adoptive transfer studies show that either T cells or dendritic cells can spark persistent autoimmunity in a naïve host, there is no guarantee that either of these cell types participate in the early stages of actual spontaneously developing clinical disease. Studies in patients are needed to support the relevance of the observations we have made in mice.
This report demonstrates for the first time that elements of the same immune responses to a rheumatic disease autoantigen can lead to divergent tissue targeting based on differences in innate immune effectors. Improved understanding of the cell and molecular biology underlying this process may allow for the development of new diagnostic and therapeutic approaches to systemic autoimmune diseases in this spectrum.
Acknowledgments
This work was supported by the Department of Veterans Affairs (ELG and RWH), the National Institutes of Health (awards AI1842 to ELG and AR43308 and AR48805 to RWH), and the Lupus Research Institute (ELG).
References
- 1.Sharp GC, Irvin WS, May CM, Holman HR, McDuffie FC, Hess EV, Schmid FR. Association of antibodies to ribonucleoprotein and Sm antigens with mixed connective-tissue disease, systematic lupus erythematosus and other rheumatic diseases. N Engl J Med. 1976;295:1149–54. doi: 10.1056/NEJM197611182952101. [DOI] [PubMed] [Google Scholar]
- 2.Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003 Oct 16;349(16):1526–33. doi: 10.1056/NEJMoa021933. [DOI] [PubMed] [Google Scholar]
- 3.Greidinger EL, Hoffman RW. The appearance of U1 RNP antibody specificities in sequential autoimmune human antisera follows a characteristic order that implicates the U1-70 kd and B'/B proteins as predominant U1 RNP immunogens. Arthritis Rheum. 2001 Feb;44(2):368–75. doi: 10.1002/1529-0131(200102)44:2<368::AID-ANR55>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Greidinger EL, Zang Y, Jaimes K, Hogenmiller S, Nassiri M, Bejarano P, Barber GN, Hoffman RW. A murine model of mixed connective tissue disease induced with U1 small nuclear RNP autoantigen. Arthritis Rheum. 2006 Feb;54(2):661–9. doi: 10.1002/art.21566. [DOI] [PubMed] [Google Scholar]
- 5.Keith MP, Moratz C, Egan R, Zacharia A, Greidinger EL, Hoffman RW, Tsokos GC. Anti-Ribonucleoprotein Antibodies Mediate Enhanced Lung Injury Following Mesenteric Ischemia/Reperfusion in Rag-1−/− Mice. Autoimmunity. 2007;40:208–16. doi: 10.1080/08916930701262986. [DOI] [PubMed] [Google Scholar]
- 6.Fatenejad S, Mamula MJ, Craft J. Role of intermolecular/intrastructural B- and T-cell determinants in the diversification of autoantibodies to ribonucleoprotein particles. Proc Natl Acad Sci USA. 1993;90:12010–4. doi: 10.1073/pnas.90.24.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang D, Hill JA, Jevnikar AM, Cairns E, Bell DA. Induction of transient arthritis by the adoptive transfer of a collagen II specific Th1 clone to HLA-DR4 (B1*0401) transgenic mice. J Autoimmun. 2002 Aug–Sep;19(1–2):37–43. doi: 10.1006/jaut.2002.0601. [DOI] [PubMed] [Google Scholar]
- 8.Greidinger EL, Zang YJ, Jaimes K, Martinez L, Nassiri M, Hoffman RW. CD4+ T cells target epitopes residing within the RNA binding domain of the U1-70kD small nuclear ribonucleoprotein autoantigen and have restricted TCR diversity in an HLA-DR4 transgenic murine model of Mixed Connective Tissue Disease. J Immunol. 2008;180:8444–8454. doi: 10.4049/jimmunol.180.12.8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoffman RW, Takeda Y, Sharp GC, Lee DR, Kaneoka H, Caldwell CW. Human T cell clones reactive against U-small nuclear ribonucleoprotein autoantigens from connective tissue disease patients and healthy individuals. J Immunol. 1993;151:6460–6469. [PubMed] [Google Scholar]
- 10.Holyst MM, Hill DL, Hoch SO, Hoffman RW. Analysis of human T cell and B cell responses against U small nuclear ribonucleoprotein 70k, B and D polypeptides among patients with systemic lupus erythematosus and mixed connective tissue disease. Arthritis Rheum. 1997;40:1493–1503. doi: 10.1002/art.1780400818. [DOI] [PubMed] [Google Scholar]
- 11.Greidinger EL, Gazitt T, Jaimes KF, Hoffman RW. Human T cell clones specific for heterogeneous nuclear ribonucleoprotein A2 autoantigen from connective tissue disease patients assist in autoantibody production. Arthritis Rheum. 2004;50:2216–2222. doi: 10.1002/art.20287. [DOI] [PubMed] [Google Scholar]
- 12.Greidinger EL, Foecking MF, Schafermeyer KR, Bailey CW, Primm SL, Lee DR, Hoffman RW. T cell immunity in connective tissue disease patients targets the RNA binding domain of the U1-70kDa small nuclear ribonucleoprotein. J Immunol. 2002 Sep 15;169(6):3429–37. doi: 10.4049/jimmunol.169.6.3429. [DOI] [PubMed] [Google Scholar]
- 13.Greidinger EL, Zang Y, Martinez L, Jaimes K, Nassiri M, Bejarano P, Barber GN, Hoffman RW. Differential Tissue Targeting of Autoimmune Manifestations by Autoantigen-Associated Y RNAs. Arthritis Rheum. 2007;56:1589–97. doi: 10.1002/art.22601. [DOI] [PubMed] [Google Scholar]
- 14.Greidinger EL, Foecking MF, Magee J, Wilson L, Ranatunga S, Ortmann RA, Hoffman RW. A major B cell epitope present on the apoptotic but not the intact form of the U1-70-kDa ribonucleoprotein autoantigen. J Immunol. 2004 Jan 1;172(1):709–16. doi: 10.4049/jimmunol.172.1.709. [DOI] [PubMed] [Google Scholar]
- 12.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, Akira S, Reis e Sousa C. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003 Apr;33(4):827–33. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 13.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006 Sep;25(3):383–92. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 14.Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Núñez G, Curtis JL, Luker GD. TLR3 Increases Disease Morbidity and Mortality from Vaccinia Infection. J Immunol. 2008 Jan 1;180(1):483–91. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato A, Favoreto S, Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol. 2007 Jul 15;179(2):1080–7. doi: 10.4049/jimmunol.179.2.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeon SG, Oh SY, Park HK, Kim YS, Shim EJ, Lee HS, Oh MH, Bang B, Chun EY, Kim SH, Gho YS, Zhu Z, Kim YY, Kim YK. TH2 and TH1 lung inflammation induced by airway allergen sensitization with low and high doses of double-stranded RNA. J Allergy Clin Immunol. 2007 Oct;120(4):803–12. doi: 10.1016/j.jaci.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 17.Bodolay E, Szekanecz Z, Dévényi K, Galuska L, Csípo I, Vègh J, Garai I, Szegedi G. Evaluation of interstitial lung disease in mixed connective tissue disease (MCTD) Rheumatology (Oxford) 2005 May;44(5):656–61. doi: 10.1093/rheumatology/keh575. [DOI] [PubMed] [Google Scholar]
- 18.Le Goffic R, Pothlichet J, Vitour D, Fujita T, Meurs E, Chignard M, Si-Tahar M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol. 2007 Mar 15;178(6):3368–72. doi: 10.4049/jimmunol.178.6.3368. [DOI] [PubMed] [Google Scholar]
- 19.Rudd BD, Smit JJ, Flavell RA, Alexopoulou L, Schaller MA, Gruber A, Berlin AA, Lukacs NW. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J Immunol. 2006 Feb 1;176(3):1937–42. doi: 10.4049/jimmunol.176.3.1937. [DOI] [PubMed] [Google Scholar]
- 20.Camateros P, Tamaoka M, Hassan M, Marino R, Moisan J, Marion D, Guiot MC, Martin JG, Radzioch D. Chronic asthma-induced airway remodeling is prevented by toll-like receptor-7/8 ligand S28463. Am J Respir Crit Care Med. 2007 Jun 15;175(12):1241–9. doi: 10.1164/rccm.200701-054OC. [DOI] [PubMed] [Google Scholar]
- 21.Demedts IK, Bracke KR, Maes T, Joos GF, Brusselle GG. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am J Respir Cell Mol Biol. 2006 Sep;35(3):387–93. doi: 10.1165/rcmb.2005-0382OC. [DOI] [PubMed] [Google Scholar]
- 22.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006 Sep;25(3):417–28. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Smolen JS, Steiner G. Mixed connective tissue disease: to be or not to be? Arthritis Rheum. 1998 May;41(5):768–77. doi: 10.1002/1529-0131(199805)41:5<768::AID-ART3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Sharp GC, Hoffman RW. Clinical, immunologic, and immunogenetic evidence that mixed connective tissue disease is a distinct entity: comment on the article by Smolen and Steiner. Arthritis Rheum. 1999 Jan;42(1):190–1. doi: 10.1002/1529-0131(199901)42:1<190::AID-ANR29>3.0.CO;2-J. author reply 193–6. [DOI] [PubMed] [Google Scholar]
- 25.Isenberg D, Black C. Naming names! Comment on the article by Smolen and Steiner. Arthritis Rheum. 1999 Jan;42(1):191–6. doi: 10.1002/1529-0131(199901)42:1<191::AID-ANR30>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]