

The conversion of simple precursors into optically active products is a continuing goal in chemistry to efficiently access complex architectures for use in total synthesis and medicinal chemistry.1 An especially important substitution pattern is a carbon atom bonded to four other carbon substituents (i.e., quaternary), and the generation of these congested centers is a significant challenge.2 The desymmetrization of achiral molecules is a powerful strategy for the synthesis of optically active materials and presents a distinct method for the construction of stereogenic quaternary carbon centers.3 In this Communication, we report the enantioselective synthesis of α,α-disubstituted cyclopentenes (2) by an intramolecular aldol reaction of achiral tricarbonyl compounds (1) catalyzed by chiral N-heterocyclic carbenes (eq 1).
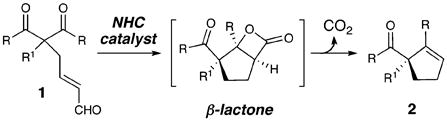 |
(1) |
In 2006, Nair demonstrated that N-heterocyclic carbenes (NHCs) catalyze the formation of cyclopentenes from unsaturated aldehydes and chalcones.4 A premise in this elegant work is that, after the homoenolate addition, a β-lactone intermediate is formed which undergoes liberation of carbon dioxide to generate the alkene product.5,6 Recently, Bode reported a route to optically active cyclopentenes using an NHC-catalyzed crossed benzoin/oxy-Cope sequence.7 Our investigations of new NHC-catalyzed reactions8 have involved the use of these unique nucleophilic catalysts to access homoenolate and enolate/enol reactivity from the same unsaturated aldehyde starting material.9,10 We had observed this interesting duality in our early investigations with NHC-derived homoenolates in which IMes11 promoted the unusual dimerization of cinnamaldehyde to afford the β-lactone 3, as determined by X-ray analysis (eq 2).
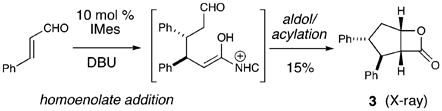 |
(2) |
This finding, combined with our recently developed NHC-catalyzed enantioselective Michael reaction,12 prompted us to examine the possibilities of carbene-catalyzed aldol reactions. Our objective was to couple a decarboxylation event to a symmetry breaking operation (e.g., an aldol reaction) in order to generate optically active alkenes.13
Our proposed reaction pathway (Scheme 1) involves initial addition of the NHC to the aldehydes. As in our Michael reaction, the key protonation of I generates an enol (II) in situ which is poised to undergo productive bond formation. By employing a chiral NHC catalyst with substrates such as 1, an enantioselective aldol reaction ensues to produce β-hydroxy ketone intermediate III. Intramolecular acylation of this tertiary alcohol releases the NHC catalyst, and the resulting β-lactone (IV) undergoes loss of CO2 to generate 2.
Scheme 1.
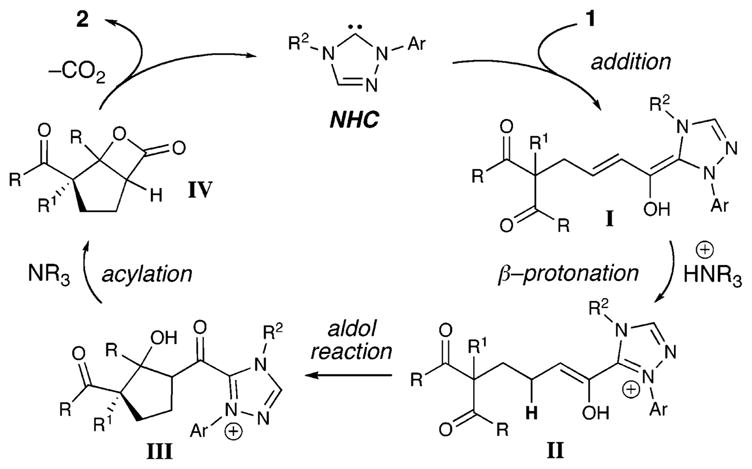
Proposed Reaction Pathway
The combination of chiral triazolium salt A derived from L-phenylalanine with unsaturated aldehyde 1a and i-Pr2EtN as base led to a selective reaction (−83% ee) with only moderate yield of cyclopentene 4 (Table 1, entry 1).14 Interestingly, the substitution on the six-membered ring of the catalyst significantly impacts the stereochemical outcome of the reaction. For example, using the geminal dimethyl catalyst B from L-phenylalanine affords −76% ee of 4 (entry 2), yet the diphenyl catalyst C with the same absolute stereochemistry as B generates 4 in 51% ee (entry 3). By switching to azolium D9c and elevating the temperature of the reaction (40 °C) to ensure complete decarboxylation, an 80% yield of 4 is obtained in excellent enantioselectivity with 10 mol % of D (entries 6 and 7).15 Notably, the exclusion of oxygen in the reaction increases the overall yields (entry 4 vs 5). For entry 4, we observe the unsaturated acid derived from 1 (not shown), apparently from oxidation of the homoenolate intermediate I. Studies to capitalize on this observation are in progress.
Table 1.
Optimization of Conditionsa
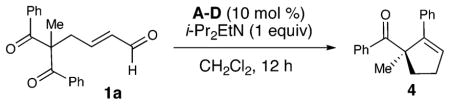 | ||||
|---|---|---|---|---|
| entry | temperature (°C) | azolium salt | yield (%)b | ee (%)c |
| 1 | 23 | A | 47 | −83 |
| 2 | 23 | B | 38 | −76 |
| 3 | 23 | C | 45 | 51 |
| 4 | 23 | D | 40 | 94 |
| 5d | 23 | D | 66 | 94 |
| 6d | 40 | D | 80 | 93 |
| 7d,e | 40 | D | 70 | 93 |
See Supporting Information for reaction details.
Isolated yield.
Determined by HPLC using Chiracel AD-H column.
Careful exclusion of oxygen.
5 mol % of D.
Once the optimal catalyst and conditions were identified, we investigated the scope of this desymmetrization (Table 2). Different aryl ketones are good substrates for the reaction (entries 1–4). Replacing the methyl group with other alkyl groups causes a slight decrease in enantioselectivity for substituents larger than ethyl (entries 6–8). Aliphatic diketones deliver the β-lactone products (12, 13) instead of alkenes. In these systems, the relative stereochemistry of the methyl group and lactone in the products depends on whether the diketone moiety is locked in a ring.
Table 2.
Substrate Scope
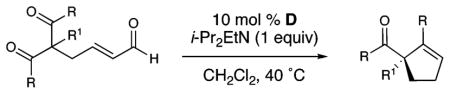 | |||||
|---|---|---|---|---|---|
| entry | R | R1 | cyclopentene | yield (%) | ee (%)a |
| 1 | Ph | Me | 4 | 80 | 93 |
| 2 | 4-Cl-Ph | Me | 5 | 76 | 94 |
| 3 | 4-Me-Ph | Me | 6 | 60 | 94 |
| 4 | 3-Me-Ph | Me | 7 | 65 | 93 |
| 5 | Ph | Et | 8 | 73 | 90 |
| 6 | Ph | allyl | 9 | 70 | 83 |
| 7 | 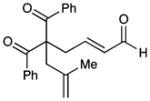 |
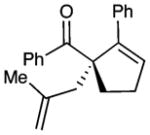 10 |
69 | 83 | |
| 8b | 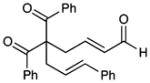 |
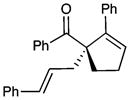 11 |
64 | 82 | |
| 9b | 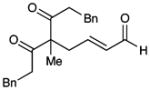 |
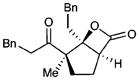 12 |
65c | 93 | |
| 10b | 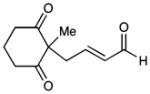 |
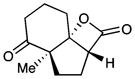 13 |
51c | 96 | |
Determined by HPLC Chiracel AD-H.
20 mol % of D.
Diastereomeric ratio = 20:1. Relative stereochemistry of 12 and 13 determined by NOE and X-ray crystallography, respectively. See Supporting Information for details.
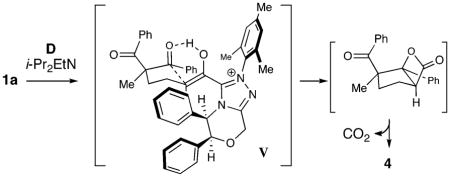
Our current model for this reaction involves the Z(O)-enol intermediate V. A six-membered hydrogen-bonded aldol reaction occurs which minimizes the nonbonding interactions between the phenyl substituents on the catalyst and the phenyl ketone not undergoing attack. The ensuing liberation of carbon dioxide from the resulting β-lactone affords optically active 4.16
In summary, we have developed a highly selective route to α,α-disubstituted cyclopentenes catalyzed by N-heterocyclic carbenes. This new process combines an enantioselective aldol reaction with a decarboxylation of a β-lactone intermediate to afford functionalized carbocycles containing a quaternary carbon stereocenter. The use of chiral triazolium salts generates chiral nucleophilic enols capable of promoting a desymmetrization event. The investigation and applications of nucleophiles generated in situ from the combination of unsaturated aldehydes and NHCs are ongoing.
Supplementary Material
Acknowledgments
Research support was generously provided by NIH/NIGMS (RO1 GM73072), Abbott Laboratories, Amgen, 3M, and Boehringer Ingelheim. We thank FMCLithium and BASF for providing reagents. Funding for the NU Analytical Services Laboratory has been furnished in part by the NSF (CHE-9871268). K.A.S. is a fellow of the A. P. Sloan Foundation.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis. 1–3 Springer; Berlin, Germany: 1999. [Google Scholar]; (b) Ojima I, editor. Catalytic Asymmetric Synthesis. 2. Wiley; New York: 2000. [Google Scholar]
- 2.(a) Corey EJ, Guzman-Perez A. Angew Chem, Int Ed. 1998;37:389–401. doi: 10.1002/(SICI)1521-3773(19980302)37:4<388::AID-ANIE388>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; (b) Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tian SK, Chen YG, Hang JF, Tang L, McDaid P, Deng L. Acc Chem Res. 2004;37:621–631. doi: 10.1021/ar030048s. [DOI] [PubMed] [Google Scholar]; (d) O’Donnell MJ. Acc Chem Res. 2004;37:506–517. doi: 10.1021/ar0300625. [DOI] [PubMed] [Google Scholar]; (e) Ramon DJ, Yus M. Angew Chem, Int Ed. 2005;44:1602–1634. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Jiang CH. Synthesis. 2006:369–396. [Google Scholar]
- 3.(a) Willis MC. J Chem Soc, Perkin Trans 1. 1999:1765–1784. [Google Scholar]; (b) Chen YG, McDaid P, Deng L. Chem Rev. 2003;103:2965–2983. doi: 10.1021/cr020037x. [DOI] [PubMed] [Google Scholar]; (c) France S, Guerin DJ, Miller SJ, Lectka T. Chem Rev. 2003;103:2985–3012. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]; (d) Garcia-Urdiales E, Alfonso I, Gotor V. Chem Rev. 2005;105:313–354. doi: 10.1021/cr040640a. [DOI] [PubMed] [Google Scholar]; (e) Liu Q, Rovis T. J Am Chem Soc. 2006;128:2552–2553. doi: 10.1021/ja058337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nair V, Vellalath S, Poonoth M, Suresh E. J Am Chem Soc. 2006;128:8736–8737. doi: 10.1021/ja0625677. [DOI] [PubMed] [Google Scholar]
- 5.For a catalytic enantioselective approach to bicyclic β-lactones, see: Cortez GS, Tennyson R, Romo D. J Am Chem Soc. 2001;123:7945–7946. doi: 10.1021/ja016134+.Purohit VC, Richardson RD, Smith JW, Romo D. J Org Chem. 2006;71:4549–4558. doi: 10.1021/jo060392d.
- 6.For an NHC-catalyzed route to β-lactones, see: Burstein, C.; Tschan, S.; Xie, X.; Glorius, F. Synthesis 2006, 2418–2439.
- 7.Chiang PC, Kaeobamrung J, Bode JW. J Am Chem Soc. 2007;129:3520–3521. doi: 10.1021/ja0705543. [DOI] [PubMed] [Google Scholar]
- 8.(a) Mattson AE, Bharadwaj AR, Scheidt KA. J Am Chem Soc. 2004;126:2314–2315. doi: 10.1021/ja0318380. [DOI] [PubMed] [Google Scholar]; (b) Mattson AE, Scheidt KA. Org Lett. 2004;6:4363–4366. doi: 10.1021/ol0481129. [DOI] [PubMed] [Google Scholar]; (c) Chan A, Scheidt KA. J Am Chem Soc. 2006;128:4558–4559. doi: 10.1021/ja060833a. [DOI] [PubMed] [Google Scholar]; (d) Maki BE, Chan A, Phillips EM, Scheidt KA. Org Lett. 2007;9:371–374. doi: 10.1021/ol062940f. [DOI] [PubMed] [Google Scholar]
- 9.(a) Chan A, Scheidt KA. Org Lett. 2005;7:905–908. doi: 10.1021/ol050100f. [DOI] [PubMed] [Google Scholar]; (b) Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334–5335. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Phillips EM, Wadamoto M, Chan A, Scheidt KA. Angew Chem, Int Ed. 2007;46:3107–3110. doi: 10.1002/anie.200605235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For recent NHC-catalyzed processes with proposed enol intermediates, see: Reynolds NT, Rovis T. J Am Chem Soc. 2005;127:16406–16407. doi: 10.1021/ja055918a.He M, Struble JR, Bode JW. J Am Chem Soc. 2006;128:8418–8420. doi: 10.1021/ja062707c.
- 11.IMes = bis(1,3-(2,4,6-trimethylphenyl)imidazol-2-ylidene).
- 12.See ref 9c.
- 13.For recent examples of catalytic reactions employing loss of carbon dioxide, see: Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x.Burger EC, Tunge JA. Org Lett. 2004;6:2603–2605. doi: 10.1021/ol049097a.Myers MC, Bharadwaj AR, Milgram BC, Scheidt KA. J Am Chem Soc. 2005;127:14675–14680. doi: 10.1021/ja0520161.Trost BM, Xu J. J Am Chem Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c.Rayabarapu DK, Tunge JA. J Am Chem Soc. 2005;127:13510–13511. doi: 10.1021/ja0542688.Magdziak D, Lalic G, Lee HM, Fortner KC, Aloise AD, Shair MD. J Am Chem Soc. 2005;127:7284–7285. doi: 10.1021/ja051759j.
- 14.For early uses of chiral triazolium catalysis, see: Enders D, Breuer K, Teles JH. Helv Chim Acta. 1996;79:1217–1221.Enders D, Breuer K, Runsink J, Teles JH. Helv Chim Acta. 1996;79:1899–1902.
- 15.(a) β-Lactones typically undergo decarboxylation at high temperatures or using acid catalysis: Mulzer J, Zippel M. Chem Commun. 1981:891–892.Mulzer J, Zippel M. Tetrahedron Lett. 1987;21:751–754.While the aromatic substituent R may facilitate the decarboxylation, current investigations are ongoing to understand the facile nature of this process.
- 16.Absolute configuration of 4 determined by X-ray crystallographic analysis. See Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.