

Abstract
Stargardt-like macular degeneration (STGD3) is an early onset, autosomal dominant macular degeneration. STGD3 is characterized by a progressive pathology, the loss of central vision, atrophy of the retinal pigment epithelium, and accumulation of lipofuscin, clinical features that are also characteristic of age-related macular degeneration. The onset of clinical symptoms in STGD3, however, is typically observed within the second or third decade of life (i.e., starting in the teenage years). The clinical profile at any given age among STGD3 patients can be variable suggesting that, although STGD3 is a single gene defect, other genetic or environmental factors may play a role in moderating the final disease phenotype. Genetic studies localized the STGD3 disease locus to a small region on the short arm of human chromosome 6, and application of a positional candidate gene approach identified protein truncating mutations in the elongation of very long chain fatty acids-4 gene (ELOVL4) in patients with this disease. The ELOVL4 gene encodes a protein homologous to the ELO group of proteins that participate in fatty acid elongation in yeast. Pathogenic mutations found in the ELOVL4 gene result in altered trafficking of the protein and behave with a dominant negative effect. Mice carrying an Elovl4 mutation developed photoreceptor degeneration and depletion of very long chain fatty acids (VLCFA). ELOVL4 protein participates in the synthesis of fatty acids with chain length longer than 26 carbons. Studies on ELOVL4 indicate that VLCFA may be necessary for normal function of the retina, and the defective protein trafficking and/or altered VLCFA elongation underlies the pathology associated with STGD3. Determining the role of VLCFA in the retina and discerning the implications of abnormal trafficking of mutant ELOVL4 and depleted VLCFA content in the pathology of STGD3 will provide valuable insight in understanding the retinal structure, function, and pathology underlying STGD3 and may lead to a better understanding of the process of macular disease in general.
Keywords: STGD3, ELOVL4, Fatty acids, Dominant negative effect, Protein accumulation, Animal models
1. Introduction
1.1. Overview of Stargardt-like macular degeneration
Autosomal dominant Stargardt-like macular degeneration, Stargardt disease 3 (STGD3), is one of many disorders that affect the central region of the retina, the macula. Macular dystrophies are the leading cause of visual impairment leading to irreversible blindness in the developed world (Leibowitz et al., 1980; O'Shea, 1996; Starr et al., 1998; Congdon et al., 2004). Some of these dystrophies constitute a genetically heterogenous group of disorders that are inherited in a simple Mendelian fashion and are initially characterized by the degeneration of the macula leading to a gradual loss of visual acuity. A number of genes throughout the genome have become associated with simple macular degenerative disease (MD) as well as age-related macular degeneration (AMD; Fig. 1A). AMD is the leading cause of blindness in individuals over the age of 60 years and is a complex multifactorial disease that can involve multiple environmental and genetic factors. The strongest risk factors of AMD that influence its manifestation are age, family history, and lifestyle such as diet, smoking, alcohol consumption, and exposure to sunlight (Seddon et al., 1996; 2001; 2003a; 2003b; 2005; Cho et al., 2000; Cho et al., 2001; Evans, 2001; Hyman and Neborsky, 2002; Clemons et al., 2005; Haddad et al., 2006). The likelihood of a genetic component contributing to AMD pathogenesis is supported by family and twin studies (Meyers, 1994; Klaver et al., 1998; De Jong et al., 2001). In addition, a number of gene variants associated with AMD, such as specific complement factor-H and B (CFH and CFB) alleles among others, have been identified that modify the risk for developing AMD (Edwards et al., 2005; Haines et al., 2005; Klein et al., 2005; Gold et al., 2006; Yates et al., 2007; Swaroop et al., 2009). Macular degeneration and AMD can be characterized by drusen, atrophy of the retinal pigment epithelium (RPE), and choroidal neovascularization. Drusen, the earliest sign of AMD, is an accumulation of extracellular deposits that contains lipofuscin and other proteins (Ben-Shabat et al., 2001; Johnson et al., 2001; Penfold et al., 2001; Ben-Shabat et al., 2002; Barthes et al., 2005; Bressler et al., 2005; Umeda et al., 2005; Crabb et al., 2002; Sakaguchi et al., 2002). The multifactorial nature and late age of onset of AMD make it difficult to study and fully understand the disease process. A more general approach to the understanding of AMD has been to study retinal degenerative diseases with simple modes of inheritance that share characteristic clinical features with the more complex AMD state. The rationale in this approach is that if AMD and MD share clinical features, then the underlying cellular processes that define the phenotype must be functionally related. In support of this approach we found that a meta-analysis of 30 genes known to underlie MD conditions and/or represent AMD-associated genes (Fig. 1) generated a single macular degeneration–associated functional interactome (Fig. 2), suggesting that many genes involved in MD and associated with AMD are functionally related. Here we review the understanding of one disorder, STGD3, examine the function of the gene (ELOVL4) that underlies it, and reflect on its relevance with respect to AMD.
Figure 1. Summary of known MD and AMD loci.
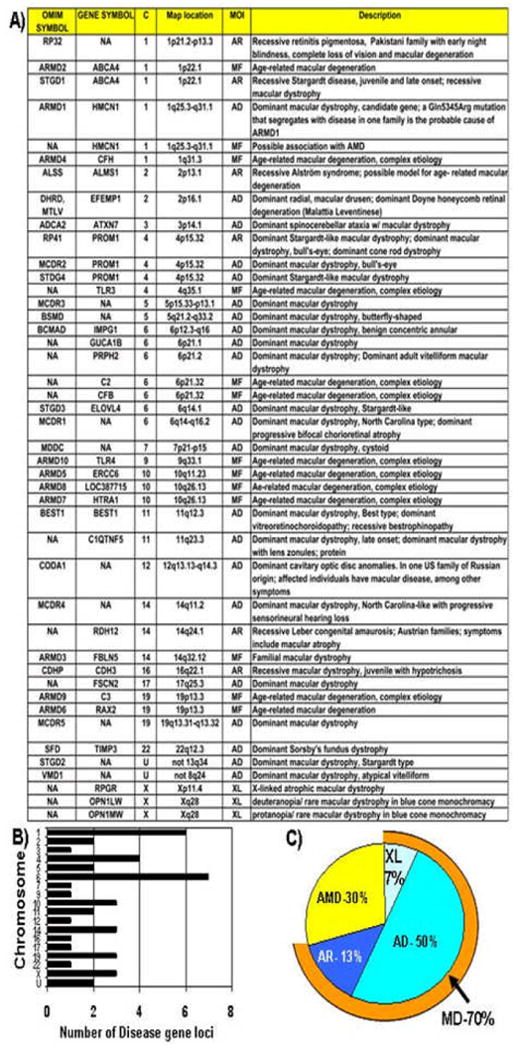
A) A table summarizing known MD and AMD loci generated from selective data mining of the Retnet database for the words macula and fundus (http://www.sph.uth.tmc.edu/retnet/). In some cases different mutations in the same gene define distinct disorders. MOI, mode of inheritance; AD, autosomal dominant; AR, autosomal recessive; XL, X-linked; MF, multifactorial.
B) Profile of macular disease genes or related genes in the human genome. Loci summarized in A are distributed throughout the genome on 18 human chromosomes. There are apparent hot spot of MD and AMD loci on chromosomes 1 and 6.
C) Distribution of MD and AMD loci according to mode of inheritance. Approximately 2/3 of the macular disease loci ascertained correspond to disorders inherited as a single gene defect by means of a standard Mendelian mode of inheritance (AD, AR, XL). One third of the loci correspond to AMD-related genes governed by a more complex mode of inheritance (MF).
Figure 2. MD and AMD interactome.
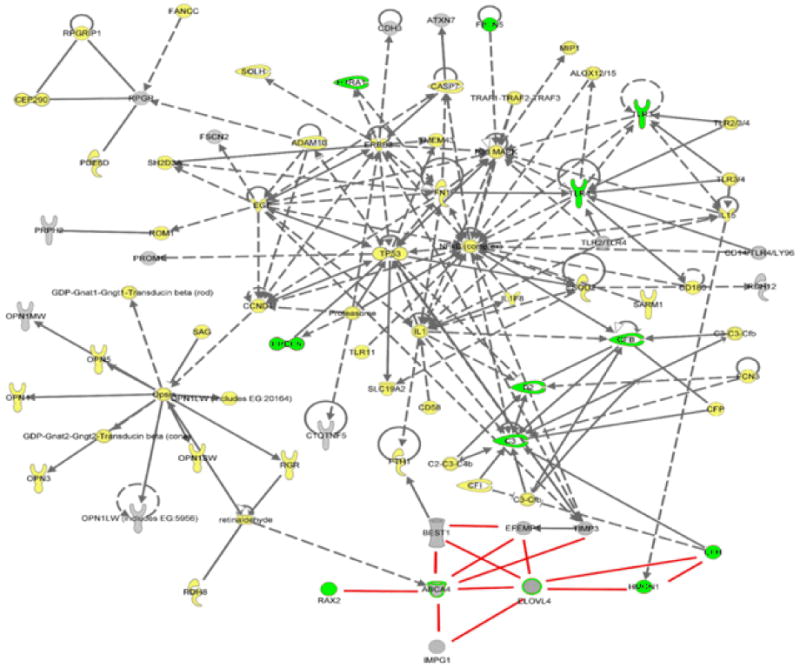
Gene loci underlying MD (Gray) and/or AMD (Green) conditions were submitted to Ingenuity (http://www.ingenuity.com/) and String databases (http://string.embl.de/) to define potential functional interacting pathways. The generation of a single interactome suggests that the MD/AMD gene loci/gene products considered are functionally related. Ingenuity incorporated additional gene loci/proteins (yellow) as part of the pathway stemming from the initial input of the disease genes. Direct interactions are shown in solid lines (for example, protein to protein binding, phosphorylation, etc.); indirect interactions are shown in dashed lines (for example, effects through signaling pathways, expression); and undefined interactions are shown as red lines. Proteins are also functional coded:
| Cytokine/growth factor | Ion channel | ||
| Chemical/toxicant | Peptidase | ||
| Enzyme | Transcription regulator | ||
| G-protein coupled receptor | Transmembrane receptor | ||
| Group/complex/ | other transporter | ||
| Growth factor | Undefined |
1.2. Clinical studies of STGD3
Stargardt-like macular degeneration (STGD3), as its name implies, has a clinical profile that is very similar to Stargardt macular degeneration (STGD1). STGD1 is the most frequent juvenile macular dystrophy and is characterized by progressive loss of central vision, starting within the first few decades of life, atrophy of the macula, degeneration of the underlying RPE, and frequent presence of prominent yellow flecks in the retina (Stargardt, 1909; Blacharski, 1988; Kaplan et al., 1990). The yellow flecks represent an accumulation of fluorescent lipofuscin deposits in the RPE (Stargardt, 1909; Hadden and Gass, 1976; Noble and Carr, 1979; Eagle et al., 1980; Lopez et al., 1990). These clinical features of STGD1 are consistent in STGD3 as well as AMD (Young, 1987). STGD1 can be distinguished from other macular degenerations by its autosomal recessive pattern of inheritance, minimal color vision abnormality, and normal electrophysiology. Moreover, a hallmark feature of STGD1 that differentiates it from most other macular dystrophies is the presentation of a dark choroid during a fluorescein angiography examination. A dark choroid occurs because STGD1 patients express an abnormal material at the level of the RPE that masks detection of the fluorescein that has been introduced into the choroidal circulation. Clinical features of STGD3 include progressive loss of central vision starting as early as the second decade of life, but onset of the disease can vary between the second and fifth decade of life. Affected STGD3 individuals can have variable visual acuities ranging from 20/50 to 20/200 (or worse); part, typical STGD3 patients experience visual acuities less than 20/200 at older ages with minimal to no color vision defects and no significant changes in electroretinogram (ERG) (Donoso and Martens, 2001). This is not an absolute STGD3 clinical phenotype, and the disease has a wide degree of variability.
The fundus defects are progressive and can be marked by atrophic macular changes with (Fig. 3D) or without flecks (Fig. 3A–C). Historically, STGD3 was first observed in a four-generation pedigree of a family reported by Klien and Krill (1967). STGD3 typically does not present with a dark choroid, a hallmark feature in patients with STGD1. A second family with 99 members, expressing an apparently dominant form of macular dystrophy with fundus flecks was reported by Cibis et al. (1980). The ERGs were reported to be normal in all the affected members of the family. Fifty percent of the affected members in this family showed normal color vision and some patients had flecks. Additional families with autosomal dominant Stargardt-like disease were subsequently reported (Donoso et al., 2001b; Lopez et al., 1990; Mansour, 1992; Aaberg, 1986).
Figure 3. Fundus photographs of two individuals from a large Canadian STGD3 pedigree, an affected STGD3 male member and his affected nephew. (Photographs taken from Lagali et al., 2000 and used with permission from the Canadian Journal of Ophthalmology).
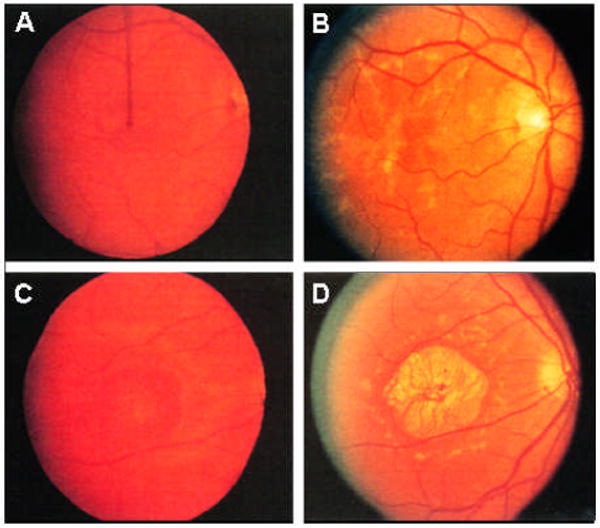
Fundus examination reveals a variety of progressive abnormalities, from RPE defects (A,C) to pattern dystrophy (B) and macular atrophy surrounded by flecks (D). Both individuals show a consistent progressive loss of visual acuity over time, but the rate at which this occurs and the degree of changes in the fundus is markedly different between the two affected individuals
Fundus photograph of the right eye of the STGD3 affected Uncle at age 39 years with a visual acuity of 20/30 (A) and again at the age of 48 with a visual acuity of 20/80 (B).
Fundus photographs of right eye of the nephew at 21 years of age with a visual acuity of 20/200 (C) and after a reexamination at age 43(D) with a visual acuity of 20/400.
1.3. Human genetic studies of STGD3
A summary of the genetic studies performed to define and map the STGD3 locus in the human genome is presented in Figure 4. Stone et al. (1994) performed a genetic linkage study in a large family with autosomal dominant Stargardt-like macular dystrophy that had clinical features consistent with the above STGD3 phenotype with known genetic markers and the disease locus in question. The logic of a linkage study is straightforward—if an unmapped locus is found to be linked to a genetic marker with a known map location then the unmapped locus must also be in the vicinity of the marker with the known location. Total LOD scores (the end metric of a linkage study) that are 3 or greater indicate that the two genetic loci or markers being tested are genetically linked at the genetic distance considered (theta value). LOD scores that fall to a value of −2 or lower lead to the conclusion that the two loci or markers being tested are not linked. Linkage analysis of the STGD3 family excluded the STGD1 locus and mapped the disease locus to chromosome 6 (Stone et al., 1994). Multipoint analysis resulted in a peak LOD score of 6.2 in the interval between markers D6S313 and D6S252 (Fig. 4). Subsequently, a common founder haplotype between D6S430 to D6S300 was found in two independently ascertained STGD3 pedigrees (Edwards et al., 1999). Linkage studies on various branches of the pedigree confirmed mapping of the STGD3 locus to chromosome 6 (Edwards et al., 2001).
Fig. 4. STGD3 maps to human chromosome 6.
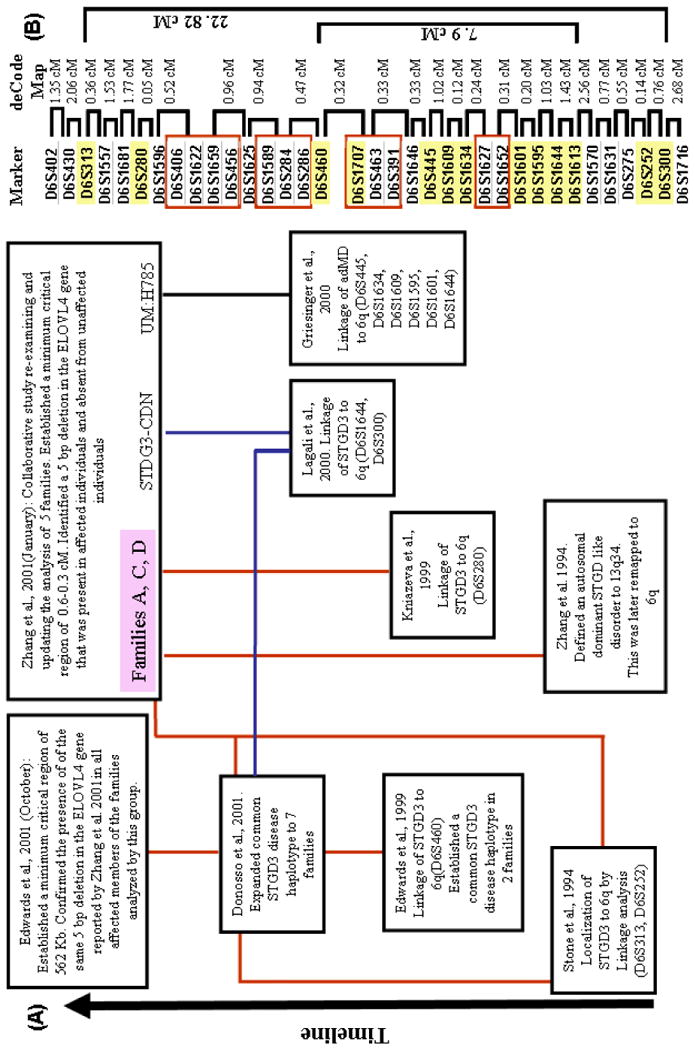
(A) Summary of linkage studies localizing the STGD3 disease locus to the long arm of human chromosome 6. (B) A list of microsatellite gene markers that defines the relative region to which STGD3 maps. Markers highlighted in yellow represent markers in which formal levels of accepting linkage were obtained in the linkage studies summarized.
In contrast to the families reported previously (Stone et al., 1994; Edwards et al., 1999; Donoso et al., 2001a; Donoso et al., 2001b), affected members in a large multigeneration Canadian family presented with a later age of disease onset, with symptoms usually appearing in the fourth or fifth decade of life. Fundus examinations of affected members showed a variety of progressive abnormalities from RPE defects to pattern dystrophy to an atrophic macular region surrounded by flecks (Lagali et al., 2000; Fig. 3). Visual acuity and color discrimination in affected patients displayed progressive deterioration with increasing age. Fluorescein angiography revealed the absence of a dark choroid. Photopic and scotopic ERG appeared normal in the majority of affected members tested. One patient did, however, present with an abnormal ERG indicative of a cone and rod dysfunction. The clinical presentation in this family, with the exception of the age of onset, was similar to STGD3. Linkage analysis of 27 members of this family with known chromosome 6 markers demonstrated a significant LOD score of 5.50 at a theta value of 0 with D6S300. Haplotype analysis of all available family members indicated that the disease locus was within a minimum critical region flanked by D6S271 and D6S1716, which overlaps the general region previously implicated (Stone et al., 1994).
A family with autosomal dominant macular dystrophy, characterized by progressive retinal pigment epithelial atrophy in the macula with flecks but without apparent peripheral changes, has been reported (Griesinger et al., 2000). Loss of visual acuity progressed with age, with higher levels of visual loss in older affected individuals. Rod and cone function were normal in all individuals tested up to the age of 61 years. Color vision in all but one of these patients was normal. A dark choroid was not reported in any of the affected members. The clinical phenotype in this family was noted to be similar to STGD3, but with a highly variable severity. The disease gene in this family was mapped to chromosome 6q to a 1.8 cM interval overlapping the STGD3 interval. Levels for the acceptance of formal linkage were obtained for D6S445, D6S1634, D6S1609, D6S1595, D6S1601, and D6S1644 (Griesinger et al., 2000). The critical region for STGD3 was minimized to a 0.6 to 0.3 cM region between markers D6S460 and D6S391 by combining the results from multiple studies (Griesinger et al., 2000; Lagali et al., 2000; Zhang et al., 2001). Further genetic analysis established a minimal critical region for STGD3 between D6S460 and D6S1707 (Edwards et al., 2001; Fig. 4). Two important observations can be made on the large number of independent STGD3 family studies. First, the localization of the disease locus for STGD3 to chromosome 6 has been confirmed a number of times. Second, there exists a widely variable clinical phenotype between affected individuals from different families and even between affected individuals in the same family.
1.4 Analysis of STGD3 candidate gene
With the STGD3 minimal critical region being less than 1 cM, a positional candidate gene approach was taken, which led to the identification of a 5-bp deletion in exon 6 (790–794 del AACTT) of the ELOVL4 (elongation of very long chain fatty acids) gene that cosegregated with the disease (Zhang et al., 2001). The deletion results in a frameshift mutation that leads to an inappropriate stop codon, loss of the C-terminal 51 amino acids, and aberrant sequence from amino acid 264 to 271 (Fig. 5). This mutation was subsequently confirmed in a large extended pedigree (Edwards et al., 2001).
Figure 5. Schematic representation of human wild type and different known mutations in ELOVL4 gene and their protein products.
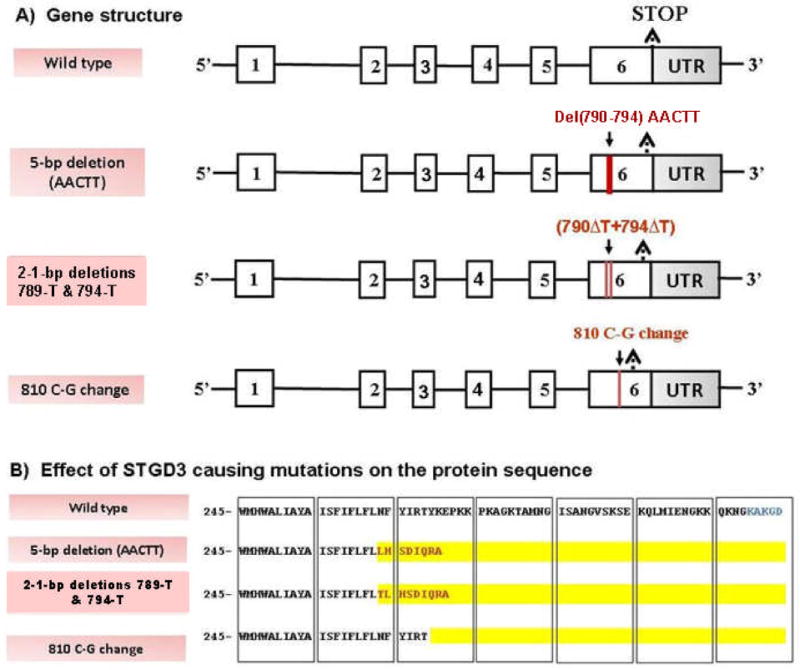
(A) ELOVL4 gene structure. Boxes depict exons and intervening lines depict introns. ELOVL4 consists of six exons and five introns and codes for a 3085-nucleotide transcript. All known STGD3-causing mutations (red lines depicting deletions or nucleotide changes) fall in exon 6 of the ELOLV4 gene. (B) Effect of STGD3-causing mutations on the protein sequence. Sequences of wild type and mutant proteins starting at amino acid 245 are shown. For the mutant ELOVL4 protein, changes in amino acid sequence as compared to wild type are indicated in red type. Amino acid regions that are deleted from the sequence are highlighted in yellow. For all three known STGD3 mutations the end effect is a premature stop to the protein sequence and a deletion of the endoplasmic reticulum retention signal (indicated in blue type in the wild type sequence).
In an independent Utah family with a STGD3-like phenotype, a complex mutation of two 1-bp deletions separated by four nucleotides (789ΔT+794ΔT) in the ELOVL4 gene was detected in all affected members of the family and was absent from all unaffected individuals screened (Bernstein et al., 2001). The mutation results in a frameshift and leads to the truncation of the ELOVL4 protein with an aberrant sequence for the last nine amino acids, similar to the effect of the 5-bp deletion mutation. In an unrelated European family with STGD3 phenotype, a point mutation (C-to-G change) at nucleotide 810 (810C→G) in exon 6 of the ELOVL4 gene was observed (Maugeri et al., 2004). The mutation results in a substitution of a stop codon for tyrosine 270 (Y270X) and leads to a truncated protein that is missing the last 45 amino acids. The discovery of three different mutations (Fig. 5) in the ELOVL4 gene segregating with a STGD3 phenotype confirmed the role of ELOVL4 in autosomal dominant macular dystrophies.
2. Molecular and Cellular Analysis of STGD3-Causing Gene, ELOVL4
2.1. Molecular analysis of ELOLV4
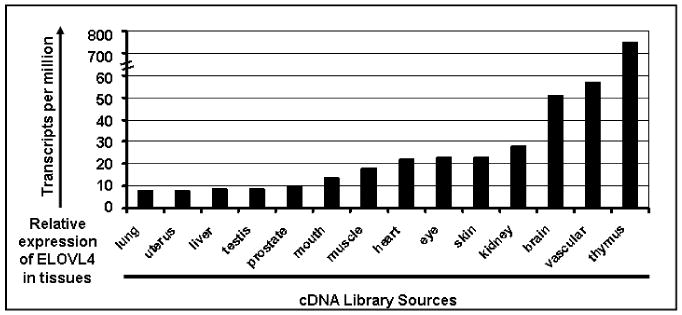
Human ELOVL4 is a 32.7-kb gene with six exons. ELOVL4 encodes a putative protein of 314 amino acids with approximately 35% sequence homology to the GNS1/SURF4 family of elongases, which are involved in fatty acid (FA) chain elongation in yeast (Oh et al., 1997; Zhang et al., 2001). The yeast ELO genes encode components of the membrane-bound FA elongation system. Human ELOVL4 shares all three characteristic features typical for members of the ELO family: five putative transmembrane segments, a single dioxy iron binding motif (HXXHH), and a carboxy terminal dilysine motif for the retention of the transmembrane protein in endoplasmic reticulum (ER), which is a known site for FA chain synthesis. In the case of all three STGD3 disease–causing alleles, the observed mutations resulted in a loss of the genetic information for the dilysine motif required for ER retention of the protein product. Characteristic features of the ELOVL4 gene and its expression are summarized in Table 1. These studies verify that ELOVL4 is an evolutionary conserved gene that is expressed in rod and cone photoreceptor cells of the retina. Although ELOVL4 is expressed in the retina, ELOVL4 expression is not retina specific (Fig. 6). Aside from the retina, high levels of ELOVL4 mRNA are also expressed in skin, brain, and testis (Mandal et al., 2004). Recently studies on pancreatic ductal adenocar-cinoma demonstrated that 68% of ELOVL4 undergoes methylation and the differ-entially methylated ELOVL4 could be used as a marker for pancreatic ductal adenocar-cinoma (Omura et al., 2008).
Table 1. Characteristic features of ELOVL4.
| Observation | Method | Reference |
|---|---|---|
| At the level of DNA, ELOVL4 is an evolutionarily conserved nucleotide sequence | Bioinformatic analysis, Southern Analysis | Zhang et al., 2001; Lagali et al., 2003 |
| ELOVL4 mRNA is expressed early in development as well as in the adult animal | qRT-PCR, In situ hybridization | Zhang et al., 2003; Mandal et al., 2004 |
| ELOVL4 mRNA is expressed in rod and cone photoreceptor cells of the monkey and mouse retina | In situ hybridization | Zhang et al., 2001 |
| ELOVL4 mRNA is expressed in the retina across several different species | Northern analysis, qRT-PCR analysis | Edwards et al., 2001; Zhang et al., 2001; Lagali et al., 2003; Mandal et al., 2004 |
| ELOVL4 mRNA is retina expressed but not retina specific Northern analysis | qRT-PCR | Lagali et al., 2003; Mandal et al., 2004 |
| At the level of the protein sequence, ELOVL4 is highly similar across species | Bioinformatic analysis | Zhang et al., 2001; Lagali et al., 2003 |
| ELOVL4 protein is present as a 37 kDa protein in mammalian retina (mouse, cow, cat, rat, human) | Western analysis | Lagali et al., 2003 |
| ELOVL4 protein is present in the inner and outer segment regions of photoreceptor cells in the retina | Immunohistochemical analysis | Lagali et al., 2003 |
| Within the cell, wild type ELOVL4 protein localizes to the endoplasmic reticulum, the site of fatty acid synthesis | Transfection of cells in culture with an ELOVL4 tagged construct | Ambasudhan et al., 2004 |
| ELOVL4 participates in the elongation of VLCFA (C> 26) | Transfection of rat neonatal cardiomyocytes with Elovl4 and supplemented with 24:0, 20:5n3, or 22:5n3 FA | Agbaga et al., 2008 |
Figure 6. Visual expression profiling based on Unigene data for identified ELOVL4 Expressed Sequence Tags (ESTs). (http://www.ncbi.nlm.nih.gov/UniGene/ESTProfileViewer.cgi? uglist= Hs.101915).
2.2. Cellular characterization of STGD3 candidate gene, ELOVL4
Immunofluorescence analysis of COS-7 cells or HEK 293 cells transfected with a green fluorescent protein (GFP) tagged or wild type (Wt) ELOVL4 construct revealed a predominant localization of the ELOVL4 protein to the endoplasmic reticulum (ER). In contrast cellular transfection with constructs representing any of the three known STGD3 mutant ELOVL4 alleles resulted in mislocalization of the mutant protein to a juxtanuclear region (Ambasudhan et al., 2004; Karan et al., 2004b). This was later verified to be mislocalization of mutant ELOVL4 into aggresomes (Grayson and Molday, 2005; Karan et al., 2005b; Vasireddy et al., 2005). ELOVL4 protein with a direct deletion or substitution of the dilysine motif that defines the ER retention signal also resulted in the loss of ER localization (Ambasudhan et al., 2004; Karan et al., 2005b). The results imply the biological significance of the ELOVL4 ER retention signal. The ER localization of Wt ELOVL4 would be essential for it to participate in the elongation of FA, since the ER is the site of FA biosynthesis and elongation (Cinti et al., 1992). With respect to the STGD3-causing 5-bp ELOVL4 deletion mutant, it was observed that transfection of HEK 293 cells with this ELOVL4 mutant construct resulted in a significantly higher level of apoptosis compared with HEK 293 cells transfected with the vector alone or a Wt ELOVL4 construct (Karan et al., 2004a). Moreover, transfection of cells with the mutant construct correlated with a cellular induction of biological markers that define the unfolded protein response (UPR) mechanism. The key steps for proper maturation and function of proteins depend on the folding of the nascent polypeptide chains and post-transcriptional modifications in the ER. If the influx of unfolded or mutant protein that cannot be folded properly exceeds the capacity of the ER resident protein-folding machinery, the normal physiology of the ER is disturbed and the UPR is activated (Pahl, 1999; Kopito and Ron, 2000; Harding et al., 2002). Neurodegenerative diseases, such as Alzheimer disease, Parkinson disease, and retinitis pigmentosa, belong to a group of protein misconformation disorders that are associated with accumulation of abnormal protein aggregates in cells (Saliba et al., 2002; Ross and Poirier, 2004). The pathogenesis of these diseases is mediated by a defect in protein folding, a saturated proteosome, activation of the UPR, and progression to active cell death. Despite progress on establishing the role of UPR and ER stress in STGD3, our knowledge of the mechanism underlying the aggregation of mutant ELOVL4 and the associated cell death remains incomplete. Further studies promise to expand our understanding of how ER stress impacts the cellular signaling pathways that define the STGD3 disease condition.
2.3. Dominant negative effect exerted by mutant ELOVL4 protein
Given that STGD3 is inherited as an autosomal dominant trait, additional cell-based studies were pursued to examine the mechanism that defines the dominant nature of the disease. COS-7 cells in culture were simultaneously transfected with both Wt and mutant ELOVL4 constructs and the co-expressed recombinant proteins were subjected to the same subcellular localization studies as described under section 2.2 (Fig. 7). These studies led to the conclusion that the mutant ELOVL4 protein physically interacts with Wt ELOVL4 and that this complex mislocalizes to form perinuclear aggregates (Grayson and Molday, 2005; Karan et al., 2005b; Vasireddy et al., 2005). Formation of Wt/mutant ELOVL4 aggresomes is evident by the reorganization of the major cytoskeletal elements, vimentin, and colocalization of ELOVL4 aggregates with γ-tubulin, a marker of the microtubule organization center (Johnston et al., 1998). Furthermore, all known ELOVL4 mutations inhibited the localization of Wt ELOVL4 to the ER and resulted in formation of juxtanuclear aggresomes. These data suggest that the STGD3-causing ELOVL4 mutations act in a dominant negative manner and cell death may be induced by the formation of aggresomes.
Figure 7. Mutant ELOVL4 protein expression results in aggresome formation.
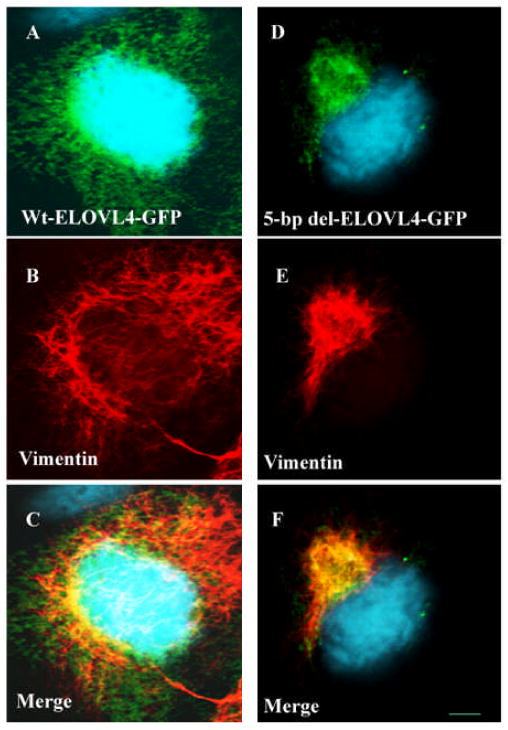
An expression construct was created to express the wild type ELOVL4 allele fused to a green fluorescent protein marker (EGFP-Wt ELOVL4). A second expression construct was created to express the STGD3-causing 5-bp deletion ELOVL4 allele, also fused to a green fluorescent protein marker (EGFP- 5-bp del-ELOVL4). Cos-7 cells grown on dual chambers were transfected with EGFP-Wt ELOVL4 or EGFP-mut ELOVL4 using lipofectamine plus reagent. Post-transfected cells were fixed with methanol and processed for immunocytochemistry using antibodies specific to vimentin and visualized with Alexa fluor 555 probe–tagged secondary antibody. The Alexa fluor 555 probe fluoresces under a different wavelength than GFP. Fluorescence images were acquired using appropriate filters and lasers. GFP is imaged as green fluorescence, vimentin is detected as red fluorescence, and blue fluorescence results from 4′,6-diarnidino-2-phenylindole (DAPI) staining of the nucleus. GFP-ELOVL4 fluorescence is demonstrated in panels A D, and vimentin distribution is shown in panels B,E. Panels C,F are the merged image of panels A,B and D,E. Formation of aggresomes is associated with the reorganization of intermediary filament protein, vimentin. In Wt-ELOVL4– transfected cells, vimentin was found to be distributed in a reticulate pattern all over the cells, where as in cells expressing mutant-ELOVL4 protein, distribution of vimentin was found to be altered. Vimentin labeling in these cells was observed to be relocated to the perinuclear region where mutant ELOVL4 is accumulated as an aggregate. Formation of aggresome in mutant ELOVL4 transfected cells is visualized by this reorganization of vimentin. Scale bar is 5 μM.
3. Mouse Models of Human STGD3
The effectiveness of a specific gene mutation to cause disease is often evaluated in an animal model. Progress in genetic engineering has led to an increase in the generation of genetically modified animals. The mouse is one of the more popular choices to evaluate whether the incorporation of a specific disease gene results in a disease phenotype. If a specific gene is important to tissue function then the ablation of this gene or incorporation of a defective version of the gene in question should result in a defect in that tissue. The advantages of using a mouse model include the facts that the mouse genome has a 90% overall similarity to the human genome, the lifespan of a mouse is relatively short (about 2 years), and mice are inexpensive to maintain. A common criticism of a mouse model system with respect to human macular degeneration is that the mouse retina does not have a macula. However, there is a growing acknowledgment that human macular disease affects the entire retina and not just the macular region, and there have been a number of successes in showing that incorporation of known human macular disease genes or mutations into mice result in retinal disease (Weng et al., 1999; Mears et al., 2001; Ambati et al., 2003; Marmorstein and Marmorstein, 2007).
STGD3 is an autosomal dominant disease in humans. Typically dominant gene mutations can cause a disease phenotype through haploinsufficiency or a dominant negative effect. Haploinsufficiency occurs when a single functional copy of a normal gene does not produce enough protein to establish the Wt condition. In case of a dominant negative effect the altered mutant gene product may affect the Wt product, resulting in a decrease in the amount of functional gene product. To determine the mechanism underlying STGD3, five recombinant mouse models that carry an Elovl4 allele that either expresses a mutant Elovl4 gene product or no Elovl4 at all have been made and evaluated for retinal phenotypes (Table 2). These five mouse models can be divided into three groups: 1) knock-out mouse models generated with an Elovl4 gene deletion; 2) transgenic mouse models generated with an Elovl4 gene
Table 2. Mouse models for human STGD3.
| Reference | Li et al., 2006 | Raz-Prag et al., 2006 | Karan et al., 2005b | Vassireddy et al., 2006 | McMahon et al., 2007 |
|---|---|---|---|---|---|
| Alteration | Gene Deletion | Gene Deletion | Gene Addition | Gene Substitution | Gene Substitution |
| Genetic Change | KO1: A conditional Elovl4 knock-out was generated by inserting a pGKneo cassette into exon 2 of an endogeneous mouse Elovl4 gene. | KO2: A conditional Elovl4 knock-out was generated by inserting a LacZ-pGKneo cassette into exon 2 of an endogeneous mouse Elovl4 gene. | Gene Addition of a human 790-794 delAACTT ELOVL4 gene construct driven with an IRBP promoter into a mouse background. 3 transgenic (TG) lines defined by the relative level of human ELOVL4 expression (TG3>TG2>TG1) were established. | KI mice: Knock in of “mouse 790-794 delAACTT ELOVL4” by homologous recombination into an endogenous mouse ELVOL4 gene. KI mice | KI+2 mice: Knock in of “mouse 790-794 delAACTT ELOVL4 + 2 point mutations” by homologous recombination into an endogenous mouse ELVOL4 gene. |
| Fundus | NA | KO2 +/- mice at 17-19 mo had a normal gross appearance. | 1yo: Severity of fundus abnormalities (depigmented sub-retinal spots and geographic atrophy) correlated with levels of human mutant ELOVL4. | NA | NA |
| ERG Response | KO1 +/- mice at 9 mo showed a slightly altered ERG response within a defined region of light intensities. | KO2 +/- mice at 16 mo showed no significant difference to Wt | Increased loss in ERG response correlated with increased mutant human human ELOVL4 expression (TG3>TG2>TG1). The b-wave response was not detectable at 35 wks in TG3, by 84 wks in TG2 animals, and In 84 wk old TG1 animals b-wave levels were a third of Wt levels. Cone b-waves showed similar patterns. | KI +/-: at 8 mo both b and a-wave amplitudes were significantly higher than Wt mice. By 15 mo the same pattern existed but the levels were no longer significantly different from Wt. | KI+2 +/- mice at 8 mo showed a reduced ERG response as compared to Wt. |
| Neural Retina/PR | LM: KO1 +/- mice, up to 11 mo, presented with a normal retina. At 11 mo ROS appeared shorter and less organized as compared to Wt, although the ONL thickness remained equivalent to Wt. | LM: KO2 +/- mice at 17-19 mo showed shortening of OS and a minor loss of PR. Regions of apoptotic PR were observed. EM: KO2 +/- mice showed disrupted ROS, ROS membrane whorls and irregular gaps between stacks of discs at a higher incidence than age matched Wt. | LM: There is loss of PR 200-300 urn dorsal and ventral from the optic nerve head (severity of PR loss TG3>TG2>TG1). 50% loss of PR cells in TG3 at 6 wks, TG2 at 16 wks, and TG3 at 18mo. EM: ROS morphology is compromised in animals with higher levels of human mutant ELOVL4 expression by 3 wks(TG3). | LM: Progressive changes seen in the ONL from 2-15 mo KI+/- mice. A decrease in cone cell number begin as early as 2mo. Rod loss is apparent by 10 mo. The OPL contained gaps. At the EM level these gaps were surrounded by rod sherules. BA: A significant decrease in FA levels, 20:5n3, 22:5n3, and 24:6n3 was seen in 6 mo KI +/- animals compared to Wt | LM: No retinal degeneration was observed in KI+2 +/-mice at 8 mo. |
| RPE | NA | LM: KO2 +/- mice showed some RPE irregularities (wavy appearance of the basal RPE-Bruch's memberane interface, and occasional presence of more than one layer of RPE cells), Otherwise the RPE appeared normal. | LM/EM: Accumulation of lipofuscin in the RPE could be observed in TG1 and TG2. By 6 mo in TG3 there is marked RPE atrophy. BA: High levels of A2E and all-trans retinal were observed in TG2 RPE as compared to age matched Wt. | LM: KI +/- mouse abnormalities include thickened RPE and vacuoles, packets of OS, and irregular shaped dense pigment granules (lipofuscin-like) in the RPE cytoplasm. KI +/-animals 8 mo and older show sub-retinal debris. BA: A2E and lso-A2E levels were significantly increased by 4 mo. | BA: KI+2 +/- A2E levels were comparable to Wt, but significant increases in A2E precursor levels were seen in eyecups of 9 mo KI+2 +/-animals. |
Abbreviations: TG, transgenic; KI, knock in; KO, knock out; del, deletion; Wt, wild type; yo, year old; mo, months old; wk, weeks; LM, light microscopy; EM, electron microscopy; BA. biochemical analysis; PR, photoreceptor cells; ROS, rod outer segments; ONL: Outer nuclear layer; OS: Outer segments; OPL, outer plexiform layer; RPE, retinal pigment epithelium; HZ, heterozygous; NA, not applicable.
3.1. Retinal phenotype in heterozygous Elovl4 knock-out mouse models
Two Elovl4 knock-out mouse models (KO1 and KO2) have been generated by a targeted deletion of Elovl4 in exon 2 in order to determine if haploinsufficency is responsible for a disease phenotype (Raz-Prag et al., 2006; Li et al., 2007a). The result of the targeted deletion within exon 2 of Elovl4 is the production of an Elovl4 allele that does not lead to the expression of ELOVL4 protein. In both models only minor differences were detected in the retinal morphology and ultrastructure of heterozygous knockout animals when compared to Wt control mice, suggesting that haploinsufficiency is not the molecular mechanism underlying STGD3.
3.2. Retinal phenotype in ELOVL4 transgenic mouse model
A number of mouse models for STGD3 using either gene addition or gene substitution methods have been engineered to test if a dominant negative effect might underlie the STGD3 disease phenotype. Three different transgenic animal lines (transgenic line 1, 2, and 3 [TG1, TG2, and TG3]) were generated by integration of copies of the human ELOVL4 5-bp deletion mutant transgene driven by a photoreceptor-specific promoter, IRBP, into the mouse genome (Karan et al., 2005a). The three different transgenic lines vary with respect to the expression level of the human ELOVL4 transgene. Mice from all three transgenic lines developed a progressive retinal degeneration. The severity of degeneration of photoreceptors was proportional to the expression levels of the mutant ELOVL4 transgene. In addition, lipofuscin accumulation was observed. In this model, addition of the human defective ELOVL4 gene into a mouse genetic background resulted in retinal degeneration.
3.3. Retinal phenotype in heterozygous ELOVL4 5-bp deletion knock-in mouse models
In two independently generated knock-in mouse models, the 5-bp deletion that defines a human STGD3 mutation was introduced into a single copy of the mouse Elovl4 homolog by homologous recombination (Vasireddy et al., 2006; McMahon et al., 2007b). In the model reported by McMahon et al. (2007b) the 5-bp deletion and two additional point mutations downstream of the 5-bp deletion were introduced into a copy of the mouse homolog of ELOVL4 so that the sequence in this region was identical to the DNA sequence of the human STGD3 allele. This model will be referred as the KI+2 mouse line here (Table 2). The mouse model with the introduction of just the 5-bp deletion will be referred to as the KI mutant (Vasireddy et al., 2006).
The heterozygous KI mouse developed progressive cone photoreceptor degeneration and significant morphological changes between ages 2 and 18 months. Accumulation of lipofuscin in RPE was detected as early as 2 months of age, and subretinal deposits were observed at later ages. The ERGs of 8-month-old heterozygous STGD3 (+/−) mice were slightly higher than the ERG response of age-matched control mice, whereas the ERGs of these animals at 15 and 24 months were similar to Wt littermate controls. In contrast, 8-month-old heterozygous KI+2 animals did not demonstrate significant photoreceptor cell degeneration at the ages examined in the study, but did show accumulation of lipofuscin and reduced visual function. ERG response of 8-month-old heterozygous KI+2 mice showed a reduced light adapted response. In both models (KI and KI+2), features of human STGD3 were reproduced, although total correlation to the human STGD3 phenotype was not realized. A direct comparison of the KI mouse with the KI+2 mouse is somewhat controversial because the same retinal phenotype was not obtained even though essentially the same 5-bp deletion STGD3-causing mutation was introduced into each model system. Accumulation of lipofuscin and altered ERG amplitudes observed in KI+2 animals were similar to the retinal changes observed in ELOVL4 transgenic animals. The phenotype of the KI mouse best resembles the clinical features reported in STGD3 patients (Table 3). The STGD3 KI mice showed a mild phenotype, with progressive loss of cones, normal ERG, accumulation of subretinal debris, and late onset retinal degeneration. Phenotypic variation was, however, evident between individual KI mice. Taking the data in whole, we can conclude that the introduction of the known 5-bp STGD3 disease allele results in a dominant negative effect of the mutated allele on the retinal phenotype in Elovl4 mouse models. Besides these two 5-bp deletion knock-in mouse models, there is a third model generated by the introduction of Y270X deletion mutation. The retinal phenotype of the Y270X model, however, has not been reported.
Table 3. Retinal degeneration phenotype of different ELOVL4 animal models in comparison to human STGD3 patients.
| Feature | Human STGD3 | Gene Deletion KO1 (Rax-Prag et al., 2006) | Gene Deletion-KO2 (Li et al., 2007a) | Gene insertion KI (Vasireddy et al., 2006) | Gene insertion KI+2 (McMahon et al., 2006) | Gene substitution (Karan et al., 2005a,b) |
|---|---|---|---|---|---|---|
| Retinal degeneration | + | - | - | + | - | + |
| Color vision defects | + | NA | NA | NA | NA | NA |
| EOG abnormalities | - | NR | NR | NR | NR | NR |
| ERG changes- scotopic and photoic | (-) Most of the time | - | Both | - | Photopic | Both |
| Macular of fundus changes | + | NR | NR | NR | NR | NR |
| RPE changes | + | - | NR | + | - | + |
| Lipofuscin accumulation | + | - | - | + | + | + |
EOG, electro-oculogram; ERG, electroretinograrn, RPE, retinal pigment epithelium; +, observed; -, not observed, NA, not applicable, NR, not reported.
3.4. Phenotype of homozygous Elovl4 mutant mice
Mice homozygous for an Elovl4 knock-out allele, STGD3-causing 5-bp deletion Elovl4 knock-in allele, or the Elovl4 Y270X deletion knock-in allele were found to die within a few hours after birth (Fig. 8) (Cameron et al., 2007; Li et al., 2007b; McMahon et al., 2007a; Vasireddy et al., 2007). Developmental analysis of Elovl4 knock-out, 5-bp deletion knock-in, or Y270X knock-in pups revealed abnormalities in skin and skin development. Since the identification of the first STGD3 mutation, it has been assumed that ELOVL4 plays a role in FA and lipid metabolism because of its sequence similarities to known FA elongase enzymes. With respect to skin, lipids are key components of the epidermis. Of the different types of lipids, ceramides play an important role in maintaining the water retention property of the epidermal permeability barrier. A decrease in total ceramide content and alterations in the ceramide profile is noted in most skin disorders that have a diminished barrier function (Motta et al., 1994; Bouwstra et al., 2001; Pilgram et al., 2001; Proksch et al., 2008; Uchida and Holleran, 2008).
Figure 8. Phenotype of homozygous ELOVL4 5-bp deletion knock-in pups (E_mut +/+).
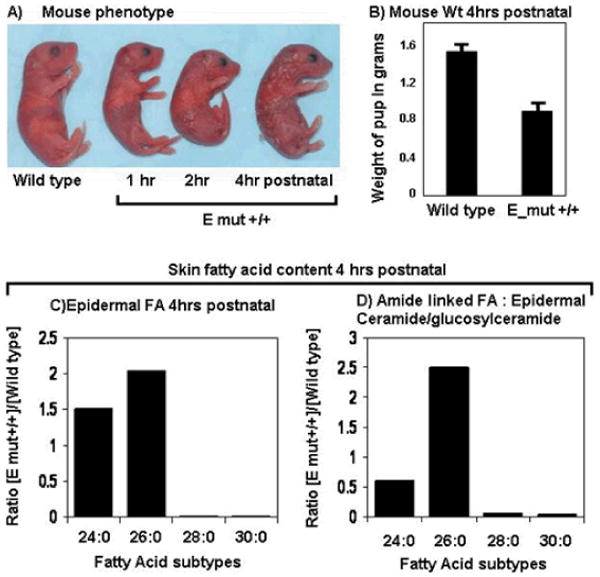
A) Skin phenotype. Comparison of the skin abnormality in E_mut +/+ pups from birtth to 4 h postnatal (compared to wild type) demonstrates a progression towards a scaly, dry, wrinkled skin phenotype followed by death.
B) At birth, the weight of control and E_mut+/+ pups is not significantly different from the wild type, but within the first 4 h of birth, the weight of E_mut +/+ pups decreases drastically. (C,D) Measurements of long chain FA in the epidermal free FA pool (C) and of the amide linked FA of epidermal ceramide/glucosylceramide. (D) show a similar pattern of elevated levels of C>26 related FA in E_mut+/+ samples as compared to wild type. Moreover there is a drastic decrease in levels of C>28 related FA in E_mut+/+ samples as compared to the wild type. (Modified after Vasireddy et al., 2007)
Analysis of the skin lipid profiles of homozygous Elovl4 knock-out or mutant Elovl4 5-bp deletion knock-in mice demonstrated that ω-O-acyl ceramides, its precursors ω-O-glucosyl ceramides, and FA with a chain length longer than 28 carbons (C>28) were depleted. In contrast to the levels of C>28 FA, there was an increase in the content of FA with chain length C>26 (Fig. 8). Therefore, C>26 FA could be serving as the substrate for ELOVL4. The lack of functional ELOVL4 might affect synthesis of C>26 FA and this could be leading to the neonatal lethality observed in the mice.
4. Role of ELOVL4 in Fatty acid Metabolism
The synthesis of FA from acetyl CoA and malonyl CoA is carried out by an enzyme called fatty acid synthase (FAS); steps in FA metabolism pertinent to the current discussion is provided in Figure 9 (Wakil, 1989). All the reactions catalyzed by FAS are carried out by the multi-enzyme complex of FAS. The primary FA synthesized by the FAS complex is palmitic acid, which has 16 carbon atoms. Elongation and unsaturation of the FA occur in both mitochondria and the ER membrane. These long chain FA and the FA obtained from the diet are further converted to very long chain fatty acids (VLCFA). Of the known FA of the retina, docosahexaenoic acid (DHA, 22:6n3) is the most abundant long chain FA (Anderson et al., 1974; Jeffrey et al., 2001). The majority of vertebrate retinal 22:6n3 is concentrated in the phosphatidyl ethanolamine and phosphatidyl serine fractions of the phospholipid pools of rod photoreceptor outer segments. Based on the expression of ELOVL4 in the photoreceptors of the retina, where 22:6n3 was predominant, it was initially proposed that ELOVL4 might be involved in the synthesis of retinal 22:6n3 (Zhang et al., 2001).
Figure 9. Fatty acid elongation in the cells and possible role of ELOVL4 in the elongation of long chain fatty acid.
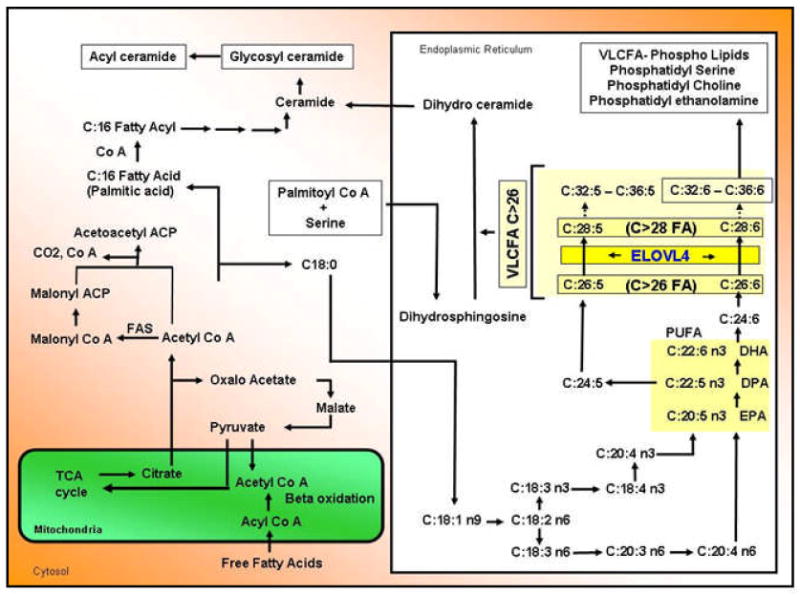
(Modified after Sprong et al., 2001; Hubbard, 2006; Agbaga et al., 2008; Katz and Minke, 2009; Oda et al., 2009). Enzymatic steps involving ELOVL4 are indicated (large yellow rectangle). Both steps are independent of the synthesis of DHA, the most abundant long chain FA in the retina. DHA is also known as C22:6n3 (“C:22” indicates that the molecule is 22 carbons long; the “:6” indicates that the structure has 6 cis double bonds; and “n3” indicates that the first double bond is located at the third carbon from the omega end of the structure). The function of ELOVL4 is to mediate the synthesis of very long chain FA (VLFA). A select number of polyunsaturated fatty acids (PUFA) that are mentioned in the text are shown. In general a PUFA is a FA that contains more than one double bond in its molecular structure. Abbreviations: ACP, acyl carrier protein; C, carbon chain; Co A, coenzyme A; DHA, docosahexaenoic acid; DPA, docosapentaenoic acid; EPA, eicosapentaenoic acid; FA, fatty acid; FAS, fatty acid synthase; TCA cycle, tricarboxylic acid cycle; VLCFA, very long chain fatty acids.
Assessment of red blood cell membrane lipids of individuals from the Utah family affected by a 2 bp deletion in the ELOVL4 gene (789ΔT+794ΔT) revealed a significant inverse relationship between the severity of the retinal phenotype and the 22:6n3 levels of red blood cells (Hubbard et al., 2006). Affected individuals with severe phenotype had average 22:6n3 and eicosapentaenoicacid (EPA, 20:5n3) levels while affected individuals with mild to moderate phenotypes had high 22:6n3 and 20:5n3 level. These results suggested that supplementation of 22:6n3 might help in slowing the progression of STGD3 (Hubbard et al., 2006). Improvement in the visual function of a patient with the STGD3-causing ELOVL4 5-bp deletion mutation undergoing 22:6n3 supplementation further supported the potential beneficial role of this FA supplementation for STGD3 patients (MacDonald et al., 2004). However, our studies on the FA profile of retinal tissue from both knock-in and knock-out mouse models for STGD3 revealed no alteration in the levels of 22:6n3, ruling out a role for ELOVL4 in 22:6n3 synthesis (Raz-Prag et al., 2006; Vasireddy et al., 2008). Several studies that demonstrate the involvement of ELOVL4 in elongation of VLCFA (C>26) and metabolism of C32–C36 acyl phosphotidyl cholines further support that ELOVL4 may not be involved in 22:6n3 synthesis (Fig. 9) (Li et al., 2007b; McMahon et al., 2007a; Vasireddy et al., 2007; McMahon et al., 2007b; McMahon and Kedzierski, 2009).
Recent studies using rat cardiomyocytes and ARPE-19 cells transduced with adenovirus carrying ELOVL4 and supplemented with various FA substrates like lignoceric acid (24:0), 20:5n3/docosapentaenoic acid (DPA, 22:5n3) further established that ELOVL4 catalyzes the steps involved in the elongation of 24:0 to 28:0 and 20:5n3/22:5n3 to a series of C28–C30 polyunsaturated fatty acids (PUFA) (Agbaga et al., 2008). ELOVL4 therefore is involved in FA chain elongation for the biosynthesis of both saturated and unsaturated C26–C28 FA (Fig. 9). Although the function of VLCFA and phosphatidyl choline in photoreceptors is not known, their presence in photoreceptors has been reported (Aveldano and Bazan, 1974; Aveldano and Sprecher, 1987; Aveldano, 1988). The assumption then is that a reduction in the synthesis of these types of FA might lead to the progressive photoreceptor degeneration observed in STGD3. The specific role of these VLCFA in the retina is yet to be determined.
5. Future Directions and Conclusions
Considerable progress has been made in understanding the autosomal dominantly inherited disorder STGD3. Mutations in the ELOVL4 gene that lead to a truncation and a loss of the ER retention signal in the protein synthesized underlie the STGD3 disease phenotype. We know that haploinsufficiency is not the genetic mechanism underlying the degeneration of photoreceptor cells in STGD3 since heterozygous ELOVL4 knock-out mice do not develop retinal degeneration (Raz-Prag et al., 2006; Li et al., 2007a). Moreover, presence of a mutant STGD3-causing Elov4 allele in animal models leads to an abnormal accumulation of ELOVL4 protein in the form of cellular aggresomes, lipofuscin, and subsequent photoreceptor degeneration. An interesting observation in the transgenic STGD3 animals is that the disease severity is correlated with increased levels of mutant ELOVL4 gene expression. This suggests that retinal degeneration in STGD3 is a consequence of the mutant ELOVL4 disease allele. When mutations in the ELOVL4 gene were first identified as the genetic cause of STGD3, the finding heralded in a new era in macular degeneration research by bringing to the forefront the concept that FA metabolism is an important cellular process in the homeostasis of the macula. Because of the high DNA sequence homology of human ELOVL4 with the ELO family of elongases that are involved in FA chain elongation in yeast, it was predicted that it would also function in FA metabolism (Oh et al., 1997; Zhang et al., 2001). Moreover because 22:6n3 represented the most abundant long chain FA in photoreceptor cells it was presumed that ELOVL4 was the elongase that led to the synthesis of 22:6n3. We now know that ELOVL4 does not participate in the 22:6n3 synthesis pathway but rather participates in the synthesis of other VLCFA in the retina and that decreased levels of these VLCFA are present in the retinae of heterozygous KI mice that show a retinal degeneration phenotype (Agbaga et al., 2008; Agbaga et al., 2009).
One aspect of the clinical phenotype of STGD3 that is not addressed by knowing the genetic lesion is the highly variable clinical profile observed, including age of disease onset. Although STGD3 has an autosomal dominant mode of inheritance, other cellular parameters may underlie the variability in the disease phenotype. In retrospect, AMD is similar in that it is also characterized by a widely variable clinical phenotype, with the exception that the genetic components contributing to the risk of developing disease in combination with other factors that underlie the disease are not as well defined. Identification of factors that may delay the onset or modify the severity of STGD3 will provide valuable clues in developing therapeutic strategies not only for STGD3 but also for other retinal degenerations.
Because of the presumed function of ELOVL4, a number of studies were performed to define the effect of FA intake on the STGD3 condition. A significant inverse relationship between STGD3 severity and blood levels of 20:5n3, 22:6n3, and dietary fat was reported (Hubbard et al., 2006). Moreover, in a 22:6n3 supplementation study with a single STGD3 patient from the Canadian STGD3 pedigree who had very minor macular fundus changes, increased plasma 22:6n3 levels correlated consistently with increased visual acuity (MacDonald et al., 2004). Since ELOVL4 does not mediate 22:6n3 synthesis (22 carbons in length) it is a curiosity how elevated blood 22:6n3 levels can mediate a change in the STGD3 phenotype. One could hypothesize that 22:6n3 might be converted to an alternative FA form that may substitute for the missing FA subtype depleted in STGD3 or it alters the cellular environment for a period of time during which the STGD3 mutant consequence is tolerated. Long chain FA such as 22:6n3 could have an overall effect on the clinical phenotype at very early stages of the disorder but not be able to overcome the long-term underlying defect. In the absence of a cure for STGD3, a timely question is how effective would FA supplementation be, in delaying the progression from a mild to a severe STGD3 phenotype.
Fatty acid (22:6n3) supplementation has also been extended into the treatment of patients with AMD and the overall results indicate that FA metabolism plays a role in the development of AMD (Seddon et al., 2001; Seddon et al., 2003b; Conley et al., 2005; Chua et al., 2006; Chong et al., 2009). Given this precedence it would be interesting to examine if other factors that have already been associated with the AMD phenotype (light, cigarette smoke, etc.) might also influence the STGD3 clinical phenotype (Seddon et al., 1996; Cho et al., 2000; Cho et al., 2001; Evans, 2001; Seddon et al., 2001; Hyman and Neborsky, 2002; Seddon et al., 2003b; Seddon et al., 2003a; Clemons et al., 2005; Seddon et al., 2005; Haddad et al., 2006). STGD3 transgenic or disease ELOVL4 allele knock-in mouse lines could be used as model systems to examine the effects of these parameters to generate a more severe or mild retina phenotype. Moreover, the STGD3 animal models could be analyzed by comparative analyses to define additional putative secondary cellular and functional pathways that may affect the progression of the disease phenotype.
Development of successful treatments for inherited retinal degenerations caused by gene mutations is always a major challenge and will vary on one's perspective as to the functional cause of the disease (Fig. 10). A common explanation for the manifestation of age-related onset disorders is that the disease defect leads to a disruption in a cellular function that results in the accumulation of a substrate or metabolite that becomes toxic at significant cellular quantities (cellular stress) and leads to the demise of the cells involved. A current hypothesis is that the abnormal cellular accumulation of the ELOVL4 protein could alter normal cellular homeostasis and induce degeneration of photoreceptor cells that express the ELOVL4 mutant protein by activating the process of apoptosis (Karan et al., 2004a). ELOVL4 mRNA and protein are expressed in fetal and adult mouse retinal tissues (Mandal et al., 2004). Data mining of the UniGene database reveals that ELOVL4 mRNA is expressed in human fetal, neonate, juvenile, and adult tissues. In most cases onset of STGD3 can start as early as in the second decade of life (Stargardt, 1909; Blacharski, 1988; Kaplan et al., 1990). This means that for a certain period of life the presence of the mutant ELOVL4 in STGD3 patients is tolerated. If the formation of ELOVL4 aggresomes is the cellular factor that mitigates cell loss then it is important to determine what cellular processes allow cells carrying the STGD3-causing mutations to tolerate the cell stress prior to onset of STGD3 clinical phenotypes. Inevitably, cell processes that function to confine the effects of misfolded proteins or remove these proteins by shuttling them to the proteasome are in play. Pharmacological agents that can target the mutant ELOVL4 protein for removal or increase the activity of chaperones that might mediate the same effect might prove useful in preventing formation of ELOVL4 aggresomes. Moreover a comparison of gene profiles between an individual carrying a STGD3-causing mutation but without onset of a clinical phenotype with one that is showing early signs of the disease could be integral in defining the changes that mitigate the switch from toleration to susceptibility to the effects of the mutation. A more fundamental question is, if a switch exists, is it a consistent cell process switch that plays a role in onset of macular disease in general.
Figure 10. Points of possible therapeutic strategies to treat retinal degeneration in STGD3 patients.
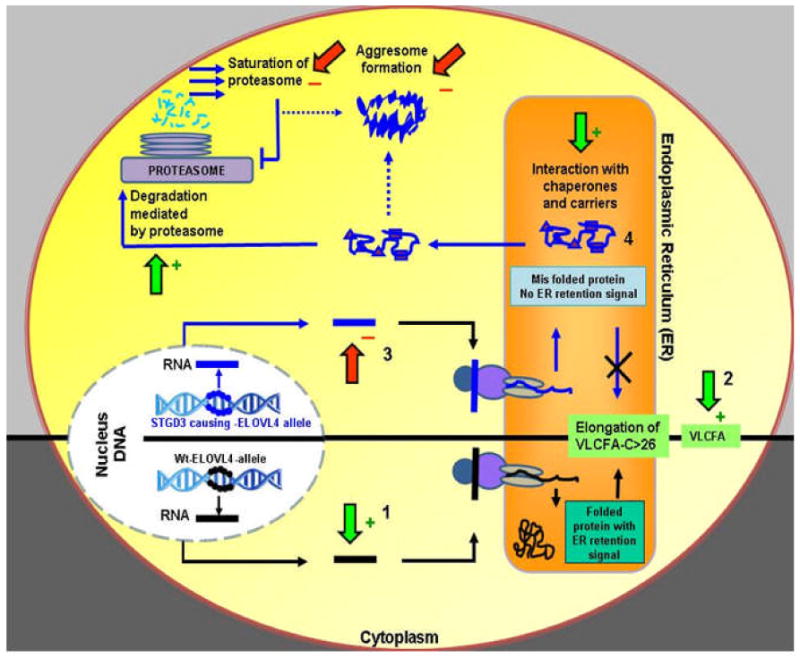
Illustrated is a cell expressing ELOVL4 from a heterozygote STGD3-affected patient. The view is simplified because events are shown for the expression of the wild type (Wt) ELOVL4 allele (DNA, mRNA, and protein illustrated in black) separate from the STGD3-causing ELOVL4 allele (DNA, mRNA and protein illustrated in blue). In the affected heterozygous condition the aggresome consists of both mutant and wild type ELOVL4 protein and not just of mutant ELOVL4 protein as illustrated. Aggresome formation therefore represents an exodus of both mutant and Wt ELOVL4 out of the ER. The ideal treatment strategy would be targeted at the functional cause of the cellular dysfunction that underlies the clinical phenotype. At present the two most likely causes are the depletion of Wt ELOVL4 from the ER (and therefore no VLCFA synthesis) or the possible toxic effects of the aggresomes formed in the cytoplasm (leading to cell death). Putative therapies will either help promote (green arrow, +) an event that leads to an increased production of the ELVOL4 end product, VLCFA or inhibit (red arrow, -) functional points that lead to the negative effects of mutant ELOVL4 allele expression. Strategic points of intervention might include: 1) Enhanced expression of Wt ELOVL4; 2) VLCFA replacement; 3) removal of mutant ELOVL4 mRNA; 4) enhanced expression of chaperone expression and targeting of the mutant ELOVL4 to the proteosome. Triangles and squares indicate molecular chaperones.
If STGD3 is purely a consequence of an inborn error of lipid metabolism, then supplementation of the missing FA should remedy the proper function of the retinal photoreceptors in STGD3 patients. As ELOVL4 is involved in the elongation of very long chain polyunsaturated fatty acids (VLC-PUFA), studies should aim in supplementing these FA. VLC-PUFA having carbon chains of C>28 are present at low levels in tissues like testis, brain, sperm, and retina (Aveldano and Sprecher, 1987; Furland et al., 2007a; Furland et al., 2007c; Furland et al., 2007b; McMahon and Kedzierski, 2009). The difficulties associated with the supplementation of these FA include the inability to extract high amounts of these VLC-PUFA (C>28) from easily available sources and the fact that these FA are difficult to solubilize in vitro. Subsequently, development of cell systems that can overproduce VLC-PUFA or methods that can efficiently synthesize these types of FA in vitro could be pursued. Microorganisms such as marine bacteria and microalgae are a natural source of VLC-PUFA, and there is a current effort to transfer the VLC-PUFA synthetic pathways inherent in these species into plants to produce a recombinant source of VLC-PUFA from oilseeds (Hoffmann et al., 2008).
From a purely molecular genetics perspective, therapeutic strategies for STGD3 aimed at inhibiting the expression of the mutated ELOVL4 allele or activating the degeneration of mutant transcript or protein thereby minimizing its biological effect are desirable. Inhibition of mutant gene expression can be achieved by a variety of approaches, including antisense, ribozymes, and, more recently, RNA interference (RNAi) (Hauswirth et al., 2000; LaVail et al., 2000; Bumcrot et al., 2006; Zimmermann et al., 2006). Allele-specific inhibition of mutant ELOVL4 gene expression can be considered as one approach for dominant STGD3. The rationale for this strategy is that the expression of a single Wt ELOVL4 allele is sufficient for photoreceptor cell function (Raz-Prag et al., 2006; Li et al., 2007a). The feasibility of this approach is highly dependent on whether or not a specific small interfering RNA (siRNA construct) can be generated that would specifically target the mutant ELOVL4 allele without affecting the Wt ELOVL4 mRNA. As in all cases, parameters that need to be considered include the efficiency of siRNA delivery to the macula, the frequency at which the treatment needs to be repeated, and of course cost.
Molecular genetic studies of STGD3 have had a positive impact on further understanding the more complex disorder of AMD. Aspects of the STGD3 clinical phenotype are similar to those observed in AMD and support the notion that STGD3 could be mirrored as a simpler genetic model for AMD. Genetic studies revealed that ω-3 FA intake reduced the risk of AMD (Seddon et al., 2001; Seddon et al., 2003b; Conley et al., 2005; Chua et al., 2006; Chong et al., 2009). Our meta-analysis for interacting relationships between known MD gene loci and genes associated with AMD (Fig. 2) strongly suggest that the genes in question were functionally related. A number of association studies have also shown a relationship of some of the genes that underlie monogenic MD disease as associated factors for an AMD condition. These include associations between AMD and the genes ABCA4 and ELOVL4 (Allikmets et al., 1997; Conley et al., 2005; Haddad et al., 2006). With respect to MD disorders, mutations in ABCA4 underlie STGD1, and of course ELOVL4 mutations underlie STGD3. The controversy of the association of these genes with AMD is that the specific associations described are not observed in all studies examining independent cohorts of AMD patients. A more direct examination of the ELOVL4 and ABCA4 interactome specifically suggests that both may be a part of a functional pathway relevant to macular disease (MD and AMD) in general (Fig. 11). In total, nine MD- and/or AMD-associated genes (33% of the genes in the larger interactome analysis) are in this network. The largest interaction overlap that ELOVL4 has is with the ABCA4 gene. Given the mix of AMD (ARMD1, ARMD4, and ARMD6) and MD disorders (BCMAD, BEST1, DHRD, SFDE, STGD1, STGD3) that this network has influence over, it is reasonable to hypothesize that factors that influence these MD disorders may also influence the listed AMD disorders and vice versa. Further analysis of STGD3 with respect to its disease mechanism will allow us to also gain further insight into the complex processes that define AMD as well as other macular dystrophies.
Figure 11. A close-up of the ELOVL4/ABCA4 interactome.
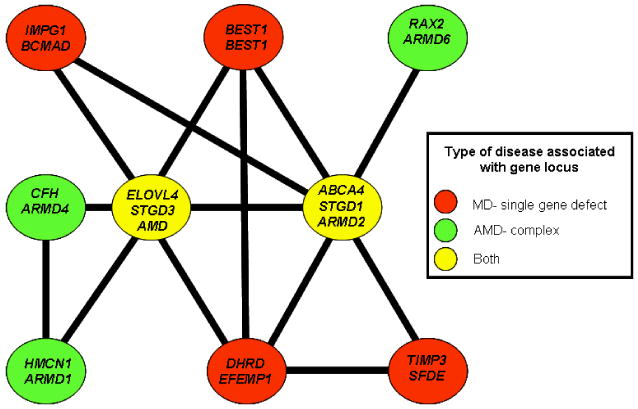
Where possible the gene symbol is given followed by the OMIM symbol for the disorders that the gene underlies. The mode of inheritance of the disorder has also been color coded.
Acknowledgments
This work was supported by grants to RA from NIH-EY13198; Foundation Fighting Blindness (RA), Research to Prevent Blindness Inc., USA (RA) and grants to PW from Knights Templar Foundation of Georgia, The Parker Foundation, Research to Prevent Blindness Inc., USA, and NIH-P30-EY06360
References
- Aaberg TM. Stargardt's disease and fundus flavimaculatus: evaluation of morphologic progression and intrafamilial co-existence. Trans Am Ophthalmol Soc. 1986;84:453–487. [PMC free article] [PubMed] [Google Scholar]
- Agbaga MP, Brush RS, Mandal MN, Henry K, Elliott MH, Anderson RE. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc Natl Acad Sci U S A. 2008;105:12843–12848. doi: 10.1073/pnas.0802607105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agbaga MP, Mandal MA, Brush RS, Zheng L, Henry K, Elliott MH, Vasireddy V, Petrukhin K, Ayyagari R, Anderson RE. Quantitative analysis of ELOVL4 protein and fatty acid products in knock-out and knock-in mouse tissues. The Association for Research in Vision and Ophthalmology; Fort Lauderdale, Florida, USA: 2009. [Google Scholar]
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- Ambasudhan R, Wang X, Jablonski MM, Thompson DA, Lagali PS, Wong PW, Sieving PA, Ayyagari R. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–625. doi: 10.1016/j.ygeno.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- Anderson RE, Benolken RM, Dudley PA, Landis DJ, Wheeler TG. Proceedings: Polyunsaturated fatty acids of photoreceptor membranes. Exp Eye Res. 1974;18:205–213. doi: 10.1016/0014-4835(74)90149-3. [DOI] [PubMed] [Google Scholar]
- Aveldano MI. Phospholipid species containing long and very long polyenoic fatty acids remain with rhodopsin after hexane extraction of photoreceptor membranes. Biochemistry. 1988;27:1229–1239. doi: 10.1021/bi00404a024. [DOI] [PubMed] [Google Scholar]
- Aveldano MI, Bazan NG. Free fatty acids, diacyl- and triacylglycerols and total phospholipids in vertebrate retina: comparison with brain, choroid and plasma. J Neurochem. 1974;23:1127–1135. doi: 10.1111/j.1471-4159.1974.tb12209.x. [DOI] [PubMed] [Google Scholar]
- Aveldano MI, Sprecher H. Very long chain (C24 to C36) polyenoic fatty acids of the n-3 and n-6 series in dipolyunsaturated phosphatidylcholines from bovine retina. J Biol Chem. 1987;262:1180–1186. [PubMed] [Google Scholar]
- Barthes A, Conrath J, Ridings B. Quantitative and spatial analysis in image processing: study of drusen distribution from the foveal center in age-related macular degeneration. J Fr Ophtalmol. 2005;28:298–302. doi: 10.1016/s0181-5512(05)81057-5. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S, Parish CA, Hashimoto M, Liu J, Nakanishi K, Sparrow JR. Fluorescent pigments of the retinal pigment epithelium and age-related macular degeneration. Bioorg Med Chem Lett. 2001;11:1533–1540. doi: 10.1016/s0960-894x(01)00314-6. [DOI] [PubMed] [Google Scholar]
- Ben-Shabat S, Parish CA, Vollmer HR, Itagaki Y, Fishkin N, Nakanishi K, Sparrow JR. Biosynthetic studies of A2E, a major fluorophore of retinal pigment epithelial lipofuscin. J Biol Chem. 2002;277:7183–7190. doi: 10.1074/jbc.M108981200. [DOI] [PubMed] [Google Scholar]
- Bernstein PS, Tammur J, Singh N, Hutchinson A, Dixon M, Pappas CM, Zabriskie NA, Zhang K, Petrukhin K, Leppert M, Allikmets R. Diverse macular dystrophy phenotype caused by a novel complex mutation in the ELOVL4 gene. Invest Ophthalmol Vis Sci. 2001;42:3331–3336. [PubMed] [Google Scholar]
- Blacharski R. Fundus flavimaculatus. New York: Raven Press; 1988. [Google Scholar]
- Bouwstra J, Pilgram G, Gooris G, Koerten H, Ponec M. New aspects of the skin barrier organization. Skin Pharmacol Appl Skin Physiol. 2001;14 1:52–62. doi: 10.1159/000056391. [DOI] [PubMed] [Google Scholar]
- Bressler NM, Silva JC, Bressler SB, Fine SL, Green WR. Clinicopathologic correlation of drusen and retinal pigment epithelial abnormalities in age-related macular degeneration. 1994. Retina. 2005;25:130–142. doi: 10.1097/00006982-200507001-00016. [DOI] [PubMed] [Google Scholar]
- Bumcrot D, Manoharan M, Koteliansky V, Sah DW. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2:711–719. doi: 10.1038/nchembio839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DJ, Tong Z, Yang Z, Kaminoh J, Kamiyah S, Chen H, Zeng J, Chen Y, Luo L, Zhang K. Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci. 2007;3:111–119. doi: 10.7150/ijbs.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E, Hankinson SE, Willett WC, Stampfer MJ, Spiegelman D, Speizer FE, Rimm EB, Seddon JM. Prospective study of alcohol consumption and the risk of age-related macular degeneration. Arch Ophthalmol. 2000;118:681–688. doi: 10.1001/archopht.118.5.681. [DOI] [PubMed] [Google Scholar]
- Cho E, Hung S, Willett WC, Spiegelman D, Rimm EB, Seddon JM, Colditz GA, Hankinson SE. Prospective study of dietary fat and the risk of age-related macular degeneration. Am J Clin Nutr. 2001;73:209–218. doi: 10.1093/ajcn/73.2.209. [DOI] [PubMed] [Google Scholar]
- Chong EW, Robman LD, Simpson JA, Hodge AM, Aung KZ, Dolphin TK, English DR, Giles GG, Guymer RH. Fat consumption and its association with age-related macular degeneration. Arch Ophthalmol. 2009;127:674–680. doi: 10.1001/archophthalmol.2009.60. [DOI] [PubMed] [Google Scholar]
- Chua B, Flood V, Rochtchina E, Wang JJ, Smith W, Mitchell P. Dietary fatty acids and the 5-year incidence of age-related maculopathy. Arch Ophthalmol. 2006;124:981–986. doi: 10.1001/archopht.124.7.981. [DOI] [PubMed] [Google Scholar]
- Cibis GW, Morey M, Harris DJ. Dominantly inherited macular dystrophy with flecks (Stargardt) Arch Ophthalmol. 1980;98:1785–1789. doi: 10.1001/archopht.1980.01020040637010. [DOI] [PubMed] [Google Scholar]
- Cinti DL, Cook L, Nagi MN, Suneja SK. The fatty acid chain elongation system of mammalian endoplasmic reticulum. Prog Lipid Res. 1992;31:1–51. doi: 10.1016/0163-7827(92)90014-a. [DOI] [PubMed] [Google Scholar]
- Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL., 3rd Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology. 2005;112:533–539. doi: 10.1016/j.ophtha.2004.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon N, O'Colmain B, Klaver CC, Klein R, Munoz B, Friedman DS, Kempen J, Taylor HR, Mitchell P. Causes and prevalence of visual impairment among adults in the United States. Arch Ophthalmol. 2004;122:477–485. doi: 10.1001/archopht.122.4.477. [DOI] [PubMed] [Google Scholar]
- Conley YP, Thalamuthu A, Jakobsdottir J, Weeks DE, Mah T, Ferrell RE, Gorin MB. Candidate gene analysis suggests a role for fatty acid biosynthesis and regulation of the complement system in the etiology of age-related maculopathy. Hum Mol Genet. 2005;14:1991–2002. doi: 10.1093/hmg/ddi204. [DOI] [PubMed] [Google Scholar]
- Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG. Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002;99:14682–14687. doi: 10.1073/pnas.222551899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong PT, Bergen AA, Klaver CC, Van Duijn CM, Assink JM. Age-related maculopathy: its genetic basis. Eye. 2001;15:396–400. doi: 10.1038/eye.2001.143. [DOI] [PubMed] [Google Scholar]
- Donoso LA, Edwards AO, Frost A, Vrabec T, Stone EM, Hageman GS, Perski T. Autosomal dominant Stargardt-like macular dystrophy. Surv Ophthalmol. 2001a;46:149–163. doi: 10.1016/s0039-6257(01)00251-x. [DOI] [PubMed] [Google Scholar]
- Donoso LA, Frost AT, Stone EM, Weleber RG, MacDonald IM, Hageman GS, Cibis GW, Ritter R, 3rd, Edwards AO. Autosomal dominant Stargardt-like macular dystrophy: founder effect and reassessment of genetic heterogeneity. Arch Ophthalmol. 2001b;119:564–570. doi: 10.1001/archopht.119.4.564. [DOI] [PubMed] [Google Scholar]
- Donoso A, Martens CC. Quantum tunneling using entangled classical trajectories. Phys Rev Lett. 2001;87:223202. doi: 10.1103/PhysRevLett.87.223202. [DOI] [PubMed] [Google Scholar]
- Eagle RC, Jr, Lucier AC, Bernardino VB, Jr, Yanoff M. Retinal pigment epithelial abnormalities in fundus flavimaculatus: a light and electron microscopic study. Ophthalmology. 1980;87:1189–1200. doi: 10.1016/s0161-6420(80)35106-3. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Donoso LA, Ritter R., 3rd A novel gene for autosomal dominant Stargardt-like macular dystrophy with homology to the SUR4 protein family. Invest Ophthalmol Vis Sci. 2001;42:2652–2663. [PubMed] [Google Scholar]
- Edwards AO, Miedziak A, Vrabec T, Verhoeven J, Acott TS, Weleber RG, Donoso LA. Autosomal dominant Stargardt-like macular dystrophy: I. Clinical characterization, longitudinal follow-up, and evidence for a common ancestry in families linked to chromosome 6q14. Am J Ophthalmol. 1999;127:426–435. doi: 10.1016/s0002-9394(98)00331-6. [DOI] [PubMed] [Google Scholar]
- Edwards AO, Ritter R, 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- Evans JR. Risk factors for age-related macular degeneration. Prog Retin Eye Res. 2001;20:227–253. doi: 10.1016/s1350-9462(00)00023-9. [DOI] [PubMed] [Google Scholar]
- Furland NE, Maldonado EN, Aresti PA, Aveldano MI. Changes in lipids containing long- and very long-chain polyunsaturated fatty acids in cryptorchid rat testes. Biol Reprod. 2007a;77:181–188. doi: 10.1095/biolreprod.106.056556. [DOI] [PubMed] [Google Scholar]
- Furland NE, Oresti GM, Antollini SS, Venturino A, Maldonado EN, Aveldano MI. Very long-chain polyunsaturated fatty acids are the major acyl groups of sphingomyelins and ceramides in the head of mammalian spermatozoa. J Biol Chem. 2007b;282:18151–18161. doi: 10.1074/jbc.M700709200. [DOI] [PubMed] [Google Scholar]
- Furland NE, Zanetti SR, Oresti GM, Maldonado EN, Aveldano MI. Ceramides and sphingomyelins with high proportions of very long-chain polyunsaturated fatty acids in mammalian germ cells. J Biol Chem. 2007c;282:18141–18150. doi: 10.1074/jbc.M700708200. [DOI] [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson C, Molday RS. Dominant negative mechanism underlies autosomal dominant Stargardt-like macular dystrophy linked to mutations in ELOVL4. J Biol Chem. 2005;280:32521–32530. doi: 10.1074/jbc.M503411200. [DOI] [PubMed] [Google Scholar]
- Griesinger IB, Sieving PA, Ayyagari R. Autosomal dominant macular atrophy at 6q14 excludes CORD7 and MCDR1/PBCRA loci. Invest Ophthalmol Vis Sci. 2000;41:248–255. [PubMed] [Google Scholar]
- Haddad S, Chen CA, Santangelo SL, Seddon JM. The genetics of age-related macular degeneration: a review of progress to date. Surv Ophthalmol. 2006;51:316–363. doi: 10.1016/j.survophthal.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Hadden OB, Gass JD. Fundus flavimaculatus and Stargardt's disease. Am J Ophthalmol. 1976;82:527–539. doi: 10.1016/0002-9394(76)90539-0. [DOI] [PubMed] [Google Scholar]
- Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Hauswirth WW, LaVail MM, Flannery JG, Lewin AS. Ribozyme gene therapy for autosomal dominant retinal disease. Clin Chem Lab Med. 2000;38:147–153. doi: 10.1515/CCLM.2000.022. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Wagner M, Abbadi A, Fulda M, Feussner I. Metabolic engineering of omega3-very long chain polyunsaturated fatty acid production by an exclusively acyl-CoA-dependent pathway. J Biol Chem. 2008;283:22352–22362. doi: 10.1074/jbc.M802377200. [DOI] [PubMed] [Google Scholar]
- Hubbard AF, Askew EW, Singh N, Leppert M, Bernstein PS. Association of adipose and red blood cell lipids with severity of dominant Stargardt macular dystrophy (STGD3) secondary to an ELOVL4 mutation. Arch Ophthalmol. 2006;124:257–263. doi: 10.1001/archopht.124.2.257. [DOI] [PubMed] [Google Scholar]
- Hyman L, Neborsky R. Risk factors for age-related macular degeneration: an update. Curr Opin Ophthalmol. 2002;13:171–175. doi: 10.1097/00055735-200206000-00007. [DOI] [PubMed] [Google Scholar]
- Jeffrey BG, Weisinger HS, Neuringer M, Mitchell DC. The role of docosahexaenoic acid in retinal function. Lipids. 2001;36:859–871. doi: 10.1007/s11745-001-0796-3. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, Bonneau D, Frezal J, Munnich A, Dufier JL. Clinical and genetic heterogeneity in retinitis pigmentosa. Hum Genet. 1990;85:635–642. doi: 10.1007/BF00193589. [DOI] [PubMed] [Google Scholar]
- Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, Thirumalaichary S, Li C, Birch DG, Vollmer-Snarr HR, Williams DS, Zhang K. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: a model for macular degeneration. Proc Natl Acad Sci U S A. 2005a;102:4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan G, Yang Z, Zhang K. Expression of wild type and mutant ELOVL4 in cell culture: subcellular localization and cell viability. Mol Vis. 2004a;10:248–253. [PubMed] [Google Scholar]
- Karan G, Yang Z, Zhang K. Annual Meeting of American Society of Human Genetics. 2004b. Expression of wild type and mutant ELOVL4 in cell culture: subcellular localization and cell viability. Abstract 1766/B577. [PubMed] [Google Scholar]
- Karan G, Yang Z, Howes K, Zhao Y, Chen Y, Cameron DJ, Lin Y, Pearson E, Zhang K. Loss of ER retention and sequestration of the wild-type ELOVL4 by Stargardt disease dominant negative mutants. Mol Vis. 2005b;11:657–664. [PubMed] [Google Scholar]
- Katz B, Minke B. Drosophila photoreceptors and signaling mechanisms. Front Cell Neurosci. 2009;3:2. doi: 10.3389/neuro.03.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaver CC, Wolfs RC, Assink JJ, van Duijn CM, Hofman A, de Jong PT. Genetic risk of age-related maculopathy. Population-based familial aggregation study. Arch Ophthalmol. 1998;116:1646–1651. doi: 10.1001/archopht.116.12.1646. [DOI] [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klien BA, Krill AE. Fundus flavimaculatus. Clinical, functional and histopathologic observations. Am J Ophthalmol. 1967;64:3–23. [PubMed] [Google Scholar]
- Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- Lagali PS, Liu J, Ambasudhan R, Kakuk LE, Bernstein SL, Seigel GM, Wong PW, Ayyagari R. Evolutionarily conserved ELOVL4 gene expression in the vertebrate retina. Invest Ophthalmol Vis Sci. 2003;44:2841–2850. doi: 10.1167/iovs.02-0991. [DOI] [PubMed] [Google Scholar]
- Lagali PS, MacDonald IM, Griesinger IB, Chambers ML, Ayyagari R, Wong PW. Autosomal dominant Stargardt-like macular dystrophy segregating in a large Canadian family. Can J Ophthalmol. 2000;35:315–324. doi: 10.1016/s0008-4182(00)80059-9. [DOI] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Drenser KA, Flannery JG, Lewin AS, Hauswirth WW. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A. 2000;97:11488–11493. doi: 10.1073/pnas.210319397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz HM, Krueger DE, Maunder LR, Milton RC, Kini MM, Kahn HA, Nickerson RJ, Pool J, Colton TL, Ganley JP, Loewenstein JI, Dawber TR. The Framingham Eye Study monograph: an ophthalmological and epidemiological study of cataract, glaucoma, diabetic retinopathy, macular degeneration, and visual acuity in a general population of 2631 adults, 1973–1975. Surv Ophthalmol. 1980;24:335–610. [PubMed] [Google Scholar]
- Li W, Chen Y, Cameron DJ, Wang C, Karan G, Yang Z, Zhao Y, Pearson E, Chen H, Deng C, Howes K, Zhang K. Elovl4 haploinsufficiency does not induce early onset retinal degeneration in mice. Vision Res. 2007a;47:714–722. doi: 10.1016/j.visres.2006.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Sandhoff R, Kono M, Zerfas P, Hoffmann V, Ding BC, Proia RL, Deng CX. Depletion of ceramides with very long chain fatty acids causes defective skin permeability barrier function, and neonatal lethality in ELOVL4 deficient mice. Int J Biol Sci. 2007b;3:120–128. doi: 10.7150/ijbs.3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez PF, Maumenee IH, de la Cruz Z, Green WR. Autosomal-dominant fundus flavimaculatus. Clinicopathologic correlation. Ophthalmology. 1990;97:798–809. doi: 10.1016/s0161-6420(90)32508-3. [DOI] [PubMed] [Google Scholar]
- MacDonald IM, Hebert M, Yau RJ, Flynn S, Jumpsen J, Suh M, Clandinin MT. Effect of docosahexaenoic acid supplementation on retinal function in a patient with autosomal dominant Stargardt-like retinal dystrophy. Br J Ophthalmol. 2004;88:305–306. doi: 10.1136/bjo.2003.024299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal MN, Ambasudhan R, Wong PW, Gage PJ, Sieving PA, Ayyagari R. Characterization of mouse orthologue of ELOVL4: genomic organization and spatial and temporal expression. Genomics. 2004;83:626–635. doi: 10.1016/j.ygeno.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Mansour AM. Long-term follow-up of dominant macular dystrophy with flecks (Stargardt) Ophthalmologica. 1992;205:138–143. doi: 10.1159/000310329. [DOI] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY. The challenge of modeling macular degeneration in mice. Trends Genet. 2007;23:225–231. doi: 10.1016/j.tig.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Maugeri A, Meire F, Hoyng CB, Vink C, Van Regemorter N, Karan G, Yang Z, Cremers FP, Zhang K. A novel mutation in the ELOVL4 gene causes autosomal dominant Stargardt-like macular dystrophy. Invest Ophthalmol Vis Sci. 2004;45:4263–4267. doi: 10.1167/iovs.04-0078. [DOI] [PubMed] [Google Scholar]
- McMahon A, Kedzierski W. Polyunsaturated extremely long chain C28-C36 fatty acids and retinal physiology. Br J Ophthalmol. 2009 Aug 9; doi: 10.1136/bjo.2008.149286. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- McMahon A, Butovich IA, Mata NL, Klein M, Ritter R, 3rd, Richardson J, Birch DG, Edwards AO, Kedzierski W. Retinal pathology and skin barrier defect in mice carrying a Stargardt disease-3 mutation in elongase of very long chain fatty acids-4. Mol Vis. 2007a;13:258–272. [PMC free article] [PubMed] [Google Scholar]
- McMahon A, Jackson SN, Woods AS, Kedzierski W. A Stargardt disease-3 mutation in the mouse Elovl4 gene causes retinal deficiency of C32-C36 acyl phosphatidylcholines. FEBS Lett. 2007b;581:5459–5463. doi: 10.1016/j.febslet.2007.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- Meyers SM. A twin study on age-related macular degeneration. Trans Am Ophthalmol Soc. 1994;92:775–843. [PMC free article] [PubMed] [Google Scholar]
- Motta S, Monti M, Sesana S, Mellesi L, Ghidoni R, Caputo R. Abnormality of water barrier function in psoriasis. Role of ceramide fractions. Arch Dermatol. 1994;130:452–456. [PubMed] [Google Scholar]
- Noble KG, Carr RE. Stargardt's disease and fundus flavimaculatus. Arch Ophthalmol. 1979;97:1281–1285. doi: 10.1001/archopht.1979.01020020023005. [DOI] [PubMed] [Google Scholar]
- Oda Y, Uchida Y, Moradian S, Crumrine D, Elias PM, Bikle DD. Vitamin D receptor and coactivators SRC2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol. 2009;129:1367–1378. doi: 10.1038/jid.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh CS, Toke DA, Mandala S, Martin CE. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem. 1997;272:17376–17384. doi: 10.1074/jbc.272.28.17376. [DOI] [PubMed] [Google Scholar]
- Omura N, Li CP, Li A, Hong SM, Walter K, Jimeno A, Hidalgo M, Goggins M. Genome-wide profiling of methylated promoters in pancreatic adenocarcinoma. Cancer Biol Ther. 2008;7:1146–1156. doi: 10.4161/cbt.7.7.6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea JG. Age-related macular degeneration: a leading cause of blindness. Med J Aust. 1996;165:561–564. [PubMed] [Google Scholar]
- Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev. 1999;79:683–701. doi: 10.1152/physrev.1999.79.3.683. [DOI] [PubMed] [Google Scholar]
- Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- Pilgram GS, Vissers DC, van der Meulen H, Pavel S, Lavrijsen SP, Bouwstra JA, Koerten HK. Aberrant lipid organization in stratum corneum of patients with atopic dermatitis and lamellar ichthyosis. J Invest Dermatol. 2001;117:710–717. doi: 10.1046/j.0022-202x.2001.01455.x. [DOI] [PubMed] [Google Scholar]
- Proksch E, Brandner JM, Jensen JM. The skin: an indispensable barrier. Exp Dermatol. 2008;17:1063–1072. doi: 10.1111/j.1600-0625.2008.00786.x. [DOI] [PubMed] [Google Scholar]
- Raz-Prag D, Ayyagari R, Fariss RN, Mandal MN, Vasireddy V, Majchrzak S, Webber AL, Bush RA, Salem N, Jr, Petrukhin K, Sieving PA. Haploinsufficiency is not the key mechanism of pathogenesis in a heterozygous Elovl4 knockout mouse model of STGD3 disease. Invest Ophthalmol Vis Sci. 2006;47:3603–3611. doi: 10.1167/iovs.05-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Miyagi M, Shadrach KG, Rayborn ME, Crabb JW, Hollyfield JG. Clusterin is present in drusen in age-related macular degeneration. Exp Eye Res. 2002;74:547–549. doi: 10.1006/exer.2002.1186. [DOI] [PubMed] [Google Scholar]
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Cote J, Davis N, Rosner B. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch Ophthalmol. 2003b;121:785–792. doi: 10.1001/archopht.121.6.785. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Cote J, Page WF, Aggen SH, Neale MC. The US twin study of age-related macular degeneration: relative roles of genetic and environmental influences. Arch Ophthalmol. 2005;123:321–327. doi: 10.1001/archopht.123.3.321. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Cote J, Rosner B. Progression of age-related macular degeneration: association with dietary fat, transunsaturated fat, nuts, and fish intake. Arch Ophthalmol. 2003a;121:1728–1737. doi: 10.1001/archopht.121.12.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon JM, Rosner B, Sperduto RD, Yannuzzi L, Haller JA, Blair NP, Willett W. Dietary fat and risk for advanced age-related macular degeneration. Arch Ophthalmol. 2001;119:1191–1199. doi: 10.1001/archopht.119.8.1191. [DOI] [PubMed] [Google Scholar]
- Seddon JM, Willett WC, Speizer FE, Hankinson SE. A prospective study of cigarette smoking and age-related macular degeneration in women. JAMA. 1996;276:1141–1146. [PubMed] [Google Scholar]
- Sprong H, van der Sluijs P, van Meer G. How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol. 2001;2:504–513. doi: 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- Stargardt K. Uber familiare, progressive degeneration in der Maculagegend des Auges. Albrecht von Graefes Arch Klin Ophthal. 1909;71:534–550. [Google Scholar]
- Starr CE, Guyer DR, Yannuzzi LA. Age-related macular degeneration. Can we stem this worldwide public health crisis? Postgrad Med. 1998;103:153–156. 161–154. doi: 10.3810/pgm.1998.05.480. [DOI] [PubMed] [Google Scholar]
- Stone EM, Nichols BE, Kimura AE, Weingeist TA, Drack A, Sheffield VC. Clinical features of a Stargardt-like dominant progressive macular dystrophy with genetic linkage to chromosome 6q. Arch Ophthalmol. 1994;112:765–772. doi: 10.1001/archopht.1994.01090180063036. [DOI] [PubMed] [Google Scholar]
- Swaroop A, Chew EY, Rickman CB, Abecasis GR. Unraveling a multifactorial late-onset disease: from genetic susceptibility to disease mechanisms for age-related macular degeneration. Annu Rev Genomics Hum Genet. 2009;10:19–43. doi: 10.1146/annurev.genom.9.081307.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Holleran WM. Omega-O-acylceramide, a lipid essential for mammalian survival. J Dermatol Sci. 2008;51:77–87. doi: 10.1016/j.jdermsci.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Umeda S, Suzuki MT, Okamoto H, Ono F, Mizota A, Terao K, Yoshikawa Y, Tanaka Y, Iwata T. Molecular composition of drusen and possible involvement of anti-retinal autoimmunity in two different forms of macular degeneration in cynomolgus monkey (Macaca fascicularis) FASEB J. 2005;19:1683–1685. doi: 10.1096/fj.04-3525fje. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Jablonski MM, Mandal MN, Raz-Prag D, Wang XF, Nizol L, Iannaccone A, Musch DC, Bush RA, Salem N, Jr, Sieving PA, Ayyagari R. Elovl4 5-bp-deletion knock-in mice develop progressive photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2006;47:4558–4568. doi: 10.1167/iovs.06-0353. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Sharon M, Salem N, Jr, Ayyagari R. Role of ELOVL4 in fatty acid metabolism. Adv Exp Med Biol. 2008;613:283–290. doi: 10.1007/978-0-387-74904-4_33. [DOI] [PubMed] [Google Scholar]
- Vasireddy V, Uchida Y, Salem N, Jr, Kim SY, Mandal MN, Reddy GB, Bodepudi R, Alderson NL, Brown JC, Hama H, Dlugosz A, Elias PM, Holleran WM, Ayyagari R. Loss of functional ELOVL4 depletes very long-chain fatty acids (> or =C28) and the unique omega-O-acylceramides in skin leading to neonatal death. Hum Mol Genet. 2007;16:471–482. doi: 10.1093/hmg/ddl480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasireddy V, Vijayasarathy C, Huang J, Wang XF, Jablonski MM, Petty HR, Sieving PA, Ayyagari R. Stargardt-like macular dystrophy protein ELOVL4 exerts a dominant negative effect by recruiting wild-type protein into aggresomes. Mol Vis. 2005;11:665–676. [PubMed] [Google Scholar]
- Wakil SJ. Fatty acid synthase, a proficient multifunctional enzyme. Biochemistry. 1989;28:4523–4530. doi: 10.1021/bi00437a001. [DOI] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357:553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- Young RW. Pathophysiology of age-related macular degeneration. Surv Ophthalmol. 1987;31:291–306. doi: 10.1016/0039-6257(87)90115-9. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ, Kakuk LE, Lagali PS, Wong PW, MacDonald IM, Sieving PA, Figueroa DJ, Austin CP, Gould RJ, Ayyagari R, Petrukhin K. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27:89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Mashima Y, Noda S, Imamura Y, Kudoh J, Shimizu N, Nishiyama T, Umeda S, Oguchi Y, Tanaka Y, Iwata T. Characterization of AOC2 gene encoding a copper-binding amine oxidase expressed specifically in retina. Gene. 2003;318:45–53. doi: 10.1016/s0378-1119(03)00753-4. [DOI] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]