

Abstract
To better understand the immunopathogenesis of chronic inflammatory lung disease, we established a mouse model of disease that develops after respiratory viral infection. The disease that develops in this model is similar to chronic obstructive lung disease in humans. Using this model we have characterized two distinct phases in the chronic disease process. The first phase appears at three weeks after viral infection and depends on type I interferon‐dependent expression and then subsequent activation of the high‐affinity IgE receptor (FcɛRI) on conventional lung dendritic cells, which in turn recruit IL‐13‐producing CD4+ T cells to the lower airways. The second phase becomes maximal at seven weeks after infection and depends on invariant natural killer T (iNKT) cells and lung macrophages. Cellular cross‐talk relies on interactions between the semi‐invariant Vα14Jα18 T‐cell receptor on lung iNKT cells and CD1d on macrophages as well as iNKT cell‐derived IL‐13 and IL‐13 receptor on macrophages. These interactions drive macrophages to a pattern of alternative activation and overproduction of IL‐13. This innate immune axis is also activated in patients with chronic obstructive lung disease, as evidenced by increased numbers of iNKT cells and IL‐13‐producing alternatively activated macrophages marked by chitinase 1 production. Together the findings identify two new immune pathways responsible for early and late phases of chronic inflammatory lung disease in experimental and clinical settings. These findings extend our understanding of the complex mechanisms that underlie chronic obstructive lung disease and provide useful targets for diagnosis and therapy of this common disorder.
Keywords: dendritic cell, natural killer T cell, NKT, alternatively activated macrophage, interleukin‐13, chronic obstructive lung disease
Introduction
Chronic inflammation is critical to the pathogenesis of many common diseases and is generally attributed to persistent activation of the immune system. This type of long‐term immune activation is often linked to aberrations of the adaptive immune system, involving antigen‐specific antibodies or effector T‐cell responses that react against endogenous stimuli. 1 , 2 It is possible, however, that components of the innate immune system also contribute to the development of chronic inflammatory disease. 3 Thus, precisely how the immune system drives chronic inflammatory disease still needs to be fully defined. In that context, lung disease offers a useful paradigm for investigating mechanisms underlying chronic inflammation as chronic obstructive lung disease represents one of the most common types of chronic illness that affects humans. In particular, asthma affects approximately 10% of the population, and chronic obstructive pulmonary disease (COPD) is the fourth leading cause of death in the United States and worldwide. 4 Although asthma and COPD are regarded as separate clinical entities, it is evident that overlapping airway pathology can occur in both diseases. 5 It is even possible that patients diagnosed initially with asthma may be at risk for developing COPD later in life. 6 Thus, asthma and COPD may constitute discrete manifestations of a disease continuum resulting from a common pathological process that should be defined in molecular, rather than clinical, terms.
Previous studies have identified genetic and environmental factors that contribute to the development of chronic obstructive lung disease. Of special interest to this review, several clinical studies have proposed a role for respiratory viral infection in precipitating as well as contributing to the pathogenesis of chronic obstructive lung disease. Perhaps the strongest disease association for virus‐induced lung disease exists for respiratory syncytial virus (RSV), 7 a member of the Paramyxoviridae family that commonly infects the bronchial airways of infants and adults, causing severe bronchiolitis in a subpopulation of individuals. 8 A number of prospective case‐controlled studies provide evidence that severe RSV‐induced bronchiolitis in early infancy constitutes a significant risk factor for the subsequent development of childhood asthma. 9 , 10 , 11 , 12 , 13 Putative etiological associations have also been made for other respiratory viruses, but most of these (especially human metapneumovirus [hMPV], rhinovirus [RV], human parainfluenza virus [hPIV], adenovirus, and coronavirus) are more likely associated with exacerbation of existing disease. 14 Furthermore, a number of clinical studies suggest that respiratory viral infections are the major trigger for exacerbations of asthma and COPD. 15 , 16 , 17 , 18 , 19 , 20 , 21 This association has been confirmed experimentally using mouse models that have demonstrated that viral infection can cause features of inflammatory lower airway disease in susceptible strains of mice. 22 , 23 However, these observations did not fully develop a causal relationship between respiratory viral infection and chronic airway disease or define the precise mechanism by which viral infections contribute to the development and persistence of this type of disease.
The purpose of this review is to summarize recent findings obtained from our laboratory that detail how the immune response to respiratory viral infection drives the development of chronic obstructive lung diseases, particularly asthma and COPD. The review is organized into five subsequent sections that address (1) immune pathways that are conventionally associated with chronic obstructive lung disease; (2) animal models that can be used to study the basis for chronic obstructive lung disease; (3) the identification of a distinct adaptive immune response that mediates the initial phase of chronic lung disease after viral infection; and (4) the identification of a novel innate immune pathway that mediates the second persistent phase of chronic lung disease after viral infection. The final section provides a summary and perspective on this field of research.
Traditional pathways for chronic obstructive lung disease
Respiratory tolerance, and mucosal immunity in general, is maintained by a complex system of host defense mechanisms aimed at eliminating aeropathogens while tolerating innocuous airborne antigens and commensal microorganisms. Presumably, this immune balance of reaction versus tolerance may become permanently skewed and thereby progress to chronic inflammation. A scheme that is commonly used to describe inflammation in the lung (and other sites) is based on a model of immune polarity for the T helper (Th) cell system. Within this system, CD4+ Th cell‐dependent responses are classified into Th type 1 (Th1) or type 2 (Th2). Th1 cells characteristically mediate delayed‐type hypersensitivity reactions and selectively produce interleukin (IL)‐2 and interferon (IFN)‐γ. This type of response is also found during respiratory viral infection. In contrast, Th2 cells promote B‐cell‐dependent humoral immunity, IgE class switching, eosinophil recruitment, mast cell proliferation, alternative activation of macrophages, and selective production of IL‐4, IL‐5, and IL‐13 that are all typical of an infection with an extracellular parasite and are commonly found during an allergic response. 24 Indeed, this type of response can be detected in allergen‐challenged mice as well as allergic asthmatics, 5 , 25 , 26 , 27 and Th2 cytokine production is often a prominent feature of asthma. 28 , 29 , 30 , 31 For example, IL‐13 is increased in the bronchoalveolar lavage fluids of allergen‐provoked asthmatic subjects 30 and IL‐13 mRNA and protein expression has been detected in bronchial biopsies obtained from allergic and nonallergic asthmatic patients. 32 , 33 In addition, polymorphisms at the IL‐13 gene locus have been shown to confer an enhanced risk of asthma and COPD. 34 , 35 , 36 Increased IL‐13 expression is also found in some but not all studies of subjects with COPD. 37 , 38 , 39 This discrepancy is likely a result of the heterogeneity of disease within and between individuals and is a challenge for current clinical trials of anti‐IL‐13 therapy in subjects with asthma. 40
Some investigators have proposed that Th2‐biased immune deviation may also influence the response to viral infection. For example, RSV infection may elicit Th2 cytokine production in allergen‐sensitized mice. 41 , 42 , 43 Whether RSV infection can drive persistent production of IL‐13 remains uncertain. Moreover, allergic inflammation is not characteristic of all forms of asthma or COPD. For example, bronchial biopsy specimens obtained from subjects with severe asthma and COPD often contain increased numbers of CD8+ T cells, macrophages, and neutrophils. 44 , 45 Moreover, subjects with even moderate asthma exhibit activation of IFN‐signaling proteins, indicative of a Th1‐type response. 46 Furthermore, studies in mice using adoptive transfer protocols indicate that pro‐inflammatory cells, such as Th1 CD4+ and CD8+ T‐cell subsets, can potentiate allergic airway disease. 47 , 48 Concerning as well is the observation that many forms of airway disease that are “Th2‐polarized” are in fact precipitated by exposure to nonallergic stimuli (such as respiratory viruses) that would ordinarily elicit a Th1 response. Lastly, the Th1 versus Th2 paradigm has been further challenged by the discovery of Th17 (IL‐17‐producing) and T regulatory or Th3 (IL‐10‐ and transforming growth factor [TGF]‐β‐producing) subsets of T cells, which appear to influence allergic inflammation of the airways. 49 , 50 Thus, the immune response underlying chronic airway disease does not strictly adhere to the Th2 paradigm as aspects of both Th1 and Th2 immune responses appear to drive disease. 51 , 52 , 53 Because the Th1 versus Th2 model of adaptive immunity did not fully explain the subsequent development and maintenance of chronic inflammatory disease, we questioned whether other aspects of immunity and inflammation might also be critical for the pathogenesis of chronic airway disease following viral infection. Accordingly, we sought to establish an experimental model that would enable us to precisely assess the role of the innate and adaptive systems in the development and maintenance of chronic obstructive lung disease.
Animal models of chronic airway disease
Animal models of inflammatory airway disease have generally relied on allergen‐driven responses that are meant to be representative of allergic asthma. The most common approach for these models involves sensitization and subsequent airway challenge using chicken ovalbumin, but others have also applied allergens derived from insects, e.g., house dust mite and cockroach, as well as infectious agents, e.g., Schistosoma mansoni, Nippostrongylus brasilensis, and Aspergillus fumigatus. Although these models do not fully replicate the features of asthma in humans, they serve to identify cellular and molecular determinants of allergen‐induced inflammation. 54 , 55 , 56 , 57 , 58 A critical drawback to acute allergen challenge models is the transient nature of the airway disease. Allergen‐driven models of airway disease therefore fundamentally limit study of the immune mechanisms that contribute to persistent inflammation found in chronic obstructive lung disease in humans. In addition, these models are unlikely to account for the nonallergic component of lung disease found after viral infection or other inflammatory stimuli, such as cigarette smoke.
Thus, to better understand how the immune response drives chronic airway disease, we developed a new model of chronic airway disease that develops after viral infection. The rationale for developing this type of model is rooted in epidemiological evidence linking acute viral infection with chronic airway disease. 9 , 10 , 11 , 12 , 13 , 15 , 16 , 17 , 18 , 19 , 20 , 21 Because infection with RSV is most strongly linked to the development of asthma, investigators have often used RSV as the etiological agent for study. In fact, RSV infection alone or in combination with allergen challenge has now been studied as a model for airway inflammation in guinea pigs, ferrets, rats, and mice. 59 , 60 , 61 , 62 , 63 On the basis of similar associations with exacerbations of asthma, others have also studied human rhinovirus (RV) and metapneumovirus (hMPV). The limitation of these models is that human pathogens often replicate poorly in other species. In the case of RSV, an extremely high inoculum of virus must be administered in order to cause disease in mice, resulting in an all‐or‐none pattern of illness. 64 Accordingly, we have advanced a model based on a natural mouse pathogen known as Sendai virus (SeV), a parainfluenza virus closely related to other human paramyxoviruses (e.g., RSV, hMPV, and hPIV). In contrast to RSV, SeV replicates at high efficiency in the lower airway of mice, resulting in acute inflammation of the small airways that maintains high fidelity to viral bronchiolitis in humans. Administration of a modest SeV inoculum (e.g., 1 × 105 plaque‐forming cells) to young immunocompetent mice (e.g., 6–10 weeks of age) elicits a top‐down pattern of infection that is limited to airway epithelial cells and neighboring macrophages. 65 , 66 , 67 This illness is therefore remarkably similar to the one observed in severe RSV infections in humans. It appears likely that this pattern of infection is necessary for the subsequent development of chronic lung disease in this model and perhaps in humans as well.
Our assessment of SeV‐induced pathology in different inbred strains of mice led to the finding that progression from acute infection to chronic disease occurred singularly in the context of the C57BL/6J (B6) genetic background. Infection of this strain with SeV triggers an acute antiviral response that is followed by a delayed but permanent switch to chronic airway disease manifested by mucous cell metaplasia (with increased lung levels of Muc5ac mRNA and Muc5ac immunostaining), immune cell filtration (with increased numbers of macrophages and neutrophils), and airway hyper‐reactivity (signified by increased pulmonary resistance in response to inhaled methacholine). 23 , 68 , 69 This chronic airway disease becomes apparent at 3 weeks after viral inoculation, reaches maximal levels at 7 weeks after inoculation, and persists indefinitely. 23 , 37 , 68 , 69 Induction of chronic disease requires live replicating virus because administration of UV‐inactivated virus or MDA5/Tlr3‐agonist polyinosinic : polycytidylic acid will not cause subsequent disease (Ref. 37 and unpublished observations).
The conventional dendritic cell–T‐cell axis in airway disease
Our analysis of the viral model in B6 mice provided the opportunity to assess gene‐targeted and transgenic mice available on this genetic background. Initial analysis of our model using mice with a targeted deletion of intercellular adhesion molecule 1 (ICAM‐1), a widely‐expressed adhesion molecule, revealed that acute disease could be genetically segregated from chronic disease. 23 In particular, ICAM‐1 −/− mice failed to develop a robust inflammatory response and concomitant airway hyper‐reactivity at 3 weeks after viral infection. Nonetheless, ICAM‐1 −/− mice remained susceptible to the development of chronic inflammatory lung disease. These observations thereby established the concept that separate pathogenic mechanisms could account for acute versus chronic inflammatory disease, laying the conceptual framework for our model of immune‐based chronic lung disease.
A significant breakthrough in our understanding of virus‐induced lung disease came with the discovery that the acute and chronic disease that developed after viral infection required the production of IL‐13 in the lung. 68 Using a combination of high‐speed fluorescence‐activated cell sorter (FACS) and real‐time PCR analysis, we were able to identify CD4+ T cells as the chief cellular source of IL‐13 production at 3 weeks after viral infection. 37 This finding raised the possibility that elements of an adaptive immune response that are characteristically involved in allergen‐induced airway disease might also contribute to virus‐induced airway disease. To address this prospect, we pursued the underlying mechanism for CD4+ T‐cell production of IL‐13 in our model and in doing so uncovered a novel immune axis that links the antiviral response to lung disease. 70 In brief, we found that IL‐13 production by CD4+ T cells following acute SeV infection is dependent upon induction of type I IFN production. Type I IFN drives expression of high‐affinity IgE receptor FcɛRI on the surface of conventional dendritic cells (cDCs). Subsequent activation of cDCs through FcɛRI cross‐linking then drives the production of CCL28 that mediates recruitment of IL‐13‐producing CD4+ T cells to the lung. These findings establish a novel mechanism whereby the antiviral adaptive immune response, which is characteristically pro‐inflammatory or Th1 in character, can be transformed into an allergic Th2 response marked by the chronic production of IL‐13 by CD4+ T cells. The next section provides some of the pertinent evidence to support this mechanism.
The antiviral immune response induces expression of FcɛRI on resident cDCs
We approached our study of the viral model by considering that respiratory viral infections are known to drive antiviral IgE production and consequent immune cell expression of FcɛRI in humans. 71 , 72 , 73 We found that total serum IgE and virus‐specific IgE were increased by 7 days and were still increased at 3 weeks after inoculation with SeV. The question as to how IgE might be involved in mediating chronic disease became the focus of further study.
What was known at the time of our studies was that the high‐affinity IgE receptor was detectable on mast cells, basophils, and eosinophils in mice and humans 74 where it is expressed as a tetrameric complex consisting of one α (FcɛRIα), one β (FcɛRIβ), and two γ (FcɛRIγ) chains. 75 However, in humans surface expression of FcɛRI was additionally described for peripheral blood cDCs, plasmacytoid DCs (pDCs), monocytes, and Langerhan cells, although in these leukocyte subsets expression is restricted to a trimeric form (FcɛR1αγγ) that lacks the β chain. 76 , 77 , 78 Given this extended profile of FcɛRI expression in humans, we questioned whether we could detect induced expression of this receptor complex on mouse lung cDCs following viral infection. Using a combination of FACS‐based forward/side scatter properties and high CD11c expression levels to identify lung cDCs (obtained following repeated bronchoalveolar lavage to remove alveolar macrophages), we observed that SeV infection leads to rapid migration of lung cDCs to the draining lymph nodes in a CCR5–CCL5‐dependent manner. 79 Furthermore, we found that the majority of the cDCs remaining in the lung after acute infection matured and differentiated, as marked by upregulation of CD86 and major histocompatibility complex (MHC)‐II surface expression.
As the function of this resident population of cDCs was uncertain, we sought to further characterize them. Real‐time PCR analysis of purified lung cDCs revealed that FcɛRIα expression was induced within 3 days and remained stable for 2–3 weeks after viral inoculation. In addition, the level of FcɛRIα expression on lung cDCs was comparable to levels on c‐kit+ lung mast cells. 70 Immunoprecipitation, immunoblotting, and real‐time PCR assays revealed that the trimeric form of the FcɛRI receptor alone was expressed in lung cDCs, consistent with the pattern of expression on human antigen‐presenting cells. Furthermore, virus‐induced expression of FcɛRIα was largely restricted to cDCs residing in the lung, and not in the spleen or draining lymph nodes, thus spatially restricting downstream effects of cDC‐specific FcɛRIα expression to events occurring in the lung parenchyma.
To determine whether FcɛRI expression was consequential to the host in the course of the acute antiviral immune response, we compared several immune parameters in wild‐type and FcɛRIα −/− mice. To this end, we observed that FcɛRIα −/− mice exhibited comparable acute morbidity and identical patterns of viral replication and clearance (as determined by plaque‐forming and real‐time PCR assays). Furthermore, comparative analysis of co‐stimulatory molecule expression on resident cDCs derived from wild‐type or FceRIa −/− mice revealed no differences in the levels of MHC‐II, CD80, or CD86, thereby indicating that the basic antigen‐presenting functions of cDCs from FceRIa −/− mice were intact. We also found that FcɛRIα‐deficient mice generated comparable numbers of antiviral cytotoxic T cells in response to virus, as determined by SeV‐specific tetramer analysis of lung immune cells. Therefore, while viral infection induces expression of FcɛRI on the surface of cDCs, this expression pattern does not influence the acute immune response to virus.
Type I IFN receptor regulates cDC expression of FcɛRI
We next assessed the mechanism responsible for induction of FceRIa gene expression in lung cDCs after viral infection. We recognized the influence of serum IgE level on the expression of the high‐affinity IgE receptor on mast cells in allergy. 80 We therefore reasoned that SeV‐specific IgE could drive FcɛRI expression on lung cDCs after SeV infection as well. However, we found that total serum IgE and SeV‐specific IgE did not appreciably increase until 7 days after infection. As the onset of expression of FcɛRIα on lung cDCs predicates the induction of IgE expression, factors other than IgE must drive FcɛRIα expression in our model. Indeed, even IgE‐deficient mice exhibit the same increase in FcɛRIα expression on lung cDCs as wild‐type mice after viral infection. 81
We therefore focused our attention on the role of type I interferon signaling in regulating FcɛRI expression as these events occur in concert after viral infection. We directly examined whether type I interferon signaling was necessary for induction of FcɛRIα expression on lung cDCs using type I IFN receptor (IFNAR)‐deficient (IFNAR −/−) mice. We found that lung cDCs from IFNAR −/− mice did not manifest induction of FcɛRIα expression after SeV infection. Adoptive transfer using either IFNAR −/− or wild‐type purified cDCs established that IFNAR expression on cDCs was not necessary for induction of FceRIa expression on lung cDCs. This finding suggested that IFN‐α/β functions through additional cellular intermediates and so we expanded our assessment to include additional adoptive transfer studies that identified neutrophils as a critical link between upstream type I interferon signaling and downstream cDC activation. 82
FcɛRI on lung cDCs drives Ccl28‐dependent recruitment of Th2 cells
We reasoned that FcɛRI activation on lung cDCs could be linked to increase in IL‐13‐producing T cells in the lung after viral infection. To assess this possibility, we applied a co‐culture system using lung cDCs and CD4+ T cells. In this system, we found that that antigen‐dependent interaction of lung cDCs and T cells can drive T‐cell proliferation and production of IL‐13; however, cross‐linking FcɛRI on the surface of cDCs did not influence this process. This finding led us to examine whether FcɛRI signaling might elicit additional responses that could account for local IL‐13 production in the lung, e.g., through recruitment of IL‐producing T cells to the lung. Indeed, we were able to demonstrate that cell supernatants derived from cross‐linked cDCs were capable of stimulating the migration of CD4+ T cells in a chemotaxis assay system. Ligand desensitization and blocking antibody assays revealed that CCL28‐CCR10 interactions were responsible for mediating this process. Of relevance to this finding, increased expression of CCL28 is associated with both human asthma and mouse models of allergic airway disease. 83 , 84 , 85 This immune axis therefore provides an additional mechanism for CCL28 production and action in the setting of inflammatory airway disease.
To extend our findings in vivo, we re‐evaluated the viral mouse model and found that lung levels of Ccl28 mRNA were significantly increased in wild‐type mice after viral infection. Furthermore, the increase in Ccl28 gene expression after infection was blocked in FcɛRI −/− mice, suggesting that FcɛRI‐mediated signals drove this process. We also found that the relative numbers of IL‐13‐producing CD4+ T cells were significantly reduced in SeV‐infected FcɛRIα −/− mice compared to wild‐type mice. In addition, FcɛRIα −/− mice demonstrated a marked decrease in the number of Muc5ac‐expressing epithelial cells and total lung Muc5ac mRNA levels at 3 weeks after viral infection. To confirm that FceRI expression on cDCs is necessary for lung disease, we performed adoptive cell‐transfer experiments using cDCs derived from wild‐type or FceRIa −/− mice transferred into FceRIa −/− recipients. We found that reconstitution with wild‐type but not FceRIa−/– cDCs restored post‐viral mucous cell metaplasia in FceRIa −/−‐recipient mice. The significance of FcɛRI‐dependent CCL28‐mediated disease was verified using mice that were treated with anti‐CCL28 blocking mAb. In these experiments, mice treated with anti‐CCL28 mAb were prevented from developing mucous cell metaplasia relative to control IgG2b‐treated mice. Together these findings establish a direct role for FcɛRI in mediating the airway disease that develops after viral infection. These findings support the proposal that mucous cell metaplasia after viral infection depends on an adaptive immune response that begins with IFN‐mediated expression of FcɛRI on resident cDCs and continues to activation of FcɛRIα, presumably through SeV‐specific IgE, to transduce signals in these cDCs to drive production of Ccl28 and subsequent recruitment of IL‐13‐producing CD4+ T cells to the lungs. This model of disease is relevant to a clinical understanding of post‐viral inflammatory airway disease, a condition that is marked by airway hyper‐reactivity and is sometimes associated with elements of atopy, including elevated levels of serum IgE. 86 This condition often resolves by several weeks after viral infection (in humans and in the mouse model), but it is possible that a chronic inflammatory condition might develop in the face of ongoing infectious or allergic stimuli.
The natural killer T cell–macrophage axis in airway disease
Our discovery of the cDC–T‐cell axis helps explain how the adaptive immune response can drive inflammatory disease in the 3‐week period after viral infection. However, mice deficient in FceRIa −/− still develop chronic disease that peaks at 7 weeks after infection (unpublished observations, L.A. Benoit, D. Byers, M.H. Grayson, and M.J. Holtzman), thus suggesting that this adaptive immune axis does not mediate the later phase of chronic disease. We therefore speculated that components of the innate immune system might drive the continued disease process that also depends on IL‐13‐expressing macrophages. 37 When we pursued the underlying mechanism for macrophage production of IL‐13 in this model, we uncovered a second novel immune pathway leading to chronic inflammatory lung disease. Unexpectedly, this pathway occurred independently of an adaptive immune response, as evidenced by disease progression in MHC‐II−/−, Cd4−/−, and Cd8−/− mice 37 as well as in B‐cell‐deficient (muMT) mice (unpublished observations). FACS‐based analysis of the lung immune infiltrates in chronically affected mice revealed a substantial increase in the number of natural killer T (NKT) cells. Further assessment revealed that CD4− NKT cells were directly responsible for the production of IL‐13 by macrophages. Assessment of lung tissue from patients with severe asthma and COPD indicated that this innate immune response axis was also activated in humans with chronic obstructive lung disease. Collectively these findings indicate that the switch between acute antiviral immune responses to chronic inflammatory disease requires the persistent activation of a novel macrophage–NKT‐cell innate immune response axis and thereby provides mechanistic insight into the pathogenesis of chronic inflammatory disease that is distinct from other reports of NKT‐cell activation in asthma and asthma models. 87 , 88 , 89 , 90 , 91 We present an outline of these findings in this section of the review.
Persistence disease depends on macrophage production of IL‐13
As indicated above, B6 mice infected with SeV develop chronic airway disease that is first detected at 3 weeks and is maximal in severity at 7 weeks after viral inoculation. Coinciding with the disease time course, significant IL‐13 production is observed at 3 weeks and is maximal at 7 weeks after inoculation. A direct causative role for IL‐13 in mediating chronic airway disease was established when mice treated with an IL‐13 decoy receptor (sIL‐13‐Rα2‐Fc) and IL‐13 gene‐targeted mice failed to develop mucous cell metaplasia or airway hyper‐reactivity after viral infection. We detected comparable levels of remnant viral RNA in Il‐13 −/− and wild‐type mice, indicating that chronic lung disease depends on immune cell production of IL‐13 rather than differences in viral RNA levels between mouse strains.
We next investigated the cellular sources of chronic IL‐13 production in the lung after viral infection. Whereas CD4+ T cells contributed the highest amount of Il‐13 mRNA at 3 weeks, we found that macrophages became the predominant source of Il‐13 mRNA at 7 weeks after inoculation. In each case, the individual cellular contribution to total lung Il‐13 mRNA levels was based on an increased number of cells recruited to the lung as well as increased production of Il‐13 mRNA per cell. Secondary sources of Il‐13 mRNA included NK cells and NKT cells, although no detectable Il‐13 mRNA was found in mast cells, basophils, neutrophils, DCs, B cells, or CD8+ T cells. We also found that lung macrophages isolated from SeV‐infected mice produced corresponding increases in IL‐13 protein. In addition, immunohistochemical analysis detected IL‐13+ cells with typical macrophage morphology and CD68 expression in alveolar, interstitial, and epithelial locations.
To determine whether IL‐13‐producing macrophages are required for persistent lung disease, we next examined macrophage deficient mice. We first assessed op/op mice that are macrophage deficient due to a loss of function mutation in the Csf1 (or M‐csf) gene. Compared to wild‐type mice, the op‐op mice exhibited markedly decreased levels of Il‐13 and Muc5ac gene expression at 7 weeks after infection. However, there are notable immune defects in these mice that could complicate the interpretation of our results, and so we applied a clodronate‐containing liposome delivery protocol to our model to selectively deplete lung macrophages. 66 Clodronate treatment was initiated after clearance of infectious virus to avoid any possible effects on the acute infection process. Using this system, we found no differences in the levels of Il‐13 or Muc5ac mRNA at 3 weeks after viral inoculation, consistent with a lack of IL‐13 production by macrophages at this early time. However, macrophage‐depleted mice demonstrated significantly decreased levels of Il‐13 and Muc5ac gene expression compared to control mice at 7 weeks after inoculation. Furthermore, immunohistochemical analysis of lung sections indicated that IL‐13‐producing macrophages were no longer found in clodronate‐treated mice. We therefore concluded that IL‐13+‐producing macrophages were contributing to the chronic disease in this model.
Natural killer T‐cell‐mediated activation of macrophages
In addressing the issue of macrophage activation in our model, we reasoned that an additional aspect of the immune system was driving persistent IL‐13 production by lung macrophage. As noted above, this type of chronic pressure is generally attributed to activation of the adaptive immune system. However, strain‐based analysis revealed that mice genetically deficient in CD4+ T cells, CD8+ T cells, or B cells continued to develop mucous cell metaplasia in concert with increased Il‐13 and Muc5ac mRNA levels after viral infection. These findings suggested that elements unrelated to the adaptive immune system might drive persistent macrophage activation. To assess this issue more thoroughly, we performed T‐cell depletion studies using antibody treatment that was initiated following clearance of infectious virus to circumvent the possibility that these primary immune deficiencies could somehow be interfering with the acute antiviral response. Similar to mice with genetic T‐cell deficiencies, mice that were depleted of CD4+ T cells, CD8+ T cells, or both CD4+ and CD8+ T cells demonstrated increased numbers of IL‐13‐producing macrophages, significant induction of Il‐13 and Muc5ac gene expression, and mucous cell metaplasia after viral infection. These findings indicated that additional components outside of the adaptive immune system must contribute to the chronic disease that develops after viral infection.
This unexpected finding in animals deficient in elements of the adaptive immune system us to consider whether innate immune cells could be responsible for chronic macrophage activation. This possibility was supported when we discovered that NKT cells were recruited to the lung (on the basis of increased numbers of cells) and activated (on the basis of increased Il‐13 mRNA production) at 7 weeks after SeV infection. Analysis of whole lung NKT‐cell populations indicated that CD4− predominated over CD4+ cells and that both populations of NKT cells produced Il‐13 without a corresponding increase in Il‐4 mRNA. Although the relative contribution of NKT cells to the overall levels of Il‐13 mRNA in the lung was relatively small compared to macrophages, the persistent production of Il‐13 mRNA by CD4− NKT cells might still regulate downstream macrophage activation and production of IL‐13. In fact, mice that were genetically deficient in NKT cells as a result of Cd1d or J α 18 gene targeting 92 , 93 were largely protected from the usual increase in IL‐13‐expressing macrophages as well as Il‐13 and Muc5ac mRNA and airway hyper‐reactivity at 7 weeks after viral inoculation. There was no influence of NKT‐cell deficiency at 3 weeks after inoculation, consistent with the time course for macrophage production of IL‐13 in the viral model. These findings support a role for invariant NKT (iNKT) cells in the chronic activation of macrophage production of IL‐13 after viral infection.
Direct and indirect mechanisms of NKT‐cell‐mediated activation of macrophages
NKT cells could promote macrophage activation through recruitment (similar to the role of cDCs in the first phase of disease) as well as activation of macrophages in the lung. Evidence for NKT‐cell‐dependent recruitment of macrophages was substantiated by the observation that Cd1d −/− mice exhibited decreased numbers of lung macrophages relative to wild‐type mice at 7 weeks after viral inoculation. Moreover, purified lung CD4− NKT cells released significant levels of monocyte/macrophage chemotaxins Ccl3 and, to a lesser extent, Ccl5, after in vitro stimulation with phorbol 12‐myristate 13‐acetate (PMA)‐ionomycin. Similarly, this cell population produced increased levels of Ccl3 mRNA when isolated from mice at 7 weeks after inoculation compared to uninfected control mice. Increased Ccl3 levels were localized to the CD4− NKT compartment, consistent with the increased activity of CD4− NKT cells in chronic lung disease after viral infection.
To determine whether NKT cells might directly stimulate macrophage production of IL‐13, we established a co‐culture system using purified populations of NKT cells and macrophages. Cells used in this assay system were principally derived from the lungs because NKT cells and macrophages isolated from other compartments proved less capable of inducing IL‐13 production. In addition, the cells were derived from uninfected mice to avoid downregulation of T‐cell receptor (TCR) function and background production of IL‐13. Using this co‐culture system, we found that direct NKT‐cell–macrophage interaction resulted in production of Il‐13 mRNA and IL‐13 protein by adherent macrophages within 24 h of co‐culture. The NKT‐cell effect on macrophage production of IL‐13 was relatively specific for lung NKT cells as naive CD4+ T cells failed to induce IL‐13 production by macrophages. Furthermore, NKT‐cell‐induced production of IL‐13 by macrophages was significantly inhibited by treatment with an anti‐CD1d‐blocking mAb and markedly increased by addition of α‐GalCer, a synthetic CD1d ligand. These results indicated that NKT‐cell‐dependent activation of macrophages required interaction of Vα14‐Jα18 invariant TCR on the NKT cell with CD1d on the macrophage. We also found that NKT cells from Il‐13 −/− mice were no longer capable of driving macrophage production of IL‐13 in the co‐culture system. This finding established a new role for IL‐13–IL‐13 receptor (IL‐13R) signaling in controlling the NKT‐cell–macrophage interaction. Co‐culture analysis also indicated that NK1.1+ NKT cells were primarily responsible for driving IL‐13 production by macrophages (unpublished observations). Analysis of liver NKT cells indicated that these cells were less effective than lung NKT cells for macrophage activation, requiring the addition of α‐GalCer to stimulate macrophage production of IL‐13. Nonetheless, preparations of either CD4+ or CD4− liver NKT cells were equally capable of activating lung macrophages to produce IL‐13 following addition of α‐GalCer. Thus, while CD4+ NKT cells have been the focus for studies of the response to allergens in the lung, 88 it appears that CD4 expression is not necessary for NKT cells to drive macrophage‐dependent disease after viral infection. Instead, the NKT‐cell–macrophage pathway is primarily dependent on invariant iTCR–CD1d and IL‐13–IL‐13R interactions as well as NK1.1 expression.
Positive feedback for IL‐13 production
We further characterized the downstream events in the NKT‐cell–macrophage–IL‐13 immune axis to better define whether this pathway could upregulate expression of additional cytokines and/or cytokine receptors. Oligonucleotide microarray analysis of lung mRNA identified Il‐13 receptorα (Il‐13ra1) as the predominant cytokine‐related gene that was significantly upregulated after viral infection. Increased Il‐13ra1 expression no longer developed in Cd1d −/− mice, suggesting that NKT cells stimulate an increase in Il‐13ra1 mRNA. The increase in lung Il‐13ra1 mRNA expression after viral infection was relatively small, further suggesting that increased expression of Il‐13ra1 might be restricted to a subpopulation of cells. In fact, Il‐13ra1 mRNA expression was restricted mainly to lung macrophages, and this expression was significantly decreased in macrophages isolated from Cd1d −/− mice. These observations suggested that the interaction of IL‐13 produced by NKT cells with IL‐13R on the surface of the macrophage could serve to amplify macrophage production of IL‐13. Indeed, we found that blockade of IL‐13 signaling by delivery of sIl‐13Rα2‐Fc caused a marked decrease in the levels of IL‐13‐producing macrophages, which in turn caused a decrease in total Il‐13 mRNA levels in the lung. Together these findings suggest a positive feedback loop whereby IL‐13 signaling induces expression of IL‐13R and IL‐13 by macrophages in an NKT‐cell‐dependent manner.
Alternative activation of macrophage
Further analysis of whole genome microarrays also revealed a pattern of gene expression that is characteristic of an alternative pathway for activation of macrophages. 94 , 95 We found especially prominent upregulation of genes encoding chitinase‐like proteins (Chi3l3/4), resistin‐like molecules (Rtn1a or Fizz1), arginase (Arg1), matrix metalloproteinases (Mmp12), and arachidonate 12‐lipoxygenase (Alox12e) at 7 weeks after viral inoculation. Our results were corroborated by real‐time PCR analysis, which further revealed that expression of these transcripts was completely blocked in Il‐13 −/− mice and largely blocked in Cd1d −/− and Ja18 −/− mice. Immunostaining and FACS analysis indicated that expression of Chi3l3/4 proteins were principally restricted to CD68+ macrophages. Previous work established that alternative activation of macrophages may occur in the setting of parasitic infection or allergy, 24 but our results provide the initial evidence that this type of response also occurs in response to viral infection. The biological consequences of alternative activation remain uncertain. There is some evidence that arachidonate 12‐lipoxygenase and arginase 1 may promote while resistin‐like molecule α may protect against allergic airway inflammation. 96 , 97 However, the role of these gene products as well as chitinase and chitinase‐like proteins for the development of chronic lung disease after viral infection in experimental and clinical settings still needs to be defined. Nonetheless, as developed in the next section, these proteins can still be useful biomarkers of the NKT‐cell–alternatively activated macrophage immune axis in humans.
NKT cell–macrophage pathway in humans
Excessive mucus production is a common feature of acute respiratory infection as well as chronic obstructive lung disease. Autopsies performed on asthmatic patients who died in status asthmaticus demonstrate postmortem findings of mucus plugging and inspissation in approximately 75% of cases. Similarly, occlusion of the small airways by mucus‐containing inflammatory exudates is associated with early death in patients with COPD. 98 Given the involvement of NKT cells and macrophages in the development of mucous cell metaplasia after viral infection, we sought to extend our analysis of this immune axis to patients with chronic lung disease that is also characterized by excess mucus production.
Although our analysis of human samples is at an early stage, we have obtained significant evidence that the NKT‐cell–macrophage immune axis is activated in severe asthma and COPD in humans. In particular we have found that bronchoalveolar lavage samples derived from severe asthmatics contain an increased number of IL‐13+CD68+ macrophages compared to samples obtained from healthy control subjects. In addition, we observed that lung samples obtained from patients with severe COPD exhibit significant mucous cell metaplasia and a corresponding increase in IL‐13 mRNA and protein relative to control lungs. IL‐13+ cells in the lungs of COPD subjects were identified as lung macrophages on the basis of typical cell morphology and CD68 immunostaining. The increase in IL‐13+CD68+ macrophages is accompanied by an increased number of invariant Vα24‐TCR NKT cells in lung samples from COPD subjects relative to non‐COPD controls. These findings are remarkably similar to our findings in the mouse model of chronic lung disease after viral infection.
Despite the high‐fidelity of our mouse‐to‐man observations, we also recently demonstrated that mouse models of chronic obstructive lung disease may serve to guide but may not fully predict human lung behavior. 99 For example, as noted above, Chit3l3/4 is the chitinase and chitinase‐like family member that is most inducible during the lung disease that develops after viral infection or allergen challenge in the mouse model. However, there is no homologue for Chi3l3/4 in the human genome. Thus, the mouse model cannot predict which chitinase or chitinase‐like family member may be equivalently increased in humans with corresponding lung disease. This circumstance, therefore, requires a comprehensive analysis of this family in humans with chronic lung disease. When this type of analysis was performed, we found instead that chitinase 1 is selectively increased in subjects with severe COPD. Similar to the mouse model, chitinase 1 expression was localized to lung macrophages. In addition, the increase in chitinase levels in the lung was reflected by an increase in circulating levels that could be detected in plasma samples from COPD subjects. In these subjects, the plasma levels of chitinase 1 were correlated significantly with disease severity. This level thereby provides a means to noninvasively track the status of alternatively activated macrophages in chronic lung disease and better differentiate molecular phenotypes in heterogeneous patient populations. In addition, these observations provide further evidence that the innate iNKT‐cell–alternatively activated macrophage immune axis may constitute a significant pathway for chronic obstructive lung disease. Additional work is needed to define the functional role of this pathway in complex lung diseases and in other chronic inflammatory diseases that may also be linked to the antiviral response.
Summary
A new mouse model has provided evidence of two distinct immune pathways that drive chronic lung disease in this model and in humans (summarized in Fig. 1). The disease that develops in this model is manifest by mucous cell metaplasia and airway hyper‐reactivity that are characteristic features of asthma and COPD in humans. The first phase of chronic disease develops at 3 weeks after viral infection and depends on type I IFN‐dependent expression of the high‐affinity IgE receptor (FcɛR1) on lung cDCs. Engagement of the FcɛRI causes the release of CCL28 to drive the recruitment of IL‐13‐producing Th2 cells to the airways, resulting in local action of IL‐13 on airway epithelial cells to produce mucous cells and on airway smooth muscle cells to promote hyper‐reactivity. This mechanism fits with the conventional proposal that the adaptive immune system is responsible for chronic inflammatory disease but raises the novel concept that an antiviral (Th1) response can be closely coupled to an allergic (Th2) response. Thus, the findings challenge the existing paradigm for separation of Th1 and Th2 responses, at least in the setting of post‐viral disease.
Figure 1.
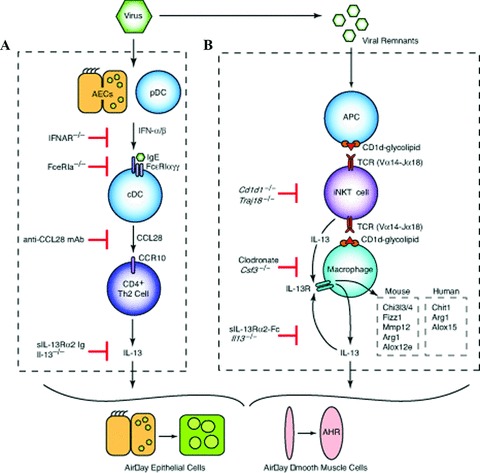
Cellular and molecular scheme for immune pathways leading to chronic lung disease after viral infection. (A) For the first phase of chronic disease, viruses cause airway epithelial cell (AEC) and plasmacytoid dendritic cell (pDC) production of type I interferon (IFN). Subsequent type I IFN receptor(IFNAR) signaling leads to upregulation of FcɛRI expression on resident lung conventional (c)DCs. In turn, FcɛRI activation by viral antigen and antiviral IgE leads to production of CCL28 and recruitment of CCR10‐expressing IL‐13‐producing T helper (Th)2 cells to the lung. The actions of IL‐13 drive differentiation of airway epithelial cell precursors toward mucous cells (mucous cell metaplasia) and airway smooth muscle cells to become more reactive to contractile agonists (airway hyper‐reactivity [AHR]). (B) For the second phase of chronic disease, viral remnants activate antigen‐presenting cells (APCs) and thereby facilitate CD1d‐dependent antigen presentation and consequent activation of invariant CD4− natural killer T (NKT) cells. NKT cells then interact directly with lung macrophages via IL‐13 production and binding to the IL‐13 receptor (IL‐13R) as well as contact between invariant Va14 T‐cell receptor (TCR) and glycolipid‐loaded CD1d. This interaction leads to increased expression of IL‐13R and production of IL‐13 that drives a positive feedback loop to amplify IL‐13 production and alternative activation of macrophages, including Chi3l3/4, Fizz1, Mmp12, Arg1, and Alox12e gene expression in mice and Chit1, Arg1, and Alox15 in humans.
The second phase of chronic disease that we have described for this mouse model is fully manifest at 7 weeks after viral infection and depends instead on an innate immune axis involving subsets of NKT cells and macrophages in the lung. These two cell types are specially programmed to allow for interactions between the semi‐invariant TCR on the NKT cell and CD1d on the macrophage as well as NKT‐cell‐derived, IL‐13 derived, and IL‐13R on the macrophage. Together these ligand–receptor interactions drive macrophages to a pattern of alternative activation and overproduction of IL‐13. This model thereby provides for how the innate immune system can be solely responsible for mediating a chronic inflammatory disease. The relevance of this NKT‐cell–macrophage immune axis is underscored by our additional findings of increased numbers of iNKT cells and IL‐13‐producing alternatively activated macrophages in the lungs of subjects with chronic obstructive lung disease. Moreover, the mouse model guided the identification of a biomarker of this innate immune pathway, i.e., macrophage‐derived chitinase 1. Initial results indicate that plasma levels of chitinase 1 correlate with disease severity in a subset of subjects with severe COPD. These findings reinforce the role of this new NKT‐cell–macrophage pathway in the development of chronic obstructive lung disease and provide a new line of investigation for linking acute viral infection to chronic inflammatory disease.
In that regard, we recognize that the present insights into new immune pathways also raise substantial questions that need to be addressed to better define the basis for chronic inflammatory disease. Perhaps the most immediate issue raised by these studies is the need to identify the mechanism for persistent activation of NKT cells after viral infection and in chronic disease. One possible explanation is that remnant virus may provide ongoing stimulation to NKT cells, perhaps through activation of the retinoic acid inducible gene protein 1‐like or Toll‐like receptor family members. In support of this possibility, we find evidence of viral persistence of SeV that is similar to reports of RSV in animal models. 37 , 100 , 101 Additional work must be directed at the possibility that viral remnants contained in cDCs, macrophages, or other cell types might drive chronic NKT‐cell activation after viral infection. A second question relates to the role of alternative activation of macrophages in chronic inflammatory disease. In that regard, our data and that of others suggest both pro‐inflammatory and anti‐inflammatory actions of alternatively activated macrophages may be found under experimental and clinical conditions. 96 , 97 , 102 At present, we view these macrophage products as potentially useful biomarkers of immune‐mediated disease, but further definition of macrophage function is needed if the same products are pursued as targets for therapeutic intervention. Thus, the role of alternatively activated macrophages in the propagation of chronic airway disease also requires further assessment. The insights obtained from the present high‐fidelity model of post‐viral disease should serve to guide these future studies.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgment
This research was supported by the National Institutes of Health (National Heart, Lung, and Blood Institute and National Institute of Allergic and Infectious Diseases), Martin Schaeffer Fund, and Alan A. and Edith L. Wolff Charitable Trust.
References
- 1. Manz, R.A. et al 2006. Immunological memory stabilizing autoreactivity. Curr. Top. Microbiol. Immunol. 305: 241–257. [DOI] [PubMed] [Google Scholar]
- 2. O’Garra, A. & Murphy K.. 1993. T‐cell subsets in autoimmunity. Curr. Opin. Immunol. 5: 880–886. [DOI] [PubMed] [Google Scholar]
- 3. Recher, M. & Lang K.S.. 2006. Innate (over)immunity and adaptive autoimmune disease. Curr. Top. Microbiol. Immunol. 305: 89–104. [DOI] [PubMed] [Google Scholar]
- 4. Murray, C.J. & Lopez A.D.. 1997. Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 349: 1498–1504. [DOI] [PubMed] [Google Scholar]
- 5. Robinson, D. et al 1993. Activation of CD4+ T cells, increased TH2‐type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J. Allergy Clin. Immunol. 92: 313–324. [DOI] [PubMed] [Google Scholar]
- 6. Silva, G.E. et al 2004. Asthma as a risk factor for COPD in a longitudinal study. Chest 126: 59–65. [DOI] [PubMed] [Google Scholar]
- 7. Ogra, P.L. 2004. Respiratory syncytial virus: the virus, the disease and the immune response. Paediatr. Respir. Rev. 5(Suppl A): S119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Domachowske, J.B. & Rosenberg H.F.. 1999. Respiratory syncytial virus infection: immune response, immunopathogenesis, and treatment. Clin. Microbiol. Rev. 12: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fjaerli, H.O. et al 2005. Acute bronchiolitis in infancy as risk factor for wheezing and reduced pulmonary function by seven years in Akershus County, Norway. BMC Pediatr. 5: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Price, J.F. 1990. Acute and long‐term effects of viral bronchiolitis in infancy. Lung 168(Suppl): 414–421. [DOI] [PubMed] [Google Scholar]
- 11. Pullan, C.R. & Hey E.N.. 1982. Wheezing, asthma, and pulmonary dysfunction 10 years after infection with respiratory syncytial virus in infancy. Br. Med. J. (Clin. Res. Ed.) 284: 1665–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sigurs, N. et al 2005. Severe respiratory syncytial virus bronchiolitis in infancy and asthma and allergy at age 13. Am. J. Respir. Crit. Care Med. 171: 137–141. [DOI] [PubMed] [Google Scholar]
- 13. Stein, R.T. et al 1999. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet 354: 541–545. [DOI] [PubMed] [Google Scholar]
- 14. Lee, W.M. et al 2007. High‐throughput, sensitive, and accurate multiplex PCR‐microsphere flow cytometry system for large‐scale comprehensive detection of respiratory viruses. J. Clin. Microbiol. 45: 2626–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Illi, S. et al 2001. Early childhood infectious diseases and the development of asthma up to school age: a birth cohort study. BMJ 322: 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kimpen, J.L. 2000. Viral infections and childhood asthma. Am. J. Respir. Crit. Care Med. 162: S108–112. [DOI] [PubMed] [Google Scholar]
- 17. Sigurs, N. et al 2000. Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am. J. Respir. Crit. Care Med. 161: 1501–1507. [DOI] [PubMed] [Google Scholar]
- 18. Glezen, W.P. et al 2000. Impact of respiratory virus infections on persons with chronic underlying conditions. JAMA 283: 499–505. [DOI] [PubMed] [Google Scholar]
- 19. Hogg, J.C. 2001. Viral infection and exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 164: 1555–1556. [DOI] [PubMed] [Google Scholar]
- 20. Smith, C.B. et al 1980. Effect of viral infections on pulmonary function in patients with chronic obstructive pulmonary diseases. J. Infect. Dis. 141: 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wiselka, M.J. et al 1993. Impact of respiratory virus infection in patients with chronic chest disease. Epidemiol. Infect. 111: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jafri, H.S. et al 2004. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long‐term airway hyperresponsiveness in mice. J. Infect. Dis. 189: 1856–1865. [DOI] [PubMed] [Google Scholar]
- 23. Walter, M.J. et al 2002. Viral induction of a chronic asthma phenotype and genetic segregation from the acute response. J. Clin. Invest. 110: 165–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3: 23–35. [DOI] [PubMed] [Google Scholar]
- 25. Kung, T.T. et al 1994. Characterization of a murine model of allergic pulmonary inflammation. Int. Arch. Allergy Immunol. 105: 83–90. [DOI] [PubMed] [Google Scholar]
- 26. Poston, R.N. et al 1992. Immunohistochemical characterization of the cellular infiltration in asthmatic bronchi. Am. Rev. Respir. Dis. 145: 918–921. [DOI] [PubMed] [Google Scholar]
- 27. Ying, S. et al 1995. Phenotype of cells expressing mRNA for TH2‐type (interleukin 4 and interleukin 5) and TH1‐type (interleukin 2 and interferon gamma) cytokines in bronchoalveolar lavage and bronchial biopsies from atopic asthmatic and normal control subjects. Am. J. Respir. Cell. Mol. Biol. 12: 477–487. [DOI] [PubMed] [Google Scholar]
- 28. Erb, K.J. & Le Gros G.. 1996. The role of Th2 type CD4+ T cells and Th2 type CD8+ T cells in asthma. Immunol. Cell. Biol. 74: 206–208. [DOI] [PubMed] [Google Scholar]
- 29. Gauvreau, G.M. , Watson R.M. & O’Byrne P.M.. 1999. Kinetics of allergen‐induced airway eosinophilic cytokine production and airway inflammation. Am. J. Respir. Crit. Care Med. 160: 640–647. [DOI] [PubMed] [Google Scholar]
- 30. Kroegel, C. et al 1996. Endobronchial secretion of interleukin‐13 following local allergen challenge in atopic asthma: relationship to interleukin‐4 and eosinophil counts. Eur. Respir. J. 9: 899–904. [DOI] [PubMed] [Google Scholar]
- 31. Lin, C.C. , Lin C.Y. & Ma H.Y.. 2000. Pulmonary function changes and increased Th‐2 cytokine expression and nuclear factor kB activation in the lung after sensitization and allergen challenge in brown Norway rats. Immunol. Lett. 73: 57–64. [DOI] [PubMed] [Google Scholar]
- 32. Humbert, M. et al 1997. Elevated expression of messenger ribonucleic acid encoding IL‐13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J. Allergy Clin. Immunol. 99: 657–665. [DOI] [PubMed] [Google Scholar]
- 33. Saha, S.K. et al 2008. Increased sputum and bronchial biopsy IL‐13 expression in severe asthma. J. Allergy Clin. Immunol. 121: 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Konstantinidis, A.K. et al 2007. Genetic association studies of interleukin‐13 receptor alpha1 subunit gene polymorphisms in asthma and atopy. Eur. Respir. J. 30: 40–47. [DOI] [PubMed] [Google Scholar]
- 35. Sadeghnejad, A. et al 2008. IL13 gene polymorphisms modify the effect of exposure to tobacco smoke on persistent wheeze and asthma in childhood, a longitudinal study. Respir Res. 9: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sadeghnejad, A. et al 2007. IL13 promoter polymorphism 1112C/T modulates the adverse effect of tobacco smoking on lung function. Am. J. Respir. Crit. Care Med. 176: 748–752. [DOI] [PubMed] [Google Scholar]
- 37. Kim, E.Y. et al 2008. Persistent activation of an innate immune response translates respiratory viral infection into chronic inflammatory lung disease. Nat. Med. 14: 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miotto, D. et al 2003. Interleukin‐13 and ‐4 expression in the central airways of smokers with chronic bronchitis. Eur. Respir. J. 22: 602–608. [DOI] [PubMed] [Google Scholar]
- 39. Saha, S. et al 2008. Induced sputum and bronchial mucosal expression of interleukin‐13 is not increased in chronic obstructive pulmonary disease. Allergy 63: 1239–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blease, K. 2008. Therapeutics targeting IL‐13 for the treatment of pulmonary inflammation and airway remodeling. Curr. Opin. Investig. Drugs 9: 1180–1184. [PubMed] [Google Scholar]
- 41. Schwarze, J. et al 1997. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J. Clin. Invest. 100: 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tripp, R.A. , Moore D. & Anderson L.J.. 2000. TH(1)‐ and TH(2)‐TYPE cytokine expression by activated t lymphocytes from the lung and spleen during the inflammatory response to respiratory syncytial virus. Cytokine 12: 801–807. [DOI] [PubMed] [Google Scholar]
- 43. Becker, Y. 2006. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy—a review. Virus Genes. 33: 235–252. [DOI] [PubMed] [Google Scholar]
- 44. Caramori, G. , Pandit A. & Papi A.. 2005. Is there a difference between chronic airway inflammation in chronic severe asthma and chronic obstructive pulmonary disease? Curr. Opin. Allergy Clin. Immunol. 5: 77–83. [DOI] [PubMed] [Google Scholar]
- 45. Jatakanon, A. et al 1999. Neutrophilic inflammation in severe persistent asthma. Am. J. Respir. Crit. Care Med. 160: 1532–1539. [DOI] [PubMed] [Google Scholar]
- 46. Sampath, D. et al 1999. Constitutive activation of an epithelial signal transducer and activator of transcription (Stat1) pathway in asthma. J. Clin. Invest. 103: 1353–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Randolph, D.A. et al 1999. Cooperation between Th1 and Th2 cells in a murine model of eosinophilic airway inflammation. J. Clin. Invest. 104: 1021–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Castro, M. et al 2000. Could asthma worsen by stimulating the T helper type 1 (Th1) response? Am. J. Respir. Cell Mol. Biol. 22: 143–146. [DOI] [PubMed] [Google Scholar]
- 49. Schnyder‐Candrian, S. et al 2006. Interleukin‐17 is a negative regulator of established allergic asthma. J. Exp. Med. 203: 2715–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lewkowich, I.P. et al 2005. CD4+CD25+ T cells protect against experimentally induced asthma and alter pulmonary dendritic cell phenotype and function. J. Exp. Med. 202: 1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saetta, M. et al 2003. Increased proportion of CD8+ T‐lymphocytes in the paratracheal lymph nodes of smokers with mild COPD. Sarcoidosis Vasc. Diffuse Lung Dis. 20: 28–32. [PubMed] [Google Scholar]
- 52. Miotto, D. et al 2003. Interleukin‐13 and ‐4 expression in the central airways of smokers with chronic bronchitis. Eur. Respir. J. 22: 602–608. [DOI] [PubMed] [Google Scholar]
- 53. Grumelli, S. et al 2004. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 1: e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shum, B.O. , Rolph M.S. & Sewell W.A.. 2008. Mechanisms in allergic airway inflammation – lessons from studies in the mouse. Expert Rev. Mol. Med. 10: e15. [DOI] [PubMed] [Google Scholar]
- 55. Tamachi, T. et al 2006. Interleukin 25 in allergic airway inflammation. Int. Arch. Allergy Immunol. 140(Suppl 1): 59–62. [DOI] [PubMed] [Google Scholar]
- 56. McLane, M.P. et al 1998. Interleukin‐9 promotes allergen‐induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am. J. Respir. Cell. Mol. Biol. 19: 713–720. [DOI] [PubMed] [Google Scholar]
- 57. Grunig, G. et al 1998. Requirement for IL‐13 independently of IL‐4 in experimental asthma. Science 282: 2261–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brusselle, G.G. et al 1994. Attenuation of allergic airway inflammation in IL‐4 deficient mice. Clin. Exp. Allergy 24: 73–80. [DOI] [PubMed] [Google Scholar]
- 59. Schwarze, J. et al 1997. Respiratory syncytial virus infection results in airway hyperresponsiveness and enhanced airway sensitization to allergen. J. Clin. Invest. 100: 226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Prince, G.A. & Porter D.D.. 1976. The pathogenesis of respiratory syncytial virus infection in infant ferrets. Am. J. Pathol. 82: 339–352. [PMC free article] [PubMed] [Google Scholar]
- 61. Prince, G.A. et al 1978. The pathogenesis of respiratory syncytial virus infection in cotton rats. Am. J. Pathol. 93: 771–791. [PMC free article] [PubMed] [Google Scholar]
- 62. Folkerts, G. , Verheyen A. & Nijkamp F.P.. 1992. Viral infection in guinea pigs induces a sustained non‐specific airway hyperresponsiveness and morphological changes of the respiratory tract. Eur. J. Pharmacol. 228: 121–130. [DOI] [PubMed] [Google Scholar]
- 63. Peebles, R.S., Jr . et al 1999. Respiratory syncytial virus infection prolongs methacholine‐induced airway hyperresponsiveness in ovalbumin‐sensitized mice. J. Med. Virol. 57: 186–192. [DOI] [PubMed] [Google Scholar]
- 64. Graham, B.S. et al 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26: 153–162. [DOI] [PubMed] [Google Scholar]
- 65. Walter, M.J. et al 2001. IL‐12 p40 production by barrier epithelial cells during airway inflammation. J. Exp. Med. 193: 339–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tyner, J.W. et al 2005. CCL5‐CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 11: 1180–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shornick, L.P. et al 2008. Airway epithelial versus immune cell Stat1 function for innate defense against respiratory viral infection. J. Immunol. 180: 3319–3328. [DOI] [PubMed] [Google Scholar]
- 68. Tyner, J.W. et al 2006. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL‐13 transdifferentiation signals. J. Clin. Invest. 116: 309–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Patel, A.C. et al 2006. Genetic segregation of airway disease traits despite redundancy of chloride channel calcium‐activated (CLCA) family members. Physiol. Genomics 25: 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grayson, M.H. et al 2007. Induction of high‐affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J. Exp. Med. 204: 2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Welliver, R.L. et al 1986. Predictive value of respiratory syncytial virus‐specific IgE responses for recurrent wheezing following bronchiolitis. J. Pediatr. 109: 776–880. [DOI] [PubMed] [Google Scholar]
- 72. Skoner, D.P. et al 1995. Effect of rhinovirus 39 (RV‐39) infection on immune and inflammatory paramenters in allergic and non‐allergic subjects. Clin. Exp. Allergy 25: 561–567. [DOI] [PubMed] [Google Scholar]
- 73. Rager, K.J. et al 1998. Activation of antiviral protein kinase leads to immunoglobulin E class switching in human B cells. J. Virol. 72: 1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Dombrowicz, D. et al 2000. Expression of a Functional Fc[epsilon]RI on Rat Eosinophils and Macrophages. J. Immunol. 165: 1266–1271. [DOI] [PubMed] [Google Scholar]
- 75. Blank, U. et al 1989. Complete structure and expression in transfected cells of high affinity IgE receptor. Nature 337: 187–189. [DOI] [PubMed] [Google Scholar]
- 76. Bieber, T. et al 1992. Human epidermal Langerhans cells express the high affinity receptor for immunoglobulin E (Fc epsilon RI). J. Exp. Med. 175: 1285–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Foster, B. , Metcalfe D.D. & Prussin C.. 2003. Human dendritic cell 1 and dendritic cell 2 subsets express FceRI: correlation with serum IgE and allergic asthma. J. Allergy Clin. Immunol. 112: 1132–1138. [DOI] [PubMed] [Google Scholar]
- 78. Schroeder, J.T. et al 2005. TLR9‐ and FceRI‐mediated responses oppose one another in plasmacytoid dendritic cells by down‐regulating receptor expression. J. Immunol. 175: 5724–5731. [DOI] [PubMed] [Google Scholar]
- 79. Grayson, M.H. et al 2007. Controls for lung dendritic cell maturation and migration during respiratory viral infection. J. Immunol. 179: 1438–1448. [DOI] [PubMed] [Google Scholar]
- 80. Yamaguchi, M. et al 1997. IgE enhances mouse mast cell Fc(epsilon)RI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE‐dependent reactions. J. Exp. Med. 185: 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Grayson, M.H. et al 2007. Induction of high‐affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J. Exp. Med. 204: 2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grayson, M.H. et al 2008. IFNAR signaling to neutrophils is necessary to induce high‐affinity receptor for IgE (FcɛRI) and activate the conventional dendritic cell (cDC) to T helper type 2 (Th2) cell immune axis after viral infection. Am. J. Respir. Crit. Care Med. 177: A84. [Google Scholar]
- 83. English, K. et al 2006. Inflammation of the respiratory tract is associated with CCL28 and CCR10 expression in a murine model of allergic asthma. Immunol. Lett. 103: 92–100. [DOI] [PubMed] [Google Scholar]
- 84. Wang, W. et al 2000. Identification of a novel chemokine (CCL28), which binds CCR10 (GPR2). J. Biol. Chem. 275: 22313–22323. [DOI] [PubMed] [Google Scholar]
- 85. John, A.E. et al 2005. Temporal production of CCL28 corresponds to eosinophil accumulation and airway hyperreactivity in allergic airway inflammation. Am. J. Pathol. 166: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Taussig, L.M. et al 2003. Tucson Children's Respiratory Study: 1980 to present. J. Allergy Clin. Immunol. 111: 661–675; quiz 676. [DOI] [PubMed] [Google Scholar]
- 87. Lisbonne, M. et al 2003. Cutting edge: invariant Va14 NKT cells are required for allergen‐induced airway inflammation and hyperreactivity in an experimental asthma model. J. Immunol. 171: 1637–1641. [DOI] [PubMed] [Google Scholar]
- 88. Akbari, O. et al 2003. Essential role of NKT cells producing IL‐4 and IL‐13 in the development of allergen‐induced airway hyper‐reactivity. Nat. Med. 9: 582–588. [DOI] [PubMed] [Google Scholar]
- 89. Akbari, O. et al 2006. CD4+ invariant T cell‐receptor+ natural killer T cells in bronchial asthma. N. Engl. J. Med. 354: 1117–1129. [DOI] [PubMed] [Google Scholar]
- 90. Sen, Y. et al 2005. Va24‐invariant NKT cells from patients with allergic asthma express CCR9 at high frequency and induce Th2 bias of CD3+ T cells upon CD226 engagement. J. Immunol. 175: 4914–4926. [DOI] [PubMed] [Google Scholar]
- 91. Vijayanand, P. et al 2007. Invariant natural killer T cells in asthma and chronic obstructive pulmonary disease. N. Engl. J. Med. 356: 1410–1422. [DOI] [PubMed] [Google Scholar]
- 92. Mattner, J. et al 2005. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434: 525–529. [DOI] [PubMed] [Google Scholar]
- 93. Cui, J. et al 1997. Requirement for Va14 NKT cells in IL‐12‐mediated rejection of tumors. Science 278: 1623–1626. [DOI] [PubMed] [Google Scholar]
- 94. Gordon, S. 2003. Alternative activation of macrophages. Nat. Rev. Immunol. 3: 23–35. [DOI] [PubMed] [Google Scholar]
- 95. Pouladi, M.A. et al 2004. Interleukin‐13‐dependent expression of matrix metalloproteinase‐12 is required for the development of airway eosinophilia in mice. Am. J. Respir. Cell Mol. Biol. 30: 84–90. [DOI] [PubMed] [Google Scholar]
- 96. Hajek, A.R. et al 2008. 12/15‐Lipoxygenase deficiency protects mice from allergic airways inflammation and increases secretory IgA levels. J. Allergy Clin. Immunol. 122: 633–639 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pesce, J.T. et al 2009. Retnla (Relma/Fizz1) suppresses helminth‐induced Th2‐type immunity. PLoS Pathog. 5: e1000393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hogg, J.C. et al 2007. Survival after lung volume reduction in chronic obstructive pulmonary disease: insights from small airway pathology. Am. J. Respir. Crit. Care Med. 176: 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Agapov, E. et al 2009. Macrophage chitinase 1 stratifies chronic obstructive lung disease. Am. J. Respir. Cell Mol. Biol. 41: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Estripeaut, D. et al 2008. Respiratory syncytial virus persistence in the lungs correlates with airway hyperreactivity in the mouse model. J. Infect. Dis. 198: 1435–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hegele, R.G. et al 1994. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest 105: 1848–1854. [DOI] [PubMed] [Google Scholar]
- 102. Shannon, V.R. et al 1993. Histochemical evidence for induction of arachidonate 15‐lipoxygenase in airway disease. Am. Rev. Respir. Dis. 147: 1024–1028. [DOI] [PubMed] [Google Scholar]