

Abstract
Analogs of nicotinic acid adenine dinucleotide phosphate (NAADP) with substitution at either the 4- or the 5-position position of the nicotinic acid moiety have been synthesized from NADP enzymatically using Aplysia californica ADP-ribosyl cyclase or mammalian NAD glycohydrolase. Substitution at the 4-position of the nicotinic acid resulted in the loss of agonist potency for release of Ca2+-ions from sea urchin egg homogenates and in potency for competition ligand binding assays using [32P]NAADP. In contrast, several 5-substituted NAADP derivatives showed high potency for binding and full agonist activity for Ca2+ release. 5-Azido-NAADP was shown to release calcium from sea urchin egg homogenates at low concentration and to compete with [32P]NAADP in a competition ligand binding assay with an IC50 of 18 nM, indicating that this compound might be a potential photoprobe useful for specific labeling and identification of the NAADP receptor.
Introduction
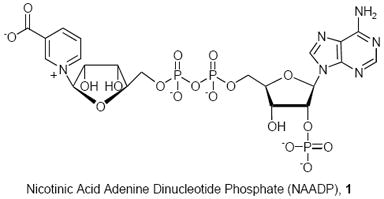
Nicotinic acid adenine dinucleotide phosphate (NAADP, 1)# was identified as a metabolite of NADP which at low concentration was shown to cause Ca2+ release from internal calcium ion stores in sea urchin eggs.1 NAADP-mediated Ca+2 release was subsequently shown to be pharmacologically distinguishable from calcium release mediated by either inositol-1,4,5-triphosphate (InsP3) or by cyclic ADP-ribose (cADPR). Additionally, NAADP mobilized Ca+2 from a different sub-cellular location than does cADPR or InsP3.2 These observations imply that NAADP releases calcium ion after binding to a unique receptor.3-6 NAADP is active in many organisms including mollusks, plants, and mammals.4 In vertebrate tissue, NAADP-mediated calcium release has been observed in pancreatic acinar cells,7 cultured human T lymphocytes,8 brain,9 heart,10 kidney,11 and liver cells.12 In some of these systems, NAADP has met the criteria for establishing it as an intracellular second messenger.3,13
Despite its’ physiological importance, the structural features of NAADP which are required for high potency calcium release are not completely understood. Lee et al. defined the pyridine-3-carboxylate, the adenosyl 2′-phosphate, and portions of the purine as important structural determinants14. Billington further defined the requirements of 3-substituted pyridine modification on agonist activity.15 Simple pyridinium-3-carboxylate salts16,17 and pyridoxalphosphate-6-azophenyl-2,4-disulfonate (PPADS)18 are reported to be low potency antagonists. A potent and selective NAADP antagonist, Ned-19, was developed recently using virtual screening19 and has been applied to studies of the NAADP-receptor20. Continued progress in the development of chemical probes for NAADP-mediated signaling and characterization of the NAADP receptor require a better understanding of the structure-activity relation (SAR) of NAADP-mediated calcium ion release. To this end, we have synthesized a series of nicotinic acid substituted NAADP derivatives using a chemoenzymatic synthesis,21,22 and characterized their ability both to mobilize calcium-ion via the NAADP receptor in sea urchin egg homogenates and to compete with NAADP in competition ligand binding assay.
Chemistry
Since the enzymes Aplysia californica ADP-ribosyl cyclase and mammalian NAD glycohydrolase are known to catalyze the exchange of the nicotinamide group of NADP with nicotinic acid (Scheme 1),21,22 we obtained nicotinic acid derivatives with substituents at either position 4 or 5 and used them as substrates (Table 1) for the enzyme catalyzed pyridine base exchange reaction to produce the corresponding NAADP analogs. The general reaction for the base exchange is shown in Scheme 1.
Scheme 1.
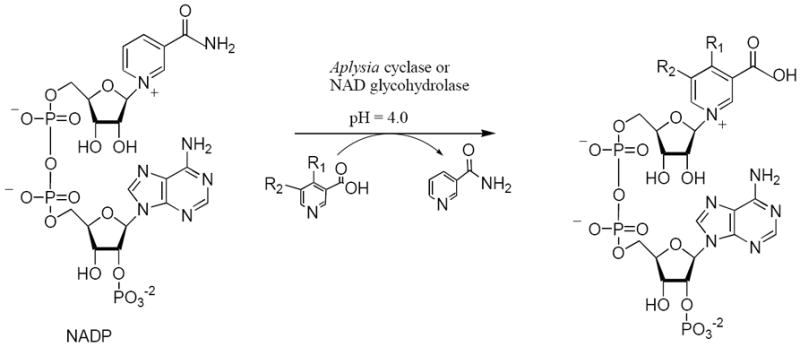
Synthesis of NAADP analogs using the enzyme catalyzed pyridine base exchange reaction.
Table 1.
4-Substituted pyridine-3-carboxylic acids (2a-d) and 5-substituted pyridine-3-carboxylic acids (3a-j) obtained and evaluated as potential substrates in the chemoenzymatic synthesis designed to produce novel pyridinium substituted NAADP derivatives.
| Structure | Cmpd | Substituent | Base-exchange product detected |
|---|---|---|---|
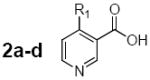 |
2a | -NH2 | no; yes for methyl ester |
| 2b | -CH3 | yes | |
| 2c | -n-C4H9 | yes | |
| 2d | -C6H5 | yes | |
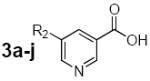 |
3a | -NH2 | yes |
| 3b | -CH3 | yes | |
| 3c | -Br | no | |
| 3d | -CO2H | yes | |
| 3e | -C≡CH | no | |
| 3f | -CH=CH2 | no | |
| 3g | -CH2CH3 | yes | |
| 3h | -C6H5 | no | |
| 3i | -N3 | yes | |
| 3j |  |
no | |
4-Substitued nicotinic acids
We sought to obtain 4-substituted nicotinic acid derivatives with alkyl, amino, or aryl groups at the 4-position. The simple 4-methylnicotinic acid (2a) as well as 4-aminonicotinic acid (2b) were both available commercially. 4-n-Butylnicotinic acid and 4-phenylnicotinic acid were synthesized by adding either n-butyllithium or phenyllithium respectively to pyridyl-3-oxazolines derived from nicotinic acid (Scheme 2)23. Addition of the organolithium reagent was shown to favor the formation of the 1,4-dihydropyridine-3-oxazoline, which was easily oxidized by oxygen in air to obtain the 4-substituted pyridine-3-oxazoline. Deprotection results in the isolation of the desired 4-substituted nicotinic acids.24
Scheme 2.
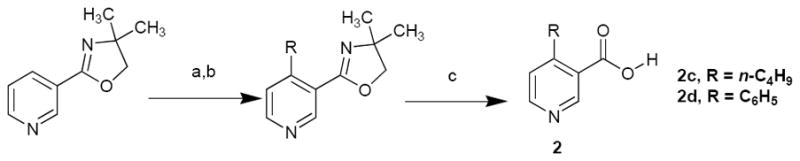
Addition of organolithium reagents to pyridine-3-oxazoline
Conditions and reagents: (a) C6H5Li or n-C4H9Li, -78°; (b) Air, 0° 3 h; (c) 3 N HCl/acetic acid 95°/36 h.
5-Substituted nicotinic acids
The simple derivatives 5-methylnicotinic acid (3a) and 5-aminonicotinic acid (3b), 5-bromonicotinic acid (3c), and pyridine-3,5-dicarboxylic acid (3d) were purchased. For the synthesis of 5-substituted nicotinic acid derivatives we started with commercially available and inexpensive 5-bromonicotinic acid (3c), and protected the carboxylic acid by its conversion into the oxazoline group (4). Transmetalation of 4 was expected to be difficult because of competing 4-addition reaction.23 Therefore we utilized intermediate 4 in palladium catalyzed C-C coupling reactions with appropriate coupling partners to produce 5-substituted pyridine-3-oxazolines.
The Sonogashira coupling has previously been used successfully to produce ethynylpyridines,25,26 and we expected that cross-coupling of terminal acetylenes with 4 (Scheme 3) would provided an approach to 5-ethynylnicotinic acid (3e). Coupling of 4 with commercially available triethylsilylacetylene (TES-acetylene), under conditions using a mixed aqueous-organic solvent and sodium carbonate at 90° C, led to formation of disubstituted acetylene 5 instead of the desired product as shown in Scheme 3. Formation of 5 probably occurred because in the presence of aqueous base and high temperature the triethylsilyl group was removed from initially formed 6, and the free acetylene liberated in situ reacted with another molecule of 4.
Scheme 3.
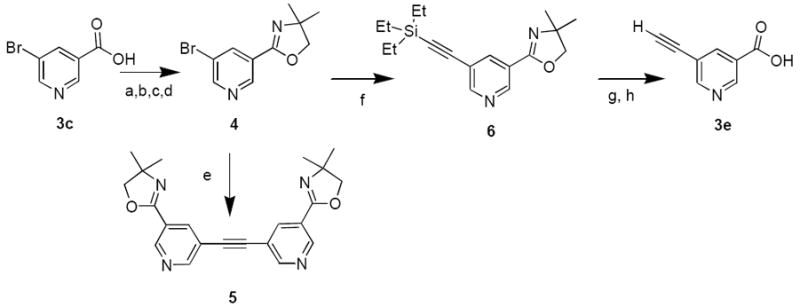
Conditions and Reagents: (a) SOCl2, reflux, 18 h; (b) 2-amino-2-methylpropanol, 48 h; (c) SOCl2, 24 h; (d) diisopropylethylamine (DIPEA), 60°C, 24 h; (e)Pd(Ph3)4, Na2CO3, CuI, TES-acetylene, dimethylformamide (DMF)/ethyl alcohol (EtOH)/H2O at 90° for 1 h; (f) Pd(OAc)2, PPh3, benzyltributylammonium bromide, TES-acetylene, DIPEA and water, room temperature, 1 h; (g) K2CO3, tetrahydrofuran (THF)/ CH3OH, room temperature, 1 h; (h) 3 N HCl, AcOH, 95° 1 h.
When the reaction was conducted at room temperature and using a phase transfer catalyst with a minimum amount of water in the reaction,27 we were able to obtain 6 in high yield. The silyl group in 6 was removed by treatment with excess aqueous potassium carbonate28 followed by acid hydrolysis of the oxazoline group29 to give free acid derivative 3e (Scheme 3).
Compound 3f was obtained by partial reduction of 3e in presence of poisoned Lindlar’s catalyst.30 The acetylene of compound 3e was completely reduced to an ethyl group using ammonium formate in presence of Pd on carbon to obtain 5-ethylnicotinic acid (3g).
For the synthesis of the 5-arylnicotinic acids, Pd-catalyzed Suzuki reaction between aryl halides and boronic acids has been used (Scheme 4).26 We synthesized 9 by means of Suzuki coupling between the boronate of oxazoline substituted nicotinic acid 7 and phenyl bromide. The boronate of the protected nicotinic acid could not be completely characterized by itself because it was a mixture of several boronate forms (boronic acids and boronic esters) that have been reported to exist.31 Characterization of the boronate was accomplished by converting the boronate into its pinacol ester (8).31
Scheme 4.
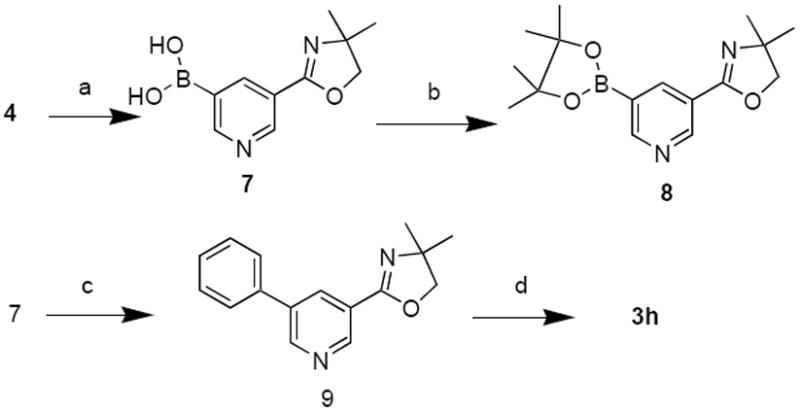
Conditions and Reagents: (a) toluene/THF, (i-C3H7O)3B, then n-BuLi, -70°; (b) (CH3)2COHCOH(CH3)2, Ph-CH3, reflux; (c) Pd(Ph3)4, Na2CO3, C6H5-Br, DMF & EtOH 90° for 4h; (d) 3N HCl, acetic acid, 95° 36 h.
The borate 7 was coupled to phenyl bromide in the presence of Pd catalyst and aqueous base at 90 °C to give the C-C coupled product 9. The deprotection of oxazoline under acidic conditions gave 5-phenylnicotinic acid 3h (Scheme 4).
5-Azidopyridine-3-carboxylate (3i) was an important precursor both to subsequently establish the SAR of NAADP derivatives and for developing high potency photoaffinity probes for receptor identification.32 Literature provided the precedent of diazotization reaction followed by nucleophilic displacement by sodium azide.33,34 This approach was used to synthesize 5-azidonicotinic acid 3i starting from 5-aminonicotinic acid 3a (Scheme 5). We started with protection of the acid group in 5-aminonicotinic acid (3a) with a hydrophobic ethyl ester (10), followed by conversion of the amine into an azide using diazotization chemistry to give 11.35 The hydrophobic ester group used for acid protection aided in extracting the compound into the organic phase, while the salts from the reaction partitioned into the aqueous phase. 5-Azidonicotinic acid ethyl ester 11 was then subjected to alkaline hydrolysis to give 5-azidonicotinic acid 3i.
Scheme 5.
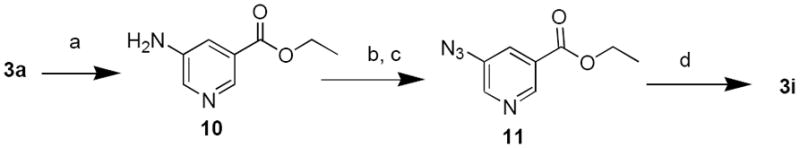
Conditions and Reagents: (a) EtOH/HCl; (b) NaNO2/HCl; (c) NaOAc/NaN3; (d) CH3OH/NaOH/H2O
Nicotinic acid derivative 3j was synthesized by click chemistry involving [1,3] dipolar cycloaddition reaction36 between the 5-azidonicotinate ethyl ester (11) and tert-butyl prop-2-ynylcarbamate 12 (Scheme 6). tert-Butyl prop-2-ynylcarbamate (12) was prepared according to procedure described by Holmes et al. (2003).25
Scheme 6.
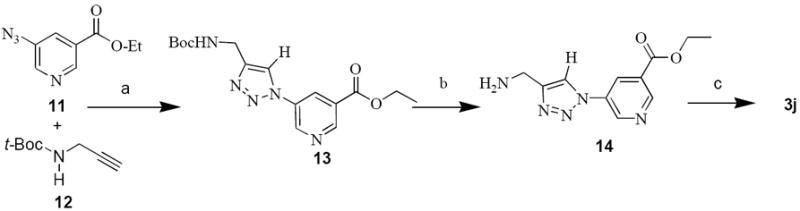
Conditions and Reagents: (a) DIPEA, CuI, 12 h; (b) trifluoroacetic acid; (c) 2 N NaOH in H2O/CH3OH
The copper catalyzed cycloaddition resulted in formation of 1,4-disubstituted triazole ring at position 5 of nicotinic acid (13). Use of copper catalyst brings regioselectivity to the reaction and catalyzed the formation of an adduct between the azide and the acetylene moiety producing only the 1,4-disubstituted isomer. The competing reaction pathway producing the 1,5-disubstitution cycloaddition product is thereby eliminated.37 Deprotection in strong anhydrous acid produced ester 14, which was saponified to the nicotinic acid derivative 3j.
Enzyme catalyzed base exchange
NAADP derivatives were synthesized from pyridine-3-carboxylic acid derivatives and NADP either using the enzymatic activity of Aplysia ADP-ribosyl cyclase or a mammalian NAD glycohydrolase (Scheme 1). We relied primarily on the Aplysia ADP-ribosyl cyclase as the catalyst because it was available as a stable, soluble, and well characterized enzyme. In cases where the Aplysia ADP-ribosyl cyclase failed to produce the expected exchange product, mammalian enzymes were also evaluated. A high concentration of a nucleophilic pyridine base is required for high yields of the exchange product. In a typical reaction, NADP was treated with about a 30-fold excess of the pyridine derivative and the enzyme catalyst at pH 4.0. Under these conditions, base exchange competes successfully with both hydrolysis and cyclization reactions, side reactions which destroy the substrate NADP by forming either ADP-ribose or cADPR by-products. The control of pH is important because it is the unionized pyridine derivative that is the substrate for the base exchange reaction. An optimal pH must be maintained to provide high concentrations of a unionized pyridine base. The exchange reactions were monitored by following the disappearance of the limiting substrate, NADP. In this assay, NADP present in the mixture was reduced using glucose-6-phosphate in the presence of glucose-6-phosphate dehydrogenase to give NADPH. The NADPH that was produced showed absorbance at 340 nm, without interference from any other reactant or bi-product.3 When the exchange reaction was complete, the catalyst was removed by ultrafiltration (membrane cut off 10 kDa). The pH of the catalyst free filtrate was adjusted to 7 and dinucleotides present in the mixture were purified by anion-exchange chromatography.
Methylated nicotinic acids such as 2b or 3b could easily be exchanged into NADP to result in the formation of NAADP derivatives 17 and 21. 5-Aminonicotinic acid (3a) was similarly an excellent exchange substrate and the exchange product 5-amino-NAADP (20) could be isolated in greater than 40% yield after purification.
We tried to exchange commercially available 4-aminonicotinic acid (2a) into NADP at pH 4.0 to enzymatically synthesize the 4-amino-NAADP (16)38 but were unsuccessful in detecting any pyridine dinucleotide derivatives as products. The likely explanation is that 2a, a vinylogous amidine, is significantly more basic than is 3a, and is largely protonated and cationic under the usual conditions of the reaction at pH 4. To circumvent this difficulty we synthesized the methyl ester of 4-aminonicotinic acid using Fischer esterification, and used the 4-aminonicotinic acid methyl ester as the co-substrate for the exchange at pH 7.5. Under these conditions we obtained exchange product 15. Use of this higher pH increased the concentration of the unprotonated and unionized 4-aminonicotinic acid methyl ester and hence it became more available to the active site of NAD glycohydrolase. The 4-amino-NAADP methyl ester was then subjected to treatment with triethylamine and water to hydrolyze the ester and give the target compound 4-amino-NAADP (16).
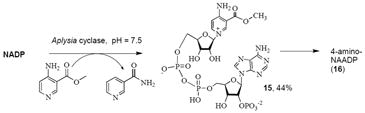
Base exchange reaction with 4-n-butylnicotinic acid (2c) or 4-phenyl nicotinic acid (2d) worked well to produce NAADP derivatives 18 and 19. Enzyme catalyzed base exchange was surprisingly more selective when substituents were placed in the 5 position. Neither 5-phenylnicotinic acid (3h) nor bromonicotinic acid (3c) formed detectable base-exchanged product. Similarly, 3e, 3f and 3j failed to react to form a substituted NAADP. It is well appreciated that electron withdrawing substituents deactivate the pyridine ring towards base exchange. Steric demands on disubstituted pyridines are less well understood, although it is apparent from our work that large substituents are better tolerated at the 4-position than at the 5-position.
Interestingly, 5-substitutions like azide and carboxylate were tolerated as cosubstrates for exchange reactions, producing reasonable yields of the corresponding NAADP analogs. In cases where the exchange reaction catalyzed by the Aplysia ADP-ribosyl cyclase was found to fail, we also tried NAD glycohydrolase enzymes from porcine brain and bovine spleen, but even using the mammalian enzyme as a catalyst the result was the same.
The results of enzyme catalyzed base exchange reactions between NADP and our set of synthetic nicotinic acids are summarized in Table 2. We also observed that all three enzymes behaved very similarly and the outcome of exchange reactions using different substrates was the same for each of the catalysts. This suggests that the active site of the three enzymes, as judged by the specificity of base exchange reactions, is very similar.
Table 2.
Result of the Pyridine Base Exchange Reaction
| Structure | R1 | R2 | # | Name | %Yield |
|---|---|---|---|---|---|
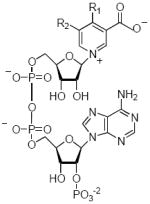 |
-NH2 | -H | 16 | 4-amino-NAADP | 44 |
| -CH3 | -H | 17 | 4-methyl-NAADP | 43 | |
| -n-C4H9 | -H | 18 | 4-n-butyl-NAADP | 60 | |
| -C6H5 | -H | 19 | 4-phenyl-NAADP | 51 | |
| -H | -NH2 | 20 | 5-amino-NAADP | 43 | |
| -H | -CH3 | 21 | 5-methyl-NAADP | 59 | |
| -H | -CO2H | 22 | 5-carboxy-NAADP | 32 | |
| -H | -CH2CH3 | 23 | 5-ethyl-NAADP | 59 | |
| -H | -N3 | 24 | 5-azido-NAADP | 61 | |
Biological Activity
Calcium Release Properties of Compounds 16-24
When tested for calcium ion-releasing activity in vitro, NAADP (1) and its analogs 16-24 were shown to elicit calcium release from sea urchin egg homogenates. Figure 1 shows the effect of treatment of calcium-ion loaded microsomes with varying concentrations of NAADP (1), 5-NH2-NAADP (20), 5-CH3-NAADP (21) and 4-CH3-NAADP (17). Each of these compounds induced a rapid release of calcium-ions which reached a concentration dependent plateau within a few seconds. Compounds 20 and 21 induce calcium-ion release at concentrations as low as 2 nM. 4-CH3-NAADP (17) was shown to be significantly less potent than was the 5-isomer 21. The maximal calcium concentration attained after treatment was found to increase in a concentration dependent manner. Saturating concentrations of compounds 20 and 21 elicited the same amount of calcium release as did the full agonist NAADP. Figure 2 shows concentration response curves for compounds 16-24 compared to that of NAADP. Compounds 20 and 21 were shown to be approximately equipotent with NAADP, compounds 23 and 24 possessing larger 5-substituents were ca. 10 to 100-fold less potent, while the 4-substituted NAADP derivatives 16 and 17 were from two hundred to two thousand fold less potent (Table 3). EC50 values for 18, 19, and 22 were higher still, and could not be accurately determined in this experiment. Compounds 16, 20, and 21 elicit maximal calcium release at high concentration and therefore are considered to possess full-agonist activity. The maximal Ca2+ release elicited at high concentrations of compounds 23, 24, and probably also 17 was show to plateau at concentrations significantly less than that elicited by NAADP, indicating that these compounds are behaving as partial agonists. Complete concentration response curves could not be determined for the low potency compounds 18, 19, and 22.
Figure 1.
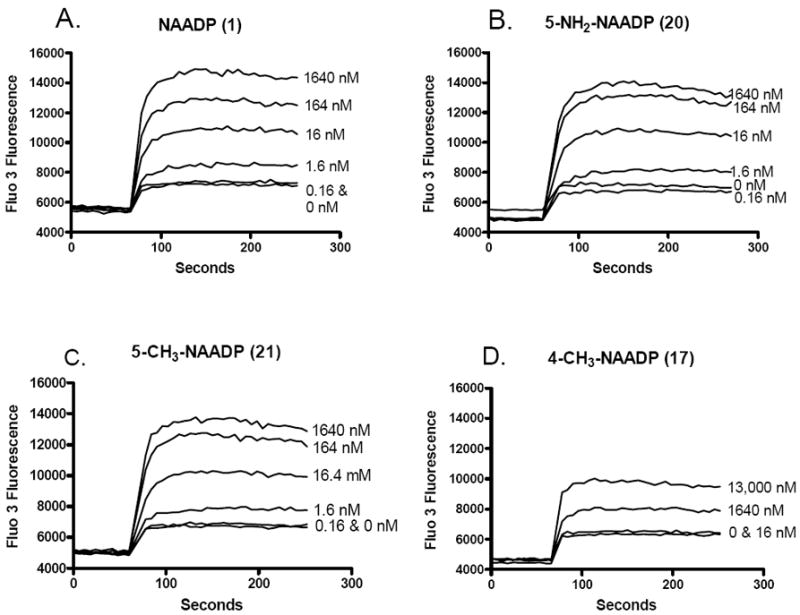
Fluorimetric calcium release traces from sea urchin egg homogenates induced by the addition of increasing concentrations of NAADP and three selected NAADP analogs. Calcium-ion concentration was measured fluorimetrically using the calcium sensitive dye Fluo-3. The effect of the addition of increasing concentrations of NAADP (1)(A), 5-NH2-NAADP (20)(B), 5-CH3-NAADP (21)(C), and 4-CH3-NAADP (17)(D) on fluorescence are shown.
Figure 2.
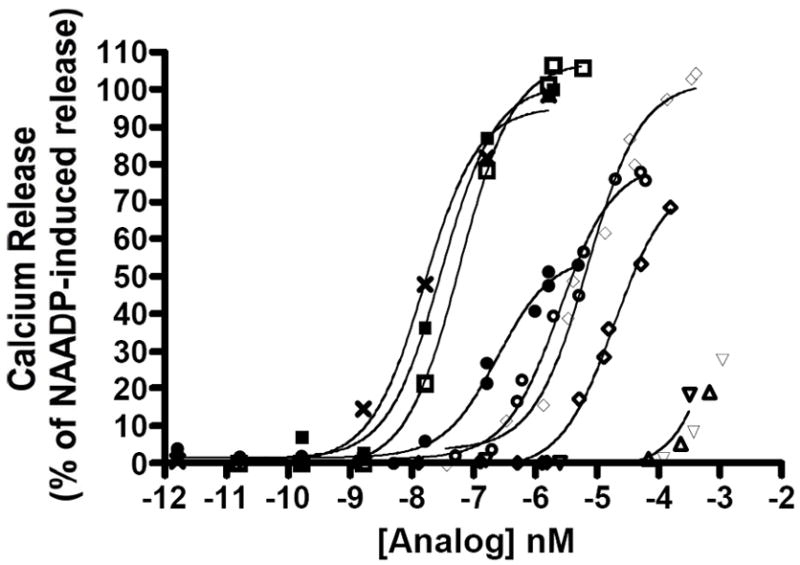
Concentration response curves depicting maximum Ca+2 concentration attained after treatment of calcium loaded sea urchin egg homogenates by the addition of varying concentrations of NAADP (1)(■), 5-NH3-NAADP (20)(□), 5-CH3-NAADP (21)(X), 5-CH2CH3-NAADP (23)(●), 5-N3-NAADP (24)(○), 4-NH3-NAADP (16)(◊), 4-CH3-NAADP (17)(◇), 4-n-butyl-NAADP (18)(▽), 4-phenyl-NAADP (19)(△), and 5-CO2H-NAADP (22)(▽). Each plotted point represents the mean of a minimum of three determinations.
Table 3.
EC50 values for Ca2+ release induced by NAADP (1) and NAADP analogs (16-24) measured fluorometrically from Ca2+ loaded sea urchin egg homogenates.
| Compound | EC50 ± s.d., nM (n) | Fold increase in EC50 |
|---|---|---|
| NAADP (1) | 18.7 ± 6.9 (19) | 1 |
| 5-amino-NAADP (20) | 27.4 ± 13.9 (9) | 1.5 |
| 5-methyl-NAADP (21) | 43.0 ± 25.3 (11) | 2.3 |
| 5-ethyl-NAADP (23) | 352 ± 219 (3) | 18 |
| 5-azido-NAADP (24) | 1689 ± 1349 (9) | 90 |
| 4-amino-NAADP (16) | 4413 ± 2794 (8) | 235 |
| 4-methyl-NAADP (17) | 42231 ± 28870 (6) | 2200 |
| 4-n-butyl-NAADP (18) | >1,000,000 (5) | > 50,000 |
| 4-phenyl-NAADP (19) | >1,000,000 (6) | > 50,000 |
| 5-carboxy-NAADP (22) | > 1,000,000 (3) | > 50,000 |
Receptor Desensitization
NAADP and other related agonists characteristically induce desensitization of the Ca2+ release mechanism when the receptor is subjected to pretreatment for a few minutes using subthreshold agonist concentrations.3 Subsequent treatment with a much higher concentration of NAADP is then unable to release calcium. Such desensitization is indicative of involvement of a receptor mediated response, and is also a useful tool to demonstrate that an agonist is releasing calcium through the NAADP receptor. It was found that pretreatment with 1 nM 5-CH3-NAADP (21) produced the same characteristic receptor desensitization as does NAADP itself (Figure 3, Panel A) and hence that 21 must be releasing Ca2+ through the same receptor as NAADP itself. Other tested compounds (16-24) were shown to be similarly effective in desensitization when used for pretreatment at a concentration of 1 nM or higher (Figure 3, Panel B). The concentrations of analogs used in Figure 3B were chosen based on previous findings3,39 that pretreatment with NAADP at sub-threshold concentrations (for calcium release) were able to completely desensitize calcium release by a maximal concentration of NAADP. In the experiment shown in Figure 3B, we attempted to use analog concentrations that were just below the threshold concentration necessary to induce measurable calcium release, approximately 20-100 fold lower than the observed EC50.
Figure 3.
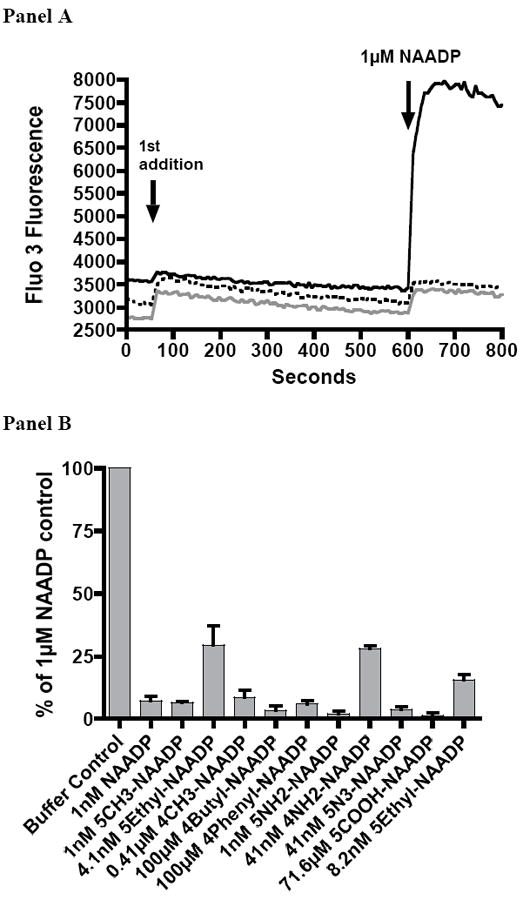
Desensitization of sea urchin egg homogenate Ca2+ release by pretreatment with subthreshold concentrations of NAADP or NAADP analogs.
Panel A: Effect of a 9 min pretreatment with buffer (solid line), 1 nM NAADP (1) (dotted line), or 1 nM 5-CH3-NAADP (21) (gray line) on calcium release in response to treatment with 1 μM NAADP. The data has been adjusted along the Y-axis to prevent overlap.
Panel B: Comparison of the abilities of a 9 min preincubation with NAADP (1) or NAADP analogs (16-24) at the stated concentrations to reduce Ca2+ release in response to the addition of 1 μM NAADP. Values shown are mean±SEM (n = 3).
Competition ligand binding studies
The ability of NAADP (1) and its analogs 16-24 to compete with [32P]NAADP for specific binding to sea urchin microsomes was determined using a competition ligand binding assay according to the procedure of Aarhus et al. (1996).3 The concentration response curves for the 10 tested compounds are shown in Figure 4, and the IC50 values derived from the fitted data derived from determinations made in two or more additional experiments in addition to that depicted in Figure 4 are presented in Table 4. The binding studies show that NAADP (1), 5-NH2-NAADP (20) and 5-CH3-NAADP (21) inhibit the binding [32P]NAADP by 50% at a concentration range of 1-2 nM, whereas other analogs are able to compete with [32P]NAADP only at much higher concentrations. Of the compounds tested, 5-carboxy-NAADP (22) was observed to be the least potent with an EC50 of > 100,000 nM, > 60,000-fold higher than the EC50 of NAADP.
Figure 4.
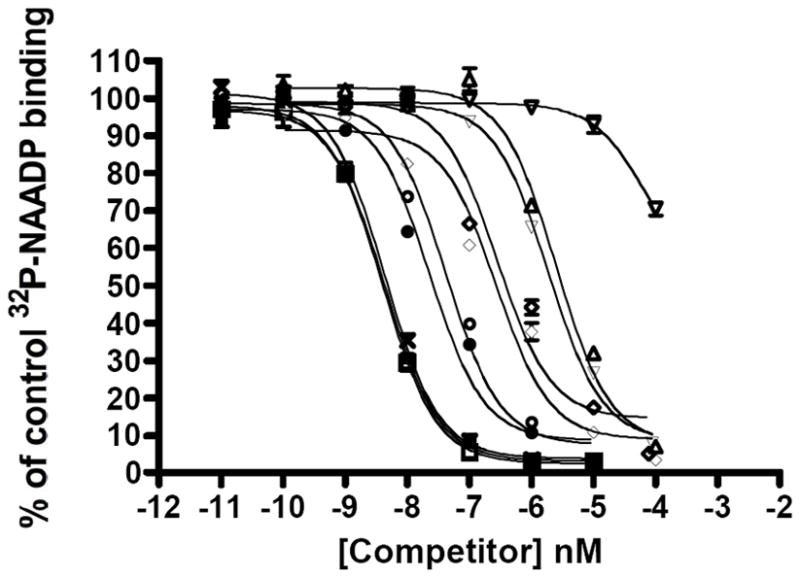
Competition radioligand binding curves for NAADP (1)(■) and the nine analogs (16-24) in sea urchin egg homogenates: 5-NH3-NAADP (20)(□), 5-CH3-NAADP (21)(×), 5-CH2CH3-NAADP (23)(●), 5-N3-NAADP (24)(○), 4-NH3-NAADP (16)(◊), 4-CH3-NAADP (17)(◇), 4-n-butyl-NAADP (18)(▽), 4-phenyl-NAADP (19)(△), and 5-CO2H-NAADP (22)(▽). Each plotted point represents the mean of a minimum of three determinations.
Table 4.
IC50 values determined for compeition ligand binding between [32P]NAADP and NAADP analogs (16-24)
| Compound (structure #) | IC50± s.d., nM (n) | Fold increase in IC50 |
|---|---|---|
| NAADP (1) | 1.6 ± 1.5 (9) | 1. |
| 5-amino-NAADP (20) | 1.7 ± 1.3 (6) | 1.1 |
| 5-methyl-NAADP (21) | 1.9 ± 1.5 (5) | 1.2 |
| 5-ethyl-NAADP (23) | 37 ± 30 (4) | 23. |
| 5-azido-NAADP (24) | 18 ± 14 (5) | 11. |
| 4-amino-NAADP (16) | 141 ± 116 (3) | 88. |
| 4-methyl-NAADP (17) | 149 ± 99 (6) | 93. |
| 4-n-butyl-NAADP (18) | 465 ± 221 (5) | 303. |
| 4-phenyl-NAADP (19) | 859 ± 896 (6) | 540 |
| 5-carboxy-NAADP (22) | > 100,000 (4) | >60000. |
Discussion
Although approaches to the chemical synthesis of NAADP and its derivatives have been developed,40 the process of producing NAADP derivatives synthetically is still lengthy and relatively difficult. Chemoenzymatic syntheses of NAD, NADP, and NAADP derivatives have been effectively used to produce novel pyridine dinucleotides since the discovery of the NAD glycohydrolase catalyzed base exchange reaction in the 1950s. Successful base exchange was found to require a uncharged species, a sufficiently nucleophilic pyridine base (pKa > 3)41, and usually the absence of sterically interfering substituents ortho- to the pyridine nitrogen. A variety of monosubstituted pyridines with large groups in the 3- or 4- positions were found to successfully exchange into the pyridine dinucleotides using the enzyme catalyzed reaction. Disubstituted nicotinamides were studied less often, but 5-methyl-NAD and 5-amino-NAD had been synthesized using the porcine brain NAD-glycohydrolase as a catalyst.42
We find that 4-substituted nicotinic acids, even those with large 4-substituents, are excellent exchange substrates. One exception is a cationic compound such as 2a, which must be converted into an ester before it can be successfully exchanged into the pyridine dinucleotide at a high pH under conditions where the uncharged base is again present at high concentration. We encountered an unexpected limitation of the pyridine base exchange reaction in this study with respect to the synthesis of the 5-substituted NAADP derivatives. Nicotinic acids containing large groups at the 5-position (3h or 3j) or those containing groups which are both electron withdrawing and hydrophobic (3c, 3e [σm for acetylene is 0.21 indicating electron withdrawal], or 3f) fail to exchange. The interesting NAADP derivatives that would be derived from these compounds must therefore be produced by an alternate method. It is interesting that 5-azidonicotinic acid was shown to exchange with the nicotinic acid base of NAADP in good yield to produce 24, (5-azido-NAADP), a potential photoaffinity label for the NAADP binding protein.
When tested as competitive ligands for NAADP binding or for calcium-ion mobilizing activity we find that NAADP derivatives containing a nicotinic acid moiety with 4-substituents are associated with loss of binding potency. Even small 4-substituents (as in 16 and 17) are associated with a 100 to 200-fold loss of potency. The 4-substituted compounds are apparently low potency agonists and we have not yet encountered any 4-substituted NAADP derivatives that behave as antagonists. Sub-threshold concentrations of the 4-substituted derivatives 16-19 desensitize the NAADP receptor, indicating that these compounds act similarly to the other agonists.
We find that the 5-position of the nicotinic acid ring is tolerant of substitution. Introduction of an amino group (20) or a methyl group (21) at the 5-position results in the production of derivatives which are only slightly less potent than is NAADP itself. An ethyl group is tolerated with only a 20-fold loss of potency, and the azide (24)—a dipolar group containing three linearly disposed heavy atoms—is associated with only a 11-fold increase in IC50 as measured in the competition ligand binding assay. The concentration response for Ca2+ release induced by 5-ethyl-NAADP (23) (Figure 2) apparently plateaus at a significantly lower per cent calcium ion release than does the full agonist NAADP indicating that 23 exhibits partial antagonist activity. Similarly the 5-azide 24 may also be a partial agonist. Most reported modifications of NAADP have been observed to result in the loss of receptor binding potency, and our observation suggests that further modification at the nicotinic acid 5-position might produce potent agonists that will be informative in future studies of receptor function, receptor localization, and receptor isolation. The EC50 for 5-azido-NAADP (24) is low enough to suggest that when 24 is appropriately labeled it should prove useful as a photoaffinity label for the identification of NAADP binding proteins (Tables 3 and 4).
The EC50 values derived from Ca2+ release (Table 3) and the IC50 values derived from competition ligand binding (Table 4) are well correlated (see Supporting Information) but the IC50 values are consistently lower. This may be due to the fact that the measurements are made under different experimental conditions. Alternately, it has been suggested recently that the NAADP receptor possesses two binding sites for NAADP, a high affinity inhibitory site and the stimulatory site with a relatively lower affinity.20 The IC50 values measured using the competition ligand binding assay might in such a case reflect the binding potency of the high affinity inhibitory site.
Experimental Section
General Procedures
The following procedures were used in all reactions unless otherwise noted. Oxygen- and moisture-sensitive reactions were carried out in oven-dried (T > 100°C) glassware sealed under a positive pressure of dry nitrogen supplied from a manifold. Moisture or air sensitive liquids and solutions were transferred under a nitrogen atmosphere by syringe through rubber septa. Reactions were stirred with a Teflon-covered magnetic stirring bar. Reagents for reactions were obtained from commercial sources and were used without further purification, except as indicated. Either sure-seal bottles of anhydrous solvents were purchased from Aldrich or they were purified according to Perrin’s Purification of Laboratory Chemicals.73 Proton (1H) NMR spectra and carbon-13 (13C) NMR spectra were recorded at 600 MHz or at 400 MHz. Chemical shifts are reported in ppm (δ) and are referenced to the residual proton signal of the deuterated solvent. 31P NMR spectra were recorded on Gemini 200 MHz spectrometer with 85 per cent H3PO4 as external reference. The pH of the solutions was adjusted to pH 7 prior to the determination of the 31P NMR spectra. The chemical shifts for 13C NMR peaks are reported to the first decimal place while the peaks which are very close are reported to the second decimal place. The following abbreviations are used to describe spin multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, b = broad, dd = doublet of doublets, dt = doublet of triplets, dq = doublet of quartets, and ddd = doublet of doublet of doublets; other combinations derive from those listed. Coupling constants (J) are reported in Hertz (Hz). Melting points were determined on an Electrothermal digital melting point apparatus and are uncorrected. Mass spectral analyses were done at University of Toledo Instrumentation centre. Electrospray ionization mass spectra (ESI) were acquired on FT/ICR spectrometer. High resolution mass spectroscopies with accurate mass analyses (HRMS) were performed at Ohio State University Mass Spectrometry and Proteomics facility. Analytical thin layer chromatography (TLC) was performed using 0.25 mm silica gel 60 glass plates with a 254 nm fluorescent indicator from Analtech. Plates were developed in a covered chamber and visualized by examination under short wavelength ultra-violet (UV) light, iodine stain, sulfuric acid charring or ninhydrin stain. Flash chromatography was done using Fisher silica gel 60, 200-425 mesh (40-60 mm) as stationary phase. For flash column purification the amount of stationary phase used was about 40 times the quantity of compound being purified. The silica gel columns were packed in non-polar solvent followed by sample application. The column was always washed with 2-3 column volumes of non-polar solvent to remove highly non-polar impurities. Solvent evaporation was done under reduced pressure using a Buchi rotavapor either at water aspirator vacuum or at vacuum achieved with a regular vacuum oil pump. Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, Georgia), and are regarded as being acceptable when within ± 0.4% of the theoretical values.
Anion exchange chromatography was performed using diethylaminoethyl substituted cellulose (DE-52 cellulose from Whatman Inc.). The dimensions of the column used for anion exchange was 1.5 cm × 40 cm. Dowex AG1-X2 resin was obtained in the Cl- form from Bio-Rad Laboratories (Hercules, CA) was converted from the Cl- form into acetate form by passing 1 M sodium acetate through the column until the column effluent tested negative for chloride ions. The cation exchange resin, Dowex 50W-X8 was purchased in H+ form and was converted to tetrabutylammonium form by treating it with an excess of 40% tetrabutylammonium hydroxide.
For base exchange enzymes, 1 unit represents the amount enzyme required to hydrolyze 1 μmol of NAD/min. For the base exchange reaction, the pH of the solution was buffered by the nicotinic acid analog and was not readjusted after the addition of NADP disodium salt. The catalyst present in base exchange reactions was removed by filtration of the mixture using Amicon Ultra centrifugal filter device with molecular weight cut of 10 kDa. The centrifugal filter device was bought from Millipore Corporation (Bedford, MA). The yields of 4 or 5 substituted NAADP derivatives were calculated with respect to NADP.
Materials
4-Aminonicotinic acid was purchased from AKSci, 5-aminonicotinic acid was bought from Combi-Blocks, Inc., 4-methylnicotinic acid was bought from Maybridge and 3,5-pyridine-dicarboxylate was purchased from Acros Organics. NADP used in pyridine base exchange reactions was bought as the disodium salt in 98% purity from Roche Diagnostics (Indianapolis, IN). (4,5-Dihydro-4,4-dimethyl-2-oxazolyl)pyridine was kindly provided by our colleague Chris Trabbic (University of Toledo).
Synthesis of 4-substituted nicotinic acid analogs
4-n-Butylnicotinic acid (2c)
The immediate precursor of 2c, 4-n-butyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine, was synthesized by the addition of n-butyllithium to the pyridyl-3-oxazoline as follows: 3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (0.72 g, 4.10 mmol) was taken in a flask and N2 was flushed through the flask. Anhydrous THF (8.0 mL) was added and the mixture was stirred at room temperature until a solution was formed. The flask was cooled in a -20 °C bath and stirred using a magnetic stirrer. The reaction temperature was regulated using a thermostatically controlled mechanical cryostat. n-Butyllithium (1.6 M solution in hexanes, 2.88 mL) was added dropwise over a period of 10 min. The reaction was allowed to stir at -20 °C for 2 h. O2 gas was bubbled into the reaction for 3 h at 0 °C (bath temperature). The reaction was quenched by adding water (25 mL) and product was extracted into diethylether (3 × 20 mL). The organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to an oily residue. This was purified by column chromatography (20-50% ethyl acetate (EtOAc) in hexanes) to obtain product as clear oil (0.67 g, 71%): TLC Rf 0.35 in EtOAc: hexanes (2:3), 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.47 (d, 1H, J=5.2), 7.11 (d, 1H, J=5.2), 4.05 (s, 2H), 2.94 (t, 2H, J=8.0), 1.58-1.48 (m, 2H), 1.4-1.3 (m, 2H), 1.36 (s, 6H), 0.89 (t, 3H, J=7.2).
4-n-Butyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (0.30 g, 1.29 mmol) was dissolved in 3 N HCl (100 mL) and acetic acid (50 mL) and was refluxed at 95 °C for 36 h. The solution was concentrated under reduced pressure and then dissolved in water (20 mL) and neutralized using 2 N NaOH. The aqueous layer was lyophilized and the solid was purified by flash chromatography (silica gel, CH2Cl2: CH3OH: acetic acid, 90:9:1) to give 2c as white solid (0.16 g, 69 %): m.p. 121-123 °C; TLC Rf 0.62 in CH2Cl2: CH3OH: Acetic acid (90:9:1), 1H NMR (400 MHz, CD3OD) δ 8.94 (s, 1H), 8.52 (d, 1H, J=3.6), 7.40 (d, 1H, J=3.6), 3.05 (t, 2H, J=5.2), 1.64-1.58 (m, 2H), 1.46-1.38 (m, 2H), 0.96 (t, 3H, J=5.2).
4-Phenylnicotinic acid (2d)
The immediate precursor of 2d, 4-phenyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine, was synthesized by the addition of phenyllithium to pyridyl-3-oxazoline as follows: 3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (0.57 g, 3.26 mmol) was taken in a flask and N2 was flushed through the flask. Anhydrous THF (9.5 mL) was added and the mixture was stirred at room temperature until it formed a solution. The flask was cooled in an acetone/dry ice bath (-78 °C) with stirring. Phenyllithium (2 M solution in ether, 1.6 mL) was added dropwise over a period of 10 min. The reaction was allowed to stir at -78 °C for 1 h and then was stirred at 0 °C for another hour. O2 gas was bubbled into the reaction for 3 h at 0 °C (bath temperature). The reaction was quenched by adding water (15 mL) and product was extracted in EtOAc (3×15 mL). The organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to oily residue. This was purified by column chromatography (50% EtOAc in hexanes) to obtain product as colorless oil (612.0 mg, 75%): TLC Rf 0.32 in EtOAc: hexanes (8:7), 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.67 (d, 1H, J=5.2), 7.39 (s, 5H), 7.30 (d, 1H, J=5.2), 3.83 (s, 2H), 1.30 (s, 6H).
4-Phenyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (0.35 g, 1.38 mmol) was dissolved in 3 N HCl (100 mL) and acetic acid (50 mL) and was refluxed at 95 °C for 36 h. The solution was concentrated under reduced pressure and then dissolved in water (20 mL) and neutralized using 2 N NaOH. The aqueous layer was lyophilized and the solid was purified by flash chromatography (CH2Cl2: CH3OH: acetic acid, 90:9:1) to give 2d as sticky solid (0.20 g, 73%): TLC Rf 0.58 in CH2Cl2: CH3OH: acetic acid (90:9:1), 1H NMR (600 MHz, CD3OD) δ 8.90 (s, 1H), 8.67 (d, 1H, J=4.8), 7.49 (d, 1H, J=4.8), 7.46-7.42 (m, 5H).
Synthesis of 5-substituted nicotinic acid analogs
5-Ethynylnicotinic acid (3e)
The immediate precursor of 3e, 5-ethynyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine, was produced from 6 by removal of the TES-protecting group. Compound 6 (0.56 g, 2.06 mmol) and anhydrous K2CO3 (1.46 g, 10.5 mmol) were added to a flask under N2. Anhydrous CH3OH (2 mL) and anhydrous THF (2 mL) were added to the flask. The mixture was stirred for 1h and monitored by TLC. After the reaction was complete, water (15 mL) was added to the flask and product was extracted into EtOAc (2×30 mL). The organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and the oily residue was purified by flash chromatography (20-50 % EtOAc in hexanes) to obtain 5-ethynyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine as white solid (0.26 mg, 62 %): m.p. 97-98 °C; TLC Rf 0.30 in EtOAc: hexanes (1:1), 1H NMR (600 MHz, CD3OD) δ 8.99 (d, 1H, J= 1.8), 8.77 (d, 1H, J=2.4), 8.31 (t, 1H, J=1.8) 4.26 (s, 2H), 3.92 (s, 1H), 1.39 (s, 6H); 13C NMR (100 MHz, CD3OD) δ 161.6, 155.5, 149.1, 139.8, 125.2, 121.3, 84.2, 80.8, 80.0, 69.1, 28.5 ; LC-MS (ESI) m/z 201.2 (M+1).
5-Ethynyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (0.8 g, 4 mmol) was dissolved in 3 N HCl (150 mL) and acetic acid (75 mL) and the mixture was refluxed at 95 °C for 36 h. The solution was concentrated under reduced pressure and then dissolved in water (25 mL) and neutralized using 2 N NaOH. The aqueous layer was lyophilized and the solid was purified by flash chromatography to give 3e as white solid (0.43 g, 74 %): m.p. 210-211 °C with decomposition; TLC Rf 0.60 in CH2Cl2: CH3OH: acetic acid (90:9:1), 1H NMR (400 MHz, CD3OD) δ 9.00 (d, 1H, J= 1.6), 8.62 (d, 1H, J=2.0), 8.34 (t, 1H, J=2.0), 3.79 (s, 1H); 13C NMR (100 MHz, CD3OD) δ 167.0, 156.4, 150.7, 141.6, 128.3, 121.4, 84.0, 80.0; analysis calcd. for C8H5NO2 0.1 H2O: C 64.52, H 3.52, N 9.40, found: C 64.29, H 3.33, N 9.31, LC-MS (ESI) m/z 148.3 (M+1).
5-Ethenylnicotinic acid (3f)
To a cooled solution of 5-ethynyl nicotinic acid (3e) (0.1 g, 0.68 mmol) in CH3OH (10 mL) were added Lindlar’s catalyst (5 mg, 5%) and 2,2’-(ethylenedithio)diethanol (50 μg, 10 parts per thousand of Pd catalyst). The mixture was shaken under H2 atmosphere (30 psi) at room temperature. The reaction was monitored by TLC and was filtered through a celite pad after 24 h. The filtrate was concentrated under reduced pressure at room temperature and the crude product was purified by column chromatography in CH2Cl2: CH3OH: acetic acid (90:9:1) to give 3f (27.4 mg, 27%): m.p. 154-156 °C; TLC Rf 0.55 in CH2Cl2: CH3OH: acetic acid (90:9:1); 1H NMR (600 MHz, CD3OD) δ 8.98 (s, 1H), 8.76 (s, 1H), 8.45 (s, 1H), 6.87-6.82 (dd, 1H, 2J= 18, 3J= 11.4), 6.02 (d, 1H, J=17.4), 5.50 (d, 1H, J=10.8); 13C NMR (150 MHz, CD3OD) δ 166.6, 150.5, 148.9, 134.3, 133.9, 132.4, 127.5, 117.3; LC-MS (ESI) m/z 150.1 (M+1).
5-Ethylnicotinic acid (3g)
In a flask under N2 5-ethynylnicotinic acid (3e) (0.04 g, 0.3 mmol) and ammonium formate (0.063 g, 1 mmol) were added. To this flask a mixture of 10% Pd-C (50 mg) and cold CH3OH (5 mL) was added and the resulting mixture was stirred for 12 h. The Pd-C was filtered off through a bed of celite and the clear filtrate was evaporated. The residue was washed with methylene chloride several times and the product went into the methylene chloride. The solvent was evaporated to give product 3g as sticky residue (25 mg, 78%): 1H NMR (600 MHz, CD3OD) δ 8.93 (s, 1H), 8.57 (s, 1H), 8.25 (s, 1H), 2.76 (q, 2H, 2J= 15.0, 3J= 7.2), 1.29 (t, 3H, J=7.2); 13C NMR (150 MHz, CD3OD) δ 167.6, 151.6, 147.4, 140.3, 137.2, 128.4, 25.5, 14.4; LC-MS (ESI) m/z 152.1 (M+1).
5-Phenylnicotinic acid (3h)
5-Phenyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (9) (0.35 g, 1.38 mmol) was dissolved in 3 N HCl (100 mL) and acetic acid (50 mL) and was refluxed at 95 °C for 36 h. The solution was concentrated under reduced pressure and then dissolved in water (20 mL) and neutralized using 2 N NaOH. The aqueous layer was lyophilized and the solid was purified by flash chromatography (CH2Cl2: CH3OH: acetic acid , 90:9:1) to give product as white solid (215 mg, 78 %): m.p. 256-257 °C (reported m.p. 267-269 °C44); TLC Rf 0.58 in CH2Cl2: CH3OH: acetic acid (90:9:1), 1H NMR (600 MHz, CD3OD) δ 9.10 (d, 1H, J=1.8), 9.00 (d, 1H, J=2.4), 8.60 (t, 1H, J=1.8), 7.73 (d, 2H, J=7.2), 7.55-7.45 (m, 3H); 13C NMR (100 MHz, CD3OD) δ 150.6, 148.6, 137.3, 136.5, 135.8, 129.2, 128.7, 128.0, 127.7,127.0.
5-Azidonicotinic acid (3i)
5-Azidonicotinic acid ethyl ester (11) (2.5 g, 13.02 mmole) was dissolved in CH3OH (15 mL). The solution was stirred at room temperature for 10 min and 2 N NaOH (10 mL) was added to it. This solution was then stirred at room temperature and the reaction was monitored by TLC for disappearance of starting material (24 h). The solution was concentrated under reduced pressure and was diluted with water (15 mL). The basic layer was then carefully neutralized with 2 N HCl resulting in precipitation of white solid. The solid was filtered and dried under vacuum to give product (1.69 g, 79 %): m.p.178-180 °C; TLC Rf 0.53 in CH2Cl2: CH3OH: acetic acid (90:9:1), 1H NMR (600 MHz, CD3OD) δ 8.88 (d, 1H, J= 1.8), 8.48 (d, 1H, J=2.4), 8.05 (t, 1H, J=1.8); 13C NMR (150 MHz, CD3OD) δ 167.1, 147.4 (d, J= 6.9), 145.4 (d, J=6.15) 139.6, 129.4, 128.5; analysis calcd. for C6H4N4O2: C 43.91, H 2.46, N 34.14, found: C 44.00, H 2.4, N 33.88, LC-MS (ESI) m/z 165.2 (M+1).
5-(4-(Aminomethyl)-1H-1,2,3,-triazol-1-yl)nicotinic acid (3j)
A solution of ethyl 5-(4-(aminomethyl)-1H-1,2,3-triazol-1-yl)nicotinate (14) (100 mg, 0.4 mmol) in CH3OH (2 mL) and NaOH (2 M, 0.5 mL, 1 mmol) was stirred for 24 h at room temperature. When no starting material was detected on TLC (CH2Cl2: CH3OH: TEA, 8:1:1) the solution was neutralized with 2 N HCl and concentrated in vacuum. The residue was dissolved in water (10 mL) and lyophilized. The solid obtained was then suspended in CH3OH and filtered. The filtrate was then concentrated in vacuum to obtain 3j as white solid with some inorganic salts (85 mg): 1H NMR (400 MHz, CD3OD) δ 9.27 (s, 1H), 9.23 (s, 1H), 8.83 (s, 1H), 8.81 (t, 1H), 4.39 (s, 1H), 13C NMR (150 MHz, CD3OD) δ 163.1, 151.7, 141.4, 130.4, 124.4, 119.8, 116.9, 35.5.
3-Bromo-5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (4)
Compound 4 was prepared from 3c in three steps according to the procedure of Robert et al., (2006)43 and isolated as a white solid (4.92 g, 78 % over three steps): m.p. 91-93 °C (lit.43 90-91 °C); TLC Rf 0.65 in EtOAc. The NMR data was in agreement with the published data.
1,2-Bis(5-(4,4-dimethyl-4,5-dihydrooxazol-2y)pyridine-3-yl)ethyne (5)
To a 2 neck flask triethyl(ethynyl)silane (TES-acetylene, 182 mg, 1.3 mmol) was added and it was protected from air under N2 atmosphere. To this flask, a solution of tetrakis(triphenylphosphine)-palladium(0) (45 mg, 0.04 mmol) in ethanol/dimethoxyethane (1:1, 2 mL), aqueous sodium carbonate (2 M, 4 mL) and copper iodide (46 mg, 0.24 mmol) was added. The resultant solution was stirred at room temperature for 15 min and then to this solution 3-bromo-5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (4) (0.6 g, 2.5 mmol) was added under a stream of nitrogen. The flask was then stirred at 90 °C (oil bath temperature) for 1 h and the reaction was brought to room temperature. The mixture was poured into a flask containing anhydrous sodium sulfate, filtered and evaporated in vacuum. The residue was then purified by column chromatography (10-50 % EtOAc in hexanes) to give compound 5 (155 mg, 58%). TLC Rf 0.30 in EtOAc: hexanes (1.1), 1H NMR (600 MHz, CDCl3) δ 9.05 (s, 1H), 8.79 (s, 1H), 8.32 (t, 1H, J=1.8), 4.12 (s, 2H), 1.36 (s, 6H ); 13C NMR (100 MHz, CDCl3) δ 159.6, 154.0, 148.8, 138.7, 138.0, 124.1, 119.6, 89.2, 68.2, 28.6; LC-MS (ESI) m/z 375.5 (M+1).
4,4-Dimethyl-2-(5-((triethylsilyl)ethynyl)pyridine-3-yl)-4,5-dihydrooxazole (6)
A two neck flask, under N2, was charged with 3-bromo-5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (4) (1.6 g, 6.27 mmol) and benzyltributyl ammonium bromide (1.8 g, 5.05 mmol). To this mixture, degassed acetonitrile (9 mL) and degassed water (0.9 mL) were added. This was followed by addition of TES-acetylene (1.4 mL, 7.7 mmol) and DIPEA (7.2 mL). The mixture was stirred well at ambient temperature. In another flask Pd(OAc)2 (222.5 mg, 1 mmol) and PPh3 (525 mg, 2 mmol) were taken and dissolved in degassed acetonitrile (45 mL) and water (4.5 mL). The latter was added to the two neck flask and was stirred at ambient temperature for 8 h. The reaction was monitored by TLC and when the starting material had disappeared water (20 mL) was added to quench the reaction. Product was extracted into EtOAc (3 × 30 mL), the organic layer was dried over sodium sulfate, and filtered. The filtrate was concentrated under reduced pressure and the oily residue was purified by flash chromatography (20-50 % EtOAc in hexanes). Product was obtained as brown oil (1.16 g, 68 %) that solidified on freezing: m.p. 67-69 °C; TLC Rf 0.54 in EtOAc:hexanes (1:1), 1H NMR (600 MHz, CDCl3) δ 8.96 (s, 1H), 8.70 (s, 1H), 8.25 (t, 1H, J=1.8), 4.09 (s, 2H), 1.34 (s, 6H), 0.99 (t, 9H, J= 8.4), 0.63 (q, 6H, 2J= 16.2, 3J= 7.8), 13C NMR (150 MHz, CDCl3) δ 159.6, 154.4, 147. 9, 138.4, 123.5, 120.2, 101.6, 96.8, 79.3, 67.9, 28.3, 7.42 (d, J=1.95), 4.18; LC-MS (ESI) m/z 315.3 (M+1).
5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3-pyridinylboronic acid (7)
3-Bromo-5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (4) (1.5 g, 5.90 mmol) was taken in a flask and flushed with N2. Anhydrous toluene (5 mL) and anhydrous THF (6.5 mL) were added to the flask and the mixture was stirred untill the solid dissolved. The flask was then transferred to a -40 °C cooling bath and stirred rapidly. Some of the solid started to precipitate. Triisopropyl borate (1.8 mL, 7.8 mmol) was added to the flask. When the reaction mixture had been cooled for 15 min, n-butyllithium (1.6 M in hexanes, 4.5 mL, 7.2 mmol) was added dropwise over a period of 1 h. By the time butyllithium addition was complete the mixture turned into a yellowish viscous liquid. It was stirred for another 30 min at -40 °C, brought to -20 °C, and 2 N HCl (20 mL) added. The acidic layer was extracted with toluene (20 mL), the toluene extract discarded and then the aqueous layer neutralized with 2 N NaOH. The product was extracted into THF (4×15 mL). The THF layer was evaporated to dryness and the residue was dissolved in mixture of CH3OH / THF (1:1) and filtered. The filtrate was evaporated under reduced pressure and the residue crystallized from acetonitrile to obtain boronic acid 7 as yellow solid (0.69 g, 53 %): m.p. 270-272 °C with decomposition.
Pinacol ester of 5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3-pyridinyl boronic acid (pinacol ester 8)
5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3-pyridinylboronic acid (7) (0.1 g, 0.4 mmol), (CH3)2COHCOH(CH3)2, (190 mg, 1.6 mmol) and toluene (4 mL) were taken in a two neck flask equipped with N2 inlet and condenser with a Dean-Stark trap. The mixture was heated in a 120 °C oil bath and refluxed for 2.5 h. The reaction was monitored with TLC and upon completion the solution was concentrated in vacuum. The residue was purified using column chromatography (10-50 % EtOAc in hexanes) to obtain product as yellow oil (63.2 mg, 46 %): TLC Rf 0.30 in EtOAc: hexanes (1:1), 1H NMR (400 MHz, CDCl3) δ 9.14(s, 1H,), 8.97 (s, 1H,), 8.59 (d, 1H, J=1.6) 4.10 (d, 2H, J=1.6), 1.36 (d, 6H, J=1.2), 1.32 (d, 12 H, J=1.2); LC-MS (ESI) m/z 303.3 (M+1).
5-Phenyl-3-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-pyridine (9)
A two neck flask fitted with a N2 inlet was charged with tetrakis(triphenylphosphine)palladium(0) (230 mg, 0.2 mmol). Degassed dimethoxyethane (6 mL) and degassed sodium carbonate (2 M, 4 mL) were added to the flask. Bromobenzene (0.45 mL, 4.2 mmol) was added to the mixture and the suspension was stirred at room temperature for 15 min. In a separate flask 5-(4,5-dihydro-4,4-dimethyl-2-oxazolyl)-3-pyridinyl boronic acid (7) (0.7 g, 3.18 mmol) was dissolved in degassed EtOH (6 mL) and added to the catalyst mixture. The mixture was allowed to stir at room temperature for 15 min. The flask was sealed with a rubber septum and transferred to oil bath at 90 °C to stir for 4 h. The reaction was monitored by TLC and when almost all starting material disappeared the reaction mixture was filtered through celite, washing with CH2Cl2. The organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated in vacuum and the residue was purified by column chromatography (40-70% EtOAc in hexanes) to obtain 9 as oil (490 mg, 61%): TLC Rf 0.55 in EtOAc: hexanes (7:3), 1H NMR (400 MHz, CDCl3) δ 9.07(d, 1H, J=2.0), 8.89 (d, 1H, J=2.4), 8.39 (t, 1H, J=2.4), 7.6 (d, 2H, J=7.2), 7.46-7.37 (m, 3H) 4.13 (s, 2H), 1.38 (s, 6H);13C NMR (100 MHz, CDCl3) δ 160.4, 150.5, 148.2, 134.1, 129.3, 128.7, 127.5, 79.5, 68.2, 28.6.
5-Aminonicotinic acid ethyl ester (10)
5-Aminonicotinic acid (3a) (5.0 g, 0.036 mol) was dissolved in EtOH (200 mL) and conc. HCl (40 mL). The solution was then refluxed for 36 h. The reaction was monitored by TLC and was concentrated under reduced pressure upon completion. The resulting residue was dissolved in distilled water (25 mL) and neutralized using 1 N NaOH. The aqueous layer was then extracted with EtOAc (3 × 50 mL). The organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to obtain white crystalline solid. The solid was dried under vacuum to obtain 10 (4.9 g, 82 %): m.p. 95-97 °C; TLC Rf 0.66 in CH3OH: CH2Cl2 (1:9), 1H NMR (600 MHz, CD3OD) δ 8.32 (d, 1H, J= 1.8), 8.10 (d, 1H, J= 2.4), 7.59-7.60 (m, 1H), 4.36 (q, 2H, 2J= 14.4, 3J= 7.2), 1.38 (t, 3H, J= 7.2), 13C NMR (150 MHz, CD3OD) δ 167.0, 146.8, 140.6, 138.8, 128.4, 122.6 (d, J=8.25) 62.56, 14.7; analysis: calcd. for C8H10N2O2; C 57.82, H 6.07, N 16.86, found: C 57.80, H 6.17, N 16.67.
5-Azidonicotinic acid ethyl ester (11)
5-Aminonicotinic acid ethyl ester (10) (3.0 g, 18.05 mmol) was stirred with concentrated HCl (100 mL) in a 0 °C bath. A solution of NaNO2 (1.38 g, 19.86 mmol) in ice water (8 mL) was added dropwise to the stirred slurry of the hydrochloride salt, resulting in a clear solution. This solution was then added to stirred slurry of NaN3 (1.77 g, 27.09 mmole) and sodium acetate (3.9 g) in ice water (3.5 mL). After a few minutes of stirring, a red oil separated from the mixture. The mixture was made basic by the addition of concentrated ammonium hydroxide solution after 10 min and the product was extracted into EtOAc. The organic layer was dried over sodium sulfate, filtered, and the filtrate concentrated under reduced pressure. The residue was purified by column chromatography (10% EtOAc in hexanes) to give 11 as golden oil34 (1.66 g, 48 %): TLC Rf 0.48 in EtOAc: hexanes (1:5), 1H NMR (400 MHz, CDCl3) δ 8.96 (d, 1H, J=1.6), 8.47 (d, 1H, J=2.8), 7.94 (d, 1H, J=2.0), 4.41 (q, 2H, 2J= 14.4, 3J=7.2), 1.40 (t, 3H, J= 7.2), 13C NMR (100 MHz, CDCl3) δ 164.7, 147.0, 144.9, 137.5, 127.2, 126.7, 62.1, 14.4; analysis: calcd. for C8H8N4O2. 0.1 EtOAc; C 50.20, H 4.41, N 27.88, found: C 49.82, H 4.21, N 27.92.
Ethyl 5-(4-((tert-butyloxycarbonylamino)methyl)-1H-1,2,3-triazol-1-yl)nicotinate (13)
To a solution of 5-azidonicotinic acid ethyl ester (11) (150 mg, 0.78 mmol) and tert-butyl prop-2-ynylcarbamate (12) (124 mg, 0.8 mmol) in THF (1 mL) were added CuI (300 mg, 1.6 mmol) and DIPEA (0.4 mL, 2.4 mmol). The reaction was stirred at room temperature for 12 h by which time all the starting material was consumed. The reaction mixture was diluted with water (10 mL) and saturated aqueous NH4Cl (10 mL) and was extracted with CH2Cl2 (3 × 20 mL). The organic layers were combined and washed with brine, dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography to obtain impure material (160 mg) which was used for next step without further purification.
Ethyl 5-(4-(aminomethyl)-1H-1,2,3-triazol-1-yl)nicotinate (14)
To a solution of 13 (160 mg) in anhydrous CH2Cl2 (2 mL) was added triflouroacetic acid (2 mL) and it was stirred at room temperature for 1.5 h. The reaction was concentrated in vacuum and again diluted with CH2Cl2 (5 mL). This solution was then neutralized with triethylamine (TEA) and concentrated in vacuum. The residue was purified using column chromatography (CH2Cl2: CH3OH: TEA, 8:1:1) to give 14 as colorless oil (92.6 mg, 48 % over two steps with respect to 5-azidonicotinic acid ethyl ester): TLC Rf 0.65 in CH2Cl2: CH3OH: TEA (8:1:1), 1H NMR (400 MHz, CD3OD) δ 9.30 (d, 1H, J=2.4), 9.22 (d, 1H, J=1.6), 8.82 (s, 1H), 8.79 (t, 1H, J=2.0), 4.47 (q, 2H, 2J= 14.4, 3J= 7.2), 4.39 (s, 1H), 1.44 (t, 3H, J= 7.2), 13C NMR (100 MHz, CD3OD) δ 165.3, 151.3, 146.1, 142.9, 135.1, 130.0, 129.0, 124.3, 63.4, 35.5, 14.6; LC-MS (ESI) m/z 248 (M+1).
Synthesis of NAADP analogs
Enzymatic assay of NAD glycohydrolase activity45
Recombinant Aplysia ADP-ribosyl cyclase expressed in yeast was produced according to procedure of Lee et al.46 NAD glycohydrolase (NADase) from pig brain was bought from Sigma-Aldrich as an acetone powder. The dry acetone powder was hydrated and dispersed into a fine suspension each time immediately before use. The stock mixture consisted of 60 mg dry acetone powder in 2 mL distilled water. The mixture was kept at 37 °C for one half-hour and then subjected to homogenization until fine milky suspension was obtained. The NADase from beef spleen was isolated according to procedure of Kaplan and Colowick47. The NADase activity of Aplysia cyclase, NADase (pig brain) and NADase (beef spleen) were measured by determining the amount of NAD present at timed intervals in the assay, by the reduction of NAD to NADH catalyzed using yeast alcohol dehydrogenase. The NAD glycohydrolase test solution was made by addition of sodium dihydrogen phosphate pH 7.4 (0.1 M, 0.8 mL), NADase enzyme (80 μL of stock made up to 0.2 mL with buffer), NAD (0.2 mL, 6 mg/mL of buffer, 1.808 μmol). The reaction was incubated at 37 °C for 10 min and trichloroacetic acid (TCA) (25% w/v, 0.3 mL) was added to quench the reaction. The blank solution was treated exactly the same way except that TCA was added even before initiating the reaction. Both the test and the blank solutions were centrifuged at the speed of 3200 × g for 5 min and 0.3 mL of aliquot was removed from the clear supernatant. This was added to a cuvette with Tris-ethanol buffer pH 10 (2.7 mL, equal volumes of 0.5 M Tris and 0.5 M ethyl alcohol). To this solution yeast alcohol dehydrogenase was added (10 μl, 10 mg/mL) and the OD was measured at 340 nm.
Enzymatic assay for NADP estimation48
NADP, glucose-6-phosphate and yeast glucose-6-phosphate dehydrogenase (2 units in 10 mL solution) were obtained commercially. In the chemoenzymatic synthesis of NAADP analogs, NADP was incubated with huge excess of nicotinic acid in the presence of NAD glycohydrolase. The reaction was monitored by determination of the amount of NADP left in the reaction mixture. A 20 μL aliquot was removed from the reaction mixture (with concentration of about 0.01 mmole of NADP/mL) at intervals of 0.5 h and the amount of NADP determined until complete disappearance of NADP was observed. For the determination, 2-(N-morpholino)ethanesulfonic acid buffer (MES) (50 mM, 1 mL) was taken in a cuvette. To this the reaction aliquot and glucose-6-phosphate (0.1 M, 40 μL) were added. The NADP present in the reaction mixture is converted into NADPH by addition of 10 μL of a solution (5 mg/mL of enzyme) of yeast glucose-6-phosphate dehydrogenase at room temperature. The NADPH that was formed showed absorbance at 340 nm and the time when no absorbance was seen in the assay was indicative of reaction completetion. This is a very sensitive assay which is capable of detecting nM concentrations of NADP. At the time at which the starting NADP concentration was determined to be zero, all the NADP was either exchanged into NAADP or was converted into ADPRP as a result of a competing hydrolyzing reaction.
Characterization of Pyridine Dinucleotide analogs 16-24
All compounds were examined by high pressure liquid chromatography (HPLC) using AG MP-1 chromatography49 immediately prior to testing and if necessary re-purified by preparative HPLC using this same system before analysis. HPLC traces are show for all tested compounds in Supporting Information. This chromatographic system was capable of separating the pyridine dinucleotide precursor, the cyclic ADP-ribose derivative and the linear ADP-ribose derivative in each case. Pyridine dinucleotides were also judged pure by determining that the aromatic and anomeric regions of the 1H-NMR spectrum were free from extraneous signals, and that the 31P-NMR exhibited no unexplained signals besides the expected phosphate monoester and the pyrophosphoryl resonance.
4-Amino-NAADP methyl ester (15)
The requisite pyridine base, 4-aminonicotinic acid methyl ester was produced by esterification of the acid 2a. A solution of 4-aminonicotinic acid (2a) (200 mg, 1.45 mmol) in anhydrous methanol (25 mL) and conc. HCl (3 mL) was refluxed for 24 h with constant stirring. The condenser was fitted with a Drierite trap at the top. When the starting material had converted to a non-polar material, as shown by TLC, all the volatiles were removed in vacuum. The residue was again dissolved in distilled water and was neutralized with NaOH (1 M). The aqueous layer was then extracted with EtOAc (3 × 20 mL). All the organic extracts were combined, dried over anhydrous sodium sulfate and evaporated to give white crystalline material (92.5 mg, 42 %): m.p. 172-174 °C; TLC Rf 0.65 in CH3OH: CH2Cl2 (1:9), 1H NMR (600 MHz, CD3OD) δ 8.67 (s, 1H), 8.00 (d, 1H, J= 6.0), 6.68 (d, 1H, J= 6.0), 3.88 (s, 3H); 13C NMR (150 MHz, CD3OD) δ 167.6, 156.4, 151.9, 150.2, 110.9, 107.0, 51.0.
A mixture of 4-aminonicotinic acid methyl ester (30 mg, 0.19 mmol) was made in distilled water (1.5 mL) and tris-(hydroxymethyl)aminomethane (Tris) HCl buffer at pH 7.5 (10 mM, 1.5 mL). The pH of the mixture was re-adjusted to 7.5 with Tris base (0.5 M) and the mixture turned to solution on stirring at 37 °C. To this solution NADP (13 mg, 0.017 mmol) and Aplysia cyclase (60 μL, 0.25 U) enzyme were added. The solution was stirred for 4.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DEAE 52 cellulose. A gradient of 10 mM- 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 290 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (6 mg, 44%): 1H NMR: 8.70(s, 1H), 8.25(s, 1H), 8.08(s, 1H), 8.06 (d, 1H, J= 7.2), 6.79(d, 1H, J= 7.2 ), 6.09 (d, 1H, J= 5.4), 5.57 (d, 1H, J= 6.0), 4.95 (m, 1H), 4.18-4.60 (9H), 3.89 (s, 3H); 31P NMR: 0.56, -11.20.
4-Amino-NAADP (16)
4-Amino-NAADP methyl ester (15) (6 mg, 0.008 mmol) was dissolved in 3 mL of a 0.2 M solution of TEA dissolved in H2O/ CH3OH. The solution was stirred at 35 °C for precisely 12 h. The reaction mixture was diluted with distilled water and CO2 was bubbled through it till the pH went down to 7.0. This was followed by lyophilization to give white solid with some triethyl ammonium bicarbonate buffer. Repeated lyophilizations gave the product as tris(triethylammonium) salt in quantitative yields: 1H NMR: 8.37(s, 1H), 8.34(s, 1H), 8.08(s, 1H), 7.88 (d, 1H, J= 7.2), 6.63(d, 1H, J= 7.2 ), 6.13 (d, 1H, J= 5.4), 5.52 (d, 1H, J= 6.0), 5.05 (m, 1H), 4.6 (m, 1H), 4.15-4.40 (8H); 31P NMR: 2.42, -10.59.
4-Methyl-NAADP (17)
A solution of 4-methylnicotinic acid HCl salt (2b·HCl, 70 mg, 0.51mmol) was made in distilled water (4 mL). The pH of the mixture was adjusted to 4.0 with 0.5 M NaOH and a solution resulted. Then 2.5 mL of this solution was added to another vial, followed by solid NADP (20 mg, 0.027 mmol) and Aplysia cyclase enzyme (60 μL, 0.25 U). The pH of the solution was not readjusted and was buffered by the nicotinate solution. The reaction was stirred for 2.5 h at 37 °C. The reaction was monitored by an enzymatic assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 300 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (8.8 mg, 43%): 1H NMR: 8.73 (s, 1H), 8.63 (d, 1H, J= 6.6), 8.41 (s, 1H), 8.08 (s, 1H), 7.76 (s, 1H, J= 6.0), 6.14 (d, 1H, J= 6.0), 5.84 (d, 1H, J= 5.4), 5.10 (m, 1H), 4.63-4.15 (9H), 2.60 (s, 3H); 31P NMR: 0.66, -10.46; HRMS: 781.0575 (M+Na).
4-n-Butyl-NAADP (18)
A solution of 4-butylnicotinic acid (2c, 30 mg, 0.17mmol) was made in distilled water (2.5 mL). The pH of the solution was adjusted to 4.0 with 0.5 M NaOH. Then this solution was added to another vial followed by addition of NADP (14 mg, 0.02 mmol) and Aplysia cyclase enzyme (60 μL, 0.25 U). The solution was buffered by the nicotinate solution. The reaction was stirred for 1 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 360 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (9 mg, 60%): 1H NMR: 8.69 (s, 1H), 8.66 (d, 1H, J= 6.4), 8.41 (s, 1H), 8.09 (s, 1H), 7.79(d, 1H, J= 6.4), 6.14 (d, 1H, J= 5.6), 5.84 (d, 1H, J= 5.2), 5.10 (m, 1H), 4.62-4.10 (9H), 2.93 (m, 2H), 1.51 (m, 2H), 1.26 (m, 2H), 0.84 (t, 3H, J=7.6); 31P NMR: 0.66, -10.46; HRMS: 801.1229 (M+Na).
4-Phenyl-NAADP (19)
A solution of 4-phenylnicotinic acid (2d, 30 mg, 0.15 mmol) was made in distilled water (2.5 mL). The pH of the solution was adjusted to 4.0 with 0.5 M KOH solution. Then this solution was added to another vial, followed by addition of solid NADP (14 mg, 0.02 mmol) and Aplysia cyclase enzyme (60 μL, 0.25 U). The solution was stirred for 1 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion the reaction mixture was centrifuged and filtered in centrifugal filter tubes at 3200 × g for 15 min. The clear filtrate was diluted, adjusted to pH 7.0 and then applied to DEAE 52 cellulose anion exchange purification. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 400 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (8 mg, 51%): 1H NMR: 8.78 (d, 1H, J=6.4), 8.68 (s, 1H), 8.19 (s, 1H), 7.87 (s, 1H), 7.77(d, 1H, J= 6.4), 7.34-7.26 (m, 5 H) 5.86 (t, 2H, J=4.8), 5.9-5.8 (m, 1H), 4.43-4.00 (9H); 31P NMR: 0.51, -10.62; HRMS: 821.0932 (M+Na).
5-Amino-NAADP (20)
A mixture of 5-aminonicotinic acid (3a, 69 mg, 0.50 mmol) was made in distilled water (6 mL). The mixture was stirred at 37 °C for 1 h but some of the solid did not dissolve. The clear supernatant (3 mL) was taken and the pH was adjusted to 4.0 with 0.5 M KOH. To this solution solid NADP (16 mg, 0.021 mmol) was added followed by Aplysia cyclase enzyme (80 μL, 0.33 U). The reaction was buffered by the nicotinate solution. The reaction was stirred for 3.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 330 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (7 mg, 43 %): 1H NMR: 8.40(s, 1H), 8.27(s, 1H), 8.20(s, 1H), 8.13(s, 1H), 7.77(s, 1H), 6.14 (d, 1H, J=4.8), 5.80 (d, 1H, J= 4.2), 5.0 (m, 1H), 4.58-4.18 (9H); 31P NMR: 0.51, -10.81; HRMS: 760.231 (M+1).
5-Methyl-NAADP (21)
The pH of a mixture of 5-methylnicotinic acid (3b) (70 mg, 0.51 mmol) and water (4 mL) was adjusted to 4.0 with 0.5 M NaOH, and the mixture formed a solution. Two mL of this solution was added to another vial, followed by solid NADP (16 mg, 0.021 mmol) and Aplysia cyclase enzyme (60 μL, 0.25 U). The pH of the solution was not adjusted again and was buffered by the 5-methylnicotinate solution. The solution was stirred for 2.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient formed between 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted into ca. 300 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (9.6 mg, 59 %): 1H NMR: 8.91 (s, 1H), 8.80 (s, 1H), 8.58 (s, 1H), 8.41 (s, 1H), 8.13 (s, 1H), 6.15 (d, 1H, J= 4.8), 5.93 (d, 1H, J= 4.2), 5.05 (m, 1H), 4.18-4.60 (9H), 2.48 (s, 3H); 31P NMR: 0.67, -10.60; HRMS: 759.031 (M+1).
5-Carboxy-NAADP (22)
3,5-Pyridine-dicarboxylic acid (3d, 70 mg, 0.42 mmol) was mixed in distilled water (6 mL). The mixture was stirred at 37 °C for 1 h and the pH was adjusted to 4.0 with 0.5 M KOH and stirring continued at 37 °C for 0.5 h. The pH had decreased to around 3.5 as the solid of the mixture was slowly dissolving. The pH was again adjusted to 4.0 and this was repeated until the mixture had formed a clear solution. To this solution solid NADP (16 mg, 0.02 mmol) was added followed by Aplysia cyclase enzyme (100 μL, 0.42 U). The pH was not readjusted and was buffered by nicotinate solution. The reaction was stirred for 3.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 500 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (5.5 mg, 32.4%): 1H NMR: 9.41(s, 2H), 9.24(s, 1H), 8.54(s, 1H), 8.38(s, 1H), 6.22 (d, 1H, J=5.2), 6.14 (d, 1H, J= 5.6), 5.1 (m, 1H), 4.62-4.18 (9H); 31P NMR: 0.26, -10.1; HRMS 811.0353 (M+Na).
5-Ethyl-NAADP (23)
The pH of a mixture of 5-ethylnicotinic acid (3h, 20 mg, 0.13 mmol) and water (2 mL) was adjusted to 4.0 with 0.5 M NaOH, and the mixture turned into solution. This solution was then added to another vial, followed by solid NADP (8 mg, 0.01 mmol) and Aplysia cyclase enzyme (60 μL, 0.25 U). The pH of the solution was not adjusted again and was buffered by the 5-methylnicotinate solution. The solution was stirred for 2.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at a centrifugal force of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 300 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (4.0 mg, 59 %): 1H NMR: 8.92 (s, 1H), 8.75 (s, 1H), 8.63 (s, 1H), 8.40 (s, 1H), 8.07 (s, 1H), 6.1 (d, 1H, J= 4.8), 5.93 (d, 1H, J= 4.2), 5.05 (m, 1H), 4.18-4.60 (9H), 2.78 (q, 2H, 2J= 15.0, 3J= 7.2), 1.20 (t, 3H, J=7.2); 31P NMR 0.86, -10.80; LC-MS (ESI) m/z 773.1 (M+1).
5-Azido-NAADP (24)
5-Azidonicotinic acid HCl (3i·HCl, 35 mg, 0.42 mmol) was mixed in distilled water (3 mL). The mixture was stirred at 37 °C for 1 h and the pH was adjusted to 4.0 with 0.5 M KOH and again stirred at 37 °C for 0.5 h. The pH had decreased to around 3.6 as the solid phase slowly dissolved. The pH was again adjusted to 4.0 and this was repeated until the mixture had formed a clear solution. To this solution NADP (10 mg, 0.013 mmol) and Aplysia cyclase enzyme (80 μL, 0.33 U) were added. The reaction was buffered by nicotinate solution and was stirred for 3.5 h at 37 °C. The reaction was monitored by an assay to determine the amount of NADP left in the reaction. Upon completion, the catalyst was removed from the reaction solution by centrifuging in Amicon Ultra centrifugal filter device (10,000 molecular weight cut) at the speed of 3200 × g for 15 min. The filtrate contained the product and the retentate was discarded. The clear filtrate was diluted, adjusted to pH 7.0 and purified by anion exchange chromatography on DE-52 cellulose. A gradient of 10 mM - 600 mM NH4HCO3 (700 mL) was applied and the product eluted at around 320 mM NH4HCO3. The appropriate fractions were lyophilized to obtain product as white solid (6.5 mg, 61%): 1H NMR: 8.82(s, 2H), 8.60(s, 1H), 8.48(s, 1H), 8.41(s, 1H), 8.12(s, 1H), 6.12 (d, 1H, J=6.0), 5.92 (d, 1H, J= 5.4), 5.05 (m, 1H), 4.60-4.16 (9H); 31P NMR: 0.41, -10.92; LC-MS (ESI) m/z 786.1 (M+1).
Biological Testing
Testing of NAADP derivatives for Ca2+ release on cell free receptor systems were performed on homogenates (1.25% v/v) prepared from sea urchin eggs (Strongylocentrotus purpuratus) diluted with intracellular medium containing 250 mM potassium gluconate buffer (pH 7.2), 0.5 mM ATP, 4 mM creatinine phosphate, creatinine kinase and 3 μM fluorescent indicator, Fluo-3. The dilutions and all experiments were conducted at 17 °C. Fluo-3 is a calcium chelating indicator which is nonfluorescent in absence of Ca2+ ions but after binding Ca2+ it emits fluorescence. This fluorescence can be measured using a fluorescence plate reader (excitation 490 nm and emission 535 nm, suitable to avoid interference from reduced pyridine nucleotide).3,50,51
When the homogenates were treated with NAADP agonists, the Ca2+ released from the sensitive stores was chelated by the indicator leading to increase in fluorescence. This increase in fluorescence intensity was measured and was proportional to the Ca2+ concentration.
Receptor binding studies of the 4 or 5 substituted NAADP analogs
The competitive binding studies were performed according to our previously published procedures3. The binding assays were done in triplicate in 96 well filter plates containing the cell free sea urchin egg homogenate and constant concentration of radioligand [32P]NAADP (0.2 nM). Seven concentrations of each compound were tested to determine their IC50. The competitor and [32P]NAADP were incubated simultaneously with the sea urchin egg homogenate for 90 min at 4 °C. The homogenate was filtered and washed and the radioactivity retained on the filter was determined by liquid scintillation.
Supplementary Material
Acknowledgments
We thank Mr. Christopher Trabbic for preparing (4,5-dihydro-4,4-dimethyl-2-oxazolyl)pyridine for this study. LeRoy A. Perez-Haddock was supported by NIH grant R25 HL088728. Supported in part by NIH grant NIH DA11806 to Timothy F. Walseth.
Footnotes
Abbreviations: cADPR, cyclic adenosine diphosphate ribose; DIPEA, diisopropylethylamine; DMF, N,N-dimethylformamide; ESI, electrospray ionization mass spectrometry; EtOAc, ethyl acetate; EtOH, ethyl alcohol; Fluo-3, refers to the fluorescent indicator Ca2+ dye developed by Dr. Roget Y. Tsien; HPLC, high pressure liquid chromatography; HRMS, high mass resolution mass spectrometry; InsP3, inositol-1,4,5-triphosphate; LC-mass spec, liquid chromatography mass spectrometry; NAADP, nicotinic acid adenine dinucleotide phosphate; NADase, nicotinamide adenine dinucleotide glycohydrolase; PPADS, pyridoxalphosphate-6-azophenyl-2,4-disulfonate; SAR, structure-activity relation; TCA, trichloroacetic acid; TEA, triethylamine; TES, triethylsilyl group; THF, tetrahydrofuran; TLC, thin layer chromatography; Tris, tris-(hydroxymethyl)aminomethane; UV, ultraviolet light;
Supporting Information Available: Detailed information on synthetic methods, analytical, spectroscopic, and Ca2+ releasing activity. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J Biol Chem. 1995;270:2152–2157. doi: 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- 2.Lee HC, Aarhus R. Functional visualization of the separate but interacting calcium stores sensitive to NAADP and cyclic ADP-ribose. J Cell Sci. 2000;113(Pt 24):4413–4420. doi: 10.1242/jcs.113.24.4413. [DOI] [PubMed] [Google Scholar]
- 3.Aarhus R, Dickey DM, Graeff RM, Gee KR, Walseth TF, Lee HC. Activation and inactivation of Ca2+ release by NAADP+ J Biol Chem. 1996;271:8513–8516. doi: 10.1074/jbc.271.15.8513. [DOI] [PubMed] [Google Scholar]
- 4.Lee HC. Nicotinic acid adenine dinucleotide phosphate (NAADP)-mediated calcium signaling. J Biol Chem. 2005;280:33693–33696. doi: 10.1074/jbc.R500012200. [DOI] [PubMed] [Google Scholar]
- 5.Mandi M, Bak J. Nicotinic acid adenine dinucleotide phosphate (NAADP) and Ca2+ mobilization. J Recept Signal Transduct Res. 2008;28:163–184. doi: 10.1080/10799890802084085. [DOI] [PubMed] [Google Scholar]
- 6.Galione A, Ruas M. NAADP receptors. Cell Calcium. 2005;38:273–280. doi: 10.1016/j.ceca.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 7.Cancela JM, Churchill GC, Galione A. Coordination of agonist-induced Ca2+-signalling patterns by NAADP in pancreatic acinar cells. Nature. 1999;398:74–76. doi: 10.1038/18032. [DOI] [PubMed] [Google Scholar]
- 8.Berg I, Potter BV, Mayr GW, Guse AH. Nicotinic acid adenine dinucleotide phosphate (NAADP(+)) is an essential regulator of T-lymphocyte Ca(2+)-signaling. J Cell Biol. 2000;150:581–588. doi: 10.1083/jcb.150.3.581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bak J, White P, Timar G, Missiaen L, Genazzani AA, Galione A. Nicotinic acid adenine dinucleotide phosphate triggers Ca2+ release from brain microsomes. Curr Biol. 1999;9:751–754. doi: 10.1016/s0960-9822(99)80335-2. [DOI] [PubMed] [Google Scholar]
- 10.Bak J, Billington RA, Timar G, Dutton AC, Genazzani AA. NAADP receptors are present and functional in the heart. Curr Biol. 2001;11:987–990. doi: 10.1016/s0960-9822(01)00269-x. [DOI] [PubMed] [Google Scholar]
- 11.Cheng J, Yusufi AN, Thompson MA, Chini EN, Grande JP. Nicotinic acid adenine dinucleotide phosphate: a new Ca2+ releasing agent in kidney. J Am Soc Nephrol. 2001;12:54–60. doi: 10.1681/ASN.V12154. [DOI] [PubMed] [Google Scholar]
- 12.Mandi M, Toth B, Timar G, Bak J. Ca2+ release triggered by NAADP in hepatocyte microsomes. Biochem J. 2006;395:233–238. doi: 10.1042/BJ20051002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gasser A, Bruhn S, Guse AH. Second messenger function of nicotinic acid adenine dinucleotide phosphate revealed by an improved enzymatic cycling assay. J Biol Chem. 2006;281:16906–16913. doi: 10.1074/jbc.M601347200. [DOI] [PubMed] [Google Scholar]
- 14.Lee HC, Aarhus R. Structural determinants of nicotinic acid adenine dinucleotide phosphate important for its calcium-mobilizing activity. J Biol Chem. 1997;272:20378–20383. doi: 10.1074/jbc.272.33.20378. [DOI] [PubMed] [Google Scholar]
- 15.Billington RA, Tron GC, Reichenbach S, Sorba G, Genazzani AA. Role of the nicotinic acid group in NAADP receptor selectivity. Cell Calcium. 2005;37:81–86. doi: 10.1016/j.ceca.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 16.Dowden J, Berridge G, Moreau C, Yamasaki M, Churchill GC, Potter BV, Galione A. Cell-permeant small-molecule modulators of NAADP-mediated Ca2+ release. Chem Biol. 2006;13:659–665. doi: 10.1016/j.chembiol.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Dammermann W, Zhang B, Nebel M, Cordiglieri C, Odoardi F, Kirchberger T, Kawakami N, Dowden J, Schmid F, Dornmair K, Hohenegger M, Flugel A, Guse AH, Potter BV. NAADP-mediated Ca2+ signaling via type 1 ryanodine receptor in T cells revealed by a synthetic NAADP antagonist. Proc Natl Acad Sci U S A. 2009;106:10678–10683. doi: 10.1073/pnas.0809997106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Billington RA, Genazzani AA. PPADS is a reversible competitive antagonist of the NAADP receptor. Cell Calcium. 2007;41:505–511. doi: 10.1016/j.ceca.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 19.Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009;5:220–226. doi: 10.1038/nchembio.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen D, Lewis AM, Mizote A, Thomas JM, Aley PK, Vasudevan SR, Parkesh R, Galione A, Izumi M, Ganesan A, Churchill GC. Analogues of the nicotinic acid adenine dinucleotide phosphate (NAADP) antagonist Ned-19 indicate two binding sites on the NAADP receptor. J Biol Chem. 2009;284:34930–34934. doi: 10.1074/jbc.M109.016519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bernofsky C. Nicotinic Acid Adenine Dinucleotide Phosphate. Methods Enzymol. 1980;66:105–112. doi: 10.1016/0076-6879(80)66446-5. [DOI] [PubMed] [Google Scholar]
- 22.Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP(+) J Biol Chem. 1995;270:30327–30333. doi: 10.1074/jbc.270.51.30327. [DOI] [PubMed] [Google Scholar]
- 23.Hauck AE, Giam CS. Regioselective nucleophilic addition of organolithium compounds to 3-(4,4-dimethyloxazolin-2-yl)pyridine. J Chem Soc Perkin Trans 1. 1980;10:2070–2076. [Google Scholar]
- 24.Hauck AE, Giam CS. The addition-elimination mechanism in the nucleophilic heteroaromatic substitution of 3-(4’,4’-dimethyl4’,5’-dihydrooxazol-2’-yl)pyridine. solvent effect of the regiochemistry of the addition reaction. J Chem Soc Perkin Trans 1. 1984;10:2227–2231. [Google Scholar]
- 25.Holmes BT, Pennington WT, Hanks TW. Efficient synthesis of a complete donor/acceptor bis(aryl)diyne family. Synth Comm. 2003;33:2447–2461. [Google Scholar]
- 26.Denton TT, Zhang X, Cashman JR. 5-substituted, 6-substituted, and unsubstituted 3-heteroaromatic pyridine analogues of nicotine as selective inhibitors of cytochrome P-450 2A6. J Med Chem. 2005;48:224–239. doi: 10.1021/jm049696n. [DOI] [PubMed] [Google Scholar]
- 27.Nguefack J-F, Bolitt V, Sinou D. An efficient pallidium-catalyzed coupling of terminal alkynes with aryl halides under Jeffery’s conditions. Tetrahedron Lett. 1996;37:5527–5530. [Google Scholar]
- 28.Weil MK, Odermatt S, Schanen P, Seiler P, Diederich F. 1,3-Diethynylallenes:stable monomers, length -defined oligomers, asummetric synthesis, and optical resolution. Eur J Org Chem. 2007;21:3449–3462. [Google Scholar]
- 29.Serota S, Simon JR, Murray EB, Linfield WM. Novel synthesis of α-substituted acrylic acids. J Org Chem. 1981;46:4147–4151. [Google Scholar]
- 30.Lindlar H, Dubuis R. Palladium catalyst for partial reduction of acetylenes. Organic Synthesis. 1973;5:880. [Google Scholar]
- 31.Li W, Nelson DP, Jensen MS, Hoerrner RS, Cai D, Larsen RD, Reider PJ. An improved protocol for the preparation of 3-pyridyl- and some arylboronic acids. J Org Chem. 2002;67:5394–5397. doi: 10.1021/jo025792p. [DOI] [PubMed] [Google Scholar]
- 32.Liu W, Gahr AA, Bao R, Li Y. Recent progress in the synthesis and evaluation of azido and diazo derivatives of NAD+ for photoaffinity labeling experiments. Trends in Organic Chemistry. 1997;6:219–231. [Google Scholar]
- 33.Wallace JM, Soderberg BCG, Hubbard JW. Synthesis of 2-alkylsubstituted benzimidazoles by thermal decomposition of 2-azidobnezenamides in the presence of an aldelyde. Synth Comm. 2006;36 [Google Scholar]
- 34.Sawanishi H, Tajima K, Tsuchiya T. Studies on diazepines. XXVIII. Synthesis of 5H-1,3-diazepines and 2H-1,4-diazepines from 3-azidopyridines. Chem Pharm Bull. 1987;35:4101–4109. [Google Scholar]
- 35.Gahr AA, Li Y. The synthesis, identification, and kinetic characterization of the photoaffinity label: 3-azidopyridine adenosine dinucleotide: an advanced undergraduate laboratory experiment appropriate for organic, biochemistry, or bio-organic chemistry. Chemical Educator [Electronic Publication] 1997;2 [Google Scholar]
- 36.Hotha S, Kashyap S. “Click chemistry” inspired synthesis of pseudo-oligosaccharides and amino acid glycoconjugates. J Org Chem. 2006;71:364–367. doi: 10.1021/jo051731q. [DOI] [PubMed] [Google Scholar]
- 37.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 38.Tonooka S, Sasaki A, Shirahama H, Matsumoto T, Kakimoto S. Enzymatic synthesis of clitidine. Chem Letters. 1977;12:1449–1452. [Google Scholar]
- 39.Genazzani AA, Empson RM, Galione A. Unique inactivation properties of NAADP-sensitive Ca2+ release. J Biol Chem. 1996;271:11599–11602. doi: 10.1074/jbc.271.20.11599. [DOI] [PubMed] [Google Scholar]
- 40.Dowden J, Moreau C, Brown RS, Berridge G, Galione A, Potter BV. Chemical synthesis of the second messenger nicotinic acid adenine dinucleotide phosphate by total synthesis of nicotinamide adenine dinucleotide phosphate. Angew Chem Int Ed Engl. 2004;43:4637–4640. doi: 10.1002/anie.200460054. [DOI] [PubMed] [Google Scholar]
- 41.Yost DA, Anderson BM. Adenosine Diphosphoribose Transfer Reactions Catalyzed by Bungarus Fasciatus Venom NAD Glycohydrolase. J Biol Chem. 1983;258:3075–3080. [PubMed] [Google Scholar]
- 42.Walter P, Kaplan NO. Substituted Nicotinamide Analogues of Nicotinamide Adenine Dinucleotide. J Biol Chem. 1963;238:2823–2830. [PubMed] [Google Scholar]
- 43.Robert N, Bonneau AL, Hoarau C, Marsais F. Unusual sterically controlled regioselective lithiation of 3-bromo-5-(4,4’-dimethyl)oxazolinylpyridine. Straightforward access to highly substituted nicotinic acid derivatives. Org Lett. 2006;8:6071–6074. doi: 10.1021/ol062556i. [DOI] [PubMed] [Google Scholar]
- 44.Farley CP, Eliel EL. Chichibabin Reactions with Phenylacetaldehyde. J Am Chem Soc. 1956;78:3477–3484. [Google Scholar]
- 45.Zatman LJ, Kaplan NO, Colowick SP. Inhibition of spleen diphosphopyridine nucleotidase by nicotinamide, an exchange reaction. J Biol Chem. 1953;200:197–212. [PubMed] [Google Scholar]
- 46.Lee HC, Graeff RM, Munshi CB, Walseth TF, Aarhus R. Large-scale purification of Aplysia ADP-ribosylcyclase and measurement of its activity by fluorimetric assay. Methods Enzymol. 1997;280:331–340. doi: 10.1016/s0076-6879(97)80124-3. [DOI] [PubMed] [Google Scholar]
- 47.Kaplan NO, Colowick SP. Animal tissue DPNase (pyridine transglycosidase) Methods in Enzymology. 1955;2:660–663. [Google Scholar]
- 48.Blomquist CH. Partial purification and characterization of nicotinamide adenine dinucleotide kinase from sea urchin eggs. J Biol Chem. 1973;248:7044–7048. [PubMed] [Google Scholar]
- 49.Axelson JT, Bodley JW, Walseth TF. A volatile liquid chromatography system for nucleotides. Anal Biochem. 1981;116:357–360. doi: 10.1016/0003-2697(81)90371-7. [DOI] [PubMed] [Google Scholar]
- 50.Wong L, Aarhus R, Lee HC, Walseth TF. Cyclic 3-deaza-adenosine diphosphate ribose: a potent and stable analog of cyclic ADP-ribose. Biochim Biophys Acta. 1999;1472:555–564. doi: 10.1016/s0304-4165(99)00161-0. [DOI] [PubMed] [Google Scholar]
- 51.Xu L, Walseth TF, Slama JT. Cyclic ADP-ribose analogues containing the methylenebisphosphonate linkage: effect of pyrophosphate modifications on Ca2+ release activity. J Med Chem. 2005;48:4177–4181. doi: 10.1021/jm049469l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.