

Abstract
Cholecystokinin (CCK) is an abundant neuropeptide involved in normal behaviour and pathophysiological conditions. Recently, CCK was shown to act as a molecular switch for perisomatic inhibition in the hippocampus, by directly depolarizing parvalbumin expressing (PV+) basket cells while indirectly depressing GABA release from CCK expressing (CCK+) basket cells. However, whether these two CCK-mediated effects are causally related is controversial, with one hypothesis proposing that the CCK-induced firing of PV+ basket cells increases the release of GABA, which, in turn, heterosynaptically inhibits GABA release from neighbouring CCK+ basket cell terminals through presynaptic GABAB receptors. Our present data from paired recording experiments from presynaptic basket cells and postsynaptic CA1 pyramidal cells in acute rat brain slices show that the P/Q Ca2+ channel antagonist agatoxin TK (250 nm) abolished GABA release from PV+ basket cells, but it had no effect on the CCK-induced depression of GABA release from CCK+ basket cells. Furthermore, CCK decreased GABA release from CCK+ basket cells even in the presence of the GABAB receptor antagonist CGP55845 (2 μm). In contrast, cannabinoid type-1 (CB1) receptor blockade with AM251 (10 μm) prevented the action of CCK on GABA release both from CCK+ basket cells and dendritically projecting, CCK+ Schaffer collateral-associated interneurons. These results demonstrate that CCK-mediated inhibition of GABA release from CCK+ cells requires no cross-talk between PV+ and CCK+ synapses, but that it critically depends on CB1 receptor-mediated endocannabinoid signalling at both perisomatic and dendritic inputs.
Non technical summary
Cholecystokinin (CCK) is a widely expressed neuropeptide known to play key roles in both normal behaviour and pathophysiological conditions. Previous reports suggested that CCK serves as a unique molecular switch that differentially regulates the output of distinct types of inhibitory interneurons in the hippocampus. The current study focused on the underlying mechanisms of the CCK actions on hippocampal interneurons, and the results show that CCK modulates transmitter release from a major class of interneurons through cannabinoid type-1 receptor signalling mechanisms. Taken together, the present data will help us better understand how CCK modulates neuronal circuits in the brain.
Introduction
Cholecystokinin (CCK) is expressed at unusually high levels (in microgram quantities) in neuronal circuits in the brain, acting as a functionally important, versatile, cell type-specific neuromodulator acting through G-protein coupled CCKB receptors (for reviews, see Noble & Roques, 2006; Lee & Soltesz, 2011). The effects of CCK on principal cells include the enhancement of glutamate release through presynaptic mechanisms, often without direct excitatory actions on somato-dendritic domains (Cox et al. 1995; Miller et al. 1997; Földy et al. 2007; Chung & Moore, 2009; Deng et al. 2010; but see Boden & Hill, 1988). In addition, CCK shows diverse, highly cell type-specific actions on GABAergic neurons. Of particular interest in this regard are the effects of CCK on basket cells (BCs) that are strategically placed to regulate the excitability of the perisomatic region of postsynaptic neurons and perform a variety of key operations in the network (Freund & Katona, 2007; Bartos et al. 2007; Ellender & Paulsen, 2010).
In the hippocampus and neocortex, there are two major, non-overlapping, functionally distinct types of BCs, the parvalbumin expressing (PV+) and the CCK expressing (CCK+) basket cells (BCs). PV+ BCs appear to be well-suited to entrain network oscillations, because they fire non-accommodating, fast action potentials, have fast membrane time constants, and respond reliably to excitatory inputs, and their GABA release is tightly coupled to intracellular Ca2+ rises through P/Q Ca2+ channels (Wilson et al. 2001; Hefft & Jonas, 2005; Glickfeld & Scanziani, 2006). In contrast, CCK+ BCs are thought to primarily serve as modulators that adapt circuit activity to behavioural states, as they fire accommodating spikes, express receptors for various neuromodulators, possess slower membrane time constants, and receive weak excitatory inputs that integrate over long time windows, and GABA release from their terminals is regulated by N-type Ca2+ channels with a looser coupling between the Ca2+ source and sensor (Wilson et al. 2001; Hefft & Jonas, 2005; Klausberger et al. 2005; Glickfeld & Scanziani, 2006). Although these distinctions imply that these two perisomatic GABAergic synaptic control systems function largely independently and in parallel, there is increasing evidence for coordination between PV+ BCs and CCK+ BCs. In addition to the existence of synaptic contacts between PV+ BCs and CCK+ BCs (Acsady et al. 2000; Karson et al. 2009), CCK itself has been shown by recent studies to act as an agent of coordination between PV+ BCs and CCK+ BCs by gating their outputs in a diametrically opposite fashion (Földy et al. 2007). Namely, CCK has been demonstrated to act as a switch that determines the source of perisomatic inhibition for hippocampal principal cells by powerfully increasing the output of PV+ BCs through a strong depolarizing effect on these neurons, while inhibiting GABA release from CCK+ BCs. Because PV+ BCs appear to be especially suited to serve functions related to hippocampal network oscillations and memory (Glickfeld & Scanziani, 2006; Freund & Katona, 2007), the CCK-mediated gating of perisomatic inhibition may favour tasks requiring precise timing and integration over narrow time windows.
However, exactly how CCK attenuates GABA release from CCK+ BCs is not fully understood (Fig. 1). According to one view (referred to as the ‘GABAB receptor hypothesis’ in this paper), the CCK-induced robust increase in PV+ BC discharges is a key step in leading to the CCK-mediated inhibition of GABA-release from CCK+ BCs (Karson et al. 2008). Specifically, GABA released from PV+ BCs, resulting from the powerful depolarization and action potential discharge of PV+ BCs by CCK, is proposed to reach and activate GABAB receptors located on the axon terminals of CCK+ BCs, leading to heterosynaptic inhibition of GABA release. In contrast, there is also evidence for an alternative mechanism (referred to as the ‘CB1 receptor hypothesis’) where the CCK-induced depolarization and action potential discharge in PV+ BCs is independent of the CCK-mediated inhibition of GABA release from CCK+ BCs (Földy et al. 2007). In the latter scenario, CCK would activate Gq/11-coupled CCKB receptors on postsynaptic pyramidal cells, leading to the synthesis and release of endocannabinoids that retrogradely cross the synaptic cleft and bind to presynaptic CB1 receptors, resulting in inhibition of GABA release from CCK+ BCs (Fig. 1).
Figure 1. Schematic drawing illustrating two alternative pathways to explain the mechanisms underlying the CCK-induced suppression of GABA release from CCK+ basket cells.
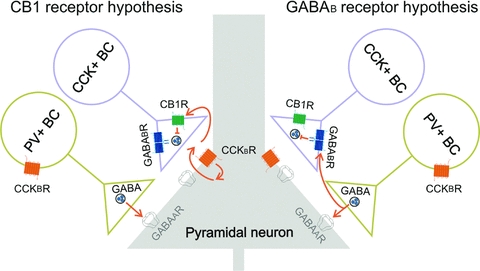
Left, according to the ‘CB1 receptor hypothesis’, CCK binds to CCKB receptors on pyramidal cells (Földy et al. 2007), leading to endocannabinoid synthesis. The postsynaptically released endocannabinoid would then bind to presynaptic CB1 receptors located on the axon terminals of CCK+ BCs, resulting in suppression of GABA release. In a separate series of events, CCK is proposed to bind to CCK receptors on PV+ BCs, causing strong depolarization and firing of PV+ BC, leading to sharp increases in the spontaneous IPSC frequency recorded from the CA1 pyramidal cells (e.g. Fig. 1 in Földy et al. 2007). Right, according to the ‘GABAB receptor hypothesis’, there is only a single direct action of CCK, namely, the activation of excitatory CCK receptors on PV+ BCs, resulting in action potential discharges in PV+ BCs. The spiking in PV+ BCs, in turn, leads to the release of GABA that reaches and activates presynaptic GABAB receptors on CCK+ BCs, resulting in a suppression of GABA release from the CCK+ BCs.
Because of the potential importance of the CCK-mediated regulatory mechanisms gating distinct perisomatic inhibitory streams in both normal functions and diseased states (Lee & Soltesz, 2010), we carried out paired recording experiments to test the mechanisms underlying the CCK-induced attenuation of GABA release from CCK+ BCs and other CCK+ non-BC interneurons within the CA1 region. Our results show that CCK is able to decrease GABA release from CCK+ BCs even in the absence of GABA release from PV+ BCs and in the presence of GABAB, but not CB1, receptor blockade, demonstrating that the action of CCK on PV+ BCs is mechanistically separate from the CCK-effects on CCK+ BC output.
Methods
All protocols were approved by the Institutional Animal Care and Use Committee of the University of California.
Electrophysiology
Sprague–Dawley rats (2–3 weeks old; in one set of experiments, 7 weeks old, see text) were decapitated under isoflurane anaesthesia. The brains were transferred rapidly to an ice-cold, sucrose-containing artificial cerebrospinal fluid (ACSF) containing (mm) 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2 and 24 NaHCO3, and transverse hippocampal slices (350 μm) were cut. Slices were incubated in the sucrose-containing ACSF for an hour and then transferred for electrophysiological recordings to a chamber continuously perfused with standard ACSF containing (mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4 and 10 glucose. All electrophysiological recordings were made at 33°C. Slices were visualized with an upright microscope (Olympus, BX61WI) with infrared differential interference contrast optics. Whole-cell recordings were obtained from the interneurons with patch-pipettes (3–5 MΩ) filled with internal solution containing (mm) 126 potassium gluconate, 4 KCl, 10 Hepes, 4 ATP-Mg, 0.3 GTP-Na, 10 phosphocreatine and 0.2% biocytin (pH 7.2, 280–290 mosmol l−1). All interneurons were located in the stratum radiatum or the pyramidal cell layer of CA1 (for examples of soma distributions for presynaptic CCK+ BCs and Schaffer collateral-associated cells (SCAs) from previous paired recording experiments, see Lee et al. 2010). Pyramidal cells (whole-cell voltage clamp configuration, the holding potential was −70 mV) were recorded with internal solution containing (mm) 40 CsCl, 90 potassium gluconate, 1.8 NaCl, 1.7 MgCl2, 3.5 KCl, 0.05 EGTA, 10 Hepes, 2 ATP-Mg, 0.4 GTP-Na and 10 phosphocreatine (pH 7.2, 280–290 mosmol l−1). All drugs were obtained from Tocris. Recordings of unitary IPSCs between the presynaptic interneurons and the postsynaptic pyramidal cells were made using MultiClamp 700B amplifiers (Molecular Devices, Sunnyvale, CA, USA). Signals were filtered at 3 kHz using a Bessel filter and digitized at 10 kHz with a Digidata 1440A analog–digital interface (Molecular Devices). Series resistances were carefully monitored, and the recordings were discarded if the series resistance changed >20% or reached 25 MΩ. The recorded traces were analysed using the Clampfit 10.2 software (Molecular Devices). During paired recordings, the frequency of presynaptic stimulation was 10 Hz. Values for the so-called ‘effective’ unitary IPSC (euIPSC; Neu et al. 2007) amplitudes include both successful events and failures (i.e. euIPSC amplitude is the average of all postsynaptic responses to presynaptic spikes). Spontaneous IPSCs (sIPSCs) were recorded from CA1 pyramidal cells in the presence of the ionotropic glutamate receptor antagonists 10 μm APV and 5 μm NBQX. Because CCK actions on GABAergic transmission are powerful but transient (Deng & Lei, 2006; Földy et al. 2007; note that the transiency is most likely due to receptor desensitization that was not studied here), quantification of CCK effects was carried out at three distinct time-points: (1) immediately before CCK application (referred to as ‘Control’ in the text and figures); (2) at the time of the maximal effect during CCK application (‘Peak’; typically observed about 3 min after the starting of the CCK application, with the peak effect lasting for about 1 min, see Fig. 1 in Földy et al. 2007); and (3) more than 5 min after the start of the CCK application, when the response returns to baseline even in the continued presence of CCK (‘Postpeak’; e.g. Fig. 2b in Földy et al. 2007 and Fig. 2B of the current paper). Data are presented as means ±s.e.m. Student's t test for paired or unpaired data was used for statistical analysis and significant differences were at the level of P < 0.05.
Figure 2. GABA release from PV+ basket cells is not required for the CCK-induced suppression of GABA release from CCK+ basket cells.
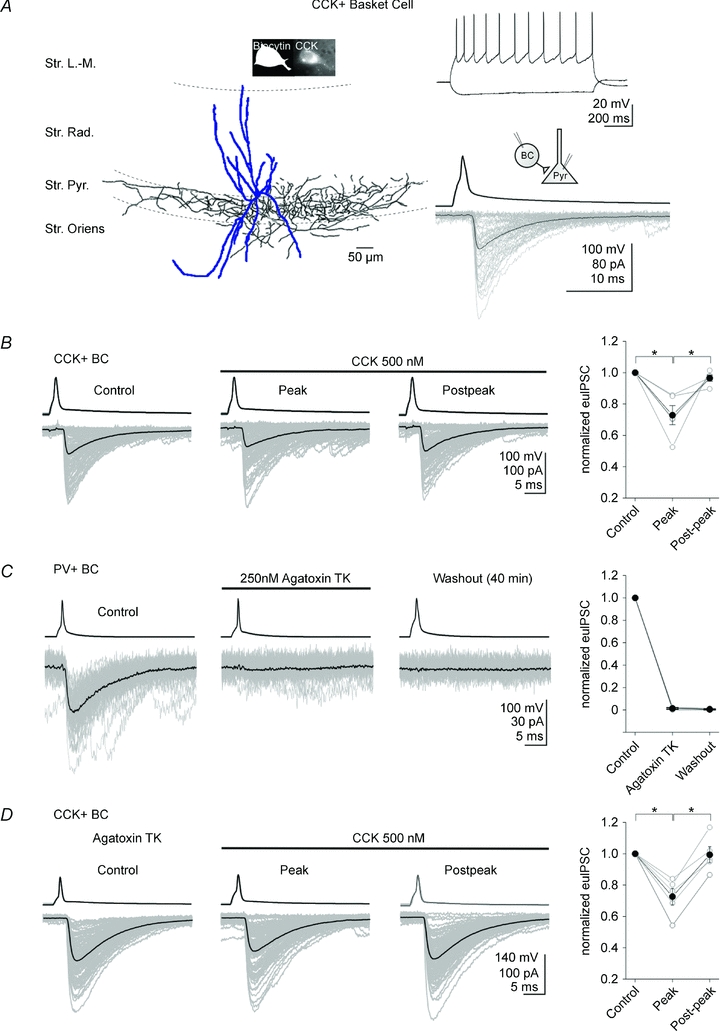
A, left, camera lucida drawing of a representative CCK+ basket cell recorded in this study; note that the axons were located within or adjacent to the CA1 pyramidal cell layer. The insets show photomicrographs indicating the immunopositivity of the cell for CCK. Right, upper panel, example traces of current-clamp recordings of the CCK+ BC in response to current steps (−100 and 160 pA, from −60 mV); note the spike frequency adaptation. Right, lower panel, averaged traces of presynaptic action potentials (top) elicited in the CCK+ BC, and the respective postsynaptic responses (bottom; voltage clamp configuration) in the pyramidal cell. Note the fast and large unitary IPSCs; 100 postsynaptic responses are shown in grey, together with their averages (black). Abbreviations: Str. L.-M.: stratum lacunosum-moleculare; Str. Rad.: stratum radiatum; Str. Pyr.: stratum pyramidale; Str. Oriens: stratum oriens. B, CCK (500 nm) significantly inhibited the amplitudes of the effective unitary IPSCs (euIPSC, including both successful events and failures; see Methods) in the pyramidal cell (lower traces) evoked by the presynaptic CCK+ BC (upper traces). The individual IPSCs are grey and the averages are black. Summary data are shown on the right (open circles: individual pairs; filled circles: means). C, agatoxin TK (250 nm) produced a complete suppression of the unitary IPSCs in pyramidal cells evoked by presynaptic PV+ BCs. Note the irreversible suppression. D, in slices preincubated (>1 h) in agatoxin TK (250 nm), 500 nm CCK still attenuated the amplitude of unitary IPSCs in pyramidal cells evoked by presynaptic CCK+ BCs. Note that the degree of CCK-induced suppression of unitary IPSCs is similar between control (B) and agatoxin TK preincubated slices (D). Error bars represent s.e.m., and the asterisks denote P < 0.05.
Immunochemistry and cell identification
After recording, slices were transferred into a fixative solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 m phosphate buffer. Slices were resectioned into 50 μm thin sections and immunoreactivity for CCK was revealed with a mouse monoclonal antibody (mAb 9303, generously provided by the CURE Digestive Diseases Research Center, Antibody RIA Core, Los Angeles (NIH grant no. DK41301); diluted 1:1000); immunoreactivity for PV was tested with a rabbit polyclonal antibody (PV-28; Swant, Bellinzona, Switzerland; diluted 1:1,000 in Tris-buffered saline containing 2% normal goat serum). The reactions were visualized with a goat anti-rabbit IgG conjugated to Alexa 488 (diluted 1:500 in Tris-buffered saline containing 2% normal goat serum; Molecular Probes/Invitrogen) and a goat anti-mouse IgG conjugated to Alexa 594 (diluted 1:500), streptavidin conjugated to Alexa-350 for biocytin (diluted 1:500). The sections were then mounted in Vectashield (Vector Laboratories, Inc., Burlingame, CA, USA) and analysed with a fluorescence microscope. In order to reveal the axonal and dendritic arbours of the interneurons in detail, the biocytin-filled cells were subsequently visualized with 3,3′-diaminobenzidine tetrahydrochloride (0.015%) using a standard ABC kit (Vector Laboratories). The identification of CCK+ BCs, CCK+ SCA interneurons, and PV+ BCs was based on the distinct axonal morphology (CCK+ BCs and PV+ BCs: a large proportion of the axons were located within or adjacent to the pyramidal cell layer; SCAs: axons ramify predominantly in the stratum radiatum, and to a lesser extent in the oriens) and the immunopositivity to CCK and immunonegativity to PV for CCK+ BCs and SCAs, and the immunopositivity to PV and immunonegativity to CCK for PV+ BCs (Vida et al. 1998; Cope et al. 2002; Pawelzik et al. 2002; Hefft & Jonas, 2005; Glickfeld & Scanziani, 2006; Ali, 2007; Földy et al. 2007; Lee et al. 2010; see Figs 2A and 4A in the current study). In paired recordings from CCK+ BCs or PV+ BCs and postsynaptic CA1 pyramidal cells, presynaptic interneuronal firing evoked unitary IPSCs that had relatively fast rise times and large amplitudes (e.g. Neu et al. 2007; Földy et al. 2007), while presynaptic SCA firing evoked IPSCs in pyramidal cells that had slower rise times and smaller amplitudes (for a quantitative comparison of unitary IPSCs from CCK+ BCs and SCAs, see Fig. 2 in Lee et al. 2010; see also Daw et al. 2009), in agreement with the location of the GABAergic input synapses along the somato-dendritic axis of the principal cells.
Figure 4. CCK attenuates GABA release from CCK+ Shaffer collateral associated cells in a CB1 receptor-dependent manner.
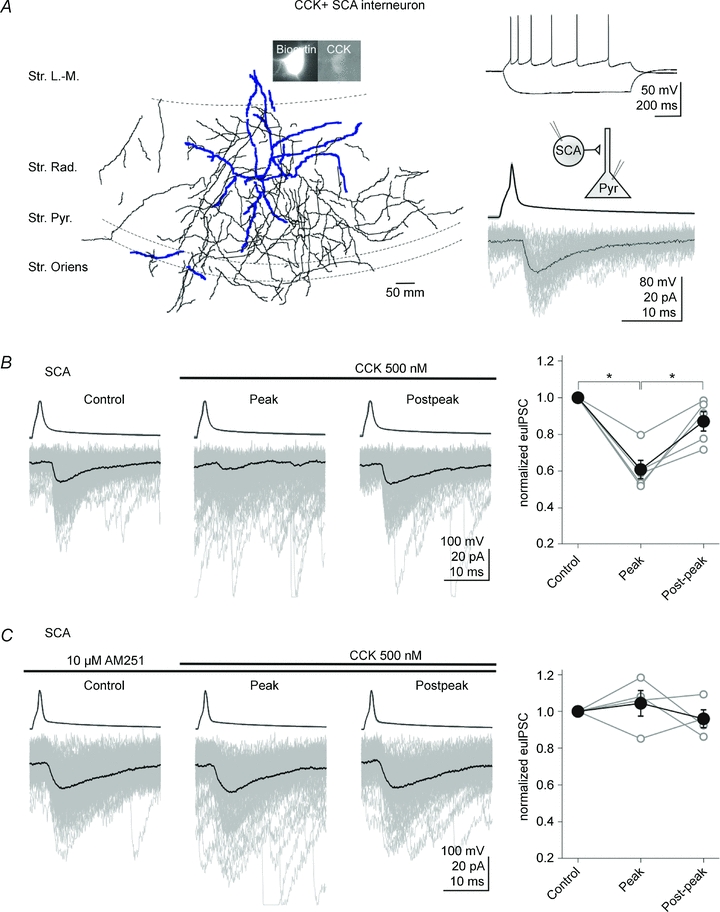
A, left, camera lucida drawing of a CCK+ Schaffer collateral-associated (SCA) cell; note the axons ramifying predominantly in the stratum radiatum. The insets illustrate photomicrographs showing the immunopositivity of the cell for CCK. Right, upper panel, example traces of current-clamp recordings of the SCA in response to current steps (−100 and 80 pA, from −56 mV); note the spike frequency adaptation. Right, lower panel, averaged traces of presynaptic action potentials (top) elicited in the SCA, and the respective postsynaptic responses (bottom) in a pyramidal cell. Note the slower and smaller unitary IPSCs evoked by the dendritically projecting SCAs (compare with Fig. 2A for the BC responses). One hundred postsynaptic responses are shown in grey, together with their averages (black). Abbreviations: Str. L.-M.: stratum lacunosum-moleculare; Str. Rad.: stratum radiatum; Str. Pyr.: stratum pyramidale; Str. Oriens: stratum oriens. B, CCK (500 nm) depressed the evoked unitary IPSCs in pyramidal cells (lower traces) elicited by the presynaptic SCA (upper traces). The individual IPSCs are grey and the averages are black. Summary data are shown on the right (open circles: individual pairs; filled circles: means). C, the CB1 receptor antagonist AM251 (10 μm) abolished the CCK induced inhibition of GABA release from SCAs. Error bars represent s.e.m., and the asterisks denote P < 0.05.
Results
Data in this study were obtained using paired whole-cell patch-clamp recordings from post hoc identified presynaptic CCK+ BCs or PV+ BCs or CCK+ SCA interneurons and postsynaptic CA1 pyramidal cells (in addition, for one set of experiments associated with the measurement of sIPSCs in older animals, single whole-cell patch clamp recordings from CA1 pyramidal cells were employed). All presynaptic interneurons in this paper were identified both anatomically (e.g. perisomatically projecting BCs versus dendritically projecting SCAs) and immunocytochemically (e.g. CCK+ or PV+ cells). Note that in the current study aiming to test two alternative hypotheses about mechanisms underlying CCK actions on GABAergic transmission, responses to CCK application were tested at only one (high) concentration (500 nm) of CCK, in order to evoke maximal effects on GABAergic transmission (for data on the qualitatively similar nature of the CCK effects on BC transmissions at low and high doses, including a dose–response curve, see Földy et al. 2007).
GABA release from PV+ BCs is not required for the CCK-induced suppression of GABA release from CCK+ BCs
First, we confirmed that, as reported previously (Földy et al. 2007), bath application of CCK (500 nm) attenuated the amplitude of euIPSCs at CCK+ BC (Fig. 2A) to CA1 pyramidal cell synapses (Fig. 2B; pre-CCK control: −59.1 ± 13.3 pA; CCK peak: −44.2 ± 12.1 pA; postpeak: −56.6 ± 12.4 pA; reduction by CCK: 27.1 ± 6.1%, n= 5; P= 0.01). Next, we tested the validity of a central assumption of the ‘GABAB receptor hypothesis’, namely, that GABA release from PV+ BCs is required for the CCK-mediated suppression of GABA release from CCK+ BCs. PV+ BCs, not CCK+ BCs, express P/Q-type Ca2+ channels at their terminals, and selective blockade of P/Q-type Ca2+ channels with agatoxin TK decreases GABA release from PV+ BCs, but not CCK+ BCs, in an irreversible manner (Wilson et al. 2001; Hefft & Jonas, 2005; Zaitsev et al. 2007). Indeed, even relatively brief (5 min) bath application of agatoxin TK (250 nm) caused a complete suppression of euIPSCs at PV+ BC to pyramidal cell synapses, and the effect was not reversible even after prolonged (40 min) washout (Fig. 2C; pre-agatoxin TK: −45.7 ± 3.9 pA; agatoxin TK: −0.7 ± 0.3 pA; washout: −0.2 ± 0.2 pA; n= 3; P= 0.007). Importantly, in slices pre-incubated for >1 h in agatoxin TK (250 nm), CCK (500 nm) still attenuated the amplitude of euIPSCs at CCK+ BC to pyramidal cell synapses (Fig. 2D; pre-CCK control: −104.8 ± 42.3 pA; CCK peak: −76.7 ± 29.8 pA; CCK post-peak: −101.8 ± 40.7 pA; reduction by CCK: 27.3 ± 5.3%, n= 5; P= 0.01). The degree of CCK-induced suppression of GABA release did not differ between control slices and slices pre-incubated in agatoxin TK (CCK-mediated decrease in euIPSC amplitude, with respect to pre-CCK control responses: control slices: 27.1 ± 6.1%, n= 5; slices pre-incubated with agatoxin TK: 27.3 ± 5.3%, n= 5; P= 0.9; note that the CCK-induced increase in the proportion of failures after presynaptic spikes was also not different under the two conditions: control slices: 16.2 ± 1.8%, n= 5; slices pre-incubated with agatoxin TK: 16.9 ± 8.2%, n= 5; P= 0.94; CCK did not significantly decrease the amplitude of the successfully evoked events in either group: decrease in amplitude, with respect to pre-CCK baseline: control slices: 14.8 ± 6.8%, n= 5; slices pre-incubated with agatoxin TK: 12.1 ± 5.7%, n= 5; P= 0.76). Thus, our current data so far suggest that GABA release from PV+ BCs is not required for the CCK-induced suppression of GABA release from CCK+ BCs.
A possible explanation for the differences between the results that gave rise to the two alternative hypotheses concerning CCK action on CCK+ BCs (Fig. 1) is a potential age dependency of the effects on the PV+ BCs. Namely, CCK-induced increase in GABA release associated with the direct CCK-mediated excitation of PV+ BCs was reported to be transient (lasting about 3 min) in juvenile rats (postnatal 2–3 weeks; Földy et al. 2007), but longer lasting (>25 min) in young adult rats (postnatal 5–7 weeks; Karson et al. 2008). Since the potentially longer lasting CCK-effects on PV+ BCs in the young adults may result in a more pronounced ‘spill-over’ of GABA from the PV+ BC synapses to the CCK+ BC synapses on pyramidal cells, we carried out additional experiments to address the issue of age dependency. The data showed that CCK (500 nm) produced a significant but still transient increase in sIPSC frequency recorded in CA1 pyramidal cells from 7-week-old rats (pre-CCK control: 4.5 ± 0.7 Hz, n= 5; Peak: 12.2 ± 5.2 Hz, n= 5; Post-peak: 4.5 ± 0.9 Hz, n= 5). In fact, the CCK-induced increase in sIPSC frequency diminished to the pre-CCK control level within about 3 min in the young adult rats (sIPSC frequency 3–4 min after the peak: 6.48 ± 1.8 Hz; P= 0.19, with respect to pre-CCK control), similarly to what was reported for juvenile rats (see Földy et al. 2007, Fig. 1B; note that the magnitude of the effect was actually smaller in the older animals: young adults: 2.7-fold increase in sIPSC frequency; juveniles: 6.5-fold increase, Földy et al. 2007). Therefore, these data suggest that it is unlikely that a more pronounced or prolonged CCK-mediated increase in GABA release from PV+ BCs in the older animals caused the discrepancy between our current findings and previous reports (Karson et al. 2008) with regard to the underlying mechanisms of CCK-mediated decrease in GABA release from CCK+ BCs (note that the CCK-induced decrease in GABA release from CCK+ BCs was also not larger in postnatal 4- to 5-week-old animals compared to juveniles; see Földy et al. 2007).
Lack of involvement of GABAB receptors
According to the ‘GABAB receptor hypothesis’ (Karson et al. 2008), GABA released from PV+ BCs in response to CCK-mediated depolarization activates presynaptic GABAB receptors on CCK+ BCs, leading to suppression of GABA release from CCK+ BCs. Therefore, we next tested the role of GABAB receptors in the CCK effect on CCK+ BC to pyramidal cell transmission. Consistent with previous studies (Neu et al. 2007), bath application of GABAB receptor agonist, baclofen (20 μm), caused a strong suppression of euIPSCs at CCK+ BC to pyramidal cell synapses that was fully reversible after application of the GABAB receptor antagonist CGP 55845 (2 μm) (Fig. 3A; pre-baclofen: −29.9 ± 9.5 pA; baclofen: −0.4 ± 0.7 pA; CGP 55845: −48.3 ± 11.5 pA; n= 3; reduction by baclofen: 95.6 ± 1.3%, P= 0.001). Note the interesting increase in euIPSC amplitude in CGP55845 and baclofen, compared to the pre-baclofen control. As shown in Fig. 7 in Neu et al. (2007), we previously had found no GABAB receptor-dependent tonic inhibition present at the CCK+ BC to pyramidal cell synapse; indeed, additional experiments confirmed that CGP 55845 alone did not produce a change in euIPSCs (pre-CGP: 38.2 ± 17.1 pA; CGP: −37.4 ± 16.6 pA; n= 3, P= 0.30, data not shown). Therefore, CGP 55845 applied after baclofen appeared to have a different effect from when applied alone; the mechanism of these differential effects was not investigated in this paper.
Figure 3. GABAB receptors are not required for the CCK-induced suppression of GABA release from CCK+ basket cells.
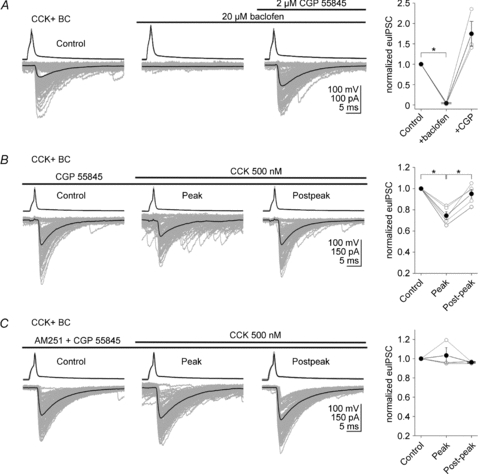
A, baclofen (20 μm; GABAB receptor agonist) caused a strong suppression of the unitary IPSCs (bottom traces) evoked by a presynaptic CCK+ BC in the pyramidal cell; the effect was fully reversible after application of 2 μm CGP 55845 (GABAB receptor antagonist). The individual IPSCs are grey and the averages are black. Summary data are shown on the right (open circles: individual pairs; filled circles: means). B, in 2 μm CGP 55845, 500 nm CCK still attenuated the amplitude of the evoked unitary IPSCs in pyramidal cells elicited by the presynaptic CCK+ BCs. Note that the degree of CCK induced suppression of GABA release did not differ in the absence (Fig. 2B) and presence of CGP 55845 (Fig. 3B). C, the CB1 receptor antagonist AM251 (10 μm) was able to block the CCK-induced reduction in evoked unitary IPSCs even in the presence of 2 μm CGP 55845 (CCK: 500 nm). Error bars represent s.e.m., and the asterisks denote P < 0.05.
Next, we examined whether CGP 55845 blocks the CCK-induced reduction in GABA release from CCK+ BCs. In CGP 55845 (2 μm), CCK (500 nm) was still able to decrease the amplitude of euIPSCs (Fig. 3B; pre-CCK control: −59.4 ± 22.3 pA; CCK peak: −42.3 ± 13.8 pA; CCK post-peak: −54.1 ± 17.5 pA; reduction by CCK: 25.5 ± 3.6%, n= 5; P= 0.01). Furthermore, the degree of CCK-induced suppression of GABA release did not differ between slices perfused with ACSF lacking or containing CGP 55845 (CCK-induced decrease in euIPSC amplitude: control, without CGP 55845: 27.1 ± 6.1%, n= 5; CGP 55845: 25.5 ± 3.6%, n= 5; P= 0.82). In addition, the CB1 receptor antagonist AM251 (10 μm) abolished the CCK-induced reduction in euIPSCs at CCK+ BC to pyramidal cell synapses not only in the absence (Földy et al. 2007), but also in the presence of CGP 55845 (Fig. 3C: pre-CCK control: −72.9 ± 46.6 pA; CCK peak: −71.6 ± 43.7 pA; CCK post-peak: −70.5 ± 45.2 pA; reduction by CCK: 3.3 ± 7.9%, n= 3; P= 0.7). These data show that GABABRs are not involved in CCK-induced suppression of GABA release from CCK+ BCs.
CCK attenuates GABA release at SCA synapses in a CB1 receptor dependent manner
Taken together, the data presented above are not consistent with the ‘GABAB receptor hypothesis’ of CCK action on CCK+ BC to CA1 pyramidal cell transmission. In the final series of experiments, we verified that, as predicted by the ‘CB1 receptor hypothesis’ and reported previously (Földy et al. 2007), the CCK-induced decrease in the synaptic transmission between CCK+ BC and CA1 pyramidal cell pairs is sensitive to CB1 receptor blockade. The experiments showed that CB1 receptors were also required for the CCK-mediated modulation of GABAergic transmission at dendritic synapses between CCK+ interneurons and CA1 pyramidal cells. Specifically, paired recordings demonstrated that CCK (500 nm) attenuated the amplitude of euIPSCs at CCK+ SCA (Fig. 4A) to pyramidal cell synapses (Fig. 4B; pre-CCK control: −33.9 ± 7.8 pA; CCK peak: −22.0 ± 6.9 pA; CCK post-peak: −29.8 ± 7.7 pA; reduction by CCK: 39.5 ± 5.2%, n= 5; P= 0.001), and AM251 (10 μm) was able to abolish the CCK-induced reduction in euIPSCs at SCA to pyramidal cell synapses as well (Fig. 4C; euIPSC amplitude in the presence of CCK and AM251, with respect to control: CCK peak, 104.4 ± 6.9%; n= 4; P= 0.57). Therefore, GABA release from both the perisomatically projecting CCK+ BCs and the dendritically projecting CCK+ SCAs is attenuated by CCK through a CB1 receptor-dependent mechanism.
Discussion
The main findings of this study are as follows: (1) blockade of P/Q-type Ca2+ channels that abolished GABA release from PV+ BCs did not inhibit the CCK induced suppression of GABA release from CCK+ BCs; (2) GABAB receptor antagonist did not reduce the CCK effect on GABA release from CCK+ BCs; (3) CB1 receptor antagonist abolished the CCK-induced suppression of GABA release from CCK+ BCs; and (4) CCK attenuated GABA release from dendritically projecting CCK+ SCAs, and the latter effect also required CB1 receptors. Taken together, these results indicate that CCK modulates GABA release from the presynaptic terminals of CCK+ interneurons along the somato-dendritic axis of postsynaptic CA1 pyramidal cells through endocannabinoid signalling, and not through heterosynaptic GABAB receptor mediated action requiring GABA release from PV+ BCs.
CCK attenuates GABA release from CCK expressing interneurons through a CB1 receptor dependent pathway
CCK is one of the most abundant and widely distributed neuropeptides in the brain, involved in a diversity of behaviours such as learning and memory, feeding, nociception, and satiety, and it is also implicated in several brain disorders including anxiety, panic attacks, deficits in fear extinction, schizophrenia and epilepsy (Noble & Roques, 2006; Lee & Soltesz, 2011). In spite of its abundance, recent results indicate that CCK regulates perisomatic GABAergic inhibition in a highly cell type specific manner through the actions of G-protein coupled CCKB receptors (Földy et al. 2007; Karson et al. 2008), by enhancing the rate of GABA release from PV+ BCs while inhibiting GABA release from CCK+ BCs. Therefore, CCK, beyond serving as a convenient marker for one of the two types of BCs, also acts as a finely tuned molecular switch regulating BC functions.
However, the precise mechanisms underlying the dual effects of CCK on BC outputs are not understood. There is general agreement that CCK, starting at low nanomolar concentrations, increases the firing rates of PV+ BCs. The increased action potential discharge of PV+ BCs in response to CCK is known to result in an enhancement of the frequency of spontaneous IPSCs (sIPSCs) in the CA1 pyramidal cells (for a quantification of the CCK induced increase in sIPSC frequency, see Fig. 1b in Földy et al. 2007; see also the sIPSCs visible on the example traces in Figs 2B and 4B in the current study, panels ‘Peak’versus‘Control’). The effect of CCK on sIPSC frequency in CA1 pyramidal cells can be abolished by P/Q channel antagonists (Figs. 1a and b in Földy et al. 2007 and Fig. 4a in Karson et al. 2008), in agreement with the involvement of PV+ BCs as the major sources of GABA underlying the increased sIPSC. However, in contrast to the direct actions of CCK on PV+ BCs, the inhibitory effect of CCK on GABA release from CCK+ BCs is controversial, with two alternative, albeit not necessarily mutually exclusive, pathways proposed to explain the cellular mechanisms underlying the effect (Fig. 1). Our paired recording data presented in this paper are consistent with the ‘CB1 receptor hypothesis’ of CCK action. CCKB receptors are thought to act typically through the Gq/11 second messenger pathway, and thus, similarly to other Gq/11-linked receptors including metabotropic glutamate and M1/3 muscarinic receptors, their activation would be predicted to enhance the synthesis of endocannabinoids and the retrograde inhibition of GABA release through the activation of presynaptic CB1 receptors (Kano et al. 2009). In agreement with the latter prediction, we confirmed our previous results (Földy et al. 2007) that CB1 receptor blockade abolishes the CCK effect on GABA release from the synaptic terminals of pair recorded presynaptic CCK+ BCs, and our experiments with SCAs extend the requirement for CB1 receptors for CCK action to dendritic CCK+ synapses as well. Furthermore, consistent with the suggestion that the source of the endocannabinoids is likely to be the postsynaptic pyramidal cells themselves, inhibition of G-protein coupled receptor function with the postsynaptic injection of the non-hydrolysable GDP analogue GDP-βS was previously shown to result in a blockade of the CCK induced suppression of GABA release from the CCK+ BCs (Földy et al. 2007).
In contrast, our data are not consistent with the ‘GABAB receptor hypothesis’ of CCK action on CCK+ BC to pyramidal cell transmission. Indeed, the lack of effect of either agatoxin TK or CGP 55845 on the CCK induced suppression of GABA release from CCK+ BCs in this study suggests that the excitatory action of CCK on PV+ BCs is mechanistically unrelated to the inhibitory CCK action on the CCK+ BC to CA1 pyramidal cell transmission. It should be noted that the ‘GABAB receptor hypothesis’ was originally proposed based on a series of pharmacological observations on CCK actions on endocannabinoid sensitive and insensitive sIPSCs in single cell recording experiments from CA1 pyramidal cells in the presence of carbachol in the perfusing medium (Karson et al. 2008). While it is difficult to discern precisely which experimental condition was responsible for the observations that led to the ‘GABAB receptor hypothesis’ by Karson and colleagues (2008), it seems likely that the differences are mainly due to the employment of single pyramidal cell recording methods versus paired recording techniques from anatomically and immunocytochemically identified interneurons, as the latter approach allows for a better isolation and more direct control of the GABAergic synaptic transmission from PV+ or CCK+ BCs onto CA1 pyramidal cells. Our results do not exclude that GABA released from PV+ terminals may be able to reach CCK+ terminals under some circumstances. Indeed, the two types of BC synapses can be in close proximity (Karson et al. 2009), although it is important to note that the overall somato-dendritic distribution patterns of PV+versus CCK+ terminals from BCs on the perisomatic regions of pyramidal cells are not identical (Földy et al. 2010).
In summary, our data support the requirement for CB1 receptors in the CCK mediated regulation of GABA release from somatic and dendritic CCK+ synapses. It is interesting in this regard that both CB1 and CCK receptors have been implicated in mood-related disorders (Freund & Katona, 2007; Lee & Soltesz, 2011), and endocannabinoid and CCK signalling systems have been reported to be able to closely interact to modulate anxiety, stress and fear extinction (Kurrikoff et al. 2008; Chhatwal et al. 2009). Future studies will be needed to determine the precise role of hippocampal CCK+ interneuronal microcircuits in limbic system functions influenced by interacting CCK and CB1 receptor-dependent pathways.
Acknowledgments
We thank Ms R. Zhu for technical assistance and Drs Raphael Winkels, Csaba Földy, Esther I. Krook-Magnuson and Soo Yeun Lee for discussion. This work was supported by the US National Institutes of Health grant NS38580 (to I.S.).
Author contributions
S.-H.L. and I.S. designed the experiments. S.-H.L. performed the experiments and analysed the data. S.-H.L. and I.S. interpreted the data and wrote the manuscript.
References
- Acsady L, Katona I, Martinez-Guijarro FJ, Buzsaki G, Freund TF. Unusual target selectivity of perisomatic inhibitory cells in the hilar region of the rat hippocampus. J Neurosci. 2000;20:6907–6919. doi: 10.1523/JNEUROSCI.20-18-06907.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AB. Presynaptic inhibition of GABAA receptor-mediated unitary IPSPs by cannabinoid receptors at synapses between CCK-positive interneurons in rat hippocampus. J Neurophysiol. 2007;98:861–869. doi: 10.1152/jn.00156.2007. [DOI] [PubMed] [Google Scholar]
- Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- Boden PR, Hill RG. Effects of cholecystokinin and pentagastrin on rat hippocampal neurones maintained in vitro. Neuropeptides. 1988;12:95–103. doi: 10.1016/0143-4179(88)90037-6. [DOI] [PubMed] [Google Scholar]
- Chhatwal JP, Gutman AR, Maguschak KA, Bowser ME, Yang Y, Davis M, et al. Functional interactions between endocannabinoid and CCK neurotransmitter systems may be critical for extinction learning. Neuropsychopharmacology. 2009;34:509–521. doi: 10.1038/npp.2008.97. [DOI] [PubMed] [Google Scholar]
- Chung L, Moore SD. Cholecystokinin excites interneurons in rat basolateral amygdala. J Neurophysiol. 2009;102:272–284. doi: 10.1152/jn.90769.2008. [DOI] [PubMed] [Google Scholar]
- Cope DW, Maccaferri G, Márton LF, Roberts JD, Cobden PM, Somogyi P. Cholecystokinin-immunopositive basket and Schaffer collateral-associated interneurones target different domains of pyramidal cells in the CA1 area of the rat hippocampus. Neuroscience. 2002;109:63–80. doi: 10.1016/s0306-4522(01)00440-7. [DOI] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Cholecystokinin depolarizes rat thalamic reticular neurons by suppressing a K+ conductance. J Neurophysiol. 1995;74:990–1000. doi: 10.1152/jn.1995.74.3.990. [DOI] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, McBain CJ. Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci. 2009;9:11112–11122. doi: 10.1523/JNEUROSCI.5760-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Lei S. Bidirectional modulation of GABAergic transmission by cholecystokinin in hippocampal dentate gyrus granule cells of juvenile rats. J Physiol. 2006;572:425–442. doi: 10.1113/jphysiol.2005.104463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng PY, Xiao Z, Jha A, Ramonet D, Matsui T, Leitges M, et al. Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. J Neurosci. 2010;30:5136–5148. doi: 10.1523/JNEUROSCI.5711-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Paulsen O. The many tunes of perisomatic targeting interneurons in the hippocampal network. Front Cell Neurosci. 2010;4:26. doi: 10.3389/fncel.2010.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Lee SH, Morgan RJ, Soltesz I. Regulation of fast-spiking basket cell synapses by the chloride channel ClC-2. Nat Neurosci. 2010;13:1047–1049. doi: 10.1038/nn.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Földy C, Lee SY, Szabadics J, Neu A, Soltesz I. Cell type-specific gating of perisomatic inhibition by cholecystokinin. Nat Neurosci. 2007;10:1128–1130. doi: 10.1038/nn1952. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I. Perisomatic inhibition. Neuron. 2007;56:33–42. doi: 10.1016/j.neuron.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat Neurosci. 2006;9:807–815. doi: 10.1038/nn1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci. 2005;8:1319–1328. doi: 10.1038/nn1542. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Karson MA, Tang AH, Milner TA, Alger BE. Synaptic cross talk between perisomatic-targeting interneuron classes expressing cholecystokinin and parvalbumin in hippocampus. J Neurosci. 2009;29:4140–4154. doi: 10.1523/JNEUROSCI.5264-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karson MA, Whittington KC, Alger BE. Cholecystokinin inhibits endocannabinoid-sensitive hippocampal IPSPs and stimulates others. Neuropharmacology. 2008;54:117–128. doi: 10.1016/j.neuropharm.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O’Neill J, Huck JH, Dalezios Y, Fuentealba P, et al. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J Neurosci. 2005;25:9782–9793. doi: 10.1523/JNEUROSCI.3269-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurrikoff K, Inno J, Matsui T, Vasar E. Stress-induced analgesia in mice: evidence for interaction between endocannabinoids and cholecystokinin. Eur J Neurosci. 2008;27:2147–2155. doi: 10.1111/j.1460-9568.2008.06160.x. [DOI] [PubMed] [Google Scholar]
- Lee SH, Földy C, Soltesz I. Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J Neurosci. 2010;30:7993–8000. doi: 10.1523/JNEUROSCI.6238-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Soltesz I. Cholecystokinin: A multi-functional molecular switch of neuronal circuits. Dev Neurobiol. 2011;71:83–91. doi: 10.1002/dneu.20815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KK, Hoffer A, Svoboda KR, Lupica CR. Cholecystokinin increases GABA release by inhibiting a resting K+ conductance in hippocampal interneurons. J Neurosci. 1997;17:4994–5003. doi: 10.1523/JNEUROSCI.17-13-04994.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu A, Földy C, Soltesz I. Postsynaptic origin of CB1-dependent tonic inhibition of GABA release at cholecystokinin-positive basket cell to pyramidal cell synapses in the CA1 region of the rat hippocampus. J Physiol. 2007;578:233–247. doi: 10.1113/jphysiol.2006.115691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble F, Roques BP. Cholecystokinin peptides in brain function. In: Lajtha A, editor. Handbook of Neurochemistry and Molecular Neurobiology. 3rd edn. New York: Springer Science + Business Media; 2006. pp. 545–571. [Google Scholar]
- Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. J Comp Neurol. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- Vida I, Halasy K, Szinyei C, Somogyi P, Buhl EH. Unitary IPSPs evoked by interneurons at the stratum radiatum–stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol. 1998;506:755–773. doi: 10.1111/j.1469-7793.1998.755bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Zaitsev AV, Povysheva NV, Lewis DA, Krimer LS. P/Q-type, but not N-type, calcium channels mediate GABA release from fast-spiking interneurons to pyramidal cells in rat prefrontal cortex. J Neurophysiol. 2007;97:3567–3573. doi: 10.1152/jn.01293.2006. [DOI] [PubMed] [Google Scholar]