

Abstract
The renin–angiotensin system (RAS) regulates blood pressure mainly via the actions of angiotensin (Ang)II, generated via angiotensin converting enzyme (ACE). The ACE homologue ACE2 metabolises AngII to Ang1-7, decreasing AngII and increasing Ang1-7, which counteracts AngII activity via the Mas receptor. However, ACE2 also converts AngI to Ang1-9, a poorly characterised peptide which can be further converted to Ang1-7 via ACE. Ang1-9 stimulates bradykinin release in endothelium and has antihypertrophic actions in the heart, attributed to its being a competitive inhibitor of ACE, leading to decreased AngII, rather than increased Ang1-7. To date no direct receptor-mediated effects of Ang1-9 have been described. To further understand the role of Ang1-9 in RAS function we assessed its action in cardiomyocyte hypertrophy in rat neonatal H9c2 and primary adult rabbit left ventricular cardiomyocytes, compared to Ang1-7. Cardiomyocyte hypertrophy was stimulated with AngII or vasopressin, significantly increasing cell size by approximately 1.2-fold (P < 0.05) as well as stimulating expression of the hypertrophy gene markers atrial natriuretic peptide, brain natriuretic peptide, β-myosin heavy chain and myosin light chain (2- to 5-fold, P < 0.05). Both Ang1-9 and Ang1-7 were able to block hypertrophy induced by either agonist (control, 186.4 μm; AngII, 232.8 μm; AngII+Ang1-7, 198.3 μm; AngII+Ang1-9, 195.9 μm; P < 0.05). The effects of Ang1-9 were not inhibited by captopril, supporting previous evidence that Ang1-9 acts independently of Ang1-7. Next, we investigated receptor signalling via angiotensin type 1 and type 2 receptors (AT1R, AT2R) and Mas. The AT1R antagonist losartan blocked AngII-induced, but not vasopressin-induced, hypertrophy. Losartan did not block the antihypertrophic effects of Ang1-9, or Ang1-7 on vasopressin-stimulated cardiomyocytes. The Mas antagonist A779 efficiently blocked the antihypertrophic effects of Ang1-7, without affecting Ang1-9. Furthermore, Ang1-7 activity was also inhibited in the presence of the bradykinin type 2 receptor antagonist HOE140, without affecting Ang1-9. Moreover, we observed that the AT2R antagonist PD123,319 abolished the antihypertrophic effects of Ang1-9, without affecting Ang1-7, suggesting Ang1-9 signals via the AT2R. Radioligand binding assays demonstrated that Ang1-9 was able to bind the AT2R (pKi= 6.28 ± 0.1). In summary, we ascribe a direct biological role for Ang1-9 acting via the AT2R. This has implications for RAS function and identifying new therapeutic targets in cardiovascular disease.
Non-technical summary
The renin–angiotensin system regulates the function of the cardiovascular system via the peptide hormone angiotensin II through a cellular receptor, the angiotensin type 1 receptor. Angiotensin II can become overactive and contribute to the development of cardiovascular disease, resulting in hypertension and other disorders such as enlargement of the heart muscle cells, termed cardiomyocyte hypertrophy. Here we demonstrate, for the first time, that an alternative angiotensin peptide, called angiotensin 1-9, is able to inhibit cardiomyocyte hypertrophy induced by angiotensin II by binding and activating an alternative angiotensin receptor, the type 2 receptor. This has implications for our understanding of the renin–angiotensin system in normal cardiovascular function and in cardiovascular disease.
Introduction
Essential hypertension is one of the most significant risk factors for cardiovascular disease (CVD) and one of its most common triggers is overactivity of the renin–angiotensin system (RAS). The main RAS mediator is angiotensin II (AngII) generated by angiotensin converting enzyme (ACE), which acts in heart, blood vessels, brain, kidney and adrenal cortex through its two receptors, AT1R and AT2R, to regulate blood pressure, sodium handling and cardiovascular homeostasis. AngII also contributes to CVD pathophysiology through stimulating inflammation, fibrosis, hypertrophy and oxidative stress. The AT1R is the main mediator of AngII's signals, both physiological and pathophysiological, including sodium reabsorption, vasoconstriction, proliferation, inflammation, oxidative stress and extracellular matrix remodelling (Bunkenburg et al. 1992; Brilla et al. 1997; Nakagami et al. 2003; Bai et al. 2004; Swaney et al. 2005; Igarashi et al. 2007). The AT2R is only 34% homologous to the AT1R (Wang et al. 1995) and the signalling mechanisms differ (Kurisu et al. 2003; Ritter et al. 2003; Yayama & Okamoto, 2008). Certain studies have suggested that AngII signalling via this receptor acts as a negative feedback loop on AT1R signalling as, for example, blocking AT2R activation can promote cardiomyocyte hypertrophy (Bartunek et al. 1999), while lentiviral-mediated overexpression of the AT2R in stroke prone spontaneously hypertensive rat hearts leads to protection from increases in left ventricular mass index (Metcalfe et al. 2004).
The ACE homologue ACE2 cleaves AngI and AngII to form angiotensin 1-9 (Ang1-9) and angiotensin 1-7 (Ang1-7), respectively (Donoghue et al. 2000; Crackower et al. 2002). Ang1-9 can also be formed by carboxypeptidase activity (Garabelli et al. 2008). Ang1-7 blocks the effects of AngII in cardiovascular tissues including heart, kidney and blood vessels (Grobe et al. 2007; Mercure et al. 2008; De Mello, 2009; Pinheiro et al. 2009) via the G protein-coupled receptor Mas (Santos et al. 2003). For example, co-infusion of Ang1-7 into AngII-infused rats attenuates fibrosis, cardiac hypertrophy and hypertension (Grobe et al. 2006) and Ang1-7 also reduces re-entrant arrhythmias (De Mello et al. 2007). Conversely, little is currently known about Ang1-9. It reduces AngII levels through acting as a competitive inhibitor of ACE activity and increases Ang1-7 levels and has previously been shown to stimulate bradykinin release in endothelial cells (Jackman et al. 2002). Most recently Ang1-9 was demonstrated to block cardiac hypertrophy in a rat myocardial infarction model (Ocaranza et al. 2010). This was not dependent on Ang1-9 increasing Ang1-7 activity via the Mas receptor, but was thought to be through competitive ACE inhibition decreasing AngII levels. Moreover, the authors demonstrated significant upregulation in endogenous plasma Ang1-9 levels in animals placed on angiotensin receptor antagonists or ACE inhibitors suggesting that, like Ang1-7, Ang1-9 may be an endogenous component of the counter-regulatory RAS. In the present study, we have further investigated Ang1-9 and Ang1-7 function in cardiomyocyte hypertrophy in rat neonatal (H9c2) and primary adult rabbit left ventricular cardiomyocytes. We demonstrate that Ang1-9 is an active RAS hormone with actions distinguishable from its merely being a substrate for Ang1-7 generation or a competitive inhibitor of ACE. Importantly, we show that Ang1-9 directly binds the AT2R and antagonises cardiomyocyte hypertrophy.
Methods
Ethical approval
The isolation of primary rabbit cardiomyocytes was approved by the University of Glasgow Animal Procedures and Ethics Committee and performed in strict accordance with UK Home Office guidelines.
Materials
All tissue culture reagents were purchased from Lonza (Braine-L’Alleud, Belgium) unless otherwise indicated. Angiotensin peptides were purchased from Sigma-Aldrich (Poole, UK) or Phoenix Pharmaceuticals (Karlsruhe, Germany: 125I-labelled AngII). Pharmacological receptor antagonists were purchased from Sigma (losartan, captopril, PD123,319) or Bachem (Rhein, Germany: A779). Phalloidin-fluorescein isothiocyanate was purchased from Sigma.
Cell culture
H9c2 cells are an immortalised cardiomyocyte cell line derived from neonatal rat cardiomyocytes and were obtained from the European Collection of Animal Cell Cultures (Porton Down, UK). H9c2 cardiomyocytes were cultured at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 2 mm l-glutamine, streptomycin (100 microg/mL) and penicillin (100 IU/mL) (Invitrogen, Paisley, UK).
Primary cardiomyocyte isolation
Adult rabbit left ventricular cardiomyocytes were isolated by cardiac retrograde aortic reperfusion. The heart was digested with collagenase type I (400 U ml−1). Cells were recovered in Krebs solution containing NaCl (20 mm), Hepes (20 mm), KCl (5.4 mm), NaH2PO4 (0.52 mm), MgCl2 (3.5 mm), taurine (20 mm), creatine (10 mm), glucose and bovine serum albumin at 37°C, pH 7.4. Cells were washed 4 times consecutively with Krebs solution supplemented with CaCl2 at 100 μm, 400 μm, 700 μm and 1 mm, respectively. Cells were pelleted by gravity, followed by gentle re-suspension in M199 medium supplemented with taurine, carnitine, creatine and antibiotics.
Hypertrophy model
H9c2 cells were seeded at 3 × 104 cells per well 24 h prior to initiation of experiments. Cells were quiesced in serum free medium for 24 h, followed by stimulation with 100 nm AngII (Sigma) and incubated for a further 96 h at 37°C in order to induce hypertrophy. We measured cell size with Image ProPlus 4.1 software (Media Cybernetics, Basingstoke, UK). For each condition we measured 100 cells (10 fields of view in each condition). Alternatively, cells were stimulated with 1 μm arginine vasopressin in a similar manner to that described above. To investigate the role of Ang1-7 and Ang1-9 in hypertrophy, we incubated H9c2 or adult rabbit primary left ventricular cardiomyocytes with either Ang1-7 or Ang1-9 30 min before addition of AngII or arginine vasopressin. To assess receptor specificity the following antagonists were used: AT1R (losartan; 1 μm), AT2R (PD123,319; 500 nm) Mas receptor (A779; 10 μm), bradykinin type 2 receptor (B2R) antagonist HOE140 (1 μm) and the ACE inhibitor (captopril; 1 μm). Antagonists were added 15 min before Ang1-7 and Ang1-9. Left ventricular rabbit cardiomyocytes were seeded as above following extraction and then stimulated with AngII or arginine vasopressin, followed by incubation for 24 h at 37°C. In H9c2 cardiomyocytes cell length was measured. In primary cardiomyocytes length and midpoint width were measured. Ang1-7 and Ang1-9 stimulation was performed as described above. Receptor specificity was assessed as described above.
Real-time quantitative polymerase chain reaction assays
H9c2 cardiomyocytes were plated in six-well plates at 1 × 105 cells per well and either left non-stimulated or stimulated with angiotensin peptides at 100 nm as described previously, and RNA was isolated at 96 h using the RNeasy mini kit (Qiagen, Crawley, UK) following the manufacturer's protocols. Following DNase treatment (Applied Biosystems, Warrington, UK), RNA was reverse transcribed to cDNA using the Applied Biosystems Reverse Transcription Kit following the manufacturer's instructions. Real-time quantitative polymerase chain reaction (QPCR) was performed using inventoried SYBR green gene expression assays (Qiagen) for atrial natriuretic peptide (ANP) brain natriuretic peptide (BNP), β-myosin heavy chain (β-MHC) and myosin light chain (MLC) and normalised to the expression of 18S RNA (Applied Biosystems) using an Applied Biosystems 7900HT Sequence Detection System following the manufacturer's instructions.
Phalloidin staining
H9c2 cardiomyocytes were seeded at 3 × 104 cells per well on coverslips. Hypertrophy was stimulated as described above. After 4 days cells were washed, fixed with 4% paraformaldehyde and permeabilised with 0.1% Triton X-100. Cells were washed in phosphate-buffered saline (PBS) and stained with phalloidin solution (5 μg ml−1 (v/v) phalloidin-FITC, 1% (w/v) bovine serum albumin in PBS). Unbound phalloidin was removed by washing in PBS and coverslips mounted using Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 4′,6-diamidino-2-phenylindole (DAPI).
Cell viability
CellTiter 96® Non-radioactive Cell Proliferation Assay (Promega, Southampton, UK) was used to determine cell death in the hypertrophy assay following the manufacturer's protocol. Briefly, H9c2 cardiomyocytes were plated at a seeding density of 3 × 104 cells per well and the hypertrophy assay protocol followed. After 96 h, dye solution containing tetrazolium was added to the culture medium of each well in a 15:100 ratio and incubated for 4 hours at 37°C in 5% CO2–95% air. After incubation 1 ml of Solubilisation Solution/Stop Mix was added to each well to solubilise the formazan product. Absorbance was recorded at 570 nm using a Wallac Victor2 plate reader (Wallac, Turku, Finland).
Radioligand binding assays
HeLa cells were plated at 3 × 104 cells per well and the following day transduced with adenoviral vectors expressing either the AT1a or AT2 receptor (AdAT1aR and AdAT2R (Thomas et al. 2002; D’Amore et al. 2005) at a range of multiplicities of infection (moi) of 50, 100 and 200. AT1aR and AT2R receptor expression was determined by whole cell 125I-labelled AngII binding at 4°C, as described previously (Thomas et al. 2002; D’Amore et al. 2005) and virus was titrated such that equivalent receptor expression levels were achieved. In subsequent experiments, the affinity of 125I-labelled AngII was determined by homologous AngII competition experiments using two separate concentrations of radioligand (0.01 nm and 0.03 nm) and these values were then used to establish the apparent affinity of various unlabelled ligands in heterologous binding experiments (Thomas et al. 2002). Homologous and heterologous binding values were determined using appropriate equations in GraphPad Prism 5.02.
Statistical analysis
Experiments were performed in triplicate on three different occasions. Data are shown as the mean ± standard error of the mean (s.e.m.) of three independent experiments. Student's t test for paired data and one-way ANOVA with Bonferroni's correction for multiple comparisons were applied and statistical difference was considered to be with P values <0.05.
Results
Effect of AngII, Ang1-7 and Ang1-9 on cardiomyocyte hypertrophy
It has been reported that addition of AngII to cardiomyocytes induces an increase in hypertrophy markers as well as an increase in cell size (Thomas et al. 2002; Bai et al. 2004; D’Amore et al. 2005; Hwang et al. 2006; Laskowski et al. 2006; Majalahti et al. 2007). Stimulation of H9c2 cardiomyocytes with 100 nm AngII for 96 h induced a significant increase in cell size when compared to non-stimulated cells (232.8 ± 9.9 μm vs. 186.4 ± 5.4 μm; P < 0.001) (Fig. 1A). Assessment of the induction of the hypertrophy gene markers ANP, BNP, β-MHC and MLC revealed that addition of AngII produced a significant increase (P < 0.05) in expression, (Fig. 1B). Phalloidin-FITC staining of cells revealed that non-stimulated cardiomyocytes displayed an irregular pattern of sarcomere units throughout the cytoplasm of the cell but increased organisation of the sarcomeres was clearly evident following exposure of H9c2 cardiomyocytes to AngII (Fig. 1C). Next, we added exogenous Ang1-7 or Ang1-9, to H9c2 cardiomyocytes stimulated with AngII in order to study their effects. Both Ang1-7 and Ang1-9 were able to block the increase in cell size induced by AngII in H9c2 cardiomyocytes (Fig. 1A). Importantly, the cell length of AngII-stimulated cardiomyocytes incubated with Ang1-7 or Ang1-9 was not significantly different from that observed in control non-stimulated cells (Ang1-7, 198.3 ± 6.0 μm; Ang1-9, 95.9 ± 2.1 μm, P < 0.001). These effects were also apparent when gene expression levels of ANP, BNP, β-MHC or MLC were quantified, with Ang1-7 and Ang1-9 abrogating the increase in each marker's gene expression induced by AngII (Fig. 1B). Phalloidin staining of actin filaments revealed that AngII + Ang1-7 or Ang1-9-stimulated H9c2 cardiomyocytes showed the same irregular sarcomeric pattern as non-stimulated cells in contrast to the organised phenotype apparent with AngII alone (Fig. 1C). In rabbit primary cardiomyocytes cell length and width were measured separately and exposure to 500 nm AngII induced a significant increase in cell width (non-stimulated, 29.6 ± 1.4 μm; AngII-stimulated, 38.8 ± 1.6 μm; P < 0.001) but not cell length (Fig. 1D). This suggested AngII induced concentric hypertrophy in these cells. When exogenous Ang1-7 and Ang1-9 were added to AngII-stimulated left ventricular rabbit primary cardiomyocytes we observed similar results. AngII induced a significant increase in the width of the cell and both Ang1-7 and Ang1-9 were able to block AngII-induced hypertrophy at all concentrations (P < 0.01) (Fig. 1D).
Figure 1. Role of Ang1-7 and Ang1-9 in AngII-induced hypertrophy.
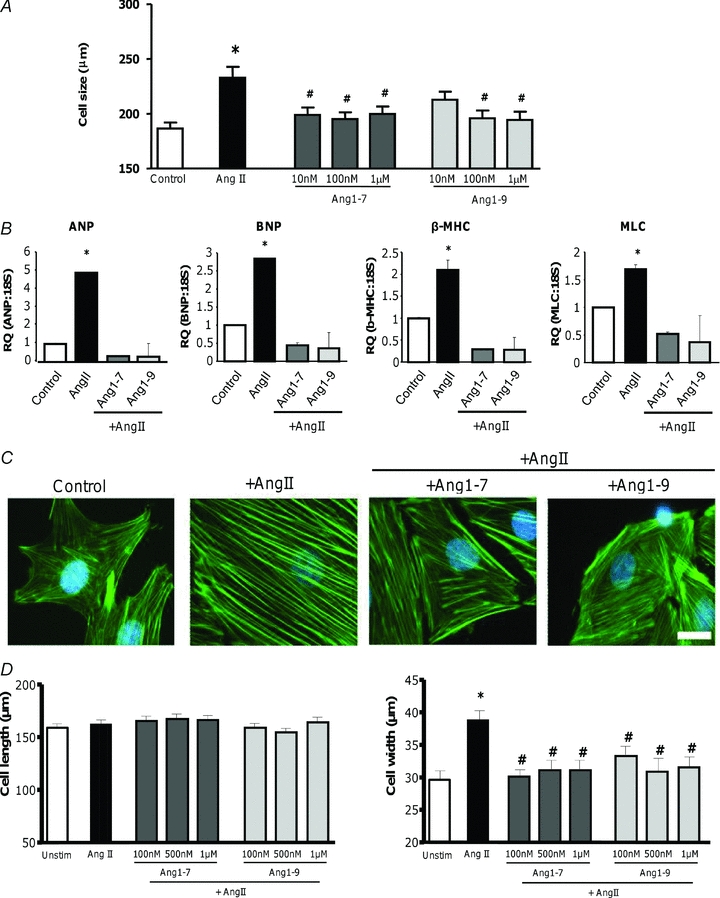
A, H9c2 cardiomyocytes were incubated with Ang1-7 or Ang1-9 at 10, 100 nm or 1 μm 30 min before stimulation with 100 nm AngII. Cells were then incubated for a further 96 h, fixed, stained with crystal violet and cell size measured using ImageProPlus. *P < 0.001 vs. control non-stimulated cells. #P < 0.05 vs. AngII-stimulated cells. B, RNA was isolated from H9c2 cardiomyocytes following 96 h exposure to 100 nm AngII and/or 100 nm Ang1-7 or Ang1-9, reverse transcribed and ANP, BNP, β-MHC and MLC expression quantified via real-time QPCR and normalised to 18S RNA expression. *P < 0.05 vs. control non-stimulated cells. C, following 96 h exposure to angiotensin peptides, α-actin filament reorganisation in H9c2 cardiomyocytes was evaluated by staining with FITC-labelled phalloidin. Magnification 100×, scale bar = 100 μm. D, left ventricular primary cardiomyocytes were extracted from adult rabbits via collagenase digestion, plated and immediately stimulated with Ang1-7 or Ang1-9 at 100 nm, 500 nm or 1 μm 30 min before stimulation with 500 nm AngII. Cells were then incubated for a further 24 h before measurement. Cell length and width were measured using ImageProPlus. *P < 0.01 vs. control non-stimulated cells. #P < 0.05 vs. AngII-stimulated cells.
Effect of ACE inhibition
Ang1-9 has been described as a substrate for Ang1-7 generation via ACE cleavage (Schluter & Wenzel, 2008) and therefore to assess if conversion to Ang1-7 was necessary for the anti-hypertrophic effects of Ang1-9 we assessed hypertrophy in the presence of the ACE inhibitor captopril. In the presence and absence of captopril Ang1-9 was still able to block AngII-induced hypertrophy (non-stimulated, 135.7 ± 5.3 μm; AngII-stimulated, 169.4 ± 7.9 μm; Ang1-9, 126.4 ± 4.6 μm; Ang1-9 + captopril, 121.2 ± 6.0 μm; P < 0.001) (Fig. 2A). The increase in cell width induced by AngII in rabbit primary left ventricular cardiomyocytes was also normalised (non-stimulated, 22.4 ± 0.6 μm; AngII-stimulated, 35.4 ± 0.9 μm; Ang1-9, 26.3 ± 0.8 μm; Ang1-9 + captopril, 24.8 ± 0.8 μm; P < 0.01) (Fig. 2B). These results imply that Ang1-9 acts independently of Ang1-7 and that it has anti-hypertrophic effects in cardiomyocytes.
Figure 2. Ang1-9 mediated inhibition of hypertrophy in the presence of ACE inhibition.
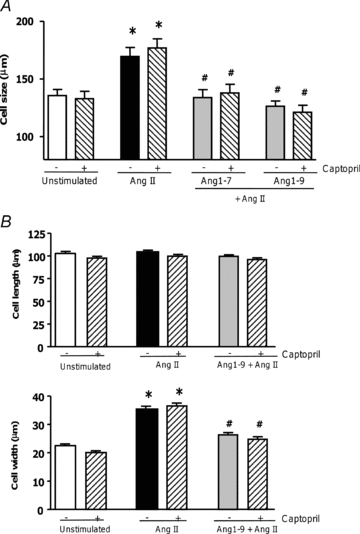
A, H9c2 cardiomyocytes were incubated with captopril at 100 μm 15 min before addition of Ang1-9 (100 nm) and AngII. B, rabbit primary cardiomyocytes were incubated with captopril at 1 μm 15 min before addition of 500 nm Ang1-9. *P < 0.001 vs. control non-stimulated cells; #P < 0.001 vs. AngII stimulated cells.
The role of the Mas, AT1 and AT2 receptors
Ang1-7 has been described to antagonise AngII-induced hypertrophy by engaging the Mas receptor (Santos et al. 2003, 2006; Dias-Peixoto et al. 2008; Pinheiro et al. 2009). To determine whether Ang1-9 signalled via Mas we used the specific Mas antagonist A779. Pre-incubation of AngII-stimulated H9c2 cardiomyocytes with 10 μm A779 prior to the addition of Ang1-7 blocked its anti-hypertrophic effect (non-stimulated, 191.1 ± 6.6 μm; AngII-stimulated, 288.6 ± 12.3 μm; Ang1-7, 218.3 ± 9.8 μm; Ang1-7 + A779, 275.5 ± 14.0 μm; P < 0.001) (Fig. 3A). However, A779 did not block the anti-hypertrophic effects of Ang1-9 (Ang1-9, 242.4 ± 10.7 μm; Ang1-9 + A779, 223.5 ± 8.4 μm; P < 0.001) (Fig. 3A). In adult left ventricular rabbit primary cardiomyocytes, A779 also blocked the anti-hypertrophic effects of Ang1-7, without affecting the actions of Ang1-9 (Fig. 3B). These results confirmed that Ang1-9 has an independent effect on cardiomyocytes from that of Ang1-7 and that it signals via a different receptor from Mas.
Figure 3. The role of the Mas receptor in Ang1-7 and Ang1-9 signalling in cardiomyocytes.
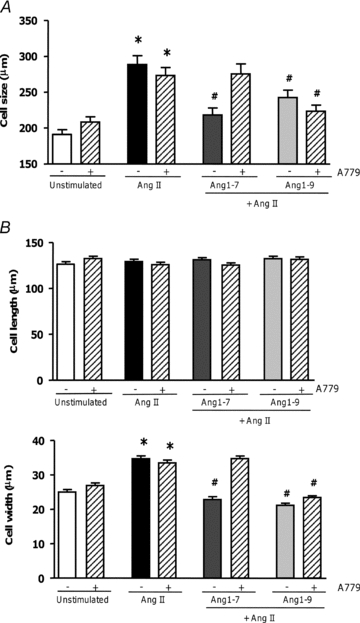
A, H9c2 cells were pre-incubated with AngII (100 nm), prior to addition of the Mas antagonist A779 (10 μm) 15 min before addition of 100 nm Ang1-7 or Ang1-9. B, addition of A779 (10 μm) in adult rabbit left ventricular primary cardiomyocytes, prior to addition of 500 nm Ang1-7 or Ang1-9. *P < 0.001 vs. control non-stimulated cells; #P < 0.001 compared to AngII stimulated cells.
Next, to determine which receptor Ang1-9 was signalling via we blocked the AT1R with losartan (Fig. 4). As AngII signals through the AT1R, we induced hypertrophy with arginine vasopressin. First, we confirmed that pre-incubation with losartan blocked AngII-induced hypertrophy (Fig. 4). Arginine vasopressin, however, was still able to induce cardiomyocyte hypertrophy in the presence of either 1 or 10 μm losartan (non-stimulated, 179.2 ± 5.4 μm; AngII-stimulated, 253.4 ± 8.4 μm; AngII + losartan 1 μm, 201.0 ± 6.1 μm; Arg-vasopressin, 227.4 ± 9.8 μm; Arg-vasopressin + losartan 1 μm, 247.8 ± 7.2 μm; P < 0.001). Next, we pre-incubated arginine vasopressin stimulated H9c2 cardiomyocytes with losartan, and Ang1-7 or Ang1-9. Blocking the AT1R with losartan did not inhibit the anti-hypertrophic effects of either Ang1-7 or Ang1-9. This confirmed that while AngII induced hypertrophy via the AT1R, both Ang1-7 and Ang1-9 did not antagonise AngII signalling by competition for the AT1R. In addition, both Ang1-7 and Ang1-9 were able to block pro-hypertrophic signals induced by alternative stimuli.
Figure 4. The role of the AT1R in Ang1-7 and Ang1-9 signalling in cardiomyocytes.
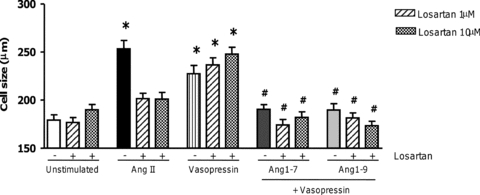
H9c2 cardiomyocytes were stimulated with arginine vasopressin (1 μm) to induce hypertrophy. Losartan (1 and 10 μm) was added to block AngII-induced hypertrophy but not arginine vasopressin's action. Cells were pre-incubated with losartan and arginine vasopressin before addition of 100 nm Ang1-7 or Ang1-9. (VSP: arginine vasopressin). *P < 0.001 vs. control non-stimulated cells; #P < 0.001 vs. AngII stimulated cells.
We next addressed the role of the AT2R, as the other classical RAS receptor mediating the effects of AngII. First we confirmed that AT2R agonism blocked AngII induced hypertrophy using the selective agonist CGP42112 (Ohkubo et al. 1997) (Supplemental Fig. S1). To assess the role of the AT2R in the effects of Ang1-7 and Ang1-9, the AT2R antagonist PD123,319 was utilised. Addition of PD123,319 at 100 nm, 500 nm or 1 μm alone did not affect AngII-induced hypertrophy, or result in inhibition of either ANP or BNP expression, confirming that AngII-induced hypertrophy in this model was via the AT1R and did not involve the AT2R (Fig. 5A and B). Additionally, the anti-hypertrophic effect of Ang1-7 was not abolished in the presence of PD123,319 (Ang1-7, 221.1 ± 6.4 μm; Ang1-7 + PD123,319 500 nm, 216.2 ± 7.0 μm; P < 0.001). However, when PD123,319 was added to Ang1-9-stimulated cardiomyocytes, the anti-hypertrophic effect of Ang1-9 on both cell size and inhibition of ANP and BNP gene expression was blocked (Ang1-9, 216.3 ± 6.1 μm; Ang1-9 + PD123,319 500 nm, 290.8 ± 8.3 μm; P < 0.001) (Fig. 5B). These results were additionally replicated in rabbit primary cardiomyocytes (Fig. 5C). These results suggest that Ang1-9 signals through the AT2R receptor. Since AngII has been described to stimulate apoptosis in cardiomyocytes (Cigola et al. 1997; Goldenberg et al. 2001) we next examined cell death following exposure to peptides. We observed no differences in cell death between non-stimulated cells and cells exposed to peptides (Supplemental Fig. S2). To begin to understand the downstream signalling pathways from Ang1-9 we focused on the classical cardiomyocyte hypertrophy pathways via protein kinase C (PKC) translocation and activation of ERK1/2 by phosphorylation (Zou et al. 1996; Vijayan et al. 2004; Pan et al. 2005) using Western phospho-immunoblotting and immunofluorescence and observed no differences between AngII and Ang1-7 or Ang1-9 stimulated cells (data not shown).
Figure 5. The role of the AT2R in Ang1-7 and Ang1-9 signalling in cardiomyocytes.
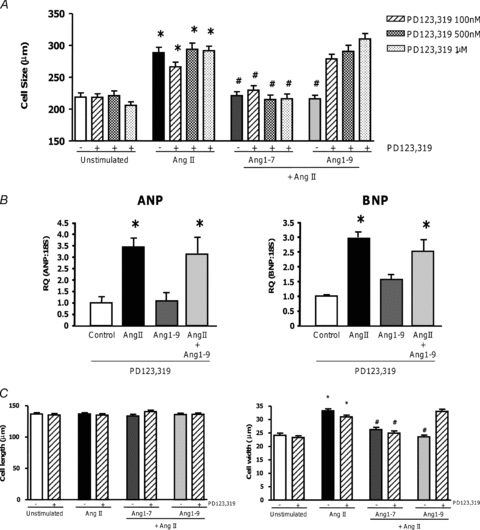
A, AngII-stimulated H9c2 cardiomyocytes were pre-incubated with the AT2R antagonist PD123,319 15 min before addition of Ang1-7 (100 nm) or Ang1-9 (100 nm). B, RNA was isolated from H9c2 cardiomyocytes following 96 h exposure to AngII (100 nm) and/or Ang1-7 (100 nm) or Ang1-9 (100 nm) in the presence or absence of the AT2R antagonist PD123,319 (500 nm) before RNA was isolated, reverse transcribed and ANP and BNP expression quantified via real-time PCR. *P < 0.05 vs. control non-stimulated cells. C, in adult rabbit left ventricular primary cardiomyocytes PD123,319 was added 15 min prior to addition of Ang1-7 (500 nm) or Ang1-9 (500 nm). (PD123 = PD123,319.) *P < 0.001 vs. control non-stimulated cells; #P < 0.001 vs. AngII stimulated cells.
Cross talk with the bradykinin type 2 receptor
Since the B2R has been reported to heterodimerise with the AT2R and Mas, leading to cellular effects (Kurisu et al. 2003), we next investigated whether it contributed to Ang1-7 and/or Ang1-9 function using the specific B2R blocker HOE140. H9c2 cardiomyocytes were incubated with either A779 or PD123,319 and HOE140 before addition of Ang1-7 or Ang1-9. HOE140 was unable to affect AngII-induced hypertrophy, indicating there was no cross-talk between the B2R and the AT1R (non-stimulated, 174.7 ± 5.5 μm; non-stimulated + HOE140, 161.6 ± 4.4 μm; AngII, 246.5 ± 9.8 μm; AngII + HOE140, 248.3 ± 7.5 μm; P < 0.001) (Fig. 6). However, blockade of the B2R in cardiomyocytes stimulated with Ang1-7 partially inhibited the anti-hypertrophic effect of Ang1-7 (Fig. 6). Although there was no significant difference between AngII-stimulated cells and Ang1-7 + HOE140 stimulated cells, there was a significant increase in cell size observed between HOE140 and Ang1-7 co-incubated cardiomyocytes versus Ang1-7 alone (Ang1-7, 167.8 ± 4.2 μm; Ang1-7 + A779, 226.4 ± 7 μm; Ang1-7 + HOE, 209.1 ± 6.2 μm; P < 0.05) (Fig. 6). However, HOE140 did not inhibit the anti-hypertrophic effect of Ang1-9 (Ang1-9, 174.7 ± 4.5 μm; Ang1-9 + PD123,319, 228.2 ± 8.0 μm; Ang1-9 + HOE140, 185.5 ± 5.5 μm) (Fig. 6) suggesting that crosstalk occurred for Ang1-7 Mas/B2R but not for Ang1-9 engagement at the AT2R.
Figure 6. The role of the B2R in Ang1-7 and Ang1-9 signalling in cardiomyocytes.
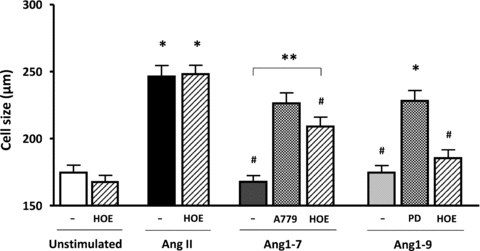
H9c2 cardiomyocytes were incubated for 24 h in serum free medium before adding HOE140 (1 μm), A779 (10 μm) or PD123,319 (500 nm). Cells were incubated for 15 min before adding Ang1-7 or Ang1-9 (100 nm) followed by AngII 30 min later. Following 96 h incubation, cells were fixed and stained with crystal violet and cell size measured with ImageProPlus. *P < 0.001 vs. non-stimulated cells; #P < 0.001 vs. AngII stimulated cells; **P < 0.001 vs. Ang1-7 stimulated cells. (PD = PD123,319; HOE = HOE140.)
Determination of Ang1-9 affinity at the AT2R
Our inhibitor studies suggested that Ang1-9 exerted its anti-hypertrophic effects via the AT2R. To confirm unequivocally that Ang1-9 was capable of binding to the AT2R, we performed radioligand binding experiments. Homologous (AngII) and heterologous competition curves were generated for AT1R and AT2R in HeLa cells exogenously expressing the receptors via adenovirus-mediated gene transfer. For AngII competition experiments, data were best fitted by a one-site homologous competition equation that accounted for radioligand depletion, as receptor binding exceeded 10% of total 125I-labelled AngII added to the assay at both concentrations of radioligand (0.01 and 0.03 nm) (Table 1). AngII showed expected pKd values of 8.42 ± .0.3 and 8.89 ± 0.13 at the AT1R and AT2R, respectively. For remaining ligands, data were analysed using a one-site heterologous binding equation with correction for depletion. In this case, we were able to demonstrate that Ang1-9 was indeed able to compete with radiolabelled AngII at both receptors with pKi values of 6.61 ± 0.32 and 6.28 ± 0.10 at the AT1R and AT2R, respectively (Table 1).
Table 1.
Apparent affinities of ligands at AT1 and AT2 receptors
| AT1 receptor pKi*s.e.m. | AT2 receptor pKi*s.e.m. | |
|---|---|---|
| AngII | 8.42 ± 0.03 | 8.89 ± 0.13 |
| Ang1-9 | 6.61 ± 0.32 | 6.28 ± 0.10 |
| Ang1-7 | 6.14 ± 0.25 | 7.02 ± 0.14† |
| PD123319 | no fit | 8.67 ± 0.34 |
| Losartan | 7.74 ± 0.05 | not determined |
pKi except for AngII which is pKd,
n= 2 at AT2 receptor.
Discussion
Here we define a novel function for the angiotensin metabolite Ang1-9 as an antagonist of cardiomyocyte hypertrophy. Ang1-9 antagonises pro-hypertrophic signals in cardiomyocytes from different species, representing both neonatal and adult lineages. Additionally, Ang1-9 utilises the AT2R to mediate its signals, segregating its actions from those of Ang1-7, which engages the Mas receptor.
Both Ang1-7 and Ang1-9 are the main products of the ACE–ACE2 axis in the heart. Ang1-7 antagonises the pathophysiological signalling of AngII, which leads to adverse cardiac remodelling such as fibrosis and left ventricular hypertrophy (Grobe et al. 2006, 2007; Mercure et al. 2008), highlighting its importance in the counter-regulatory arm of the RAS. Although Ang1-7 may interact with the AT1R and AT2R (Walters et al. 2005; Silva et al. 2007), Ang1-7 mainly utilises a unique receptor, Mas (Santos et al. 2003; Santos et al. 2006; Sampaio et al. 2007; Dias-Peixoto et al. 2008). In this study Ang1-7's effects in cardiomyocytes are mediated through Mas.
Ocaranza et al. (2010) recently demonstrated an anti-hypertrophic effect for Ang1-9 in rat myocardial infarction as a result of Ang1-9 inhibiting ACE, leading to decreased AngII. In this study ACE inhibition did not block the effects of Ang1-9. This suggests that while Ang1-9 may mediate some effects via ACE inhibition, its effects are also produced via direct AT2R activity. The AT2R is proposed to counter-regulate AngII signalling at the AT1R since AT2R activation by AngII releases bradykinin and NO (Abadir et al. 2006). The B2R has been reported to heterodimerise with the AT2R and Mas (Kurisu et al. 2003; Soares de Moura et al. 2004; Peiro et al. 2007), and Jackman et al. showed that Ang1-9 potentiated B2R activity in endothelial cells (Jackman et al. 2002). However, here B2R activity did not affect Ang1-9, in contrast to Ang1-7, as the B2R antagonist HOE140 partially blocked the effects of Ang1-7. This is in agreement with previous reports that Mas receptor and B2R cross-talk occurs to mediate effects of Ang1-7 (Ferreira et al. 2002). Although the circulating concentrations of Ang1-9 are reported to be in the 2–6 fmol ml−1 range (Campbell et al. 1993; Ocaranza et al. 2006) it has been reported that they rise following cardiac injury such as myocardial infarction (MI) (Ocaranza et al. 2010). However, the tissue concentrations in the heart are still relatively unexplored. It has been shown that in kidney tissue Ang1-9 levels are 100-fold higher than plasma and are 3 times higher than Ang1-7 concentration, reaching half the concentration of AngI (Campbell et al. 1993). Moreover, in human heart failure patients, myocardium forms in the region of 1 nm min−1 mg−1 Ang1-9 and furthermore the tissue contains as much as 170 nm AngI, of which 85% is rapidly converted to equivalent levels of Ang1-9 and AngII (Kokkonen et al. 1997). This provides evidence that in vivo tissue concentrations of Ang1-9 in (patho)physiological settings in both experimental models and patients may reach concentrations reflecting those used here.
Radioligand binding showed that Ang1-9 could bind both the AT1R and AT2R. However, Ang1-9 and not AngII affected hypertrophy through the AT2R, as PD123,319 did not alter AngII-mediated growth but did block the effects of Ang1-9. Despite having ∼100-fold lower affinity than AngII for the AT2R, the selective AT2R activity of Ang1-9 is not inconsistent with current pharmacological models of G protein-coupled receptor signalling and activation. Indeed, the concept of functional selectivity, where individual ligands at a receptor have the capacity to selectively stabilise conformations which lead to distinct signalling outcomes (Galandrin et al. 2007; Kenakin, 2007, 2008; Smith et al. 2010), is supported by a previous study in which the critical amino acids and the mode of binding of ligands at the AT1R and AT2R were investigated (Miura & Karnik, 1999). While agonist activation of the AT1R was particularly sensitive to peptide modifications that disrupted contact points between the octapeptide and receptor, substitutions within AngII were far better tolerated by the AT2R (Miura & Karnik, 1999). The authors concluded that the AT2R exists in a relaxed conformation and that AngII therefore binds at multiple indistinct contact points (Miura & Karnik, 1999). Since Ang1-9 contains the entire AngII sequence plus a C-terminal histidine, our observations indicate that this difference may stabilise the AT2R in a conformation able to counteract hypertrophic signalling in cardiomyocytes. Since we did not observe functional competition between AngII and Ang1-9 at the AT2R, Ang1-9 may be a preferential ligand for the AT2R. What remains to be determined is the downstream signalling effects from Ang1-9; preliminary studies indicate that the classical pathways via PKC translocation and ERK1/2 activation (Zou et al. 1996; Vijayan et al. 2004; Pan et al. 2005) are not different between AngII-, Ang1-7- and Ang1-9-stimulated cells. Since the downstream signalling from the AT2R is unclear at present, future studies will be required to delineate these mechanisms.
In summary, here we have demonstrated that Ang1-9 is able to antagonise AngII signalling in cardiomyocytes selectively via the AT2R, highlighting that Ang1-9, along with Ang1-7, makes up part of the counter-regulatory arm of the RAS.
Acknowledgments
We would like to thank Professor Walter Thomas (University of Queensland, Australia) for the kind gift of recombinant adenoviral vectors overexpressing rat angiotensin receptors, Dr Tony Workman and Ms Julie Russell (University of Glasgow) for the kind gift of rabbit left ventricular tissue and Dr Margaret Ann Craig (University of Glasgow) for advice and guidance in the isolation and culture of primary cardiomyocytes. We thank Professor Andrew H. Baker for critical discussions regarding the manuscript and Nicola Britton and Gregor Aitchison for technical assistance. The authors would like to thank the Medical Research Council (Grant number G0901161), University of Glasgow Postgraduate Scholarship Scheme and CONACYT for funding. Nicola J. Smith is an Australian NHMRC/NHF Overseas Fellow.
Glossary
Abbreviations
- Ang
angiotensin
- ACE
angiotensin converting enzyme
- AT1R
angiotensin type 1 receptor
- AT2R
angiotensin type 2 receptor
- B2R
bradykinin type 2 receptor
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- CVD
cardiovascular disease
- RAS
renin–angiotensin system
Author contributions
M.F.-M. performed the majority of research and analysed data, N.F.S. designed the receptor binding experiments, performed research and analysed data, C.H. performed real-time QPCR experiments and analysed data, G.M. devised experiments and analysed data and provided reagents, and S.A.N. devised research, supervised the project and wrote the manuscript. All authors approved the final version for publication.
Supplementary material
Figure S1
Figure S2
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors
References
- Abadir PM, Periasamy A, Carey RM, Siragy HM. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension. 2006;48:316–322. doi: 10.1161/01.HYP.0000228997.88162.a8. [DOI] [PubMed] [Google Scholar]
- Bai H, Wu LL, Xing DQ, Liu J, Zhao YL. Angiotensin II induced upregulation of Gαq/11, phospholipase Cβ3 and extracellular signal-regulated kinase 1/2 via angiotensin II type 1 receptor. Chin Med J (Engl) 2004;117:88–93. [PubMed] [Google Scholar]
- Bartunek J, Weinberg EO, Tajima M, Rohrbach S, Lorell BH. Angiotensin II type 2 receptor blockade amplifies the early signals of cardiac growth response to angiotensin II in hypertrophied hearts. Circulation. 1999;99:22–25. doi: 10.1161/01.cir.99.1.22. [DOI] [PubMed] [Google Scholar]
- Brilla CG, Scheer C, Rupp H. Renin-angiotensin system and myocardial collagen matrix: modulation of cardiac fibroblast function by angiotensin II type 1 receptor antagonism. J Hypertens Suppl. 1997;15:S13–19. [PubMed] [Google Scholar]
- Bunkenburg B, van Amelsvoort T, Rogg H, Wood JM. Receptor-mediated effects of angiotensin II on growth of vascular smooth muscle cells from spontaneously hypertensive rats. Hypertension. 1992;20:746–754. doi: 10.1161/01.hyp.20.6.746. [DOI] [PubMed] [Google Scholar]
- Campbell DJ, Kladis A, Duncan AM. Nephrectomy, converting enzyme inhibition, and angiotensin peptides. Hypertension. 1993;22:513–522. doi: 10.1161/01.hyp.22.4.513. [DOI] [PubMed] [Google Scholar]
- Cigola E, Kajstura J, Li B, Meggs LG, Anversa P. Angiotensin II activates programmed myocyte cell death in vitro. Exp Cell Res. 1997;231:363–371. doi: 10.1006/excr.1997.3477. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- D’Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension. 2005;46:1347–1354. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]
- De Mello WC. Opposite effects of angiotensin II and angiotensin (1-7) on impulse propagation, excitability and cardiac arrhythmias. Is the overexpression of ACE2 arrhythmogenic? Regul Pept. 2009;153:7–10. doi: 10.1016/j.regpep.2007.12.006. [DOI] [PubMed] [Google Scholar]
- De Mello WC, Ferrario CM, Jessup JA. Beneficial versus harmful effects of Angiotensin (1-7) on impulse propagation and cardiac arrhythmias in the failing heart. J Renin Angiotensin Aldosterone Syst. 2007;8:74–80. doi: 10.3317/jraas.2007.015. [DOI] [PubMed] [Google Scholar]
- Dias-Peixoto MF, Santos RA, Gomes ER, Alves MN, Almeida PW, Greco L, Rosa M, Fauler B, Bader M, Alenina N, Guatimosim S. Molecular mechanisms involved in the angiotensin-(1-7)/Mas signaling pathway in cardiomyocytes. Hypertension. 2008;52:542–548. doi: 10.1161/HYPERTENSIONAHA.108.114280. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Ferreira AJ, Santos RA, Almeida AP. Angiotensin-(1-7) improves the post-ischemic function in isolated perfused rat hearts. Braz J Med Biol Res. 2002;35:1083–1090. doi: 10.1590/s0100-879x2002000900009. [DOI] [PubMed] [Google Scholar]
- Galandrin S, Oligny-Longpre G, Bouvier M. The evasive nature of drug efficacy: implications for drug discovery. Trends Pharmacol Sci. 2007;28:423–430. doi: 10.1016/j.tips.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Garabelli PJ, Modrall JG, Penninger JM, Ferrario CM, Chappell MC. Distinct roles for angiotensin-converting enzyme 2 and carboxypeptidase A in the processing of angiotensins within the murine heart. Exp Physiol. 2008;93:613–621. doi: 10.1113/expphysiol.2007.040246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Grossman E, Jacobson KA, Shneyvays V, Shainberg A. Angiotensin II-induced apoptosis in rat cardiomyocyte culture: a possible role of AT1 and AT2 receptors. J Hypertens. 2001;19:1681–1689. doi: 10.1097/00004872-200109000-00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grobe JL, Mecca AP, Lingis M, Shenoy V, Bolton TA, Machado JM, Speth RC, Raizada MK, Katovich MJ. Prevention of angiotensin II-induced cardiac remodeling by angiotensin-(1-7) Am J Physiol Heart Circ Physiol. 2007;292:H736–742. doi: 10.1152/ajpheart.00937.2006. [DOI] [PubMed] [Google Scholar]
- Grobe JL, Mecca AP, Mao H, Katovich MJ. Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt model of hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H2417–2423. doi: 10.1152/ajpheart.01170.2005. [DOI] [PubMed] [Google Scholar]
- Hwang GS, Oh KS, Koo HN, Seo HW, You KH, Lee BH. Effects of KR-31378, a novel ATP-sensitive potassium channel activator, on hypertrophy of H9c2 cells and on cardiac dysfunction in rats with congestive heart failure. Eur J Pharmacol. 2006;540:131–138. doi: 10.1016/j.ejphar.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Hirata A, Nozaki H, Kadomoto-Antsuki Y, Tominaga M. Role of angiotensin II type-1 and type-2 receptors on vascular smooth muscle cell growth and glucose metabolism in diabetic rats. Diabetes Res Clin Pract. 2007;75:267–277. doi: 10.1016/j.diabres.2006.06.032. [DOI] [PubMed] [Google Scholar]
- Jackman HL, Massad MG, Sekosan M, Tan F, Brovkovych V, Marcic BM, Erdos EG. Angiotensin 1-9 and 1-7 release in human heart: role of cathepsin A. Hypertension. 2002;39:976–981. doi: 10.1161/01.hyp.0000017283.67962.02. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Kenakin T. What systems can and can't do. Br J Pharmacol. 2008;153:841–843. doi: 10.1038/sj.bjp.0707677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkonen JO, Saarinen J, Kovanen PT. Regulation of local angiotensin II formation in the human heart in the presence of interstitial fluid. Inhibition of chymase by protease inhibitors of interstitial fluid and of angiotensin-converting enzyme by Ang-(1-9) formed by heart carboxypeptidase A-like activity. Circulation. 1997;95:1455–1463. doi: 10.1161/01.cir.95.6.1455. [DOI] [PubMed] [Google Scholar]
- Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension. 2003;41:99–107. doi: 10.1161/01.hyp.0000050101.90932.14. [DOI] [PubMed] [Google Scholar]
- Laskowski A, Woodman OL, Cao AH, Drummond GR, Marshall T, Kaye DM, Ritchie RH. Antioxidant actions contribute to the antihypertrophic effects of atrial natriuretic peptide in neonatal rat cardiomyocytes. Cardiovasc Res. 2006;72:112–123. doi: 10.1016/j.cardiores.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Majalahti T, Suo-Palosaari M, Sarman B, Hautala N, Pikkarainen S, Tokola H, Vuolteenaho O, Wang J, Paradis P, Nemer M, Ruskoaho H. Cardiac BNP gene activation by angiotensin II in vivo. Mol Cell Endocrinol. 2007;273:59–67. doi: 10.1016/j.mce.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Mercure C, Yogi A, Callera GE, Aranha AB, Bader M, Ferreira AJ, Santos RA, Walther T, Touyz RM, Reudelhuber TL. Angiotensin(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res. 2008;103:1319–1326. doi: 10.1161/CIRCRESAHA.108.184911. [DOI] [PubMed] [Google Scholar]
- Metcalfe BL, Huentelman MJ, Parilak LD, Taylor DG, Katovich MJ, Knot HJ, Sumners C, Raizada MK. Prevention of cardiac hypertrophy by angiotensin II type-2 receptor gene transfer. Hypertension. 2004;43:1233–1238. doi: 10.1161/01.HYP.0000127563.14064.FD. [DOI] [PubMed] [Google Scholar]
- Miura S, Karnik SS. Angiotensin II type 1 and type 2 receptors bind angiotensin II through different types of epitope recognition. J Hypertens. 1999;17:397–404. doi: 10.1097/00004872-199917030-00013. [DOI] [PubMed] [Google Scholar]
- Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol. 2003;35:851–859. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- Ocaranza MP, Godoy I, Jalil JE, Varas M, Collantes P, Pinto M, Roman M, Ramirez C, Copaja M, Diaz-Araya G, Castro P, Lavandero S. Enalapril attenuates downregulation of angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension. 2006;48:572–578. doi: 10.1161/01.HYP.0000237862.94083.45. [DOI] [PubMed] [Google Scholar]
- Ocaranza MP, Lavandero S, Jalil JE, Moya J, Pinto M, Novoa U, Apablaza F, Gonzalez L, Hernandez C, Varas M, Lopez R, Godoy I, Verdejo H, Chiong M. Angiotensin-(1-9) regulates cardiac hypertrophy in vivo and in vitro. J Hypertens. 2010;28:1054–1064. doi: 10.1097/hjh.0b013e328335d291. [DOI] [PubMed] [Google Scholar]
- Ohkubo N, Matsubara H, Nozawa Y, Mori Y, Murasawa S, Kijima K, Maruyama K, Masaki H, Tsutumi Y, Shibazaki Y, Iwasaka T, Inada M. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation. 1997;96:3954–3962. doi: 10.1161/01.cir.96.11.3954. [DOI] [PubMed] [Google Scholar]
- Pan J, Singh US, Takahashi T, Oka Y, Palm-Leis A, Herbelin BS, Baker KM. PKC mediates cyclic stretch-induced cardiac hypertrophy through Rho family GTPases and mitogen-activated protein kinases in cardiomyocytes. J Cell Physiol. 2005;202:536–553. doi: 10.1002/jcp.20151. [DOI] [PubMed] [Google Scholar]
- Peiro C, Vallejo S, Gembardt F, Azcutia V, Heringer-Walther S, Rodriguez-Manas L, Schultheiss HP, Sanchez-Ferrer CF, Walther T. Endothelial dysfunction through genetic deletion or inhibition of the G protein-coupled receptor Mas: a new target to improve endothelial function. J Hypertens. 2007;25:2421–2425. doi: 10.1097/HJH.0b013e3282f0143c. [DOI] [PubMed] [Google Scholar]
- Pinheiro SV, Ferreira AJ, Kitten GT, da Silveira KD, da Silva DA, Santos SH, Gava E, Castro CH, Magalhaes JA, da Mota RK, Botelho-Santos GA, Bader M, Alenina N, Santos RA, Simoes e Silva AC. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int. 2009;75:1184–1193. doi: 10.1038/ki.2009.61. [DOI] [PubMed] [Google Scholar]
- Ritter O, Schuh K, Brede M, Rothlein N, Burkard N, Hein L, Neyses L. AT2 receptor activation regulates myocardial eNOS expression via the calcineurin-NF-AT pathway. FASEB J. 2003;17:283–285. doi: 10.1096/fj.02-0321fje. [DOI] [PubMed] [Google Scholar]
- Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension. 2007;49:185–192. doi: 10.1161/01.HYP.0000251865.35728.2f. [DOI] [PubMed] [Google Scholar]
- Santos RA, Castro CH, Gava E, Pinheiro SV, Almeida AP, Paula RD, Cruz JS, Ramos AS, Rosa KT, Irigoyen MC, Bader M, Alenina N, Kitten GT, Ferreira AJ. Impairment of in vitro and in vivo heart function in angiotensin-(1-7) receptor MAS knockout mice. Hypertension. 2006;47:996–1002. doi: 10.1161/01.HYP.0000215289.51180.5c. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, Heringer-Walther S, Pinheiro SV, Lopes MT, Bader M, Mendes EP, Lemos VS, Campagnole-Santos MJ, Schultheiss HP, Speth R, Walther T. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter KD, Wenzel S. Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol Ther. 2008;119:311–325. doi: 10.1016/j.pharmthera.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Silva DM, Vianna HR, Cortes SF, Campagnole-Santos MJ, Santos RA, Lemos VS. Evidence for a new angiotensin-(1-7) receptor subtype in the aorta of Sprague-Dawley rats. Peptides. 2007;28:702–707. doi: 10.1016/j.peptides.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Bennett KA, Milligan G. When simple agonism is not enough: Emerging modalities of GPCR ligands. Mol Cell Endocrinol. 2010 doi: 10.1016/j.mce.2010.07.009. (in press) [DOI] [PubMed] [Google Scholar]
- Soares de Moura R, Resende AC, Emiliano AF, Tano T, Mendes-Ribeiro AC, Correia ML, de Carvalho LC. The role of bradykinin, AT2 and angiotensin 1-7 receptors in the EDRF-dependent vasodilator effect of angiotensin II on the isolated mesenteric vascular bed of the rat. Br J Pharmacol. 2004;141:860–866. doi: 10.1038/sj.bjp.0705669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A. 2005;102:437–442. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WG, Brandenburger Y, Autelitano DJ, Pham T, Qian H, Hannan RD. Adenoviral-directed expression of the type 1A angiotensin receptor promotes cardiomyocyte hypertrophy via transactivation of the epidermal growth factor receptor. Circ Res. 2002;90:135–142. doi: 10.1161/hh0202.104109. [DOI] [PubMed] [Google Scholar]
- Vijayan K, Szotek EL, Martin JL, Samarel AM. Protein kinase C-α-induced hypertrophy of neonatal rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H2777–2789. doi: 10.1152/ajpheart.00171.2004. [DOI] [PubMed] [Google Scholar]
- Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension. 2005;45:960–966. doi: 10.1161/01.HYP.0000160325.59323.b8. [DOI] [PubMed] [Google Scholar]
- Wang C, Jayadev S, Escobedo JA. Identification of a domain in the angiotensin II type 1 receptor determining Gq coupling by the use of receptor chimeras. J Biol Chem. 1995;270:16677–16682. doi: 10.1074/jbc.270.28.16677. [DOI] [PubMed] [Google Scholar]
- Yayama K, Okamoto H. Angiotensin II-induced vasodilation via type 2 receptor: role of bradykinin and nitric oxide. Int Immunopharmacol. 2008;8:312–318. doi: 10.1016/j.intimp.2007.06.012. [DOI] [PubMed] [Google Scholar]
- Zou Y, Komuro I, Yamazaki T, Aikawa R, Kudoh S, Shiojima I, Hiroi Y, Mizuno T, Yazaki Y. Protein kinase C, but not tyrosine kinases or Ras, plays a critical role in angiotensin II-induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J Biol Chem. 1996;271:33592–33597. doi: 10.1074/jbc.271.52.33592. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.