

Abstract
We have previously shown that insulin increases muscle total carnitine (TC) content during acute i.v. l-carnitine infusion. Here we determined the effects of chronic l-carnitine and carbohydrate (CHO; to elevate serum insulin) ingestion on muscle TC content and exercise metabolism and performance in humans. On three visits, each separated by 12 weeks, 14 healthy male volunteers (age 25.9 ± 2.1 years, BMI 23.0 ± 0.8 kg m−2) performed an exercise test comprising 30 min cycling at 50%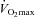 , 30 min at 80%
, 30 min at 80%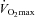 , then a 30 min work output performance trial. Muscle biopsies were obtained at rest and after exercise at 50% and 80%
, then a 30 min work output performance trial. Muscle biopsies were obtained at rest and after exercise at 50% and 80%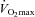 on each occasion. Following visit one, volunteers ingested either 80 g of CHO (Control) or 2 g of l-carnitine-l-tartrate and 80 g of CHO (Carnitine) twice daily for 24 weeks in a randomised, double blind manner. All significant effects reported occurred after 24 weeks. Muscle TC increased from basal by 21% in Carnitine (P < 0.05), and was unchanged in Control. At 50%
on each occasion. Following visit one, volunteers ingested either 80 g of CHO (Control) or 2 g of l-carnitine-l-tartrate and 80 g of CHO (Carnitine) twice daily for 24 weeks in a randomised, double blind manner. All significant effects reported occurred after 24 weeks. Muscle TC increased from basal by 21% in Carnitine (P < 0.05), and was unchanged in Control. At 50%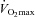 , the Carnitine group utilised 55% less muscle glycogen compared to Control (P < 0.05) and 31% less pyruvate dehydrogenase complex (PDC) activation compared to before supplementation (P < 0.05). Conversely, at 80%
, the Carnitine group utilised 55% less muscle glycogen compared to Control (P < 0.05) and 31% less pyruvate dehydrogenase complex (PDC) activation compared to before supplementation (P < 0.05). Conversely, at 80%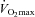 , muscle PDC activation was 38% higher (P < 0.05), acetylcarnitine content showed a trend to be 16% greater (P < 0.10), muscle lactate content was 44% lower (P < 0.05) and the muscle PCr/ATP ratio was better maintained (P < 0.05) in Carnitine compared to Control. The Carnitine group increased work output 11% from baseline in the performance trial, while Control showed no change. This is the first demonstration that human muscle TC can be increased by dietary means and results in muscle glycogen sparing during low intensity exercise (consistent with an increase in lipid utilisation) and a better matching of glycolytic, PDC and mitochondrial flux during high intensity exercise, thereby reducing muscle anaerobic ATP production. Furthermore, these changes were associated with an improvement in exercise performance.
, muscle PDC activation was 38% higher (P < 0.05), acetylcarnitine content showed a trend to be 16% greater (P < 0.10), muscle lactate content was 44% lower (P < 0.05) and the muscle PCr/ATP ratio was better maintained (P < 0.05) in Carnitine compared to Control. The Carnitine group increased work output 11% from baseline in the performance trial, while Control showed no change. This is the first demonstration that human muscle TC can be increased by dietary means and results in muscle glycogen sparing during low intensity exercise (consistent with an increase in lipid utilisation) and a better matching of glycolytic, PDC and mitochondrial flux during high intensity exercise, thereby reducing muscle anaerobic ATP production. Furthermore, these changes were associated with an improvement in exercise performance.
Non-technical summary
After 30 years of endeavour, this is the first study to show that muscle carnitine content can be increased in humans by dietary means and, perhaps more importantly, that carnitine plays a dual role in skeletal muscle fuel metabolism that is exercise intensity dependent. Specifically, we have shown that increasing muscle total carnitine content reduces muscle carbohydrate use during low intensity exercise, consistent with an increase in muscle lipid utilisation. However, during high intensity exercise muscle carnitine loading results in a better matching of glycolytic, pyruvate dehydrogenase complex and mitochondrial flux, thereby reducing muscle anaerobic energy generation. Collectively, these metabolic effects resulted in a reduced perception of effort and increased work output during a validated exercise performance test. These findings have significant implications for athletic performance and pathophysiological conditions where fat oxidation is impaired or anaerobic ATP production is increased during exercise.
Introduction
More than 95% of the body's carnitine pool is confined to skeletal muscle, where it fulfils two major metabolic roles. Firstly, in mitochondrial fatty acid translocation carnitine is a substrate for carnitine palmitoyl-transferase 1 (CPT1) (Fritz & McEwen, 1959; Fritz & Yue, 1963). Secondly, during high intensity exercise, the formation of acetylcarnitine is essential for the maintenance of a viable pool of free co-enzyme A (CoASH), thereby enabling PDC and TCA flux to continue (Childress & Sacktor, 1966; Harris et al. 1987; Constantin-Teodosiu et al. 1991a). Not surprisingly therefore, oral carnitine feeding has been advocated as an ergogenic aid, the main premise being that increasing muscle carnitine content will increase muscle fat oxidation and delay muscle glycogen depletion. However, we are aware of no study that has unequivocally shown carnitine feeding can impact on muscle fuel metabolism or exercise performance, which is undoubtedly attributable to carnitine ingestion (or indeed i.v. carnitine infusion) per se failing to increase muscle carnitine content (Barnett et al. 1994; Vukovich et al. 1994; Wächter et al. 2002; Stephens et al. 2006a).
In a series of i.v. infusion studies, we demonstrated that elevating serum insulin concentration in the presence of hypercarnitinaemia (550–600 μmol l−1) acutely increased muscle total carnitine (TC) content by ∼15% in humans (Stephens et al. 2006a,b;). Furthermore, this increase in carnitine retention occurred only when serum insulin concentration was elevated above 50 mU l−1, which we confirmed could be achieved by combined carbohydrate (CHO) and l-carnitine feeding (94 g and 3 g, respectively), albeit at a much lower rate of retention (equating to a projected ∼0.1% increase in the muscle TC pool per day; Stephens et al. 2007). Assuming this effect of combined CHO and carnitine feeding on carnitine retention is sustainable and cumulative, it was calculated that about 24 weeks of feeding would be needed to increase skeletal muscle TC content to the same extent as acute i.v. carnitine and insulin infusion.
During low intensity exercise, when PDC activation (PDCa) and flux are relatively low, the principal role of carnitine will most likely be mitochondrial fatty acid translocation. Although it has been suggested that free carnitine only limits fat oxidation at exercise intensities >70% of maximal oxygen consumption (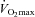 ; van Loon et al. 2001), we demonstrated that an acute 15% increase in muscle carnitine content reduced insulin-mediated muscle glycolytic flux and PDCa compared to control. Furthermore, this was accompanied by a subsequent increase in muscle glycogen and long-chain acyl-co-enzyme A accumulation (Stephens et al. 2006b), pointing to a carnitine-mediated increase in muscle fatty acid oxidation and CHO storage. Free carnitine availability may therefore be limiting to mitochondrial fatty acid translocation even at rest and during low intensity exercise, and an increase in skeletal muscle TC content would be expected to augment fatty acid oxidation and decrease PDCa and glycogen use during low intensity exercise.
; van Loon et al. 2001), we demonstrated that an acute 15% increase in muscle carnitine content reduced insulin-mediated muscle glycolytic flux and PDCa compared to control. Furthermore, this was accompanied by a subsequent increase in muscle glycogen and long-chain acyl-co-enzyme A accumulation (Stephens et al. 2006b), pointing to a carnitine-mediated increase in muscle fatty acid oxidation and CHO storage. Free carnitine availability may therefore be limiting to mitochondrial fatty acid translocation even at rest and during low intensity exercise, and an increase in skeletal muscle TC content would be expected to augment fatty acid oxidation and decrease PDCa and glycogen use during low intensity exercise.
During high intensity exercise, the primary functional role of carnitine shifts towards acetyl group buffering (i.e. forming acetylcarnitine) and maintaining a pool of free CoASH which is essential for mitochondrial flux to continue (including the PDC reaction). However, during exercise of this nature there is still an increase in the acetyl-co-enzyme A (acetyl-CoA)/CoASH ratio, possibly due to the significant depletion (to <6 mmol (kg dry muscle)−1) of the free carnitine pool caused by acetylcarnitine formation (Harris et al. 1987; van Loon et al. 2001). Therefore, it is plausible that an increase in skeletal muscle TC content would provide more effective buffering of acetyl-CoA production during high intensity exercise, offsetting the increase in the acetyl-CoA/CoASH ratio, and thereby increasing PDC flux and mitochondrial ATP production. This in turn would reduce the contribution from glycolysis and PCr hydrolysis to ATP production, particularly during the rest to exercise transition period when inertia in mitochondrial ATP production is known to reside at the level of PDC activation and flux (Timmons et al. 1997, 1998; Howlett et al. 1999; Roberts et al. 2002, 2005). In addition, increasing PDC flux during high intensity exercise would be expected to decrease muscle lactic acid production which could translate to a positive effect on exercise performance by reducing muscle acidosis (Sahlin, 1992). In support of this stance, previous work from our laboratory has shown that pharmacological activation of PDC at rest using the pyruvate dehydrogenase kinase inhibitor, dichloroacetate, markedly reduced muscle lactate production and PCr hydrolysis during a subsequent 20 min period of intense muscle contraction in isolated and perfused canine skeletal muscle, resulting in a substantial improvement in tension development (Timmons et al. 1997).
Based upon the above information, it is logical to conclude that increasing skeletal muscle TC content could alter muscle fuel metabolism in at least two different ways during exercise, with the dominating role being dictated by the exercise intensity employed. With this in mind, we first aimed to determine whether chronic l-carnitine and CHO feeding to healthy male volunteers could increase skeletal muscle TC content in a manner similar to that which we have observed acutely under i.v. l-carnitine infusion and hyperinsulinaemic clamp conditions. Secondly, we hypothesised that any increase in muscle TC content would result in a blunting of PDCa and flux during low intensity exercise, causing a corresponding decrease in muscle glycogen utilisation. Thirdly, during high intensity exercise, when the primary functional role of carnitine switches to acetyl group buffering, we proposed that increasing muscle carnitine content would increase muscle PDC flux (and mitochondrial ATP delivery), thereby reducing anaerobic ATP production and muscle lactic acidosis during exercise. Finally, we hypothesised that these positive metabolic effects of muscle carnitine loading would improve high intensity exercise performance.
Methods
Human volunteers
Fourteen healthy, non-smoking, non-vegetarian recreational athletes (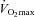 51.6 ± 2.5 ml (kg body mass)−1, training 3–5 times per week in triathlon, cycling, running or swimming), aged 25.9 ± 2.1 years and with a body mass index (BMI) of 23.0 ± 0.8 kg m–2 participated in this study. Moderately trained recreational athletes were recruited as they were accustomed to ingesting CHO supplements. The study was approved by the University of Nottingham Medical School Ethics Committee in accordance with the Declaration of Helsinki. Prior to the study, each subject completed a routine medical screening and a general health questionnaire to ensure their suitability to take part. All gave their informed written consent to participate in the study and were aware that they were free to withdraw from the experiment at any point.
51.6 ± 2.5 ml (kg body mass)−1, training 3–5 times per week in triathlon, cycling, running or swimming), aged 25.9 ± 2.1 years and with a body mass index (BMI) of 23.0 ± 0.8 kg m–2 participated in this study. Moderately trained recreational athletes were recruited as they were accustomed to ingesting CHO supplements. The study was approved by the University of Nottingham Medical School Ethics Committee in accordance with the Declaration of Helsinki. Prior to the study, each subject completed a routine medical screening and a general health questionnaire to ensure their suitability to take part. All gave their informed written consent to participate in the study and were aware that they were free to withdraw from the experiment at any point.
Pre-testing
Fourteen days before the trial, each subject's 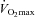 was measured using an electronically braked cycle ergometer (Lode NV Instrumenten, Groningen, the Netherlands) and a continuous and incremental, exhaustive exercise protocol. Oxygen consumption was measured using an online gas analyser (Vmax; SensorMedics, Anaheim, CA, USA) and
was measured using an electronically braked cycle ergometer (Lode NV Instrumenten, Groningen, the Netherlands) and a continuous and incremental, exhaustive exercise protocol. Oxygen consumption was measured using an online gas analyser (Vmax; SensorMedics, Anaheim, CA, USA) and 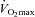 was confirmed during a repeat test 3 days later.
was confirmed during a repeat test 3 days later. 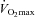 was accepted when a plateau in oxygen consumption was achieved despite a further increase in workload. Once
was accepted when a plateau in oxygen consumption was achieved despite a further increase in workload. Once 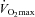 had been obtained, workloads to be used in subsequent experimental visits were calculated that would elicit 50% and 80% of
had been obtained, workloads to be used in subsequent experimental visits were calculated that would elicit 50% and 80% of 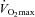 . Subjects were familiarised with the experimental exercise protocol (which also allowed confirmation that the workloads were at the appropriate intensity to elicit 50% and 80%
. Subjects were familiarised with the experimental exercise protocol (which also allowed confirmation that the workloads were at the appropriate intensity to elicit 50% and 80%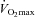 ) at least 1 week prior to the beginning of subsequent experimental visits. A repeat
) at least 1 week prior to the beginning of subsequent experimental visits. A repeat 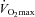 test was also performed at least 1 week after the completion of the study to confirm no significant change in aerobic capacity had occurred over the course of the study.
test was also performed at least 1 week after the completion of the study to confirm no significant change in aerobic capacity had occurred over the course of the study.
Experimental protocol
Volunteers reported to the laboratory at 08.30 h on three occasions over a 24 week period, each visit being separated by 12 weeks. Subjects arrived after an overnight fast having abstained from strenuous exercise and alcohol consumption for at least 48 h, and caffeine for at least 24 h. On each visit, subjects performed the following experimental protocol. On arrival at the laboratory, subjects were weighed and then rested in a semi-supine position whilst a resting blood sample was collected from an antecubital vein (venipuncture) for blood glucose, serum insulin and plasma TC concentration measurements. Volunteers then exercised for 30 min on the cycle ergometer at a workload corresponding to 50%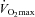 followed immediately by 30 min of exercise at a predetermined workload of 80%
followed immediately by 30 min of exercise at a predetermined workload of 80%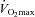 . During both bouts of exercise, a rating of perceived exertion (Borg Scale) was obtained every 10 min. Finally, immediately following the completion of exercise at 80%
. During both bouts of exercise, a rating of perceived exertion (Borg Scale) was obtained every 10 min. Finally, immediately following the completion of exercise at 80%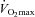 , subjects performed a 30 min work output performance test. This ‘all-out’ performance test involved using the ergometer hyperbolic mode function, where work output is dependent upon volitional cycling cadence. This performance test has been shown to be a more reliable measurement of endurance exercise performance than cycling at a fixed exercise workload to volitional exhaustion (Jeukendrup et al. 1996), and has been used previously in our laboratory to measure performance (Stephens et al. 2008).
, subjects performed a 30 min work output performance test. This ‘all-out’ performance test involved using the ergometer hyperbolic mode function, where work output is dependent upon volitional cycling cadence. This performance test has been shown to be a more reliable measurement of endurance exercise performance than cycling at a fixed exercise workload to volitional exhaustion (Jeukendrup et al. 1996), and has been used previously in our laboratory to measure performance (Stephens et al. 2008).
Supplementation protocol
After the first experimental visit, subjects were allocated in a randomised, double blind manner to two experimental treatment groups. One group (n= 7) was instructed to consume 700 ml of a solution containing 80 g of orange-flavoured CHO polymer (Vitargo; Swecarb AB, Stockholm, Sweden) on two occasions each day for 168 days (Control), whilst the remaining group consumed 80 g of orange-flavoured CHO polymer containing 2.0 g of l-carnitine tartrate (1.36 g of l-carnitine; Carnipure™, Lonza Group Ltd, Basel, Switzerland) in the same volume of solution and at the same frequency (Carnitine). Volunteers were instructed to ingest the first supplement at breakfast time and the second 4 h later. This feeding protocol was based upon a regimen we have previously shown to increase whole-body carnitine retention over a 14 day period (Stephens et al. 2007). Volunteers were informed of the caloric content of the drinks (∼600 calories per day) and advised to replace their customary CHO supplement with the prescribed supplement and/or amend their diet accordingly to try and avoid weight gain. Volunteers were requested to record any side effects associated with supplementation over the 24 week protocol. None were reported by either group.
Sample collection and analysis
On each experimental study day, venous blood samples were collected whilst subjects rested in a semi-supine position. Following collection, blood glucose concentrations were determined immediately using an autoanalyser (YSI 2300 STATplus, Yellow Springs Instruments, Yellow Springs, OH, USA). Two millilitres of each basal resting blood sample was collected into lithium heparin containers, and following centrifugation (22,000 RCF at +4°C for 2 min) the plasma was snap frozen in liquid nitrogen and stored at −80°C until used to determine plasma TC concentration using a radioenzymatic assay (Cederblad et al. 1982). Finally, a further 2 ml of each basal resting blood sample was allowed to clot and following centrifugation (1,400 RCF at +4°C for 10 min) the serum was stored frozen at −80°C until used to determine insulin concentration using a commercially available radioimmunoassay kit (Coat-a-Count Insulin, Diagnostics Products Corporation, Los Angeles, CA, USA).
On each experimental visit, muscle biopsy samples were obtained from the vastus lateralis muscle at rest and within 5 s of the end of exercise at 50% and 80%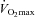 (whilst subjects were seated on the cycle ergometer) using the percutaneous needle biopsy technique (Bergström, 1975). Muscle samples were immediately snap frozen in liquid nitrogen after removal from the limb. One portion of each biopsy sample was freeze dried and stored at −80°C, whilst the remainder was stored ‘wet’ in liquid nitrogen. Freeze-dried muscle was dissected free of visible blood and connective tissue, powdered and used for the determination of muscle free carnitine, acetylcarnitine and long-chain acylcarnitine using the radioenzymatic method described previously by Cederblad (Cederblad et al. 1990). Muscle ATP, phosphocreatine (PCr), free creatine, lactate and glycogen were also determined on freeze-dried muscle using the spectrophotometric method of Harris (Harris et al. 1974). Muscle TC was calculated as the sum of the three carnitine moieties, and was normalised for the highest total creatine content from each individual's three biopsies of that visit, a procedure routinely carried out to minimise variability from non-muscle constituents (Stephens et al. 2006a,b;). Muscle total creatine content was calculated as the sum of free creatine and PCr. Approximately 10 mg of the ‘wet’ muscle was used to determine PDCa, expressed as the rate of acetyl-CoA formation (mmol min−1 (kg wet muscle)−1 at 37°C) using methodology described previously by Constantin-Teodosiu et al. (1991b). In addition, maximal citrate synthase activity was determined spectrophotometrically on whole muscle homogenates based on the methods of Opie & Newsholme (1967); Zammit & Newsholme (1976) and expressed as mmol min−1 (kg wet muscle)−1.
(whilst subjects were seated on the cycle ergometer) using the percutaneous needle biopsy technique (Bergström, 1975). Muscle samples were immediately snap frozen in liquid nitrogen after removal from the limb. One portion of each biopsy sample was freeze dried and stored at −80°C, whilst the remainder was stored ‘wet’ in liquid nitrogen. Freeze-dried muscle was dissected free of visible blood and connective tissue, powdered and used for the determination of muscle free carnitine, acetylcarnitine and long-chain acylcarnitine using the radioenzymatic method described previously by Cederblad (Cederblad et al. 1990). Muscle ATP, phosphocreatine (PCr), free creatine, lactate and glycogen were also determined on freeze-dried muscle using the spectrophotometric method of Harris (Harris et al. 1974). Muscle TC was calculated as the sum of the three carnitine moieties, and was normalised for the highest total creatine content from each individual's three biopsies of that visit, a procedure routinely carried out to minimise variability from non-muscle constituents (Stephens et al. 2006a,b;). Muscle total creatine content was calculated as the sum of free creatine and PCr. Approximately 10 mg of the ‘wet’ muscle was used to determine PDCa, expressed as the rate of acetyl-CoA formation (mmol min−1 (kg wet muscle)−1 at 37°C) using methodology described previously by Constantin-Teodosiu et al. (1991b). In addition, maximal citrate synthase activity was determined spectrophotometrically on whole muscle homogenates based on the methods of Opie & Newsholme (1967); Zammit & Newsholme (1976) and expressed as mmol min−1 (kg wet muscle)−1.
Statistical analysis
A two-way ANOVA for repeated measures (time and treatment effects) was performed to detect differences within and between treatment groups separately for the three conditions (rest, 50%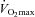 and 80%
and 80%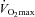 ). When a significant time or treatment effect was observed a one-way ANOVA or t test was performed, respectively, to locate individual differences. Statistical significance was declared at P < 0.05. All the values presented in text, tables and figures represent mean ± the standard error of the mean (s.e.m.).
). When a significant time or treatment effect was observed a one-way ANOVA or t test was performed, respectively, to locate individual differences. Statistical significance was declared at P < 0.05. All the values presented in text, tables and figures represent mean ± the standard error of the mean (s.e.m.).
Results
Subject characteristics
Subject characteristics are displayed in Table 1. Body mass was not different between groups before supplementation. However, there was a 2.4 kg increase in body mass from basal in the Control group after 12 weeks of supplementation (P < 0.01), which remained elevated after 24 weeks (P < 0.05). The Carnitine group showed no change in body mass over the course of the study. Fasting venous blood glucose and serum insulin concentrations at baseline were not different between groups and did not change throughout the study (Table 1). Fasting plasma TC concentration was also not different between groups before supplementation. However, plasma TC concentration in the Carnitine group was greater after 12 and 24 weeks of supplementation when compared to the Control group (P < 0.05; Table 1). Perceived exertion during exercise at 50%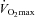 did not differ between groups on any visit. The same was also true during exercise at 80%
did not differ between groups on any visit. The same was also true during exercise at 80%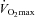 before and after 12 weeks of supplementation. However, after 24 weeks of supplementation perceived exertion was lower in the Carnitine group when compared to baseline (14.0 vs. 15.0, respectively; P < 0.05) and Control at 24 weeks (14.0 vs. 16.2, respectively; P < 0.05).
before and after 12 weeks of supplementation. However, after 24 weeks of supplementation perceived exertion was lower in the Carnitine group when compared to baseline (14.0 vs. 15.0, respectively; P < 0.05) and Control at 24 weeks (14.0 vs. 16.2, respectively; P < 0.05).
Table 1.
Subject characteristics before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7)
| 0 | 12 | 24 | ||||
|---|---|---|---|---|---|---|
| Control | Carnitine | Control | Carnitine | Control | Carnitine | |
| Age (years) | 24.6 ± 2.0 | 27.1 ± 2.2 | — | — | — | — |
| Height (cm) | 181.4 ± 2.5 | 176.2 ± 1.4 | — | — | — | — |
| Body mass (kg) | 73.1 ± 3.7 | 74.2 ± 2.5 | 75.5 ± 3.7 †† | 73.9 ± 2.5 | 75.8 ± 4.3 † | 74.4 ± 2.3 |
| BMI (kg m−2) | 22.2 ± 0.7 | 23.9 ± 0.9 | 22.9 ± 0.7 | 23.8 ± 0.8 | 23.0 + 0.9 | 24.0 ± 0.8 |
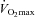 (ml kg−1 min−1) (ml kg−1 min−1) |
50 ± 2.6 | 54 ± 2.2 | — | — | 51 ± 2.7 | 56 ± 2.5 |
| Fasting blood glucose (mmol l−1) | 4.1 ± 0.1 | 4.3 ± 0.1 | 4.2 ± 0.1 | 4.3 ± 0.1 | 4.1 ± 0.1 | 4.4 ± 0.1 |
| Fasting serum insulin (mU l−1) | 10 ± 0.4 | 10 ± 0.4 | 11 ± 0.1 | 10 ± 0.4 | 10 ± 0.4 | 11 ± 0.4 |
| Fasting plasma carnitine (μmol l−1) | 43 ± 2.5 | 44 ± 4.2 | 38 ± 3.3 | 52 ± 4.0* | 41 ± 3.6 | 54 ± 3.3* |
All values are means ±s.e.m. Significantly different from Control:
P < 0.05.
Significantly different from before supplementation (0):
P < 0.05
P < 0.01.
Skeletal muscle total carnitine content
Resting muscle TC content over the course of the study is shown in Fig. 1. There was no difference between or within groups before or after 12 weeks of supplementation. However, after 24 weeks muscle TC content was 30% greater in the Carnitine group compared to Control (P < 0.05), which represented a 21% increase from baseline in the Carnitine group (P < 0.05).
Figure 1.
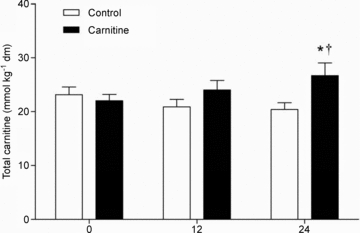
Total skeletal muscle carnitine content (calculated as the mean of 3 biopsies taken from each individual during a given visit) before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7). All values are means ±s.e.m.). Significantly different from Control: *P < 0.05. Significantly different from before supplementation (0): †P < 0.05.
Skeletal muscle metabolites
Absolute muscle metabolite values at rest and during exercise are presented in Table 2. From similar resting values, muscle PCr, glycogen, lactate, acetylcarnitine and free carnitine content changed by a similar magnitude during exercise at 50 and 80%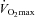 before and after 12 weeks of supplementation in Control and Carnitine groups. However, following 24 weeks of supplementation there was a trend (P= 0.09) for resting free carnitine content to be 30% greater in the Carnitine group compared to Control, and there was a significant difference between groups in the metabolic response to both low and high intensity exercise. Following exercise at 50%
before and after 12 weeks of supplementation in Control and Carnitine groups. However, following 24 weeks of supplementation there was a trend (P= 0.09) for resting free carnitine content to be 30% greater in the Carnitine group compared to Control, and there was a significant difference between groups in the metabolic response to both low and high intensity exercise. Following exercise at 50%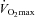 , muscle glycogen content was 35% greater in the Carnitine group compared to Control (P < 0.05), which equated to 55% less glycogen being utilised during exercise (P < 0.05; Fig. 2A), and free carnitine was 78% greater in the Carnitine group when compared to Control (P < 0.01). Following exercise at 80%
, muscle glycogen content was 35% greater in the Carnitine group compared to Control (P < 0.05), which equated to 55% less glycogen being utilised during exercise (P < 0.05; Fig. 2A), and free carnitine was 78% greater in the Carnitine group when compared to Control (P < 0.01). Following exercise at 80%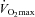 , muscle glycogen content was 71% greater in the Carnitine group compared to Control; however, this was attributable to the reduction in glycogen utilisation during the preceding exercise at 50%
, muscle glycogen content was 71% greater in the Carnitine group compared to Control; however, this was attributable to the reduction in glycogen utilisation during the preceding exercise at 50%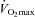 (see above), and accordingly there was no difference between groups in glycogen utilisation during exercise at 80%
(see above), and accordingly there was no difference between groups in glycogen utilisation during exercise at 80%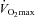 (Fig. 2A). However, muscle lactate content was 44% lower in the Carnitine group compared to Control (P < 0.05) following exercise at 80%
(Fig. 2A). However, muscle lactate content was 44% lower in the Carnitine group compared to Control (P < 0.05) following exercise at 80%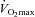 , which translated into a marked reduction in muscle lactate accumulation during exercise (P < 0.05, Fig. 2B) and was accompanied by a trend (P < 0.10) for muscle acetylcarnitine and free carnitine content to be greater in the Carnitine group when compared to Control (16% and 63%, respectively). In addition, after 24 weeks of supplementation, the muscle PCr/ATP ratio in the Carnitine group was significantly greater than Control (P < 0.05) and baseline (P < 0.05) following exercise at 80%
, which translated into a marked reduction in muscle lactate accumulation during exercise (P < 0.05, Fig. 2B) and was accompanied by a trend (P < 0.10) for muscle acetylcarnitine and free carnitine content to be greater in the Carnitine group when compared to Control (16% and 63%, respectively). In addition, after 24 weeks of supplementation, the muscle PCr/ATP ratio in the Carnitine group was significantly greater than Control (P < 0.05) and baseline (P < 0.05) following exercise at 80%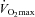 .
.
Table 2.
Skeletal muscle metabolites at rest and following 30 min of exercise at 50 and 80%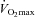 before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7)
before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7)
| Rest | 50%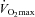
|
80%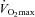
|
|||||
|---|---|---|---|---|---|---|---|
| Control | Carnitine | Control | Carnitine | Control | Carnitine | ||
| 0 | PCr | 79.3 ± 8.6 | 90.0 ± 5.2 | 71.0 ± 8.0 | 64.8 ± 12.2 | 41.9 ± 5.2 | 38.1 ± 5.3 |
| PCr/ATP ratio | 3.5 ± 0.4 | 3.6 ± 0.2 | 3.1 ± 0.1 | 2.9 ± 0.3 | 1.6 ± 0.2 | 1.4 ± 0.2 | |
| Glycogen | 430.2 ± 29.2 | 436.3 ± 20.5 | 380.7 ± 28.4 | 386.4 ± 10.1 | 235.2 ± 29.8 | 260.5 ± 22.7 | |
| Lactate | 3.7 ± 1.4 | 3.0 ± 0.8 | 7.1 ± 2.4 | 7.5 ± 2.5 | 29.0 ± 4.7 | 27.5 ± 5.5 | |
| Acetylcarnitine | 2.6 ± 0.4 | 2.5 ± 0.6 | 6.7 ± 0.8 | 8.1 ± 1.6 | 15.8 ± 0.8 | 17.4 ± 0.9 | |
| Free carnitine | 17.9 ± 1.8 | 18.0 ± 1.8 | 14.0 ± 1.4 | 14.5 ± 1.5 | 9.4 ± 1.3 | 5.1 ± 0.9 | |
| Acyl-carnitine | 0.9 ± 0.1 | 0.9 ± 0.1 | 0.8 ± 0.1 | 1.0 ± 0.1 | 1.3 ± 0.1 | 1.0 ± 0.1 | |
| 12 | PCr | 87.8 ± 4.5 | 88.7 ± 5.8 | 63.6 ± 10.6 | 69.5 ± 6.3 | 47.9 ± 5.9 | 51.9 ± 8.6 |
| PCr/ATP ratio | 3.6 ± 0.2 | 4.1 ± 0.5 | 3.1 ± 0.3 | 2.8 ± 0.2 | 1.8 ± 0.3 | 2.0 ± 0.3 | |
| Glycogen | 441.9 ± 34.0 | 393.2 ± 10.0 | 384.5 ± 40.4 | 346.4 ± 15.8 | 206.0 26.8 | 181.6 ± 17.6 | |
| Lactate | 4.5 ± 0.6 | 3.2 ± 0.5 | 7.9 ± 2.3 | 5.5 ± 0.9 | 37.9 ± 5.0 | 32.0 ± 5.8 | |
| Acetylcarnitine | 3.2 ± 0.6 | 4.4 ± 1.1 | 7.8 ± 1.7 | 7.0 ± 1.1 | 12.6 ± 1.3 | 15.6 ± 2.0 | |
| Free carnitine | 16.2 ± 1.9 | 19.5 ± 2.0 | 12.7 ± 1.5 | 15.8 ± 1.3 | 7.9 ± 1.0 | 6.5 ± 1.0 | |
| Acyl-carnitine | 0.7 ± 0.1 | 0.9 ± 0.2 | 0.9 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.1 | 1.6 ± 0.4 | |
| 24 | PCr | 70.3 ± 9.6 | 83.5 ± 9.0 | 70.3 ± 5.0 | 68.4 ± 13.0 | 33.3 ± 6.5 | 48.6 ± 6.3 |
| PCr/ATP ratio | 3.7 ± 0.1 | 3.4 ± 0.3 | 2.5 ± 0.4 | 3.0 ± 0.3 | 1.3 ± 0.2 | 2.7 ± 0.3*† | |
| Glycogen | 386.4 ± 33.8 | 467.6 ± 24.8 | 326.1 ± 33.9 | 440.7 ± 24.1* | 146.8 ± 17.4 | 251.7 ± 37.7* | |
| Lactate | 4.4 ± 0.8 | 4.5 ± 1.0 | 7.7 ± 1.2 | 5.5 ± 0.6 | 44.4 ± 6.9 | 25.0 ± 4.1* | |
| Acetylcarnitine | 2.8 ± 0.7 | 2.7 ± 0.8 | 7.7 ± 1.7 | 6.7 ± 1.5 | 15.8 ± 1.4 | 18.4 ± 1.7 | |
| Free carnitine | 15.9 ± 1.6 | 20.7 ± 2.2 | 11.0 ± 1.3 | 19.6 ± 2.4**† | 5.4 ± 0.7 | 8.8 ± 1.7 | |
| Acyl-carnitine | 0.9 ± 0.2 | 0.8 ± 0.1 | 0.7 ± 0.1 | 1.0 ± 0.2 | 0.9 ± 0.1 | 1.2 ± 0.4 | |
All values are means ±s.e.m. and expressed as mmol (kg dry muscle)−1. Significantly different from Control:
P < 0.05
P < 0.01.
Significantly different from before supplementation (0):
P < 0.05.
Figure 2.
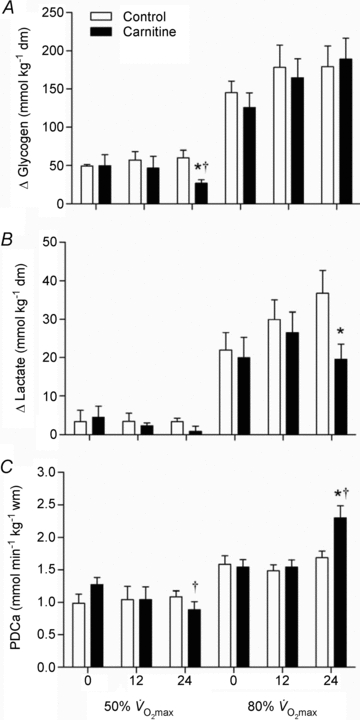
Skeletal muscle glycogen utilisation (A), lactate accumulation (B) and pyruvate dehydrogenase complex activation status (C) during 30 min of exercise at 50 and 80%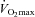 before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7). All values are means ±s.e.m. Significantly different from corresponding Control: *P < 0.05. Significantly different from before supplementation (0): †P < 0.05.
before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7). All values are means ±s.e.m. Significantly different from corresponding Control: *P < 0.05. Significantly different from before supplementation (0): †P < 0.05.
Muscle PDCa
Muscle pyruvate dehydrogenase activation status (PDCa) following exercise is shown in Fig. 2C. Resting muscle PDCa was not different between treatment groups at any time-point, being maintained at ∼0.4 mmol acetyl-CoA min−1 (kg wet muscle)−1. Similarly, muscle PDCa following exercise at 50%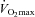 was not different between treatment groups at baseline and 12 weeks; however, at 24 weeks PDCa was 31% lower than baseline in the Carnitine group (P < 0.05). PDCa following exercise at 80%
was not different between treatment groups at baseline and 12 weeks; however, at 24 weeks PDCa was 31% lower than baseline in the Carnitine group (P < 0.05). PDCa following exercise at 80%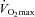 was not different between treatment groups at baseline or after 12 weeks, but was 38% greater in the Carnitine group at 24 weeks when compared with Control (P < 0.05).
was not different between treatment groups at baseline or after 12 weeks, but was 38% greater in the Carnitine group at 24 weeks when compared with Control (P < 0.05).
Muscle citrate synthase activity
Muscle citrate synthase activity was not different between Control or Carnitine groups at baseline (5.6 ± 0.9 and 5.0 ± 0.4 mmol min−1 (kg wet muscle)−1, respectively) or after 24 weeks of supplementation (5.4 ± 0.1 and 5.9 ± 0.7 mmol min−1 (kg wet muscle)−1, respectively), and there were also no differences over time.
Exercise performance
Work output (kJ) achieved in the exercise performance test is presented in Fig. 3. Performance was not different between groups before or after 12 weeks of supplementation. However, after 24 weeks work output was 35% greater in the Carnitine group compared to Control (P < 0.05), which represented an 11% increase from baseline (P < 0.05).
Figure 3.
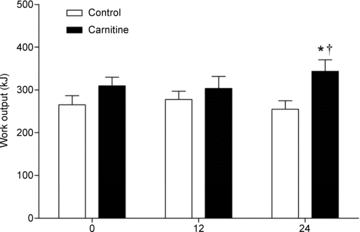
Work output generated during a 30 min ‘all-out’ exercise performance test performed immediately following 30 min of exercise at 50 and 80%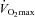 before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7). All values are means ±s.e.m. Significantly different from Control: *P < 0.05. Significantly different from before supplementation (0): †P < 0.05.
before (0) and 12 and 24 weeks after twice daily oral ingestion of either 80 g of carbohydrate (Control; n= 7) or 80 g of carbohydrate containing 2 g l-carnitine tartrate (Carnitine; n= 7). All values are means ±s.e.m. Significantly different from Control: *P < 0.05. Significantly different from before supplementation (0): †P < 0.05.
Discussion
Despite over 30 years of research demonstrating the fundamental role of carnitine in regulating muscle fuel use, attempts to increase skeletal muscle TC content in humans via l-carnitine feeding have been unsuccessful (Barnett et al. 1994; Vukovich et al. 1994; Wächter et al. 2002). The present study is the first to demonstrate that muscle TC content can be increased by 21% in healthy human volunteers when l-carnitine is ingested for 24 weeks in combination with a CHO solution. Moreover, this increase in TC content had a profound effect on muscle fuel utilisation during exercise which was exercise intensity dependent and consistent with the reported dual role of carnitine in muscle fuel metabolism. Namely, during low intensity exercise muscle glycogen utilisation was halved (consistent with an increase in muscle lipid utilisation), whereas during high intensity exercise muscle lactate accumulation was substantially reduced and the muscle PCr/ATP ratio was better maintained, which probably resulted from the carnitine-mediated increase in PDC activation and flux observed at this workload. Finally, increasing skeletal muscle TC content was associated with a 35% improvement in work output over Control, which we propose resulted directly from the observed changes in muscle fuel metabolism.
A major role of carnitine in skeletal muscle is as a substrate for the CPT1-mediated translocation of fatty acids into mitochondria for subsequent β-oxidation (Fritz & McEwen, 1959; Fritz & Yue, 1963). We have previously shown that a 15% increase in muscle TC content resulted in the attenuation of insulin-induced increases in glycolytic flux and PDCa in healthy, resting volunteers, as well as a subsequent overnight increase in muscle glycogen storage. This effect was attributed to a carnitine-mediated increase in acetyl-CoA delivery from fat oxidation, which inhibited PDCa and diverted glucose uptake from oxidation towards storage, and therefore suggests that carnitine availability is limiting to the CPT1 reaction under insulin-stimulated conditions, even at rest (Stephens et al. 2006b). We therefore hypothesised that during low intensity exercise in the present study, when glycolytic and PDC flux are well matched, an increase in muscle free carnitine availability would have a similar effect, i.e. it would augment muscle lipid oxidation thereby blunting PDCa and glycolytic flux. Consistent with this hypothesis, we observed that the increase in muscle TC content after 24 weeks of supplementation was linked to a 55% reduction in muscle glycogen utilisation during exercise at 50%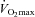 compared to Control. Furthermore, this was accompanied by muscle free carnitine content being ∼80% greater and PDCa being 31% lower during exercise compared to before supplementation, suggesting that a carnitine-mediated increase in lipid-derived acetyl-CoA inhibited PDCa (Pettit et al. 1975) and thereby reduced muscle CHO flux, which is consistent with The Randle Cycle (Randle et al. 1963). Free carnitine availability has been suggested to limit muscle fat oxidation in vivo in humans during intense exercise when its concentration declines below 6 mmol (kg dry muscle)−1 (∼2 mmol (l intracellular water)−1) (van Loon et al. 2001). However, during exercise at 50%
compared to Control. Furthermore, this was accompanied by muscle free carnitine content being ∼80% greater and PDCa being 31% lower during exercise compared to before supplementation, suggesting that a carnitine-mediated increase in lipid-derived acetyl-CoA inhibited PDCa (Pettit et al. 1975) and thereby reduced muscle CHO flux, which is consistent with The Randle Cycle (Randle et al. 1963). Free carnitine availability has been suggested to limit muscle fat oxidation in vivo in humans during intense exercise when its concentration declines below 6 mmol (kg dry muscle)−1 (∼2 mmol (l intracellular water)−1) (van Loon et al. 2001). However, during exercise at 50%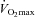 in the present study muscle free carnitine concentration was 11 mmol (kg dry muscle)−1 (∼3.5 mmol (l intracellular water)−1), which is above the value reported by van Loon et al. (2001) and well above the reported Km of CPT1 for free carnitine (0.5 mmol l−1) (McGarry et al. 1983). Therefore, either the reported Km of carnitine for CPT1, generated via in vitro experiments (McGarry et al. 1983) is not transferable to the in vivo situation or, alternatively, although the cellular carnitine pool is thought to be predominantly (90%) cytosolic (Zammit, 1999), the availability of free carnitine to CPT1 is markedly lower than suggested from determination of free carnitine in whole muscle homogenates. An explanation for this may be that the known catalytic site of CPT1 is located within the contact sites of the outer mitochondrial membrane and therefore not entirely available to the cytosolic carnitine pool. An increase in mitochondrial content over the duration of the study could also explain the apparent increase in fat oxidation and glycogen sparing observed in the Carnitine group during exercise at 50%
in the present study muscle free carnitine concentration was 11 mmol (kg dry muscle)−1 (∼3.5 mmol (l intracellular water)−1), which is above the value reported by van Loon et al. (2001) and well above the reported Km of CPT1 for free carnitine (0.5 mmol l−1) (McGarry et al. 1983). Therefore, either the reported Km of carnitine for CPT1, generated via in vitro experiments (McGarry et al. 1983) is not transferable to the in vivo situation or, alternatively, although the cellular carnitine pool is thought to be predominantly (90%) cytosolic (Zammit, 1999), the availability of free carnitine to CPT1 is markedly lower than suggested from determination of free carnitine in whole muscle homogenates. An explanation for this may be that the known catalytic site of CPT1 is located within the contact sites of the outer mitochondrial membrane and therefore not entirely available to the cytosolic carnitine pool. An increase in mitochondrial content over the duration of the study could also explain the apparent increase in fat oxidation and glycogen sparing observed in the Carnitine group during exercise at 50%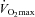 after 24 weeks. However, if this was the case it would be expected that citrate synthase activity and/or
after 24 weeks. However, if this was the case it would be expected that citrate synthase activity and/or 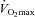 would have also increased in this group, which was not observed. These observations, together with the finding that there was no evidence of glycogen sparing during exercise at 80%
would have also increased in this group, which was not observed. These observations, together with the finding that there was no evidence of glycogen sparing during exercise at 80%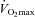 in the Carnitine group, makes it highly unlikely that an increase in mitochondrial content occurred over the 24 weeks of supplementation.
in the Carnitine group, makes it highly unlikely that an increase in mitochondrial content occurred over the 24 weeks of supplementation.
Another widely documented function of carnitine is as an acetyl group buffer during conditions of high glycolytic and PDC flux. During high intensity exercise, when acetyl group production by the PDC reaction is in excess of its utilisation by the TCA cycle, free carnitine buffers against acetyl-CoA accumulation by forming acetylcarnitine in a reaction catalysed by carnitine acetyl transferase (CAT), thereby ensuring a viable supply of free CoASH to sustain TCA cycle flux (Childress & Sacktor, 1966; Harris et al. 1987; Constantin-Teodosiu et al. 1991a). In the context of this, another major finding of the present study was the marked reduction in muscle lactate accumulation during exercise at 80%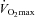 in the carnitine-loaded state after 24 weeks of supplementation when compared to Control, an effect probably mediated by the greater PDCa (38%) and flux (as evidenced by the 16% greater acetylcarnitine content) observed compared to Control. While it is clear that after 24 weeks muscle lactate did not accumulate in the Carnitine group to any lesser of an extent than seen at baseline, it is important to note that the absence of a change occurred in the face of increased glycogenolysis in both groups, which resulted in an increased lactate accumulation in the Control group only, and is explained by the carnitine-mediated increase in PDCa and flux in the Carnitine group. Furthermore, the magnitude of cellular energy disturbance (as indicated by PCr/ATP ratio) was significantly reduced during exercise at 80%
in the carnitine-loaded state after 24 weeks of supplementation when compared to Control, an effect probably mediated by the greater PDCa (38%) and flux (as evidenced by the 16% greater acetylcarnitine content) observed compared to Control. While it is clear that after 24 weeks muscle lactate did not accumulate in the Carnitine group to any lesser of an extent than seen at baseline, it is important to note that the absence of a change occurred in the face of increased glycogenolysis in both groups, which resulted in an increased lactate accumulation in the Control group only, and is explained by the carnitine-mediated increase in PDCa and flux in the Carnitine group. Furthermore, the magnitude of cellular energy disturbance (as indicated by PCr/ATP ratio) was significantly reduced during exercise at 80%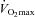 at 24 weeks in the Carnitine group when compared to baseline and Control at 24 weeks. In keeping with this observation, we, and others, have previously reported that inertia in mitochondrial ATP production during the rest to exercise transition is at least partly limited by PDC activation and flux, resulting in increased anaerobic ATP generation (Timmons et al. 1997, 1998; Howlett et al. 1999; Roberts et al. 2002, 2005).
at 24 weeks in the Carnitine group when compared to baseline and Control at 24 weeks. In keeping with this observation, we, and others, have previously reported that inertia in mitochondrial ATP production during the rest to exercise transition is at least partly limited by PDC activation and flux, resulting in increased anaerobic ATP generation (Timmons et al. 1997, 1998; Howlett et al. 1999; Roberts et al. 2002, 2005).
Pyruvate dehydrogenase activation status is principally regulated by a covalent mechanism of competing pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatases (PDP), which inactivate and activate the PDC, respectively. Pyruvate dehydrogenase kinase and PDP are themselves subject to metabolic regulation, and Ca2+ has been suggested as the principal metabolic activator of PDC during exercise by stimulating PDP (Constantin-Teodosiu et al. 2004). Considering that subjects in the present study exercised at the same work intensity (80%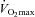 ) during each visit, it can be assumed that the increase in cellular Ca2+ concentration was equal between visits and accordingly a similar stimulatory effect on the PDC was exerted regardless of skeletal muscle TC content. However, PDC activation is also regulated by end-product inhibition, primarily by an increase in the acetyl-CoA/CoASH ratio which stimulates PDK (Cooper et al. 1975; Pettit et al. 1975). Thus, as well as blunting acetyl-CoA accumulation during intense exercise and augmenting PDC flux, increased acetyl group buffering in the Carnitine group may also have modulated a reduction in the acetyl-CoA/CoASH ratio, thereby explaining the greater PDCa in the carnitine-supplemented group at 80%
) during each visit, it can be assumed that the increase in cellular Ca2+ concentration was equal between visits and accordingly a similar stimulatory effect on the PDC was exerted regardless of skeletal muscle TC content. However, PDC activation is also regulated by end-product inhibition, primarily by an increase in the acetyl-CoA/CoASH ratio which stimulates PDK (Cooper et al. 1975; Pettit et al. 1975). Thus, as well as blunting acetyl-CoA accumulation during intense exercise and augmenting PDC flux, increased acetyl group buffering in the Carnitine group may also have modulated a reduction in the acetyl-CoA/CoASH ratio, thereby explaining the greater PDCa in the carnitine-supplemented group at 80%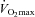 following 24 weeks of carnitine supplementation compared with Control.
following 24 weeks of carnitine supplementation compared with Control.
Given that the performance test used in this study is reproducible (Jeukendrup et al. 1996; Stephens et al. 2008), and all the volunteers were recreational athletes familiar with intense exercise, a major finding of the present study has to be that the increase in muscle TC content after 24 weeks of supplementation resulted in a 35% increase in work output compared to Control (and an 11% increase from baseline). We, and others, have previously demonstrated that complete activation of PDC in the resting state, by pharmacological inhibition of PDK, markedly reduced anaerobic energy production in canine and human skeletal muscle during subsequent intense contraction (Timmons et al. 1997, 1998; Howlett et al. 1999; Roberts et al. 2002, 2005), and resulted in a substantial improvement in muscle contractile function (Timmons et al. 1997). It would appear therefore that the 38% greater PDCa and associated flux during exercise at 80%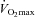 in the carnitine-loaded state in the present study, coupled with the reduction in muscle lactate accumulation and lower perceived exertion during exercise compared with Control, positively impacted upon work output during the subsequent performance trial. Indeed, it is likely that these metabolic effects observed at 80%
in the carnitine-loaded state in the present study, coupled with the reduction in muscle lactate accumulation and lower perceived exertion during exercise compared with Control, positively impacted upon work output during the subsequent performance trial. Indeed, it is likely that these metabolic effects observed at 80%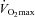 in the carnitine-loaded state continued on into the performance trial given that subjects were attempting to perform as much work as possible in the 30 min of exercise. In keeping with our observations, Brass and colleagues have shown that carnitine loading of rodent soleus muscle reduced fatigue by 25% during electrically evoked contraction (Brass et al. 1993). Furthermore, Coyle (1995) concluded that the ability to maintain a high steady-state
in the carnitine-loaded state continued on into the performance trial given that subjects were attempting to perform as much work as possible in the 30 min of exercise. In keeping with our observations, Brass and colleagues have shown that carnitine loading of rodent soleus muscle reduced fatigue by 25% during electrically evoked contraction (Brass et al. 1993). Furthermore, Coyle (1995) concluded that the ability to maintain a high steady-state 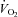 with low muscle lactate content is a prerequisite for enhanced endurance exercise performance in elite athletes. Indeed, muscle lactic acidosis has been suggested as a primary cause of fatigue during high-intensity exercise (Sahlin, 1992), hence the efforts to increase muscle buffering capacity by β-alanine feeding (Hill et al. 2007) or establish pre-exercise metabolic alkalosis by sodium bicarbonate ingestion (Wilkes et al. 1983; McKenzie et al. 1986; Bird et al. 1995) to improve high intensity exercise performance in humans. Finally, given that muscle glycogen content was 147 mmol (kg dry muscle) −1 following exercise at 80%
with low muscle lactate content is a prerequisite for enhanced endurance exercise performance in elite athletes. Indeed, muscle lactic acidosis has been suggested as a primary cause of fatigue during high-intensity exercise (Sahlin, 1992), hence the efforts to increase muscle buffering capacity by β-alanine feeding (Hill et al. 2007) or establish pre-exercise metabolic alkalosis by sodium bicarbonate ingestion (Wilkes et al. 1983; McKenzie et al. 1986; Bird et al. 1995) to improve high intensity exercise performance in humans. Finally, given that muscle glycogen content was 147 mmol (kg dry muscle) −1 following exercise at 80%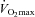 in the Control group, it is possible that glycogen availability may have limited performance in this group during the 30 min work output trial, which would not have been the case in the Carnitine group where glycogen was 250 mmol (kg dry muscle)−1. It cannot be ruled out therefore that at least some of the positive effect of muscle carnitine loading on exercise performance was attributable to the glycogen sparing that occurred during exercise at 50%
in the Control group, it is possible that glycogen availability may have limited performance in this group during the 30 min work output trial, which would not have been the case in the Carnitine group where glycogen was 250 mmol (kg dry muscle)−1. It cannot be ruled out therefore that at least some of the positive effect of muscle carnitine loading on exercise performance was attributable to the glycogen sparing that occurred during exercise at 50%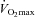 . Whether the beneficial effects of muscle carnitine loading on high intensity exercise and exercise performance led to the better maintenance of body mass in the face of 24 weeks of additional daily caloric intake when compared to control (i.e. via a regular increased energy expenditure during exercise training) is an interesting notion, but cannot be determined from the present study and remains to be explored.
. Whether the beneficial effects of muscle carnitine loading on high intensity exercise and exercise performance led to the better maintenance of body mass in the face of 24 weeks of additional daily caloric intake when compared to control (i.e. via a regular increased energy expenditure during exercise training) is an interesting notion, but cannot be determined from the present study and remains to be explored.
In summary, this is the first study to demonstrate that muscle carnitine content can be increased in humans by dietary means and, perhaps more importantly, that carnitine plays a dual role in skeletal muscle fuel metabolism that is exercise intensity dependent. Specifically, we have shown that increasing muscle TC content spares muscle glycogen during low intensity exercise (consistent with an increase in muscle lipid utilisation), but during high intensity exercise results in a better matching of glycolytic, PDC and mitochondrial flux, thereby reducing muscle anaerobic ATP production. Furthermore, these metabolic changes resulted in positive effects on perception of effort and work output using a validated exercise performance test. Collectively these findings have significant implications for athletic performance and pathophysiological conditions where fat oxidation is impaired or anaerobic ATP production is accelerated during exercise (Noland et al. 2009).
Acknowledgments
The authors would like to thank Mr Ryan Atkins for his assistance with the determination of citrate synthase activity, QinetiQ for their sponsorship of this research and Energ8 Ltd for donating the carbohydrate and l-carnitine supplements used in the study.
Glossary
Abbreviations
- CHO
carbohydrate
- CoASH
free co-enzyme A
- CPT1
carnitine palmitoyl-transferase 1
- PDC
pyruvate dehydrogenase complex
- TC
total carnitine
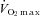
maximal oxygen uptake
Author contributions
The experiments in this paper were conducted in the School of Biomedical Sciences, University of Nottingham. All authors approved the final version of the manuscript to be published and all authors contributed to drafting the article and revising it critically for important intellectual content. All authors contributed to the conception and design, or analysis and interpretation of the data in the manuscript.
References
- Barnett C, Costill D, Vukovich M, Cole K, Goodpaster B, Trappe S, Fink W. Effect of l-carnitine supplementation on muscle and blood carnitine content and lactate accumulation during high-intensity sprint cycling. Int J Sport Nutr. 1994;4:280–288. doi: 10.1123/ijsn.4.3.280. [DOI] [PubMed] [Google Scholar]
- Bergström J. Percutaneous needle biopsy of skeletal muscle in physiological and clinical research. Scand J Clin Lab Invest. 1975;35:609–616. [PubMed] [Google Scholar]
- Bird S, Wiles J, Robbins J. The effect of sodium bicarbonate ingestion on 1500-m racing time. J Sports Sci. 1995;13:399–403. doi: 10.1080/02640419508732255. [DOI] [PubMed] [Google Scholar]
- Brass E, Scarrow A, Ruff L, Masterson K, Van Lunteren E. Carnitine delays rat skeletal muscle fatigue in vitro. J Appl Physiol. 1993;75:1595–1600. doi: 10.1152/jappl.1993.75.4.1595. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Carlin J, Constantin-Teodosiu D, Harper P, Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Anal Biochem. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- Cederblad G, Hermansson G, Ludvigsson J. Plasma and urine carnitine in children with diabetes mellitus. Clin Chim Acta. 1982;125:207–217. doi: 10.1016/0009-8981(82)90197-8. [DOI] [PubMed] [Google Scholar]
- Childress C, Sacktor B. Pyruvate oxidation and the permeability of mitochondria from blowfly flight muscle. Science. 1966;154:268–270. doi: 10.1126/science.154.3746.268. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Carlin J, Cederblad G, Harris R, Hultman E. Acetyl group accumulation and pyruvate dehydrogenase activity in human muscle during incremental exercise. Acta Physiol Scand. 1991a;143:367–372. doi: 10.1111/j.1748-1716.1991.tb09247.x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Cederblad G, Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Anal Biochem. 1991b;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- Constantin-Teodosiu D, Peirce N, Fox J, Greenhaff P. Muscle pyruvate availability can limit the flux, but not activation, of the pyruvate dehydrogenase complex during submaximal exercise in humans. J Physiol. 2004;561:647–655. doi: 10.1113/jphysiol.2004.073411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper R, Randle P, Denton R. Stimulation of phosphorylation and inactivation of pyruvate dehydrogenase by physiological inhibitors of the pyruvate dehydrogenase reaction. Nature. 1975;257:808–809. doi: 10.1038/257808a0. [DOI] [PubMed] [Google Scholar]
- Coyle E. Integration of the physiological factors determining endurance performance ability. Exerc Sport Sci Rev. 1995;23:25–63. [PubMed] [Google Scholar]
- Fritz I, McEwen B. Effects of carnitine on fatty-acid oxidation by muscle. Science. 1959;129:334–335. doi: 10.1126/science.129.3345.334. [DOI] [PubMed] [Google Scholar]
- Fritz I, Yue K. Long-chain carnitine acyltransferase and the role of acylcarnitine derivatives in the catalytic increase of fatty acid oxidation induced by carnitine. J Lipid Res. 1963;4:279–288. [PubMed] [Google Scholar]
- Harris R, Foster C, Hultman E. Acetylcarnitine formation during intense muscular contraction in humans. J Appl Physiol. 1987;63:440–442. doi: 10.1152/jappl.1987.63.1.440. [DOI] [PubMed] [Google Scholar]
- Harris R, Hultman E, Nordesjö L. Glycogen, glycolytic intermediates and high-energy phosphates determined in biopsy samples of musculus quadriceps femoris of man at rest. Methods and variance of values. Scand J Clin Lab Invest. 1974;33:109–120. [PubMed] [Google Scholar]
- Hill C, Harris R, Kim H, Harris B, Sale C, Boobis L, Kim C, Wise J. Influence of β-alanine supplementation on skeletal muscle carnosine concentrations and high intensity cycling capacity. Amino Acids. 2007;32:225–233. doi: 10.1007/s00726-006-0364-4. [DOI] [PubMed] [Google Scholar]
- Howlett R, Heigenhauser G, Hultman E, Hollidge-Horvat M, Spriet L. Effects of dichloroacetate infusion on human skeletal muscle metabolism at the onset of exercise. Am J Physiol Endocrinol Metab. 1999;277:E18–E25. doi: 10.1152/ajpendo.1999.277.1.E18. [DOI] [PubMed] [Google Scholar]
- Jeukendrup A, Saris W, Brouns F, Kester A. A new validated endurance performance test. Med Sci Sports Exerc. 1996;28:266–270. doi: 10.1097/00005768-199602000-00017. [DOI] [PubMed] [Google Scholar]
- McGarry J, Mills S, Long C, Foster D. Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J. 1983;214:21–28. doi: 10.1042/bj2140021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie D, Coutts K, Stirling D, Hoeben H, Kuzara G. Maximal work production following two levels of artificially induced metabolic alkalosis. J Sports Sci. 1986;4:35–38. doi: 10.1080/02640418608732096. [DOI] [PubMed] [Google Scholar]
- Noland R, Koves T, Seiler S, Lum H, Lust R, Ilkayeva O, Stevens R, Hegardt F, Muoio D. Carnitine insufficiency caused by aging and overnutrition compromises mitochondrial performance and metabolic control. J Biol Chem. 2009;284:22840–22852. doi: 10.1074/jbc.M109.032888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opie LH, Newsholme EA. The activities of fructose 1,6-diphosphatase, phosphofructokinase and phosphoenolpyruvate carboxykinase in white muscle and red muscle. Biochem J. 1967;103:391–399. doi: 10.1042/bj1030391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit F, Pelley J, Reed L. Regulation of pyruvate dehydrogenase kinase and phosphatase by acetyl-CoA/CoA and NADH/NAD ratios. Biochem Biophys Res Commun. 1975;65:575–582. doi: 10.1016/s0006-291x(75)80185-9. [DOI] [PubMed] [Google Scholar]
- Randle P, Garland P, Hales C, Newsholme E. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- Roberts P, Loxham S, Poucher S, Constantin-Teodosiu D, Greenhaff P. The acetyl group deficit at the onset of contraction in ischaemic canine skeletal muscle. J Physiol. 2002;544:591–602. doi: 10.1113/jphysiol.2002.021097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts P, Loxham S, Poucher S, Constantin-Teodosiu D, Greenhaff P. Acetyl-CoA provision and the acetyl group deficit at the onset of contraction in ischemic canine skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:E327–E334. doi: 10.1152/ajpendo.00441.2003. [DOI] [PubMed] [Google Scholar]
- Sahlin K. Metabolic factors in fatigue. Sports Med. 1992;13:99–107. doi: 10.2165/00007256-199213020-00005. [DOI] [PubMed] [Google Scholar]
- Stephens F, Constantin-Teodosiu D, Laithwaite D, Simpson E, Greenhaff P. Insulin stimulates l-carnitine accumulation in human skeletal muscle. FASEB J. 2006a;20:377–379. doi: 10.1096/fj.05-4985fje. [DOI] [PubMed] [Google Scholar]
- Stephens F, Constantin-Teodosiu D, Laithwaite D, Simpson E, PL G. An acute increase in skeletal muscle carnitine content alters fuel metabolism in resting human skeletal muscle. J Clin Endocrinol Metab. 2006b;91:5013–5018. doi: 10.1210/jc.2006-1584. [DOI] [PubMed] [Google Scholar]
- Stephens F, Evans C, Constantin-Teodosiu D, Greenhaff P. Carbohydrate ingestion augments l-carnitine retention in humans. J Appl Physiol. 2007;102:1065–1070. doi: 10.1152/japplphysiol.01011.2006. [DOI] [PubMed] [Google Scholar]
- Stephens F, Roig M, Armstrong G, Greenhaff P. Post-exercise ingestion of a unique, high molecular weight glucose polymer solution improves performance during a subsequent bout of cycling exercise. J Sports Sci. 2008;26:149–154. doi: 10.1080/02640410701361548. [DOI] [PubMed] [Google Scholar]
- Timmons J, Gustafsson T, Sundberg C, Jansson E, Greenhaff P. Muscle acetyl group availability is a major determinant of oxygen deficit in humans during submaximal exercise. Am J Physiol Endocrinol Metab. 1998;274:E377–E380. doi: 10.1152/ajpendo.1998.274.2.E377. [DOI] [PubMed] [Google Scholar]
- Timmons J, Poucher S, Constantin-Teodosiu D, Macdonald I, Greenhaff P. Metabolic responses from rest to steady state determine contractile function in ischemic skeletal muscle. Am J Physiol Endocrinol Metab. 1997;273:E233–E238. doi: 10.1152/ajpendo.1997.273.2.E233. [DOI] [PubMed] [Google Scholar]
- van Loon L, Greenhaff P, Constantin-Teodosiu D, Saris W, Wagenmakers A. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J Physiol. 2001;536:295–304. doi: 10.1111/j.1469-7793.2001.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukovich M, Costill D, Fink W. Carnitine supplementation: effect on muscle carnitine and glycogen content during exercise. Med Sci Sports Exerc. 1994;26:1122–1129. [PubMed] [Google Scholar]
- Wächter S, Vogt M, Kreis R, Boesch C, Bigler P, Hoppeler H, Krähenbühl S. Long-term administration of l-carnitine to humans: effect on skeletal muscle carnitine content and physical performance. Clin Chim Acta. 2002;318:51–61. doi: 10.1016/s0009-8981(01)00804-x. [DOI] [PubMed] [Google Scholar]
- Wilkes D, Gledhill N, Smyth R. Effect of acute induced metabolic alkalosis on 800-m racing time. Med Sci Sports Exerc. 1983;15:277–280. doi: 10.1249/00005768-198315040-00004. [DOI] [PubMed] [Google Scholar]
- Zammit V. Carnitine acyltransferases: functional significance of subcellular distribution and membrane topology. Prog Lipid Res. 1999;38:199–224. doi: 10.1016/s0163-7827(99)00002-8. [DOI] [PubMed] [Google Scholar]
- Zammit VA, Newsholme EA. The maximum activities of hexokinase, phosphorylase, phosphofructokinase, glycerol phosphate dehydrogenases, lactate dehydrogenase, octopine dehydrogenase, phosphoenolpyruvate carboxykinase, nucleoside diphosphatekinase, glutamate-oxaloacetate transaminase and arginine kinase in relation to carbohydrate utilization in muscles from marine invertebrates. Biochem J. 1976;160:447–462. doi: 10.1042/bj1600447. [DOI] [PMC free article] [PubMed] [Google Scholar]