

Abstract
Myostatin (MSTN) is a transforming growth factor-ß family member that plays a critical role in regulating skeletal muscle mass. Genetic studies in multiple species have demonstrated that mutations in the Mstn gene lead to dramatic and widespread increases in muscle mass as a result of a combination of increased fiber numbers and increased fiber sizes. MSTN inhibitors have also been shown to cause significant increases in muscle growth when administered to adult mice. As a result, there has been an extensive effort to understand the mechanisms underlying MSTN regulation and activity with the goal of developing the most effective strategies for targeting this signaling pathway for clinical applications. Here, I review the current state of knowledge regarding the regulation of MSTN extracellularly by binding proteins and discuss the implications of these findings both with respect to the fundamental physiological role that MSTN plays in regulating tissue homeostasis and with respect to the development of therapeutic agents to combat muscle loss.
Keywords: Myostatin, transforming growth factor-ß, skeletal muscle, tissue growth, latency, propeptide, bone morphogenetic protein-1 (BMP-1), tolloid, follistatin, FLRG, FSTL-3, GASP-1, GASP-2, activin, activin type II receptor
Introduction
Myostatin (MSTN) is a transforming growth factor-ß (TGF-ß) family member that normally acts to limit muscle mass [1]. Mstn RNA is first expressed during embryogenesis by cells of the myotome compartment of developing somites and continues to be expressed by cells of the muscle lineage throughout development as well as in adult mice. The function of MSTN was elucidated through gene targeting studies, in which Mstn knockout mice were found to have widespread increases in skeletal muscle mass, with individual muscles weighing approximately twice as much as those of control animals as a result of a combination of both increased fiber number and increased fiber size. Subsequent genetic studies in cattle [2-5], sheep [6], dogs [7], and humans [8] have all shown that the function of MSTN as a negative regulator of muscle mass has been highly conserved. Moreover, pharmacological agents capable of blocking MSTN activity have been shown to cause significant increases in muscle growth when administered systemically to adult mice [9-12], demonstrating that MSTN plays a critical role in regulating muscle homeostasis postnatally by suppressing muscle growth.
The discovery of MSTN and its biological function immediately suggested the possibility that targeting this signaling pathway may be an effective strategy for treating patients with debilitating muscle loss. Loss of muscle mass and function occurs in a wide range of diseases and physiological states, and a large number of studies have shown that inhibition of myostatin signaling can have beneficial effects in many of these disease settings (these are reviewed in detail by other articles in this collection). Loss of MSTN signaling has also been shown to have beneficial effects on fat and glucose metabolism, suggesting that targeting MSTN may also be useful for preventing or treating metabolic diseases, like obesity and type II diabetes (the metabolic roles of MSTN are reviewed in detail by other articles in this collection). As a result, there has been an extensive effort directed at understanding the molecular and cellular mechanisms underlying MSTN activity with the long-term goal of determining the most effective strategies for targeting this signaling pathway for therapeutic applications. Since the original report of the discovery of MSTN and its biological function, an enormous amount of progress has been made in terms of identifying key components of this regulatory system (Fig. 1). Here, I will review the work that led to the identification of these regulatory components as well as the current state of knowledge regarding the specific roles that each of these molecules may play in regulating MSTN activity and muscle growth.
Fig. (1).
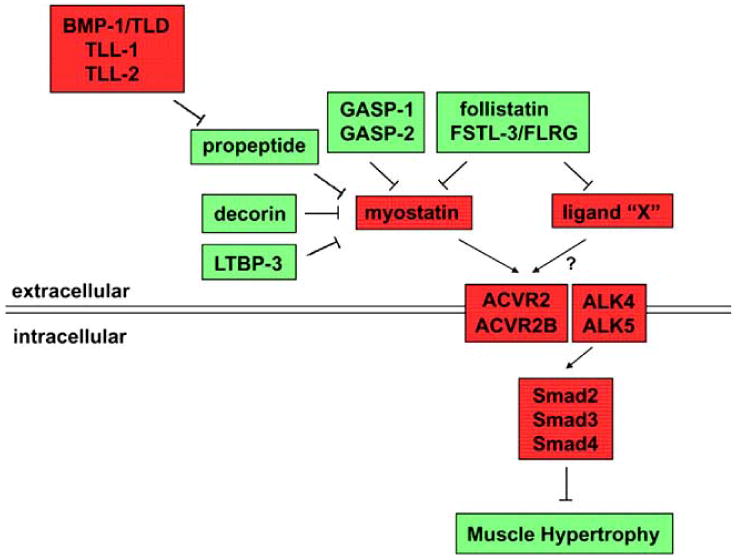
Regulation of myostatin activity and muscle growth. Myostatin is negatively regulated by various naturally-occurring binding proteins. When not bound to these inhibitory proteins, myostatin signals by binding initially to the two activin type II receptors, ACVR2 and ACVR2B, which then leads to binding and activation of the type I receptors, ALK4 and/or ALK5. Signaling through this pathway results in inhibition of muscle growth. Ligand “X” refers to the as yet unidentified TGF-ß related ligand or ligands that cooperate with myostatin to limit muscle growth. Components shown in red act to block muscle growth, and components shown in green act to promote muscle growth. As described in the text, ligand “X” can be blocked by either follistatin or FSTL-3, but whether ligand “X” can also be inhibited by other regulatory components is not known.
Biosynthesis of MSTN
Myostatin was originally identified in a screen for new members of the TGF-ß superfamily in mammals [1]. The cDNA sequence predicted a 376 amino acid protein containing all of the hallmarks present in other TGF-ß family members, including an N-terminal signal sequence for secretion, a pro region followed by an RSRR sequence representing a putative proteolytic processing site, and a C-terminal domain of 109 amino acids containing nine cysteine residues with their characteristic spacing. Expression of the full-length cDNA in Chinese hamster ovary (CHO) cells confirmed that MSTN can be secreted and processed to generate a 38 kD N-terminal propeptide and a 12.5 kD C-terminal domain [1, 13, 14]. The processing of the MSTN precursor protein (pro-MSTN) by CHO cells was shown to be relatively inefficient and could be significantly enhanced by transfecting in an expression cassette for a furin protease, sPACE-1. This processing event also seems to occur inefficiently in vivo, as Western analysis of muscle extracts using antibodies directed against the C-terminal domain has detected multiple immunoreactive species, including a major band likely to represent the full-length unprocessed MSTN precursor protein (for examples, see refs. [15-17]). At least two studies have suggested that processing of the precursor protein may be a key regulatory step in the biosynthesis of MSTN in vivo. In one study, the relative amounts of unprocessed and fully processed MSTN were found to change following differentiation of C2C12 cells in culture as well as during bovine muscle development in vivo [16]. In another study, the majority of MSTN protein detectable in adult mouse skeletal muscle was found to be in the unprocessed form, and at least a portion of this MSTN precursor protein was shown to be present extracellularly [17]. The observations that pro-MSTN can be cleaved by furin extracellularly in cultured cells, that LTBP-3 (Latent TGF-ß Binding Protein-3) is capable of binding pro-MSTN and inhibiting furin-mediated processing, and that electroporation of an LTBP-3 expression cassette directly into muscle could cause fiber hypertrophy led these authors to hypothesize that extracellular processing of MSTN may be a key regulatory step for MSTN signaling in muscle.
The biological activity of MSTN in terms of actual signaling to target cells appears to reside in the C-terminal domain, which has been shown to be capable of forming disulfide-linked homodimers [13, 14]. Recent x-ray crystallographic studies [18] have confirmed that as has been shown for other TGF-ß family members, the two subunits of the MSTN C-terminal dimer are linked via a single intermolecular disulfide bond involving the 6th cysteine residue and that the remaining cysteine residues are arranged in the classic cystine knot structure characteristic of many growth factor-like molecules, including other members of the TGF-ß superfamily [19]. Initial studies using MSTN protein purified to homogeneity from the conditioned medium of CHO cells transfected with a full-length expression construct demonstrated that the C-terminal dimer is fully active both in receptor binding and reporter gene activation assays [13, 14]. Subsequent studies have demonstrated that the purified MSTN C-terminal dimer exhibits a wide range of activities in regulating cell proliferation and differentiation in a variety of different cell types in culture. As these in vitro activities are discussed extensively in the context of the overall biological function of MSTN by other articles in this collection, here I will discuss the activity of MSTN only as it relates to the regulation of MSTN activity by binding proteins.
Role of the Propeptide and MSTN Latency
Although the signaling activity of MSTN appears to reside entirely in the C-terminal domain, the N-terminal pro domain clearly plays an important role in the biology of MSTN. As has been reported for certain other TGF-ß family members, the MSTN pro domain seems to perform at least two distinct functions. First, the presence of the pro domain appears to be important for the maturation of the C-terminal dimer, specifically with respect to the proper folding of the MSTN precursor protein. The essential role of the pro domain in folding of the precursor protein was predicted based on what had been described previously for other TGF-ß family members. In particular, earlier studies with two other family members, namely activin A and TGF-ß itself, had shown that the presence of the pro domain was essential for proper dimer formation when these ligands were expressed in mammalian cells [20]. Remarkably, this effect of the pro domain in the case of activin A and TGF-ß could be conferred in trans, as expressing the propeptide as a separate molecule could direct the dimerization and secretion of these ligands when the respective C-terminal domains were co-expressed in a separate construct using a synthetic signal sequence.
In the case of MSTN, the most convincing evidence for a similar role for the pro domain has come from studies analyzing the properties of MSTN produced in bacteria. Specifically, although the unprocessed porcine or piscine MSTN precursor proteins produced in E. coli could be readily refolded to form disulfide-linked dimers, the isolated C-terminal domains were incapable of forming dimers when subjected to the same re-folding conditions [21, 22]. In the case of piscine MSTN, attempts to re-fold the C-terminal domain in a mixture with added propeptide were unsuccessful, leading the authors to suggest that unlike the case for activin and TGF-ß, the MSTN propeptide may not be capable of acting in trans to direct dimer formation [22]. Given that analogous studies to those carried out with activin A and TGF-ß in which the propeptide and C-terminal domains were co-expressed as separate proteins in mammalian cells have not yet been reported for MSTN, however, it is possible that the trans effect of the propeptide might be observed only when it is co-synthesized with the C-terminal domain or only in mammalian cells.
In addition to playing a role in the folding and dimerization of the MSTN C-terminal dimer, the propeptide also plays a second role in regulating MSTN activity following proteolytic processing and secretion. There is now considerable evidence that the propeptide remains non-covalently bound to the MSTN C-terminal dimer following processing of the precursor protein and that the propeptide thereby maintains MSTN in an inactive, latent state. This role for the propeptide first became apparent from studies analyzing recombinant MSTN protein produced by CHO cells. Two groups working independently purified MSTN protein from the conditioned medium of CHO cells engineered to produce high levels of either mouse [13] or human [14] MSTN, and both groups observed that the propeptide co-purified with the C-terminal dimer through multiple column chromatography steps. Both groups went on to show that the propeptide remains tightly bound non-covalently to the C-terminal dimer following processing and secretion and that the two could be purified away from each other by HPLC in the presence of trifluoroacetic acid and acetonitrile. As discussed earlier, the isolated C-terminal dimer was shown to contain the biological activity of MSTN in both receptor binding and reporter gene activation assays in vitro; these activities of the C-terminal dimer, however, could be completely inhibited by the addition of purified propeptide [13, 14]. Subsequent deletion analysis revealed that the inhibitory activity of the propeptide is fully contained within a small region spanning amino acid residues 42-115 [23].
The ability of the propeptide to block MSTN activity has also been documented extensively in vivo. Three groups [13, 24, 25] independently generated transgenic mouse lines expressing the propeptide specifically in skeletal muscle by placing the expression of the propeptide under the control of myosin light chain regulatory sequences. In each case, the transgenic mice exhibited significant increases in muscle mass, thereby phenocopying Mstn null mice, although some differences were noted among the various studies with respect to the magnitude of the effect and with respect to the contribution of increased fiber numbers to the overall phenotype. Because the propeptide was expressed specifically in muscle in these transgenic lines, however, these studies left open the formal possibility that the effect of the propeptide may have been not to block the activity of the C-terminal dimer but rather to interfere with MSTN production by forming heterodimers with endogenously-expressed full length MSTN precursor molecules. Indeed, this type of dominant negative mechanism was almost certainly responsible for the increased muscling observed in two other sets of transgenic mouse lines expressing mutant forms of MSTN in which either the furin cleavage site [26] or one of the conserved cysteine residues [27] was altered.
A number of subsequent studies have provided additional evidence demonstrating that excess propeptide is capable of inducing muscle growth by blocking MSTN signaling rather than simply by interfering with MSTN production. One set of studies has been to use viral vectors to deliver propeptide expression constructs to mice [28-32]. Increases in muscle mass were observed in all of these studies, and in two cases, effects on muscle mass were apparent at sites distant from the site of propeptide production. In one case, the virus was delivered by intramuscular injection, and effects on muscle growth were observed on the contralateral side [28]. In the other case, significant muscle growth was induced when an AAV8 vector was used to deliver a propeptide expression construct intravenously even though the propeptide was found to be expressed in liver and heart but not muscle [29]. These findings suggested that in each case, at least part of the effect was being mediated by circulating propeptide protein. The most definitive studies demonstrating the ability of the propeptide to block MSTN signaling in vivo have been ones in which purified propeptide protein was administered directly to mice [11, 33]. In these experiments, the propeptide was made as a fusion protein with an Fc domain in order to enhance its stability in vivo, and systemic administration of the propeptide/Fc fusion protein to either wild type or mdx mice was shown to cause significant and widespread increases in muscle mass. As discussed below, the most potent form of the propeptide has been shown to be a mutant version in which aspartate residue 76 was changed to alanine in order to render it resistant to cleavage by members of the BMP-1/TLD family of metalloproteases [11].
Mechanisms Underlying Activation of Latent MSTN
Although both of the initial studies identifying the MSTN latent complex [13, 14] were carried out using MSTN produced by CHO cells, subsequent studies confirmed the important role that the propeptide plays in regulating MSTN activity in vivo. Specifically, MSTN was shown to circulate at relatively high levels in the blood in mice, and the circulating form of MSTN was found to exhibit properties reminiscent of those observed with CHO cell-produced MSTN [34]. By testing serum samples for their ability to activate a Smad-responsive reporter gene, these authors observed that although no MSTN activity could be detected in untreated serum samples, MSTN activity could be readily detected in serum samples that had been subjected to low pH. Because acid treatment was known to be capable of dissociating the purified MSTN latent complex, these findings were consistent with the possibility that the circulating form of MSTN might be a complex of the propeptide with the C-terminal dimer, similar to what had been observed for CHO cell-produced MSTN. This possibility was confirmed by a follow-up study analyzing proteins bound to the C-terminal dimer when affinity purified from serum using a monoclonal antibody directed against this portion of the MSTN molecule [35]. Following analysis by mass spectrometry, a major protein species identified in the bound fraction was the propeptide, and quantitative analysis suggested that perhaps the majority of the C-terminal dimer present in serum is normally bound to the propeptide.
Based on these studies, it seems clear that MSTN is produced as a latent complex of the propeptide and the C-terminal dimer in vivo and that this non-covalently-held complex represents the major circulating form of MSTN in the blood. A critical question with respect to understanding the regulation of this signaling pathway is how MSTN is activated from this latent state. Studies with purified latent complex demonstrated that MSTN can be activated artificially in vitro by heat treatment [11]. A clue to how this complex might be activated in vivo came from the analysis of MSTN produced by CHO cells [11]. Western analysis of conditioned medium prepared from MSTN-expressing CHO cells using antibodies directed against the propeptide revealed that although most of the propeptide exhibited an electrophoretic mobility consistent with the predicted molecular weight, a faster migrating species could be detected to varying levels in different preparations. Analysis of this faster migrating species identified it to be a degradation product resulting from proteolytic cleavage of the propeptide immediately N-terminal to aspartate residue 76. The finding that this truncated product could not be detected in the conditioned medium of CHO cells expressing a mutant form of MSTN in which aspartate 76 was changed to alanine suggested that this degradation product resulted from cleavage of the propeptide by specific proteases made by CHO cells.
For two reasons, members of the BMP-1/tolloid family of metalloproteases were attractive candidates for the proteases that might be responsible for cleavage of the propeptide. First, although most of the substrates that had been identified for these proteases were components of the extracellular matrix, most notably the numerous isoforms of procollagen (for review, see ref. [36]), this group of proteases had also been shown to be capable of cleaving chordin, which normally functions to bind to and inhibit the activities of certain TGF-ß family members, namely, the bone morphogenetic proteins (BMPs). Genetic and biochemical studies in Drosophila and Xenopus had demonstrated that the BMP-1/TLD family of proteases is capable of cleaving and inactivating chordin, thereby releasing the bound BMPs to allow signaling to occur [37-39]. These findings raised the possibility that this group of proteases may also be capable of similarly regulating the activities of other binding proteins for the TGF-ß family, including their respective propeptides. Second, analysis of the precise cleavage sites for many of the substrates revealed that in virtually every case, the cleavage occurs immediately N-terminal to an aspartate residue (for review, see ref. [36]).
These known properties of the BMP-1/TLD proteases taken together with the observation that the degradation product of the propeptide present in the conditioned medium of CHO cells overexpressing MSTN resulted from proteolytic cleavage immediately N-terminal to aspartate residue 76 raised the possibility that proteolytic degradation of the propeptide may have resulted from the activities of one or more members of this protease family. Indeed, studies using purified proteases and purified MSTN latent complex showed that each of the four proteases in the BMP-1/TLD family (BMP-1, TLD, TLL-1, and TLL-2) is capable of cleaving the MSTN propeptide in the latent complex precisely N-terminal to aspartate residue 76 [11]. Furthermore, analysis of the products of the reaction in reporter gene assays demonstrated that cleavage of the propeptide by each of these proteases resulted in activation of the MSTN C-terminal dimer. The actual catalytic activity of these proteases was shown to be essential for activation of the MSTN latent complex, as purified latent complex comprised of the C-terminal dimer and a mutant form of the propeptide in which aspartate 76 had been changed to alanine (D76A) was shown to be resistant to proteolytic cleavage and incapable of being activated by BMP-1/TLD proteases.
Although these experiments focused on the ability of this group of metalloproteases to activate the MSTN latent complex in vitro, there is now considerable evidence supporting a role for this activation mechanism in vivo. In the initial study describing this activation mechanism, it was shown that unlike the wild type propeptide, the D76A mutant form of the propeptide was capable of increasing muscle growth when administered systemically as an Fc fusion protein to wild type adult mice [11]. This finding suggested that the mutant propeptide was capable of binding free C-terminal dimers in vivo and forming latent complexes that could not be activated by proteolytic cleavage. The role of proteolysis in activation of latent MSTN has also been investigated using genetic approaches in mice to manipulate levels of proteolytic cleavage in vivo. One approach was to knock in the D76A mutation into the endogenous Mstn gene, thereby replacing the normal MSTN protein with the D76A mutant version [40]. As in the case of mice carrying the original Mstn deletion mutation, mice heterozygous or homozygous for the D76A point mutation exhibited dose-dependent increases in muscle mass, consistent with a critical role for proteolysis at this site in the activation of MSTN. A remarkable finding in these mice was that the circulating levels of MSTN protein were increased dramatically compared to those of wild type mice, and yet the mice behaved phenotypically as if they substantially lacked MSTN activity; the simplest interpretation of these data is that MSTN protein accumulated to very high levels in these mutant mice and that essentially all of this protein was incapable of being activated from its latent state. Although the increases in muscle mass observed in mice carrying the D76A point mutation approached those seen in mice carrying the Mstn deletion mutation, the effect was incomplete, implying that proteolytic cleavage of the propeptide is likely to be the major but not the sole mechanism for activating latent MSTN in vivo.
A second genetic approach to validating the role of these proteases in activating latent MSTN in vivo was to target the proteases themselves. In particular, mice homozygous for a deletion mutation in one of the genes in this family, namely Tll2, were shown to exhibit statistically significant increases in muscle mass, consistent with a role for TLL-2 in regulating MSTN activity [40]. The effects seen in Tll2 mutant mice, however, were relatively small compared to those seen in mice carrying the D76A point mutation in the Mstn gene, implying that the function of TLL-2 is redundant with those of other members of this protease family. In this respect, although Tll2 has been shown to be expressed in developing muscle [41], little or no TLL-2 appears to be made by adult muscle, suggesting that other proteases likely play a more prominent role in regulating MSTN activity in adult muscle. Because inactivating mutations in the other genes in this family, namely Bmp1/Tld (BMP-1 and TLD are made from a single gene by alternative splicing) and Tll1, have been reported previously to result in embryonic lethality in the homozygous state [42, 43], further genetic analysis of the role of these proteases in the biology of MSTN will await the generation and characterization of mice carrying conditional knockout alleles for each of these genes.
Role of Follistatin in Regulating MSTN Activity
In addition to the propeptide, several other proteins have been identified that appear to be involved in binding and regulating MSTN extracellularly. One of these is follistatin (FST), which was originally identified in follicular fluid for its ability to block secretion of follicle stimulating hormone (FSH) by pituitary cells [44, 45]. Follistatin was found to act not by signaling directly to target cells but rather by binding and inhibiting activins [46], which are TGF-ß family members shown to have the opposite activity to follistatin, namely, to stimulate FSH secretion [47, 48]. Subsequent studies showed that follistatin is also capable of blocking the activities of other TGF-ß family members as well, including some of the BMP isoforms [49-51]. Of particular interest with respect to MSTN was the finding that follistatin could bind and inhibit GDF-11/BMP-11 [52]. Because GDF-11/BMP-11 is highly related to MSTN (the two are greater than 90% identical at the amino acid level in the C-terminal domain) [1, 52], these findings raised the possibility that follistatin might also be capable of binding and inhibiting MSTN.
Indeed, follistatin has been shown to function as a potent inhibitor of MSTN both in vitro and in vivo. Initial studies demonstrated that purified follistatin is capable of blocking MSTN activity in vitro in both receptor binding [13] and reporter gene activation [34] assays, with IC50s in the range of about 300-500 pM. Two groups subsequently used surface plasmon resonance analysis to demonstrate that follistatin can bind directly to MSTN, although the measured dissociation constants were somewhat discrepant, ranging from 584 pM in one study [53] to 12.3 nM in the other study [54]. The nature of the binding interaction between these proteins has now been revealed in detail by the analysis of the crystal structure of the follistatin/MSTN complex [18]. In the complex, two molecules of follistatin were shown to surround the MSTN C-terminal dimer, completely blocking all of the putative sites of interaction between MSTN and its receptors. The N-terminal domains (ND) of each of the two follistatin molecules in the complex were shown to contact both MSTN subunits in the dimer and block the prehelix loops of the MSTN molecules necessary for interaction with the type I receptors. The first two follistatin domains (FSD1 and FSD2) of each follistatin molecule in the complex were shown to contact one subunit of the MSTN dimer and thereby block the putative MSTN binding site for the activin type II receptors. No direct contacts were observed between MSTN and the third follistatin domain, FSD3.
A comparison of the crystal structure of the follistatin/MSTN complex with that of the follistatin/activin A complex [55] revealed both similarities and differences in the manner in which follistatin interacts with these two ligands to regulate their activities. In general, the role of the ND in blocking the type I receptor binding interface and the role of the FSDs in blocking the type II receptor binding interface were found to be similar for the two ligands. One significant difference, however, is that the ND appears to play a more prominent role in the case of MSTN binding by making more extensive contacts with the ligand and burying more surface area. Two observations, however, still remain somewhat unexplained by these structural studies. First, despite the fact that more overall surface area seems to be buried in the follistatin/MSTN complex than in the follistatin/activin A complex, activin A has been reported to have a higher affinity than MSTN for follistatin [53, 54, 56]. Second, despite the fact that the FSD1 and FSD2 domains each make intimate contacts with the ligand in both complexes, structure-function studies have shown that the FSD1 domain seems to be more important for binding to MSTN whereas the FSD2 domain seems to be more important for binding to activin A [57]. Whatever the molecular basis for this specificity might be, the differing roles of the individual FSD domains have been exploited to engineer follistatin variants with higher affinity for one ligand versus the other. In particular, a construct consisting of the configuration ND/FSD1/FSD1 has been shown to have much higher inhibitory activity on MSTN than on activin A [54, 57], raising the possibility that neo molecules of this type may be preferable to full length follistatin in terms of being able to generate more selective targeting for clinical applications.
In this respect, there is now considerable evidence that follistatin can function as a potent inhibitor of MSTN signaling in vivo. In the initial study identifying follistatin as a MSTN antagonist, overexpression of follistatin in skeletal muscle as a transgene under the control of a myosin light chain promoter and enhancer was shown to cause dramatic increases in muscle mass, consistent with inhibition of MSTN activity [13]. The follistatin transgene phenocopied the Mstn loss-of-function mutation in that both fiber numbers and fiber sizes appeared to be affected, although the relative contribution of muscle fiber hypertrophy to the overall phenotype was much more substantial in the follistatin transgenic mice than in Mstn -/- mice [13, 58]. These findings suggested that follistatin is capable of blocking MSTN activity postnatally to induce muscle fiber growth, and this has now been documented definitively by a number of subsequent studies using a variety of different experimental approaches. Specifically, significant effects on muscle growth have been demonstrated by using viral vectors to deliver follistatin expression cassettes to adult mice [59] and to adult monkeys [60], by electroporating follistatin expression constructs directly into the muscles of adult mice [61], by treating adult mice with deacetylase inhibitors to induce endogenous follistatin expression [62, 63], and by delivering purified follistatin protein systemically to neonatal mice [64] and rats [65]. Moreover, two studies utilizing an engineered follistatin variant with enhanced specificity for MSTN have provided strong evidence that at least a substantial component of the effect of follistatin in vivo results from inhibition of MSTN activity. In particular, a follistatin variant with the configuration ND/FSD1/FSD1, which can bind MSTN but not activins [54, 57], was shown to cause significant increases in muscle mass when overexpressed either as a standard germline transgene [54] or following delivery of an expression cassette by electroporation into adult muscles [61]; importantly, in the latter study, the effect of overexpressing this altered form of follistatin was eliminated when electroporated into muscles of Mstn -/- mice, clearly demonstrating that this molecule was, indeed, targeting MSTN.
Although numerous studies have documented the ability of follistatin to promote muscle growth when overexpressed in vivo, what role follistatin plays normally in regulating muscle homeostasis is still somewhat unclear. Mice homozygous for a targeted deletion of the Fst gene have been shown to have multiple abnormalities [66], some of which can be clearly attributed to loss of inhibition of specific ligands. For example, Fst -/- mice exhibit posterior transformations of the axial skeleton, which is the opposite of what is seen in Gdf11 -/- mice [67], suggesting that at least one role for follistatin during development is to block GDF-11 signaling. Significantly, Fst -/- mice were also found to have a reduced amount of muscle tissue at birth [66], which is what one might expect for loss of inhibition of MSTN signaling. Because Fst -/- mice die immediately after birth, however, an elucidation of the normal role for follistatin in regulating muscle growth and homeostasis will await additional genetic studies in which follistatin function has been targeted in a conditional or tissue-specific manner.
Roles of Other Binding Proteins in MSTN Regulation
In addition to follistatin, a related protein, FSTL-3 (also called FLRG), has also been implicated as a regulator of MSTN activity. FSTL-3 was originally cloned from the breakpoint for a chromosomal translocation in a B-cell chronic lymphocytic leukemia [68]. FSTL-3 was found to be similar to follistatin both in its predicted sequence, which contains two follistatin domains [68], and in its ability to bind activin and inhibit its activity in vitro [69, 70]. The initial study suggesting that FSTL-3 may play a role in regulating MSTN signaling was the analysis of proteins normally bound to MSTN in serum [35]. These investigators used a monoclonal antibody directed against the C-terminal domain of MSTN to affinity purify MSTN protein complexes from serum and then identified bound proteins eluted from gel slices by mass spectrometry. As discussed earlier, one of the major proteins detected in this analysis was the MSTN propeptide. A second major protein detected in the bound fraction in both mouse and human serum was FSTL-3. Curiously, follistatin itself was not detected as one of the proteins bound to MSTN despite the fact that follistatin is known to circulate in the blood (for review, see ref. [71]). Although it is possible that these negative findings mean that follistatin does not play a role in regulating systemic MSTN activity, it is also possible that binding of follistatin to MSTN masked the epitope recognized by the anti-MSTN monoclonal antibody or that the levels of follistatin were below the limits of detection in these studies.
A variety of studies have demonstrated that FSTL-3, like follistatin, can act as a potent inhibitor of MSTN activity. In the original study identifying FSTL-3 as a MSTN binding protein, recombinant FSTL-3 was shown to be capable of blocking MSTN activity in vitro in reporter gene assays [35]. The ability of FSTL-3 to block MSTN activity in vivo has been demonstrated in two subsequent studies. In one study, transgenic mice overexpressing FSTL-3 in skeletal muscle using myosin light chain regulatory sequences were shown to exhibit significant increases in muscle mass [58]. In the other study, delivery of an FSTL-3 expression cassette to adult mice using an AAV vector was also shown to be effective in increasing muscle mass and grip strength, demonstrating the effectiveness of FSTL-3 in inducing muscle growth postnatally [59]. What role FSTL-3 plays normally in regulating MSTN activity and muscle growth, however, is unclear, as muscle mass has been reported to be normal in mice carrying a deletion of Fstl3 [72].
In addition to follistatin and FSTL-3, several other MSTN binding proteins have also been identified. One of these is GASP-1 (Growth and Differentiation Factor-Associated Serum Protein-1; also called WFIKKN2), which was the third protein, along with the MSTN propeptide and FSTL-3, that was shown to be bound to MSTN following affinity purification of MSTN complexes from serum using a monoclonal antibody directed against the MSTN C-terminal domain [73]. A full length cDNA clone encoding GASP-1 was isolated based on the peptides identified by mass spectrometry, and the predicted GASP-1 amino acid sequence was found to contain a follistatin domain as well as multiple domains associated with protease inhibitors, including a whey acidic protein domain, a Kazal domain, two Kunitz domains, and a netrin domain; in fact, it was on the basis of the predicted netrin domain that this gene had been previously identified by another group and named WFIKKN (subsequently renamed WFIKKN2) [74]. Studies with recombinant GASP-1 demonstrated that the protein is capable of binding directly to the MSTN C-terminal dimer and inhibiting its activity in vitro in reporter gene assays [73]. A related protein, GASP-2 (also called WFIKKN1), has also been shown to be capable of binding MSTN [75], although GASP-2 was not detected as one of the proteins bound to endogenous MSTN in serum [73]. Surprisingly, GASP-1 was shown to be capable of binding not only the MSTN C-terminal dimer but also the MSTN propeptide [73], and subsequent structure-function studies with GASP-2 showed that different domains of the protein are responsible for these different interactions, with the follistatin domain mediating the binding to the C-terminal region and the netrin domain mediating the binding to the propeptide [75]. GASP-1 has also been shown to be capable of inhibiting the MSTN-related protein, GDF-11/BMP-11; unlike follistatin and FSTL-3, however, GASP-1 is unable to block activin activity [73]. GASP-2 appears to be similar to GASP-1 in its ability to bind GDF-11/BMP-11 [75], but effects of GASP-2 on other TGF-ß related ligands have not yet been reported. Finally, GASP-1 also appears to be capable of blocking MSTN activity in vivo, as viral delivery of a GASP-1 expression cassette into adult muscle has been shown to induce increases in muscle mass and grip strength [59]. An elucidation of the roles that GASP-1 and GASP-2 normally play in vivo will await the characterization of mice carrying targeted mutations in these genes.
In addition to the MSTN propeptide, follistatin, FSTL-3, GASP-1, and GASP-2, several other proteins have also been identified as potential regulators of MSTN activity. One of these is Latent TGF-ß Binding Protein-3 (LTBP-3), and as discussed earlier, LTBP-3 has been proposed to play a role in regulating the processing of the MSTN precursor protein [17]. Decorin has also been shown to bind MSTN with high affinity and has been proposed to play a role in maintaining MSTN bound to the extracellular matrix [76]. Decorin also appears to be capable of regulating muscle cell differentiation and muscle regeneration, which may be mediated at least in part by effects on expression of both MSTN and follistatin [77]. Finally, two proteins, Titin-Cap [78] and hSGT [79] were identified as MSTN binding proteins using yeast two-hybrid screens and have been proposed to play roles in regulating MSTN secretion and/or activation.
Existence of Other TGF-ß Family Members with Functional Redundancy with Myostatin
Several studies investigating the effects of these various MSTN binding proteins in vivo have demonstrated that MSTN is not the sole target for these inhibitors in muscle and that other TGF-ß family members seem to function with MSTN to limit muscle mass. The first hint that other ligands may be functionally redundant with MSTN came from the original transgenic studies overexpressing follistatin in muscle [13]. A striking finding was that the increases in muscle mass observed in at least one of the founder animals appeared to surpass those seen in Mstn knockout mice, raising the possibility that the effects of follistatin were being mediated by inhibition of more than just MSTN. Indeed, this possibility was borne out by subsequent studies demonstrating that the follistatin transgene could cause significant increases in muscle mass even in mice lacking MSTN [58]; in fact, the follistatin transgene could cause yet another doubling of muscle mass (i.e. an overall quadrupling) when crossed onto a Mstn -/- background. The fact that most of the increased muscle mass in these mice could be attributed to muscle fiber hypertrophy implied that this additional ligand (or ligands) also functions to regulate postnatal growth of fibers, and this was demonstrated conclusively by electroporation studies in which delivery of a follistatin expression cassette was shown to induce muscle fiber hypertrophy not only in wild type mice but also in Mstn -/- mice [61].
The existence of additional ligands with similar activity to MSTN in suppressing muscle growth was also demonstrated in studies using a different MSTN inhibitor. The demonstration that MSTN signals by binding initially to the activin type II receptors, ACVR2 and ACVR2B [12, 13, 80], led investigators to develop and test a soluble form of one of these receptors for its ability to block MSTN signaling. Specifically, the extracellular domain of ACVR2B fused to an Fc domain (ACVR2B/Fc) was shown to be capable of inhibiting MSTN activity in vitro and promoting muscle growth when administered systemically to adult mice [12]. The effects of ACVR2B/Fc, however, were significantly greater than what had been observed with other myostatin inhibitors, raising the possibility that ACVR2B/Fc was targeting more than just MSTN to exert its effect, and indeed, the effects of ACVR2B/Fc were shown to be attenuated but not eliminated in Mstn null mice.
Finally, genetic studies analyzing the effect of a muscle-specific FSTL-3 transgene in combination with a Mstn mutation demonstrated that like follistatin, FSTL-3 was also capable of inducing muscle hypertrophy to an extent that could not be explained simply by inhibition of MSTN [58]. Taken together, all of these studies have demonstrated that muscle mass is suppressed by the coordinate activities of multiple TGF-ß family members, including MSTN, and that the capacity for increasing muscle growth by targeting this general signaling pathway is much greater than previously appreciated.
The identities of the other ligands that cooperate with MSTN to limit muscle growth are currently unknown. The TGF-ß family consists of almost 40 proteins, and only some of these can be eliminated as candidates based on the criterion that the key ligands can be blocked both by follistatin, by FSTL-3, and by the soluble ACVR2B receptor. One approach to homing in the key ligands has been to identify those that can be affinity purified from serum using ACVR2B/Fc as a ligand trap [81]. This approach identified GDF-11, BMP-9, BMP-10, and various activin isoforms as possible candidates present in both mouse and human serum. Among these, the most obvious candidate was GDF-11, which is highly related in amino acid sequence to MSTN and is known to be expressed in muscle [1, 52, 82]. Mice homozygous for a targeted deletion of the Gdf11 gene have been shown to exhibit multiple developmental abnormalities and die during the perinatal period [67, 83-88], which has precluded a detailed analysis of the effect of complete loss of GDF-11 on muscle function. Studies have been carried out, however, in which a floxed allele of Gdf11 was used to analyze the effect of targeting the gene selectively in skeletal muscle, and these studies failed to uncover any role for GDF-11 in regulating muscle development or growth [82]. Perhaps the next most obvious candidates would be the activins, as a variety of studies have demonstrated that they can regulate differentiation of muscle cells in culture [81, 89-91]. Moreover, electroporation of an activin A expression construct into muscle has been shown to induce muscle atrophy [61], analogous to the muscle atrophy that has been observed upon overexpression of MSTN in vivo [34]. To date, however, no loss-of-function data have been reported supporting a role for any of the activin isoforms in regulating muscle mass.
Implications and Speculations
As discussed in the preceding sections, an extensive volume of work over the past 13 years has led to the identification of numerous components of the MSTN regulatory system, and a variety of experimental approaches have been used to demonstrate that manipulating the activities of these components can lead to significant effects on skeletal muscle mass. A partial summary of some of the genetic data demonstrating the effects of manipulating many of these regulatory components is shown in Fig. (2), where percent increases in muscle mass in a variety of transgenic and mutant mouse lines are plotted for four different muscles (pectoralis, triceps, quadriceps, and gastrocnemius). Collectively, these genetic data illustrate a number of key features of this regulatory system. First, all of these data are consistent with the model shown in Fig. 1 that many different components can be manipulated to regulate levels of signaling through this pathway. In particular, increased activity of components shown in green or decreased activity of components shown in red all lead to decreased signaling and, consequently, to increased muscle growth. The fact that effects in muscle can be achieved by manipulating so many different components implies that there will be many alternative strategies for targeting this general signaling pathway for development of therapeutic agents. Second, the range of effects that can be generated in terms of muscle mass is enormous, with the maximal effect generated to date being a 250-350% increase in muscle weights (i.e. an overall quadrupling) in mice carrying both a follistatin transgene and a Mstn loss-of-function mutation. Third, within this large range, one can generate an almost continuous spectrum of effects depending on how the system is manipulated. The fact that the effect on muscle is titratable suggests that there is a dose-dependent effect of signaling through this pathway. In this respect, one of the striking features of this system is that many of the components exhibit haploinsufficiency, such that mice heterozygous for a given mutation have muscle weights that are intermediate between homozygous mutant and wild type mice. This dose-dependence is one of the most attractive features of this signaling system from the standpoint of targeting the pathway for the development of therapeutic agents. In particular, the dose-dependence suggests that it will not be necessary to block the pathway completely in order to produce a therapeutic effect and that it should be possible to produce different magnitudes of effects for different applications and different patient populations. Fourth, multiple TGF-ß family members, including MSTN, work coordinately to suppress muscle growth. Hence, agents capable of targeting multiple ligands have the potential to produce much larger effects than those targeting just MSTN. Of course, agents with a broader range of specificity also have a higher potential for causing side effects outside of muscle, so it will be essential to pursue multiple strategies for targeting this pathway in order to determine the optimal profile of biologics in which the appropriate balance between therapeutic benefit and off-target activity can be achieved.
Fig. (2).
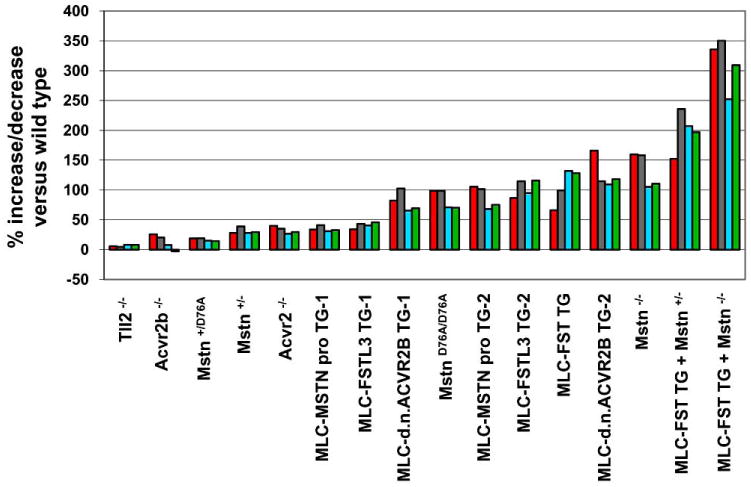
Effects of targeted mutations and/or muscle-specific transgenes for various components of the myostatin signaling pathway in mice. Percent increases in muscle weights relative to wild type mice are shown for four muscles: pectoralis (red), triceps (gray), quadriceps (blue), and gastrocnemius (green). For details, see refs. [12, 13, 40, 58].
All of these features of this signaling system relate to what is likely to be the fundamental reason that this regulatory network is so complex; that is, this system has likely evolved to include so many regulatory components in order to allow the control of growth of individual muscles to be linked both to a variety of specific local physiological stimuli and to the overall physiological state of the animal. In this respect, a conundrum with respect to the biology of MSTN has been the fact that MSTN circulates in the blood. The question is how the growth of individual muscles can be regulated via this signaling system if MSTN protein is distributed to all muscles of the body via the circulation. One possibility is that the circulating protein actually plays no regulatory role and is merely the result of leakage of MSTN from the muscle into the blood. This possibility seems unlikely for two reasons. First, studies have shown that MSTN made at one site in the body (albeit at rather high levels) can exert effects at distant sites [34]. Second, the Mstn loss-of-function mutation appears to exert a maternal effect such that effects of MSTN loss can vary to some extent depending on the genotype of the mother, consistent with the possibility that MSTN protein in the maternal circulation can cross the placenta and influence muscle development in the fetus [58].
A more likely possibility is that the circulating MSTN protein can, indeed, enter the active pool to regulate local muscle growth. In this respect, a critical aspect of this regulatory system is that most, if not all, of the circulating MSTN protein is bound to inhibitory proteins and is therefore inactive. Hence, a reasonable model is that the levels of active MSTN protein at any given site may be determined by the extent to which this latent form is activated, such as by proteolytic cleavage of the propeptide by members of the BMP-1/tolloid family of metalloproteases, and that levels of activation could be controlled locally to regulate growth of individual muscles. An appealing aspect of this model is that the levels of circulating MSTN could then be regulated in response to other physiological stimuli in order to set limits on the amount of protein that would be made available generally to skeletal muscle. Given that levels of MSTN signaling can have profound influences on both fat and glucose metabolism (metabolic functions of MSTN are reviewed in detail by other articles in this collection), I speculated previously that perhaps this regulatory system is so complex because a major role for MSTN may be to regulate the overall balance between fat and muscle [92]. According to this model, circulating levels of MSTN would be high in certain physiological states, which would favor storage of energy reserves in adipose tissue, whereas circulating levels of MSTN would be low in other physiological states, which would allow more of a metabolic shift toward muscle. Because the circulating MSTN protein exists in an inactive, latent state, MSTN can serve this dual role of globally regulating the metabolic balance between fat and muscle while at the same time serving as the local gatekeeper to regulate the growth of individual muscles. Clearly, the identification of additional components of this regulatory system and a detailed analysis of the specific roles played by each of the regulatory components under different physiological conditions and disease states will be essential not only for understanding how both local and systemic cues are integrated by this signaling system to regulate muscle growth but also for developing the most effective strategies for manipulating this regulatory system for clinical applications.
Acknowledgments
I apologize to those colleagues whose work I did not cite as a result of either space limitations or oversight. Myostatin was licensed by the Johns Hopkins University to MetaMorphix (MMI) and sublicensed to Wyeth. I am entitled to a share of sales royalty received by the University from sales of this factor. The Johns Hopkins University and I also own MMI stock, which is subject to certain restrictions under University policy. I am a consultant to MMI. The terms of these arrangements are being managed by the University in accordance with its conflict of interest policies. Work in my laboratory on myostatin is supported by grants from the National Institutes of Health (R01AR059685, R01AR060636), the Muscular Dystrophy Association, and the Jain Foundation.
Abbreviations
- AAV
Adeno-associated virus
- ACVR2
Activin receptor type II
- BMP
Bone morphogenetic protein
- cDNA
Complementary DNA
- CHO
Chinese hamster ovary
- Fc
Immunoglobulin fragment crystallizable
- FSD
Follistatin domain
- FSH
Follicle stimulating hormone
- FST
Follistatin
- FSTL-3
Follistatin-like-3
- GASP
Growth and differentiation factor-associated serum protein
- GDF
Growth/differentiation factor
- HPLC
High pressure liquid chromatography
- IC50
Inhibitory concentration-50
- kD
Kilo-Dalton
- LTBP-3
Latent TGF-ß binding protein-3
- MSTN
Myostatin
- ND
N-terminal domain
- TGF-ß
Transforming growth factor-ß
- TLD
Tolloid
- TLL
Tolloid-like
References
- 1.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-ß superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 2.Grobet L, Martin LJR, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Ménissier F, Massa-banda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- 3.Kambadur R, Sharma M, Smith TPL, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–915. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- 4.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grobet L, Poncelet D, Royo LJ, Brouwers B, Pirottin D, Michaux C, Ménissier F, Zanotti M, Dunner S, Georges M. Molecular definition of an allelic series of mutations disrupting the myostatin function and causing double-muscling in cattle. Mamm Genome. 1998;9:210–213. doi: 10.1007/s003359900727. [DOI] [PubMed] [Google Scholar]
- 6.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 7.Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet. 2007;3(5):779–786. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Kömen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 9.Bogdanovich S, Krag TOB, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 10.Whittemore LA, Song K, Li X, Aghajanian J, Davies MV, Girgenrath S, Hill JJ, Jalenak M, Kelley P, Knight A, Maylor R, O'Hara D, Pearson AA, Quazi A, Ryerson S, Tan XY, Tomkinson K, Veldman G, Widom A, Wright J, Wudyka S, Zhao L, Wolfman NM. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. BBRC. 2003;300:965–971. doi: 10.1016/s0006-291x(02)02953-4. [DOI] [PubMed] [Google Scholar]
- 11.Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspan DS, Lee SJ. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci USA. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SJ, Reed A, Davies M, Girgenrath S, Goad M, Tomkinson K, Wright J, Barker C, Ehrmantraut G, Holmstrom J, Trowell B, Gertz B, Jiang MS, Sebald S, Matzuk M, Li E, Liang L, Quattlebaum E, Stotish R, Wolfman N. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18:251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- 15.Sharma M, Kambadur R, Matthews KG, Somers WG, Devlin GP, Conaglen JV, Fowke PJ, Bass JJ. Myostatin, a transforming growth factor-ß superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J Cell Phys. 1999;180:1–9. doi: 10.1002/(SICI)1097-4652(199907)180:1<1::AID-JCP1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.McFarlane C, Langley B, Thomas M, Hennebry A, Plummer E, Nicholas G, McMahon C, Sharma M, Kambadur R. Proteolytic processing of myostatin is auto-regulated during myogenesis. Dev Biol. 2005;283(1):58–69. doi: 10.1016/j.ydbio.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 17.Anderson SB, Goldberg AL, Whitman M. Identification of a novel pool of extracellular pro-myostatin in skeletal muscle. J Biol Chem. 2008;283(11):7027–7035. doi: 10.1074/jbc.M706678200. [DOI] [PubMed] [Google Scholar]
- 18.Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. The structure of myostatin:follistatin 288: insights into receptor utilization and heparin binding. EMBO J. 2009;28(17):2662–2676. doi: 10.1038/emboj.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitt UA, Hsu SY, Hsueh AJ. Evolution and classification of cystine knot-containing hormones and related extracellular signaling molecules. Mol Endocrinol. 2001;15(5):681–694. doi: 10.1210/mend.15.5.0639. [DOI] [PubMed] [Google Scholar]
- 20.Gray AM, Mason AJ. Requirement for activin A and transforming growth factor-ß1 pro- regions in homodimer assembly. Science. 1990;247:1328–1330. doi: 10.1126/science.2315700. [DOI] [PubMed] [Google Scholar]
- 21.Jin HJ, Dunn MA, Borthakur D, Kim YS. Refolding and purification of unprocessed porcine myostatin expressed in Escherichia coli. Protein Expr Purif. 2004;35(1):1–10. doi: 10.1016/j.pep.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Funkenstein B, Rebhan Y. Expression, purification, renaturation and activation of fish myostatin expressed in Escherichia coli: facilitation of refolding and activity inhibition by myostatin prodomain. Protein Expr Purif. 2007;54(1):54–65. doi: 10.1016/j.pep.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Jiang MS, Liang LF, Wang S, Ratovitski T, Holmstrom J, Barker C, Stotish R. Characterization and identification of the inhibitory domain of GDF-8 propeptide. Biochem Biophys Res Commun. 2004;315(3):525–531. doi: 10.1016/j.bbrc.2004.01.085. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Ratovitski T, Brady J, Solomon M, Wells K, Wall R. Expression of myostatin pro domain results in muscular transgenic mice. Mol Reprod Dev. 2001;60:351–361. doi: 10.1002/mrd.1097. [DOI] [PubMed] [Google Scholar]
- 25.Pirottin D, Grobet L, Adamantidis A, Farnir F, Herens C, Daa Schroder H, Georges M. Transgenic engineering of male-specific muscular hypertrophy. Proc Natl Acad Sci USA. 2005;102(18):6413–6418. doi: 10.1073/pnas.0502426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X, Hadhazy M, Wehling M, Tidball JG, McNally EM. Dominant negative myostatin produces hypertrophy without hyperplasia in muscle. FEBS Lett. 2000;474:71–75. doi: 10.1016/s0014-5793(00)01570-2. [DOI] [PubMed] [Google Scholar]
- 27.Nishi M, Yasue A, Nishimatu S, Nohno T, Yamaoka T, Itakura M, Moriyama K, Ohuchi H, Noji S. A missense mutant myostatin causes hyperplasia without hypertrophy in the mouse muscle. Biochem Biophys Res Commun. 2002;293:247–251. doi: 10.1016/S0006-291X(02)00209-7. [DOI] [PubMed] [Google Scholar]
- 28.Bartoli M, Poupiot J, Vulin A, Fougerousse F, Arandel L, Daniele N, Roudaut C, Noulet F, Garcia L, Danos O, Richard I. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not alpha-sarcoglycan deficiency. Gene Ther. 2007;14(9):733–740. doi: 10.1038/sj.gt.3302928. [DOI] [PubMed] [Google Scholar]
- 29.Qiao C, Li J, Jiang J, Zhu X, Wang B, Li J, Xiao X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum Gene Ther. 2008;19(3):241–254. doi: 10.1089/hum.2007.159. [DOI] [PubMed] [Google Scholar]
- 30.Amthor H, Otto A, Vulin A, Rochat A, Dumonceaux J, Garcia L, Mouisel E, Hourdé C, Macharia R, Friedrichs M, Relaix F, Zammit PS, Matsakas A, Patel K, Partridge T. Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc Natl Acad Sci USA. 2009;106(18):7479–7484. doi: 10.1073/pnas.0811129106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foster K, Graham IR, Otto A, Foster H, Trollet C, Yaworsky PJ, Walsh FS, Bickham D, Curtin NA, Kawar SL, Patel K, Dickson G. Adeno-associated virus-8-mediated intravenous transfer of myostatin propeptide leads to systemic functional improvements of slow but not fast muscle. Rejuvenation Res. 2009;12(2):85–94. doi: 10.1089/rej.2008.0815. [DOI] [PubMed] [Google Scholar]
- 32.Matsakas A, Foster K, Otto A, Macharia R, Elashry MI, Feist S, Graham I, Foster H, Yaworsky P, Walsh F, Dickson G, Patel K. Molecular, cellular and physiological investigation of myostatin propeptide-mediated muscle growth in adult mice. Neuromuscul Disord. 2009;19(7):489–99. doi: 10.1016/j.nmd.2009.06.367. [DOI] [PubMed] [Google Scholar]
- 33.Bogdanovich S, Perkins K, Krag T, Whittemore LA, Khurana T. Myostatin propeptide-mediated amelioration of dystophic pathophysiology. FASEB J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- 34.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 35.Hill JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J Biol Chem. 2002;277:40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- 36.Hopkins DR, Keles S, Greenspan DS. The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 2007;26:508–523. doi: 10.1016/j.matbio.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blader P, Rastegar S, Fischer N, Strahle U. Cleavage of the BMP-4 antagonist chordin by zebrafish tolloid. Science. 1997;278:1937–1940. doi: 10.1126/science.278.5345.1937. [DOI] [PubMed] [Google Scholar]
- 38.Marques G, Musacchio M, Shimell MJ, Wunnenberg-Stapleton K, Cho KW, O'Conner MB. Production of a DPP activity gradient in the early Drosophila embryo through the opposing actions of the SOG and TLD proteins. Cell. 1997;91:417–426. doi: 10.1016/s0092-8674(00)80425-0. [DOI] [PubMed] [Google Scholar]
- 39.Piccolo S, Agius E, Lu B, Goodman S, Dale L, DeRobertis EM. Cleavage of chordin by xolloid metalloprotease suggests a role for proteolytic processing in the regulation of Spemann organizer activity. Cell. 1997;91:407–416. doi: 10.1016/s0092-8674(00)80424-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee SJ. Genetic analysis of the role of proteolysis in the activation of latent myostatin. PLoS One. 2008;3(2):e1628. doi: 10.1371/journal.pone.0001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott IC, Blitz IL, Pappano WN, Imamura Y, Clark TG, Steiglitz B, Thomsa CL, Maas SA, Takahara K, Cho KWY, Greenspan DS. Mammalian BMP-1/tolloid-related metalloproteinases, including novel family member mammalian tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev Biol. 1999;213:283–300. doi: 10.1006/dbio.1999.9383. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki N, Labosky P, Furuta Y, Hargett L, Dunn R, Fogo A, Takahara K, Peters D, Greenspan D, Hogan B. Failure of ventral body wall closure in mouse embryos lacking a procollagen C-proteinase encoded by Bmp1, a mammalian gene related to Drosophila tolloid. Development. 1996;122:3587–3595. doi: 10.1242/dev.122.11.3587. [DOI] [PubMed] [Google Scholar]
- 43.Clark T, Conway S, Scott I, Labosky P, Winnier G, Bundy J, Hogan B, Greenspan D. The mammalian Tolloid-like 1 gene, Tll1, is necessary for normal septation and positioning of the heart. Development. 1999;126:2631–2642. doi: 10.1242/dev.126.12.2631. [DOI] [PubMed] [Google Scholar]
- 44.Robertson DM, Klein R, de Vos FL, McLachlan RI, Wettenhall RE, Hearn MT, Burger HG, de Kretser DM. The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which are structurally different to inhibin. Biochem Biophys Res Commun. 1987;149(2):744–749. doi: 10.1016/0006-291x(87)90430-x. [DOI] [PubMed] [Google Scholar]
- 45.Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R. Isolation and partial characterization of follistatin: a single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc Natl Acad Sci USA. 1987;84(23):8282–8286. doi: 10.1073/pnas.84.23.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H. Activin-binding protein from rat ovary is follistatin. Science. 1990;247:836–838. doi: 10.1126/science.2106159. [DOI] [PubMed] [Google Scholar]
- 47.Ling N, Ying SY, Ueno N, Shimasaki S, Esch F, Hotta M, Guillemin R. Pituitary FSH is released by a heterodimer of the ß-subunits from the two forms of inhibin. Nature. 1986;321:779–782. doi: 10.1038/321779a0. [DOI] [PubMed] [Google Scholar]
- 48.Vale W, Rivier J, Vaughan J, McClintock R, Corrigan A, Woo W, Karr D, Spiess J. Purification and characterization of an FSH releasing protein from porcine ovarian follicular fluid. Nature. 1986;321:776–779. doi: 10.1038/321776a0. [DOI] [PubMed] [Google Scholar]
- 49.Yamashita H, ten Dijke P, Huylebroeck D, Sampath TK, Andries M, Smith J, Miyazono K. Osteogenic protein-1 binds to activin type II receptors and induces certain activin-like effects. J Cell Biol. 1995;130:217–226. doi: 10.1083/jcb.130.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fainsod A, Deißler K, Yelin R, Marom K, Epstein M, Pillemer G, Steinbeisser H, Blum M. The dorsalizing and neural inducing gene follistatin is an antagonist of BMP-4. Mech Dev. 1997;63:39–50. doi: 10.1016/s0925-4773(97)00673-4. [DOI] [PubMed] [Google Scholar]
- 51.Iemura SI, Yamamoto TS, Takagi C, Uchiyama H, Natsume T, Shimasaki S, Sugino H, Ueno N. Direct binding of follistatin to a complex of bone-morphogenetic protein and its receptor inhibits ventral and epidermal cell fates in early Xenopus embryo. Proc Natl Acad Sci USA. 1998;95:9337–9342. doi: 10.1073/pnas.95.16.9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gamer LW, Wolfman NM, Celeste AJ, Hattersley G, Hewick R, Rosen V. A novel BMP expressed in developing mouse limb, spinal cord, and tail bud is a potent mesoderm inducer in Xenopus embryos. Dev Biol. 1999;208:222–232. doi: 10.1006/dbio.1998.9191. [DOI] [PubMed] [Google Scholar]
- 53.Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R, Patel K. Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesis. Dev Biol. 2004;270(1):19–30. doi: 10.1016/j.ydbio.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 54.Nakatani M, Takehara Y, Sugino H, Matsumoto M, Hashimoto O, Hasegawa Y, Murakami T, Uezumi A, Takeda S, Noji S, Sunada Y, Tsuchida K. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008;22(2):477–487. doi: 10.1096/fj.07-8673com. [DOI] [PubMed] [Google Scholar]
- 55.Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev Cell. 2005;9(4):535–543. doi: 10.1016/j.devcel.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 56.Hashimoto O, Kawasaki N, Tsuchida K, Shimasaki S, Haya-kawa T, Sugino H. Difference between follistatin isoforms in the inhibition of activin signalling: activin neutralizing activity of follistatin isoforms is dependent on their affinity for activin. Cell Signal. 2000;12(8):565–571. doi: 10.1016/s0898-6568(00)00099-1. [DOI] [PubMed] [Google Scholar]
- 57.Schneyer AL, Sidis Y, Gulati A, Sun JL, Keutmann H, Krasney PA. Differential antagonism of activin, myostatin and growth and differentiation factor 11 by wild-type and mutant follistatin. Endocrinology. 2008;149(9):4589–4595. doi: 10.1210/en.2008-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SJ. Quadrupling muscle mass in mice by targeting TGF-ß signaling pathways. PLoS One. 2007;2(8):e789. doi: 10.1371/journal.pone.0000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C, Boue D, Martin PT, Sahenk Z, Mendell JR, Kaspar BK. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008;105(11):4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kota J, Handy C, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR, Tucker D, Shilling CJ, Therlfall WR, Walker CM, Weisbrode SE, Janssen PML, Clark KR, Sahenk Z, Mendell JR, Kaspar BK. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci Transl Med. 2009;1(6):1–8. doi: 10.1126/scitranslmed.3000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009;297:E157–E164. doi: 10.1152/ajpendo.00193.2009. [DOI] [PubMed] [Google Scholar]
- 62.Iezzi S, Di Padova M, Serra C, Caretti G, Simone C, Mak-lan E, Minetti G, Zhao P, Hoffman EP, Puri PL, Sartorelli V. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6(5):673–684. doi: 10.1016/s1534-5807(04)00107-8. [DOI] [PubMed] [Google Scholar]
- 63.Minetti GC, Colussi C, Adami R, Serra C, Mozetta C, Parente V, Fortuni S, Straino S, Sampaolesi M, Di Padova M, Illi B, Gallinari P, Steinkühler C, Capogrossi MC, Sartorelli V, Bottinelli R, Gaetano C, Puri PL. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12(10):1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 64.Rose FF, Jr, Mattis VB, Rindt H, Lorson CL. Delivery of recombinant follistatin lessens disease severity in a mouse model of spinal muscular atrophy. Hum Mol Genet. 2009;18(6):997–1005. doi: 10.1093/hmg/ddn426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suryawan A, Frank JW, Nguyen HV, Davis TA. Expression of the TGF-beta family of ligands is developmentally regulated in skeletal muscle of neonatal rats. Pediatr Res. 2006;59(2):175–179. doi: 10.1203/01.pdr.0000196718.47935.6e. [DOI] [PubMed] [Google Scholar]
- 66.Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A. Multiple defects and perinatal death in mice deficient in follistatin. Nature. 1995;374:360–363. doi: 10.1038/374360a0. [DOI] [PubMed] [Google Scholar]
- 67.McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Gen. 1999;22:260–264. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- 68.Hayette S, Gadoux M, Martel S, Bertrand S, Tigaud I, Magaud JP, Rimokh R. FLRG (follistatin-related gene), a new target of chromosomal rearrangement in malignant blood disorders. Oncogene. 1998;16:2949–2954. doi: 10.1038/sj.onc.1201807. [DOI] [PubMed] [Google Scholar]
- 69.Tsuchida K, Arai KY, Kuramoto Y, Yamakawa N, Hasegawa Y, Sugino H. Identification and characterization of a novel follistatin-like protein as a binding protein for the TGF-ß family. J Biol Chem. 2000;275:40788–40796. doi: 10.1074/jbc.M006114200. [DOI] [PubMed] [Google Scholar]
- 70.Stamler R, Keutmann HT, Sidis Y, Kattamuri C, Schneyer A, Thompson TB. The structure of FSTL3.activin A complex. Differential binding of N-terminal domains influences follistatin-type antagonist specificity. J Biol Chem. 2008;283(47):32831–32838. doi: 10.1074/jbc.M801266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Welt C, Sidis Y, Keutmann H, Schneyer A. Activins, inhibins, and follistatins: from endocrinology to signaling. A paradigm for the new millennium. Exp Biol Med (Maywood) 2002;227(9):724–752. doi: 10.1177/153537020222700905. [DOI] [PubMed] [Google Scholar]
- 72.Mukherjee A, Sidis Y, Mahan A, Raher MJ, Xia Y, Rosen ED, Bloch KD. FSTL3 deletion reveals roles for TGF-ß family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci USA. 2007;104(4):1348–1353. doi: 10.1073/pnas.0607966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hill JJ, Qiu Y, Hewick RM, Wolfman NM. Regulation of myostatin in vivo by growth and differentiation factor-associated serum protein-1: a novel protein with protease inhibitor and follistatin domains. Mol Endocrinol. 2003;17:1144–1154. doi: 10.1210/me.2002-0366. [DOI] [PubMed] [Google Scholar]
- 74.Trexler M, Banyai L, Patthy L. A human protein containing multiple types of protease-inhibitory modules. Proc Natl Acad Sci USA. 2001;98(7):3705–3709. doi: 10.1073/pnas.061028398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kondás K, Szláma G, Trexler M, Patthy L. Both WFIKKN1 and WFIKKN2 have high affinity for growth and differentiation factors 8 and 11. J Biol Chem. 2008;283(35):23677–23684. doi: 10.1074/jbc.M803025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miura T, Kishioka Y, Wakamatsu J, Hattori A, Hennebry A, Berry CJ, Sharma M, Kambadur R, Nishimura T. Decorin binds myostatin and modulates its activity to muscle cells. Biochem Biophys Res Commun. 2006;340(2):675–680. doi: 10.1016/j.bbrc.2005.12.060. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, Li J, Zhu J, Sun B, Branca M, Tang Y, Foster W, Xiao X, Huard J. Decorin gene transfer promotes muscle cell differentiation and muscle regeneration. Mol Ther. 2007;15(9):1616–1622. doi: 10.1038/sj.mt.6300250. [DOI] [PubMed] [Google Scholar]
- 78.Nicholas G, Thomas M, Langley B, Somers W, Patel K, Kemp CF, Sharma M, Kambadur R. Titin-cap associates with, and regulates secretion of, Myostatin. J Cell Physiol. 2002;193(1):120–131. doi: 10.1002/jcp.10158. [DOI] [PubMed] [Google Scholar]
- 79.Wang H, Zhang Q, Zhu D. hSGT interacts with the N-terminal region of myostatin. Biochem Biophys Res Commun. 2003;311(4):877–883. doi: 10.1016/j.bbrc.2003.10.080. [DOI] [PubMed] [Google Scholar]
- 80.Rebbapragada A, Benchabane H, Wrana J, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor ß-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23(20):7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Souza TA, Chen X, Guo Y, Sava P, Zhang J, Hill JJ, Yaworsky PJ, Qiu Y. Proteomic identification and functional validation of activins and bone morphogenetic 11 as candidate novel muscle mass regulators. Mol Endocrinol. 2008;22(12):2689–2702. doi: 10.1210/me.2008-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11. BMC Dev Biol. 2009;9:24–32. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Esquela AF, Lee SJ. Regulation of metanephric kidney development by growth/differentiation factor 11. Dev Biol. 2003;257:356–370. doi: 10.1016/s0012-1606(03)00100-3. [DOI] [PubMed] [Google Scholar]
- 84.Wu HH, Ivkovic S, Murray RC, Jaramillo S, Lyons KM, Johnson JE, Calof AL. Autoregulation of neurogenesis by GDF11. Neuron. 2003;37:197–207. doi: 10.1016/s0896-6273(02)01172-8. [DOI] [PubMed] [Google Scholar]
- 85.Harmon EB, Apelqvist AA, Smart NG, Gu X, Osborne DH, Kim SK. GDF11 modulates NGN3+ islet progenitor cell number and promotes ß-cell differentiation in pancreas development. Development. 2004;131:6163–6174. doi: 10.1242/dev.01535. [DOI] [PubMed] [Google Scholar]
- 86.Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, Calof AL. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- 87.Dichmann DS, Yassin H, Serup P. Analysis of pancreatic endocrine development in GDF11-deficient mice. Dev Dyn. 2006;235:3016–3025. doi: 10.1002/dvdy.20953. [DOI] [PubMed] [Google Scholar]
- 88.Liu JP. The function of growth/differentiation factor 11 (Gdf11) in rostrocaudal patterning of the developing spinal cord. Development. 2006;133:2865–2874. doi: 10.1242/dev.02478. [DOI] [PubMed] [Google Scholar]
- 89.Link B, Nishi R. Opposing effects of activin A and follistatin on developing skeletal muscle cells. Exp Cell Res. 1997;233:350–362. doi: 10.1006/excr.1997.3575. [DOI] [PubMed] [Google Scholar]
- 90.He L, Vichev K, Macharia R, Huang R, Christ B, Patel K, Amthor H. Activin A inhibits formation of skeletal muscle during chick development. Anat Embryol (Berl) 2005;209(5):401–407. doi: 10.1007/s00429-005-0454-1. [DOI] [PubMed] [Google Scholar]
- 91.Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- 92.Lee SJ. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol. 2004;20:61–86. doi: 10.1146/annurev.cellbio.20.012103.135836. [DOI] [PubMed] [Google Scholar]