

Abstract
The vacuolar proton-translocating ATPase (V-ATPase) plays a major role in organelle acidification and works together with other ion transporters to maintain pH homeostasis in eukaryotic cells. We analyzed a requirement for V-ATPase activity in protein trafficking in the yeast secretory pathway. Deficiency of V-ATPase activity caused by subunit deletion or glucose deprivation results in missorting of newly synthesized plasma membrane proteins Pma1 and Can1 directly from the Golgi to the vacuole. Vacuolar mislocalization of Pma1 is dependent on Gga adaptors although no Pma1 ubiquitination was detected. Proper cell surface targeting of Pma1 was rescued in V-ATPase-deficient cells by increasing the pH of the medium, suggesting that missorting is the result of aberrant cytosolic pH. In addition to mislocalization of the plasma membrane proteins, Golgi membrane proteins Kex2 and Vrg4 are also missorted to the vacuole upon loss of V-ATPase activity. Because the missorted cargos have distinct trafficking routes, we suggest a pH dependence for multiple cargo sorting events at the Golgi.
Keywords: Plasma Membrane Proton Pumps, Protein Sorting, Trafficking, Ubiquitination, Vacuolar ATPase
Introduction
The yeast plasma membrane ATPase, Pma1, is an electrogenic proton pump that regulates intracellular pH and generates the membrane potential across the plasma membrane. Pma1 is a member of the conserved P-type ATPase family. Because Pma1 is abundant and its activity at the cell surface is essential for cell viability (1), it has served as a model polytopic membrane protein to study protein sorting and quality control in the secretory pathway. Studies using misfolded Pma1 mutants have revealed disposal of conformationally defective proteins by post-endoplasmic reticulum (ER)2 mechanisms as well as the better characterized ER-associated degradation (ERAD) pathway (2). Misfolded Pma1–7 (P434A,G789S) is impaired in plasma membrane targeting and routed instead from Golgi to the endosomal/vacuolar system for degradation (3, 4). By contrast, misfolded Pma1–10 (A165G,V197I) fails to remain stable at the cell surface and undergoes vacuolar degradation through endocytosis (5). Additional studies have shown that wild-type Pma1 is associated with sphingolipid- and cholesterol-enriched microdomains, and both plasma membrane targeting of Pma1 and its stability at the cell surface require sphingolipids (6, 7).
The vacuolar proton-translocating ATPase (V-ATPase) is a second proton pump, mechanistically distinct from Pma1, that plays a major role in maintaining pH homeostasis (8, 9). The importance of V-ATPase for acidification of the vacuole/lysosomes, Golgi, and endosomes of eukaryotic cells is well established (10, 11). In contrast with Pma1, which is thought to function as a homohexamer (12), V-ATPase is composed of two multisubunit subcomplexes, the peripheral catalytic V1 subcomplex and the integral membrane proton-translocating V0 subcomplex (11). V-ATPase biogenesis is complex and incompletely understood. Assembly of V1 and V0 sector subunits require a protein complex called RAVE (regulator of the (H+)-ATPase of vacuolar and endosomal membranes), which also regulates V-ATPase activity in response to low extracellular glucose by reversible disassembly of V1 and V0 sectors (13).
A recent structure-function study noted significant vacuolar alkalization and cytosolic acidification in cells lacking the V0 subunits Vma2 or Vma3 (14). Surprisingly, Pma1 was also mislocalized to the vacuole in vma2Δ and vma3Δ cells (14). Because Pma1 and V-ATPase work together to maintain cellular pH homeostasis, we carried out studies on vacuolar sorting of Pma1 in V-ATPase-deficient cells. We find missorting is not specific to Pma1; rather, there is cargo-specific missorting from the Golgi that is induced by aberrant pH in cells defective in V-ATPase activity.
EXPERIMENTAL PROCEDURES
Media, Strains, and Plasmids
Standard yeast media and genetic manipulations were as described (15). Yeast transformations were performed by the lithium acetate method (16). Strains were grown at 30 °C unless otherwise indicated. Strains including vma3Δ, vph1Δ, vma1Δ, and rav1Δ are in the BY4741 background (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Open Biosystems). Deletion strains were confirmed by PCR. NY430 is MATa ura3–52 sec14–3 (17). CMY119 is MATα gga1Δ1::TRP1 gga2Δ1::HIS3 ura3–52 leu2–3,112 his3-Δ200 trp1–901 lys2–801 suc2-Δ9 (18). CJY35 was generated in a one-step gene replacement by transformation of wild-type cells with pCKR3A, a LEU2-marked VPS1 disruption construct (19). CJY5 was generated in a one-step gene replacement by transformation of vma3Δ cells with pSN273, a LEU2-marked construct to disrupt PEP4 (20). CJX14–1C is a vps27Δvma3Δ strain generated by a cross between CJY37 (MATa vps27Δ::LEU2 his3Δ1 met15Δ0 ura3Δ0) and CJX2–2C (MATα vma3Δ::kanMX his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0).
Pma1-GFP is under the control of its native promoter on a URA3-marked centromeric plasmid (pRS316) (21), a gift from Annick Breton, CNRS, France. pCHL571.4 is a URA3-marked centromeric plasmid (pRS416) bearing Can1-GFP, as described (22), and pGFP-CPS is a 2μ TRP1-marked plasmid (pRS424) (23); both are gifts from Scott Emr, Cornell University, Ithaca, NY. pBG2Kex2-GFP is a 2μ URA3-marked plasmid (pRS426) (24), a gift from Tom Vida, University of Texas, Houston. Vrg4-GFP is carried on a URA3-marked centromeric plasmid (YCplac33), as described (25), a gift from Ben Glick, University of Chicago, IL. pS1 and pS3 are HIS3-marked centromeric plasmids bearing MET25-HA-PMA1 and MET25-HA-pma1–7, respectively (26).
Western Blot, Protein Induction, and Protein Secretion
For Western blot, cell lysates were prepared by vortexing with glass beads in the presence of a protease inhibitor mixture including 1 mm phenylmethylsulfonyl fluoride, as described previously (27). Samples were normalized to lysate protein by Bradford assay (Bio-Rad). Samples were separated by SDS-PAGE and transferred to nitrocellulose for Western blot.
Cells bearing pMET25-HA-PMA1 or pMET25-HA-pma1–7 were grown in minimal medium supplemented with 600 μm methionine. Mid-log cells were harvested, washed once with water, and resuspended in methionine-free medium for 3 h to derepress synthesis of HA-Pma1. Cells were shifted to 37 °C during derepression as Pma1–7 is temperature-sensitive (4). For detection of ubiquitination, cells were harvested and lysed in the presence of 10 mm N-ethylmaleimide. Immunoprecipitations with anti-HA antibody (Covance, Princeton, NJ) were with 200 μg of lysate protein in RIPA buffer. Immunoprecipitations were analyzed by Western blot with anti-ubiquitin (1 μg/ml; Zymed Laboratories Inc., San Francisco, CA), as described (28). After chemiluminescence detection, blots were stripped and reprobed with anti-HA antibody.
For analysis of invertase and Bgl2 secretion, mid-log phase cells were cultured in YPD medium at 25 °C (or shifted to 37 °C for 1 h for the sec14–3 temperature-sensitive mutant). Invertase was induced by resuspending cells in low glucose medium (YEP plus 0.1% glucose) for 5 h. Cells and media were collected, and cells were spheroplasted with 0.1 mg/ml zymolyase at 37 °C for 30 min. Internal and external fractions were analyzed by Western blot with rabbit anti-invertase (Scott D. Emr, Cornell University) and anti-Bgl2 antibody (Patrick Brennwald, University of North Carolina, Chapel Hill).
Metabolic Labeling
Cells were grown overnight in minimal medium to mid-log phase. Cells were resuspended in fresh medium at a density of 1 A600/ml and incubated at room temperature for 10 min before pulse-labeling with Expre35S35S (0.4 mCi/A600 cells) (PerkinElmer Life Sciences). Cells were labeled for 5 min before chase with an equal volume of synthetic complete (SC) medium supplemented with 20 mm methionine and cysteine. At various times of chase, aliquots were removed and added to 10 mm azide on ice. Lysate was prepared by vortexing with glass beads (27), and immunoprecipitations were normalized to acid-precipitable cpm. Immunoprecipitations with anti-Pma1, anti-carboxypeptidase Y (CPY, Rockland Immunochemicals), or anti-carboxypeptidase S (CPS, Scott Emr, Cornell University) antibodies were analyzed by SDS-PAGE and fluorography.
Fluorescence Microscopy
Cells bearing GFP constructs were grown to mid-log in SC medium, harvested, and visualized with an Olympus fluorescence microscope equipped with a fluorescein isothiocyanate (FITC) filter. For indirect immunofluorescence staining, cells were fixed, permeabilized, and stained with monoclonal anti-HA (Covance) and CY3-conjuncated secondary antibody (Jackson ImmunoResearch), as described previously (29); fluorescence was visualized with a Texas Red filter. Fluorescence and differential interference contrast (DIC) images were collected with a Hamamatsu Orca CCD camera.
For glucose deprivation prior to imaging, cells were pelleted, washed with water, and resuspended in SC medium with 2% raffinose for various times. For actin depolymerization, 10 μm latrunculin A (Lat-A, Sigma) was added for 3 h. Control cells received an equal volume of dimethyl sulfoxide.
Cell Fractionation
Fractionation on Renografin density gradients was as described (30). RenoCal-76 (Bracco Diagnostics, Princeton, NJ) was substituted for Renografin-76. Fourteen fractions were collected and diluted 10-fold with buffer (50 mm Tris, pH 7.5, 1 mm EDTA), and membranes were pelleted by centrifugation at 100,000 × g for 1 h in a TLA 120.2 rotor and analyzed by Western blotting with anti-GFP (Covance). Fractionation of Gas1 and alkaline phosphatase (ALP), markers for plasma membrane and vacuole, was determined using antibodies from Howard Riezman (University of Geneva, Geneva, Switzerland) and Greg Payne (UCLA), respectively. Quantitation was performed using NIH Image on scanned Western blots.
RESULTS
Pma1 Is Mislocalized to the Vacuole in Cells Lacking V-ATPase Activity
Previous work has shown mislocalization of Pma1 to the vacuole in a V-ATPase mutant, vma3 (14). To address the molecular basis for Pma1 mislocalization, we used an HA-tagged PMA1 construct under the control of a MET25 promoter (26). As shown in Fig. 1, HA-Pma1, synthesized following derepression of the MET25 promoter, localized to the cell surface in wild-type cells, like endogenous Pma1 (26). In vma3Δ cells, however, HA-Pma1 appears predominantly coincident with the lumen of the vacuole, visualized as a crater by DIC microscopy. Similarly, a Pma1-GFP construct was observed to localize properly to the plasma membrane in wild-type cells while mislocalizing to the vacuolar lumen in vma3Δ cells (Fig. S1A). Like Pma1-GFP, Can1-GFP, the arginine permease, is also mislocalized from the plasma membrane in wild-type cells to the vacuole in vma3Δ cells (Fig. S1B).
FIGURE 1.

Mislocalization of HA-Pma1 in wild-type and vma3Δ strains. Wild-type and vma3Δ cells bearing pMET25-HA-PMA1 were derepressed by shifting into methionine-free medium, as described under “Experimental Procedures.” Cells were fixed and permeabilized for indirect immunofluorescence staining with anti-HA antibody followed by CY3-conjugated secondary antibody. Cells before (off) and after a 3-h derepression (on) are indicated.
Deletion of the Vma3 subunit of the V-ATPase results in loss of more than 95% of the proton pumping activity (31). Mislocalization of Pma1-GFP and Can1-GFP in vma3Δ cells appears to derive from loss of V-ATPase activity because mislocalization of these proteins also occurs in the absence of Vma1, the ATP-binding catalytic subunit of the V1 subcomplex, and in vph1Δ cells lacking a V0 subunit with 30-fold less proton transport activity (32) (Fig. S2). Although Pma1 is predominantly mislocalized to the vacuole in V-ATPase-deficient cells, a fraction necessarily escapes to the cell surface to sustain cell viability (1).
Proper assembly and activity of the V-ATPase holoenzyme requires the RAVE complex, composed of Rav1, Rav2, and Skp1, which associates with the V1 subcomplex (33). The V-ATPase activity in rav1Δ or rav2Δ cells is only 1–2% that of wild-type cells (13), which is similar to the level of activity loss in strains lacking one of the structural subunits of the enzyme (34). Both Pma1 and Can1 are localized to the vacuole in rav1Δ cells (Fig. S2). These results suggest a requirement for V-ATPase activity for proper sorting of Pma1 and Can1.
Pma1 Is Missorted from the Golgi to Vacuole in vma3Δ Cells
Vacuolar mislocalization of Pma1 in vma3Δ cells could occur by Pma1 endocytosis from the cell surface or by routing of newly synthesized Pma1 from Golgi to the vacuole via the endosomal system. To determine whether vacuolar localization of Pma1 in vma3Δ cells requires endocytosis, we used Lat-A, an inhibitor of actin polymerization, to block endocytosis (35). As a positive control, we visualized Ste3, the α mating factor receptor, which turns over rapidly from the plasma membrane (36). Ste3-GFP was predominantly observed in the vacuole in vma3Δ cells at steady state (Fig. 2A), as in wild-type cells (data not shown). After 3 h in the presence of Lat-A, Ste3-GFP accumulation at the plasma membrane was apparent, indicating an internalization block. By contrast, both Pma1 and Can1 were observed predominantly in the vacuole in vma3Δ cells in the presence or absence of Lat-A (Fig. 2, B and C). These results suggest a sorting defect in the absence of V-ATPase activity, causing Pma1 and Can1 to go from the Golgi to the vacuole directly without arriving at the plasma membrane.
FIGURE 2.
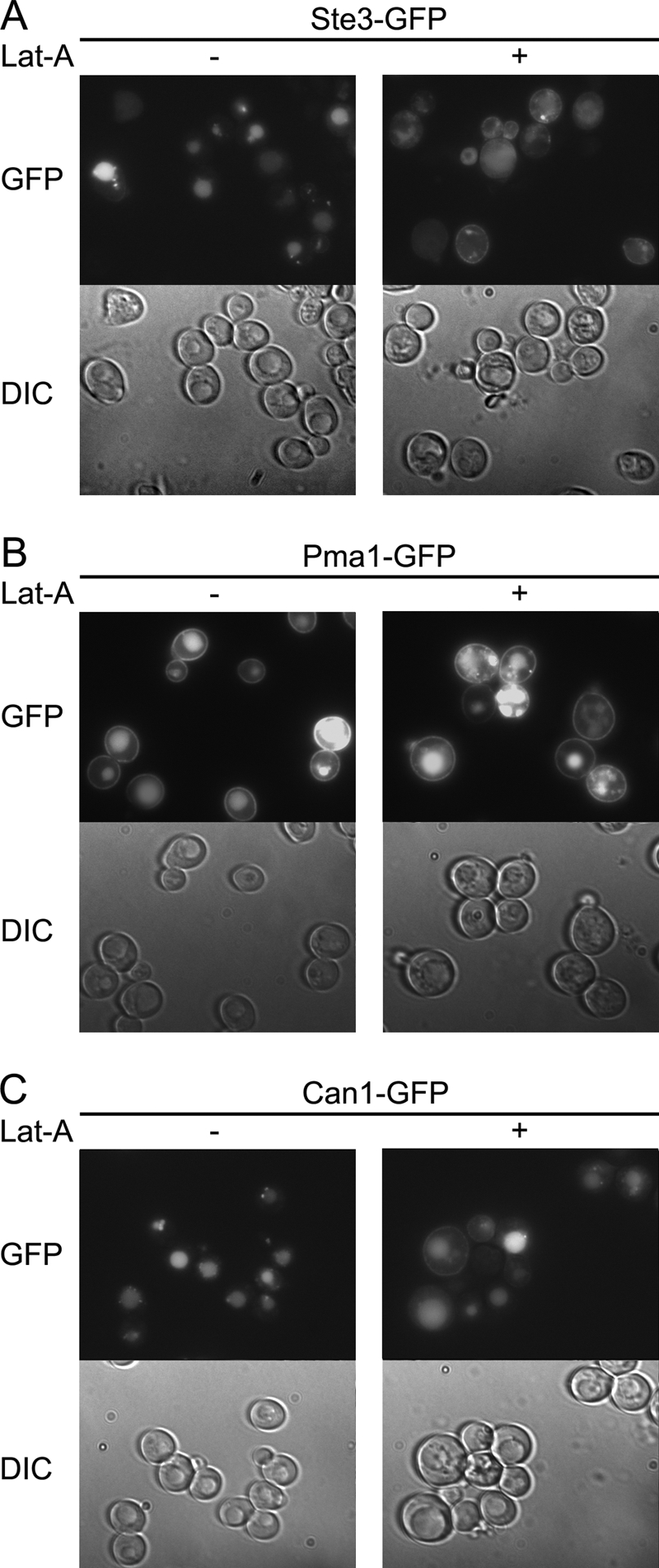
Endocytosis is not required for mislocalization of Pma1 and Can1 in vma3Δ cells. vma3Δ cells expressing Ste3-GFP (A), Pma1-GFP (B), and Can1-GFP (C) were treated with 5 μm Lat-A (+) or dimethyl sulfoxide (−). After 3 h, GFP fluorescence and DIC were imaged and shown in top and bottom panels, respectively.
Mislocalization of Pma1 Occurs Rapidly upon V-ATPase Disassembly
V-ATPase activity is regulated by disassembly of intact active V-ATPase complexes into V1 and V0 subcomplexes within 5 min of glucose deprivation, resulting in the loss of >60% ATPase activity (33, 37). Glucose readdition results in rapid reassembly of the V-ATPase (37). If Pma1 sorting is dependent on V-ATPase activity, rapid inactivation of the V-ATPase by glucose deprivation should result in Pma1 mislocalization. In Fig. 3, wild-type cells bearing Pma1-GFP were switched from growth in glucose to raffinose. After 30 min of glucose deprivation, Pma1 fluorescence was coincident with vacuoles visualized by DIC. No vacuolar localization was observed when cycloheximide was included during glucose deprivation (Fig. 3C), indicating newly synthesized Pma1 is missorted to the vacuole in V-ATPase-deficient cells. These results are consistent with direct transport of newly synthesized Pma1 from the Golgi to the vacuole; pre-existing Pma1 is not internalized from the cell surface.
FIGURE 3.

Glucose deprivation causes missorting of newly synthesized Pma1. Wild-type cells expressing Pma1-GFP were collected, washed with water, and then resuspended in SC minus uracil with 2% glucose (A), 2% raffinose (B), or 2% raffinose plus 10 μg/ml cycloheximide (C). At various times, cells were harvested for GFP fluorescence (top panels) and DIC imaging (bottom panels).
Pma1 Is Missorted to the Vacuole Due to pH Disturbance in vma3Δ Cells
In vma3Δ cells there is cytosolic acidification and vacuolar alkalization with cytosolic pH decreasing from 6.0 to 5.5 and vacuolar pH increasing from 6.0 to 6.4 (14). Indeed, vma3Δ cells, as well as other mutants defective in V-ATPase activity, have impaired growth in medium of pH 7.0 or higher (38). To determine whether Pma1 missorting is dependent on pH, cells were grown in buffered medium. Previous work has shown that buffering the culture medium at pH 7.5 can restore the cytoplasmic pH of vma3Δ cells, although vacuolar pH still remains as high as 6.6 (14). Fig. 4A shows that plasma membrane localization of Pma1 in wild-type cells is not affected by medium pH. In vma3Δ cells, however, vacuolar localization of Pma1-GFP was observed in cells grown in medium buffered to 4.2. Strikingly, vacuolar localization disappears in medium buffered to pH 6.6 (Fig. 4A), and instead, Pma1-GFP was visualized at the plasma membrane in vma3Δ cells under these conditions. Like Pma1, vacuolar localization of Can1-GFP disappears at pH 6.6 in vma3Δ cells, whereas in wild-type cells, Can1-GFP localization to the plasma membrane is not affected by medium pH (Fig. 4B). Under conditions identical to a previous report showing restoration of cytosolic pH in vma3Δ cells by buffering the medium to pH 7.5 (14), plasma membrane localization of both Pma1-GFP and Can1-GFP were restored (Fig. S3). These results suggest that missorting of Pma1 and Can1 is suppressed by buffering the medium to higher pH, increasing cytosolic pH in vma3Δ cells.
FIGURE 4.

Missorting of Pma1 and Can1 in vma3Δ cells is rescued by buffering the culture medium to alkaline pH. Wild-type and vma3Δ cells bearing Pma1-GFP (A) or Can1-GFP (B) were grown in medium buffered to pH 4.2 and pH 6.6 (with 20 mm sodium citrate). GFP fluorescence and DIC images are shown in top and bottom panels, respectively.
To confirm the restoration effect, cell fractionation was used to examine Pma1 localization in wild-type and vma3Δ cells buffered at normal and high pH. Renocal-76 density gradients were used to resolve plasma membrane from intracellular membranes (26, 39). Fig. 5A shows density gradients separating the plasma membrane marker Gas1p, maximal in fractions 10–11, from the vacuolar membrane marker ALP, maximal in fractions 3–5. In wild-type cells at both pH 4.2 and 6.6, Pma1-GFP is coincident with Gas1, maximal in fractions 9–12 (Fig. 5). In vma3Δ cells grown at pH 4.2, Pma1-GFP was observed in fractions coincident with ALP and consistent with vacuolar localization. In contrast, in vma3Δ cells grown in pH 6.6 medium, distribution of Pma1-GFP was found coincident with the plasma membrane marker Gas1p (Fig. 5). Therefore, buffering vma3Δ cells at high pH restores normal targeting of Pma1 to the plasma membrane.
FIGURE 5.

Cell fractionation on Renocal-76 density gradients. A, wild-type and vma3Δ cells bearing Pma1-GFP were grown in SC minus uracil buffered to pH 4.2 and pH 6.6. Cell lysates were separated on density gradients, fractions were collected, and membranes were analyzed by Western blot. Pma1-GFP was detected by anti-GFP antibody. Gas1 and ALP were assayed as plasma membrane and vacuolar membrane markers, respectively. Asterisk indicates nonspecific band. B, Pma1-GFP distribution was quantified and plotted.
Perturbation of Transport from the Golgi in vma3Δ Cells
A battery of marker proteins was examined to determine whether pH perturbation in vma3Δ cells results in mislocalization of other proteins. These markers, including markers for vacuolar membrane and multivesicular body, are localized normally in vma3Δ cells (data not shown). However, mislocalization of two Golgi membrane markers, Kex2 and Vrg4, was detected in vma3Δ cells (Fig. 6). Kex2 cycles between Golgi and endosome, and impaired Golgi-endosome trafficking results in delivery of Kex2 to the vacuole (40, 41). Fig. 6A shows Kex2-GFP in puncta characteristic of Golgi and endosomes in wild-type cells. In vma3Δ cells, some Kex2-GFP is present in puncta but also coincident with vacuolar indentations seen by DIC. Like Kex2-GFP, mislocalization of Vrg4-GFP to the vacuole was also observed in vma3Δ cells (Fig. 6B). Vrg4, a GDP-mannose transporter, cycles between ER and Golgi and undergoes vacuolar delivery when COPI-dependent retrograde transport is impaired (42). Other Golgi membrane markers such as Sec7, Sec21, and Sys1 have normal punctate Golgi localization in both WT and vma3Δ cells (data not shown).
FIGURE 6.
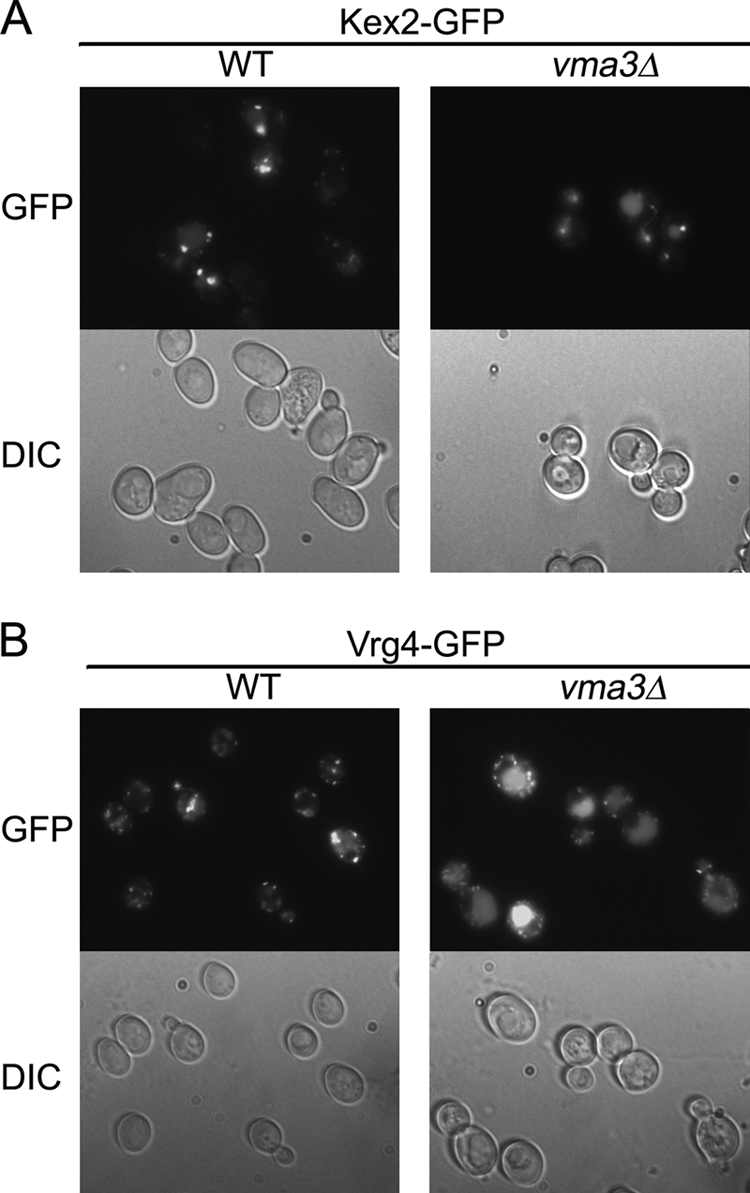
Two Golgi membrane proteins Kex2 and Vrg4 are missorted in vma3Δ cells. Wild-type and vma3Δ cells bearing Kex2-GFP (A) and Vrg4-GFP (B) were visualized. GFP fluorescence and DIC images are in top and bottom panels, respectively.
Exocytosis and ER-Golgi Transport Appear Unaffected in vma3Δ Cells
Additional vesicular transport pathways were examined to determine whether these are also affected by disturbed pH homeostasis in vma3Δ cells. To examine the exocytic pathway in vma3Δ cells, secretion of invertase and Bgl2 was assayed. Invertase and Bgl2 are secreted via different populations of post-Golgi secretory vesicles (43). No significant difference in release of invertase (upper panel) or Bgl2 (lower panel) to the extracellular space was detected in wild-type and vma3Δ cells (Fig. 7A). As a control, secretion of invertase and Bgl2 is blocked in sec14–3 cells at the restrictive temperature but not at the permissive temperature (44) (Fig. 7A).
FIGURE 7.

Exocytosis and ER-Golgi transport are unaffected in vma3Δ cells. A, secretion of invertase (upper panel) and Bgl2 (lower panel) was detected by Western blot. sec14–3 is a control for a secretory block. I and E are intracellular and extracellular fractions, respectively. B, vacuolar delivery of newly synthesized CPY. Cells were pulse-labeled with Expre35S35S for 5 min and chased for various times. CPY was immunoprecipitated from lysate and analyzed by SDS-PAGE and fluorography. ER-localized p1, Golgi-modified p2, and proteolytically processed mature forms of CPY are indicated.
Pulse-chase analysis was used to follow vacuolar delivery of newly synthesized CPY. CPY transport from ER to Golgi is accompanied by conversion from p1 to p2 forms, and proteolytic processing to the mature form occurs upon vacuolar arrival (45, 46). In wild-type cells, mature CPY is the predominant form seen by 15 min of chase, whereas p1 and p2 forms are still detectable at this time in vma3Δ cells (Fig. 7B). Either transport from Golgi to vacuole and/or proteolytic processing appear slightly delayed in vma3Δ cells, but ER-Golgi transport is not significantly affected.
Pma1 Missorting Is Dependent on GGA Adaptor Proteins Although Pma1 Is Not Ubiquitinated
We examined whether Pma1 missorting from Golgi to vacuole is dependent on GGA proteins, Gga1 and Gga2 (Golgi-localized, γ-ear-containing, ARF-binding), adaptors for clathrin-coated vesicles forming at the late Golgi (47). In gga1Δ gga2Δ cells, Pma1-GFP is properly localized at the plasma membrane (Fig. 8A). Upon glucose deprivation to impair V-ATPase, Pma1-GFP was observed predominantly at the plasma membrane in gga1Δ gga2Δ cells (Fig. 8A), suggesting rerouting of Pma1 to the cell surface when sorting into clathrin-coated vesicles is blocked. Consistently, Pma1-GFP was also rerouted to the cell surface in the absence of glucose in vps1Δ cells lacking dynamin required for clathrin-coated vesicle formation (48) (Fig. S4). The AP-1 complex is another adaptor for Golgi-derived clathrin-coated vesicles (49). In aps1Δ cells, lacking a subunit of AP-1, Pma1-GFP is not prevented from vacuolar delivery upon glucose deprivation (Fig. 8A). Thus, in cells impaired in V-ATPase activity, Pma1 appears to enter clathrin-coated vesicles via interaction with GGA adaptors for delivery to the vacuole.
FIGURE 8.

Missorting of Pma1 is blocked in gga1Δ gga2Δ cells, but Pma1 is not ubiquitinated in vma3Δ cells. A, gga1Δ gga2Δ (left panel) and aps1Δ (right panel) cells bearing Pma1-GFP were grown in SC minus uracil with 2% glucose or 2% raffinose. GFP fluorescence and DIC images are shown in top and bottom panels, respectively. B, cells bearing pMET-HA-Pma1 (pS1) or pMET-HA-Pma1–7 (pS3) were derepressed for 3 h at 37 °C to express tagged Pma1 or Pma1–7. HA-Pma1 or HA-Pma1–7 was immunoprecipitated with anti-HA antibody, and immunoprecipitations were analyzed by Western blot with anti-ubiquitin (top) followed by anti-HA (bottom). The position of HA-tagged Pma1 is indicated by the arrow.
Because GGA proteins can recognize ubiquitinated cargo for sorting to endosomes (50), we asked whether Pma1 is ubiquitinated in vma3Δ cells. For these experiments, we used a construct in which HA-tagged Pma1 is under the control of the MET25 promoter. Newly synthesized HA-Pma1 was immunoprecipitated and assayed by Western blot with anti-ubiquitin. In wild-type cells, no ubiquitin signal is associated with Pma1 (Fig. 8B, lane 1), consistent with the longevity of wild-type Pma1 (51, 52). As a positive control, Pma1–7 was analyzed because it undergoes temperature-sensitive ubiquitination and mislocalization to the vacuole (4). A series of high molecular weight bands migrating above the position of HA-Pma1–7 was observed in wild-type cells (Fig. 8B, lane 2), as well as in vma3Δ cells (Fig. 8B, lane 4). By contrast, ubiquitination of wild-type Pma1 was not detectable in vma3Δ cells (Fig. 8B, lane 3). To increase detection of ubiquitinated protein, HA-tagged Pma1 was accumulated in pep4 cells, defective in vacuolar proteolysis, and vps27 cells, defective in sorting to multivesicular bodies (53). Although Pma1–7 ubiquitination is enhanced (Fig. 8B, lanes 6 and 8), ubiquitination remains undetectable for wild-type Pma1 in vma3Δ pep4Δ and vma3Δ vps27Δ cells (Fig. 8B, lanes 5 and 7).
Proteolytic Processing Is Delayed in vma3Δ Cells
Although ubiquitinated Pma1 was not detected in vma3Δ cells, Pma1-GFP is localized to the vacuolar lumen, suggesting it undergoes internalization into multivesicular bodies for degradation. Pulse-chase was used to examine Pma1 degradation. Fig. 9A shows that Pma1 is stable at the plasma membrane in wild-type cells. Surprisingly, Pma1 in the vacuole in vma3Δ cells remains stable even after 3 h of chase. Consistent with this result, degradation of missorted Kex2-GFP is also delayed in vma3Δ cells (Fig. S5).
FIGURE 9.

Vacuolar proteolysis in vma3Δ strain. Wild-type and vma3Δ cells were pulse-labeled with Expre35S35S for 5 min and chased for various times. Pma1 (A) and CPS (B) were immunoprecipitated from lysate and analyzed by SDS-PAGE and fluorography. The position of precursor (p) and mature (m) forms of CPS are indicated by black lines. The arrow indicates an intermediate band position where pCPS and mCPS forms comigrate (55).
Maturation of CPS was analyzed to assess vacuolar proteolysis in vma3Δ cells. CPS is synthesized as a precursor (pCPS), which undergoes ubiquitination, internalization into multivesicular endosomes, and proteolytic processing to a mature form (mCPS) in the vacuolar lumen (54). In wild-type cells, mCPS is formed after an ∼40-min chase (Fig. 9B), consistent with a previous report (55). Both pCPS and mCPS migrate as two differentially glycosylated forms with the lower molecular weight form of pCPS comigrating with the higher molecular weight mCPS (55) (Fig. 9B, arrow). In vma3Δ cells, CPS processing to the mature form is delayed so that mCPS was not apparent until 2 h of chase (Fig. 9B). Localization of GFP-CPS to the vacuolar lumen is unaffected in vma3Δ cells (Fig. S6). These results are in agreement with previous findings suggesting that V-ATPase activity is important for vacuolar proteolytic activity (55, 61–64).
DISCUSSION
A major finding of this study is pH-dependent cargo sorting from the Golgi in the yeast secretory pathway. Impaired V-ATPase activity perturbs pH homeostasis, causing cytoplasmic acidification and vacuolar alkalization (14). Mislocalization of the cell surface proteins Pma1 and Can1 to the vacuole is a result of diminished V-ATPase activity, caused by loss of subunits of the V1 or V0 subcomplex, loss of RAVE complex assembly factors, or glucose deprivation (Figs. S1–S2). Cytoplasmic pH is restored upon growing V-ATPase-deficient cells in medium buffered to high pH (14). A role for cytoplasmic pH in regulating cargo sorting is supported by suppression of Pma1 mislocalization in vma3Δ cells upon buffering the medium pH (Fig. 3). Vacuolar mislocalization of Pma1 upon glucose deprivation is prevented by cycloheximide, suggesting a defect in cell surface targeting of newly synthesized Pma1.
In V-ATPase-deficient cells, missorting of Pma1 from the Golgi to the vacuole is dependent on GGA adaptor proteins in clathrin-coated vesicles (Figs. 1 and 8). Previously, we reported that two misfolded Pma1 mutants, Pma1–7 and Pma1–10, undergo ubiquitination and vacuolar delivery upon recognition by post-ER quality control mechanisms (4, 56). To address whether a change in cytoplasmic pH might produce a conformational change in wild-type Pma1, ubiquitination of Pma1 was examined in vma3Δ cells (Fig. 8B). Mutant Pma1–7 was examined as a positive control as it undergoes temperature-sensitive ubiquitination and vacuolar delivery (4). Although ubiquitination of Pma1–7 was readily observed, ubiquitination of wild-type Pma1 was not detected in vma3Δ cells. Ubiquitination of wild-type Pma1 was undetectable even when vacuolar proteolysis or sorting to multivesicular endosomes was prevented in vma3Δpep4Δ and vma3Δvps27Δ cells, respectively (Fig. 8B). These results are consistent with vacuolar sorting of Pma1 in V-ATPase-deficient cells occurring by a mechanism distinct from that involved in vacuolar targeting of misfolded mutant Pma1. GGA recognition of wild-type Pma1 in V-ATPase-deficient cells appears ubiquitin-independent (50) whereas GGA adaptors are required for recognition of ubiquitinated Pma1–7 (4). These results are consistent with substrate recognition by GGA via both ubiquitin-dependent and ubiquitin-independent binding (57).
Vacuolar mislocalization of Pma1 and Can1 suggests that V-ATPase activity is required for sorting of these cargos into post-Golgi secretory vesicles. By contrast, loss of V-ATPase activity does not affect secretion of Bgl2 and invertase (Fig. 7). Thus, post-Golgi vesicles are generated properly when V-ATPase activity is lost or decreased, but sorting of specific cargo into these vesicles is impaired.
In addition to missorting of plasma membrane proteins Pma1 and Can1, deficient V-ATPase activity also results in missorting of two Golgi membrane proteins, Kex2 and Vrg4 (Fig. 6). In wild-type cells, localization of Kex2 and Vrg4 is maintained by cycling between Golgi and endosome and Golgi and ER, respectively (40, 42). Transport of Kex2 from Golgi to endosome occurs via GGA adaptor-mediated clathrin-coated vesicles, and recycling to the Golgi occurs via retromer-coated vesicles (58, 59); Vrg4 recycles from Golgi to ER via COPI vesicles (42). When their recycling routes are impaired, Kex2 and Vrg4 are delivered to the vacuole (40, 42). Because anterograde pathways for vacuolar delivery of Kex2 and Vrg4 appear unaffected in V-ATPase-deficient cells, our results suggest that retrieval of Vrg4 from Golgi to the ER and Kex2 retrieval from endosome to Golgi are pH-dependent. Of significance to a requirement for V-ATPase activity for proper Golgi function, loss of function mutations in the a2 subunit of V-ATPase are linked to cutis laxa, or wrinkly skin syndrome, and are associated with impaired Golgi-to-ER transport (60).
V-ATPase plays a critical role in membrane trafficking by maintaining cytosolic pH homeostasis (8) as well as generating acidic intracellular compartments (10). Impaired V-ATPase activity results in increased luminal pH (14). As a consequence, vacuolar proteolysis is impaired, as revealed by delayed CPY and CPS processing in vma3Δ cells (Figs. 7B and 9) and prolonged vacuolar stability of mislocalized Pma1 and Kex2 (Figs. 9 and S5). Our results are consistent with previous work showing that luminal acidification is required for efficient processing of vacuolar proteins and sorting within endosomes and multivesicular bodies (61–64). Luminal acidity is also necessary for lipid turnover in vacuoles/lysosomes (65). At the cytosolic face of membranes, there is a requirement for V-ATPase activity for recruitment of the cytosolic regulatory proteins Arf6 and ARNO to initiate transport vesicle formation at endosomal membranes (66). Our discovery of a requirement for V-ATPase activity in cargo sorting at the Golgi suggests additional pH-regulated mechanisms in protein trafficking.
Acknowledgments
We thank Nick Davis, Ben Glick, Scott Emr, Patrick Brennwald, Howard Riezman, and Greg Payne for plasmids and antibodies.
This work was supported, in whole or in part, by National Institutes of Health Grant GM 58212.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6.
- ER
- endoplasmic reticulum
- V-ATPase
- vacuolar proton-translocating ATPase
- RAVE
- regulator of the (H+)-ATPase of vacuolar and endosomal membranes
- CPY
- carboxypeptidase Y
- CPS
- carboxypeptidase S
- DIC
- differential interference contrast
- Lat-A
- latrunculin A
- ALP
- alkaline phosphatase
- GGA
- Golgi-localized, γ-ear-containing, ARF-binding
- pCPS
- precursor CPS
- mCPS
- mature CPS.
REFERENCES
- 1. Serrano R., Kielland-Brandt M. C., Fink G. R. (1986) Nature 319, 689–693 [DOI] [PubMed] [Google Scholar]
- 2. Arvan P., Zhao X., Ramos-Castaneda J., Chang A. (2002) Traffic 3, 771–780 [DOI] [PubMed] [Google Scholar]
- 3. Chang A., Fink G. R. (1995) J. Cell Biol. 128, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pizzirusso M., Chang A. (2004) Mol. Biol. Cell 15, 2401–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gong X., Chang A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 9104–9109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bagnat M., Chang A., Simons K. (2001) Mol. Biol. Cell 12, 4129–4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Toulmay A., Schneiter R. (2007) Biochimie 89, 249–254 [DOI] [PubMed] [Google Scholar]
- 8. Catty P., de Kerchove d'Exaerde A., Goffeau A. (1997) FEBS Lett. 409, 325–332 [DOI] [PubMed] [Google Scholar]
- 9. Stevens T. H., Forgac M. (1997) Annu. Rev. Cell Dev. Biol. 13, 779–808 [DOI] [PubMed] [Google Scholar]
- 10. Forgac M. (2007) Nat. Rev. Mol. Cell Biol. 8, 917–929 [DOI] [PubMed] [Google Scholar]
- 11. Kane P. M. (2006) Microbiol. Mol. Biol. Rev. 70, 177–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kühlbrandt W., Zeelen J., Dietrich J. (2002) Science 297, 1692–1696 [DOI] [PubMed] [Google Scholar]
- 13. Smardon A. M., Tarsio M., Kane P. M. (2002) J. Biol. Chem. 277, 13831–13839 [DOI] [PubMed] [Google Scholar]
- 14. Martínez-Muñoz G. A., Kane P. (2008) J. Biol. Chem. 283, 20309–20319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sherman F., Hicks J. B., Fink G. R. (1986) Methods in Yeast Genetics, pp. 523–585, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 16. Gietz D., St. Jean A., Woods R. A., Schiestl R. H. (1992) Nucleic Acids Res. 20, 1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salminen A., Novick P. J. (1987) Cell 49, 527–538 [DOI] [PubMed] [Google Scholar]
- 18. Dell'Angelica E. C., Puertollano R., Mullins C., Aguilar R. C., Vargas J. D., Hartnell L. M., Bonifacino J. S. (2000) J. Cell Biol. 149, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rothman J. H., Raymond C. K., Gilbert T., O'Hara P. J., Stevens T. H. (1990) Cell 61, 1063–1074 [DOI] [PubMed] [Google Scholar]
- 20. Nothwehr S. F., Bryant N. J., Stevens T. H. (1996) Mol. Cell. Biol. 16, 2700–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balguerie A., Bagnat M., Bonneu M., Aigle M., Breton A. M. (2002) Eukaryot. Cell 1, 1021–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lin C. H., MacGurn J. A., Chu T., Stefan C. J., Emr S. D. (2008) Cell 135, 714–725 [DOI] [PubMed] [Google Scholar]
- 23. Odorizzi G., Babst M., Emr S. D. (1998) Cell 95, 847–858 [DOI] [PubMed] [Google Scholar]
- 24. Gerhardt B., Kordas T. J., Thompson C. M., Patel P., Vida T. (1998) J. Biol. Chem. 273, 15818–15829 [DOI] [PubMed] [Google Scholar]
- 25. Losev E., Reinke C. A., Jellen J., Strongin D. E., Bevis B. J., Glick B. S. (2006) Nature 441, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 26. Luo W., Chang A. (2000) Mol. Biol. Cell 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang A., Slayman C. W. (1991) J. Cell Biol. 115, 289–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Swerdlow P. S., Finley D., Varshavsky A. (1986) Anal. Biochem. 156, 147–153 [DOI] [PubMed] [Google Scholar]
- 29. Rose M. D., Winston F., Hieter F. (1990) Methods in Yeast Genetics, p. 198, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30. Chang A. (2002) Methods Enzymol. 351, 339–350 [DOI] [PubMed] [Google Scholar]
- 31. Umemoto N., Yoshihisa T., Hirata R., Anraku Y. (1990) J. Biol. Chem. 265, 18447–18453 [PubMed] [Google Scholar]
- 32. Perzov N., Padler-Karavani V., Nelson H., Nelson N. (2002) J. Exp. Biol. 205, 1209–1219 [DOI] [PubMed] [Google Scholar]
- 33. Seol J. H., Shevchenko A., Deshaies R. J. (2001) Nat. Cell Biol. 3, 384–391 [DOI] [PubMed] [Google Scholar]
- 34. Curtis K. K., Francis S. A., Oluwatosin Y., Kane P. M. (2002) J. Biol. Chem. 277, 8979–8988 [DOI] [PubMed] [Google Scholar]
- 35. Ayscough K. R. (2005) Protoplasma 226, 81–88 [DOI] [PubMed] [Google Scholar]
- 36. Davis N. G., Horecka J. L., Sprague G. F., Jr. (1993) J. Cell Biol. 122, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kane P. M. (1995) J. Biol. Chem. 270, 17025–17032 [PubMed] [Google Scholar]
- 38. Nelson H., Nelson N. (1990) Proc. Natl. Acad. Sci. U. S. A. 87, 3503–3507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schandel K. A., Jenness D. D. (1994) Mol. Cell. Biol. 14, 7245–7255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brickner J. H., Fuller R. S. (1997) J. Cell Biol. 139, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Conibear E., Stevens T. H. (1998) Biochim. Biophys. Acta 1404, 211–230 [DOI] [PubMed] [Google Scholar]
- 42. Abe M., Noda Y., Adachi H., Yoda K. (2004) J. Cell Sci. 117, 5687–5696 [DOI] [PubMed] [Google Scholar]
- 43. Harsay E., Bretscher A. (1995) J. Cell Biol. 131, 297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Curwin A. J., Fairn G. D., McMaster C. R. (2009) J. Biol. Chem. 284, 7364–7375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rothman J. H., Howald I., Stevens T. H. (1989) EMBO J. 8, 2057–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stevens T., Esmon B., Schekman R. (1982) Cell 30, 439–448 [DOI] [PubMed] [Google Scholar]
- 47. Bonifacino J. S. (2004) Nat. Rev. Mol. Cell Biol. 5, 23–32 [DOI] [PubMed] [Google Scholar]
- 48. Vater C. A., Raymond C. K., Ekena K., Howald-Stevenson I., Stevens T. H. (1992) J. Cell Biol. 119, 773–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yeung B. G., Phan H. L., Payne G. S. (1999) Mol. Biol. Cell 10, 3643–3659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scott P. M., Bilodeau P. S., Zhdankina O., Winistorfer S. C., Hauglund M. J., Allaman M. M., Kearney W. R., Robertson A. D., Boman A. L., Piper R. C. (2004) Nat. Cell Biol. 6, 252–259 [DOI] [PubMed] [Google Scholar]
- 51. Benito B., Moreno E., Lagunas R. (1991) Biochim. Biophys. Acta 1063, 265–268 [DOI] [PubMed] [Google Scholar]
- 52. Wang Q., Chang A. (2002) Proc. Natl. Acad. Sci. U. S. A. 99, 12853–12858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Piper R. C., Cooper A. A., Yang H., Stevens T. H. (1995) J. Cell Biol. 131, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Spormann D. O., Heim J., Wolf D. H. (1992) J. Biol. Chem. 267, 8021–8029 [PubMed] [Google Scholar]
- 55. Anand V. C., Daboussi L., Lorenz T. C., Payne G. S. (2009) Mol. Biol. Cell 20, 1592–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu Y., Chang A. (2006) J. Cell Sci. 119, 360–369 [DOI] [PubMed] [Google Scholar]
- 57. Babst M. (2004) Nat. Cell Biol. 6, 175–177 [DOI] [PubMed] [Google Scholar]
- 58. Mullins C., Bonifacino J. S. (2001) Mol. Cell Biol. 21, 7981–7994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spelbrink R. G., Nothwehr S. F. (1999) Mol. Biol. Cell 10, 4263–4281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kornak U., Reynders E., Dimopoulou A., van Reeuwijk J., Fischer B., Rajab A., Budde B., Nurnberg P., Foulquier F., Lefeber D., Urban Z., Gruenewald S., Annaert W., Brunner H. G., van Bokhoven H., Wevers R., Morava E., Matthijs G., Van Maldergem L., Mundlos S. (2008) Nat. Genet. 40, 32–34 [DOI] [PubMed] [Google Scholar]
- 61. Klionsky D. J., Nelson H., Nelson N. (1992) J. Biol. Chem. 267, 3416–3422 [PubMed] [Google Scholar]
- 62. Klionsky D. J., Nelson H., Nelson N., Yaver D. S. (1992) J. Exp. Biol. 172, 83–92 [DOI] [PubMed] [Google Scholar]
- 63. Morano K. A., Klionsky D. J. (1994) J. Cell Sci. 107, 2813–2824 [DOI] [PubMed] [Google Scholar]
- 64. Yaver D. S., Nelson H., Nelson N., Klionsky D. J. (1993) J. Biol. Chem. 268, 10564–10572 [PubMed] [Google Scholar]
- 65. Futerman A. H., van Meer G. (2004) Nat. Rev. Mol. Cell Biol. 5, 554–565 [DOI] [PubMed] [Google Scholar]
- 66. Recchi C., Chavrier P. (2006) Nat. Cell Biol. 8, 107–109 [DOI] [PubMed] [Google Scholar]