

Abstract
A recent proteomics study identified FAM129B or MINERVA as a target of the MAP kinase (Erk1/2) signaling cascade in human melanoma cells. Phosphorylation of the protein was found to promote cell invasion and the dissociation of the protein from the cell-cell junctions. Suppression of apoptosis during metastasis is a prerequisite for the survival and spread of cancer cells. During apoptosis, the adherens junctions are disassembled as the dying cell retracts, and new contacts are formed between normal neighboring cells. In this study, we show that FAM129B was cytosolic in exponentially growing HeLa cells but was translocated to the adherens junctions where it colocalized with β-catenin whenever contact between two or more cells was established. Silencing the FAM129B gene expression by specific siRNAs did not induce apoptosis or inhibit the growth of HeLa cells. However, when apoptosis was induced by exposure to TNFα/cycloheximide or other apoptotic signaling molecules, the onset of apoptosis was accelerated 3–4-fold when FAM129B was depleted. Annexin V binding, the inactivation of the DNA repair enzyme, poly(ADP-ribose) polymerase, and the activation of the caspases occurred more rapidly in the cells lacking FAM129B. The rapid induction of apoptosis in FAM129B knockdown cells was reversed by co-transfection with recombinant FAM129B, indicating that its effect on apoptosis was specific. As apoptosis proceeded, FAM129B was degraded and disappeared from the plasma membrane. Thus, one crucial facet of the mechanism by which FAM129B promotes cancer cell invasion is likely to be the suppression of apoptosis.
Keywords: Apoptosis, Caspase, Cell Junctions, Cell-Cell Interaction, Cellular Regulation, ERK, Fluorescence, Plasma Membrane, Adherens Junctions, Cancer Cell Invasion
Introduction
FAM129B or MINERVA is a member of a small family of proteins that include Niban (FAM129A) and Niban-like protein 2 (FAM129C). The function of these homologous proteins is not well understood. FAM129A is an endoplasmic reticulum stress-induced protein that is up-regulated in renal and thyroid cancer (1–3), whereas FAM129C is a B-cell membrane protein that is overexpressed in chronic lymphocytic leukemia (4). FAM129B has a predicted molecular mass of 83 kDa and includes two distinctive regions (Fig. 1). The PH or pleckstrin homology domain found near the amino end of the polypeptide of FAM129B is also present in FAM129C, whereas FAM129A has only a truncated PH domain. Near the carboxyl end of the FAM129B polypeptide chain is a region rich in proline shown in a recent proteomics study to be phosphorylated at six serine residues (5). The phosphorylation of four of these serines is catalyzed by the B-RAF/MAPKK/ERK (MAP kinase) signaling cascade. In melanoma cells, FAM129B was found dispersed throughout the cytoplasm when the MAPK cascade was active. However, exposure to the MAPKK inhibitor, UO126, which effectively shuts down the cascade, resulted in the migration of FAM129B to the cell membrane. A well established in vitro assay for invasion (6), in which cells are grown in a three-dimensional collagen matrix, was used to show that shRNA-mediated knockdown of FAM129B had no effect on growth. However, the invasion into the collagen matrix was blocked suggesting that FAM129B plays a critical role in cancer cell invasion. Mutants in which the serine residues targeted by MAPK were replaced with alanine were less invasive, whereas transfection of a wild type clone overexpressing FAM129B enhanced the invasiveness of the melanoma cells. The authors concluded that MAPK-dependent phosphorylation of FAM129B controls melanoma cell invasion and proposed that the protein be renamed MINERVA (melanoma invasion by ERK). There are no other published studies of FAM129B thus far.
FIGURE 1.

FAM129B mRNA and polypeptide. FAM129B, an 83-kDa polypeptide (residues 1–746), has a PH domain near the amino end (residues 69–192) and a proline-rich region (residues 628–730) near the carboxyl end. Four Erk1/2 phosphorylation sites (P) have been identified (5) corresponding to Ser628, Ser633, Ser679, and Ser683. The diagram also shows the FAM129B mRNA and the location where four siRNAs bind (see “Experimental Procedures”). All four oligonucleotides effectively silenced FAM129B expression, but the oligonucleotide HSS185602 exhibited some off target effects and was not used in these studies.
Apoptosis plays a crucial role in cancer progression and invasion (7). During metastasis, the cells are subjected to numerous challenges in escaping the site of the primary tumor, traversing the circulatory system and invading the distal cells, which would normally induce apoptosis (8). As a consequence, metastasis is a very inefficient process because very few metastatic cells survive to colonize other tissues (9). Thus, the survival of the cancer cell depends on the suppression of apoptosis. Many cancer related genes can disrupt apoptosis. The gene Bcl-2 does not promote cell cycle progression or cell proliferation but instead prevents induction of apoptosis (10). The expression of Bcl-2 has been shown to be associated with a poor prognosis in prostatic cancer, colon cancer, and neuroblastoma (11, 12). Moreover, there is a high frequency of apoptosis in tumors that spontaneously regress and in tumors treated with chemotherapeutic agents (7, 13). The efficacy of many anticancer agents is related to their ability to promote apoptosis (14). Moreover, drug resistance in melanoma is most likely the result of dysregulation leading to suppression of apoptosis, although other mechanisms may be involved as well (15).
Disruption of the adherens cell junctions is an early event in apoptosis (16). Adherens junctions are protein complexes at the plasma membrane responsible for establishing cell-cell adhesion (17–21). The adherens junction complex includes a transmembrane receptor, cadherin, and the associated components on the cytosolic face of the membrane, α-catenin, β-catenin, and P120 catenin that mediate the interaction of cadherin with the underlying actin cytoskeleton. During apoptosis, the adherens junction proteins are cleaved, and the junction is lost. At this stage of apoptosis, the actin cytoskeleton retracts, and new junctions are formed between neighboring, robust cells to fill the gap created by the shrinkage of the dying cell (22). This study was undertaken to explore the role of FAM129B/MINERVA in apoptosis.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
Antibodies used for this study were rabbit anti-FAM129B (catalog no. 5122), rabbit anti-caspase-3 (catalog no. 9662), rabbit anti-cleaved caspase-3 (catalog no. 9664), rabbit anti-poly(ADP-ribose) polymerase (PARP3; catalog no. 9542), rabbit anti-caspase-9 antibody (catalog no. 9502), mouse monoclonal anti-cdk6 (catalog no. 3136), mouse anti-caspase-8 (catalog no. 9746) (Cell Signaling, Beverly, MA); rabbit anti-cdk2 (catalog no. 21111) and rabbit anti-Akt (catalog no. 21054) (Signalway Antibody, Pearland, TX), mouse monoclonal antibody anti-β-catenin (catalog no. 610154) (BD Transduction Laboratories), mouse monoclonal anti-p53 (catalog no. sc-126), mouse monoclonal anti-GFP (catalog no. sc-9996), and mouse monoclonal β-tubulin (catalog no. sc-5274) (Santa Cruz Biotechnology, Inc., Santa Cruz). The general caspase inhibitor (Z-VAD-fmk) was purchased from R&D Systems (Minneapolis, MN); MG132 and cycloheximide (CHX) was from Sigma; and staurosporine (STS) and recombinant human TNFα were purchased from Invitrogen. The compounds UO126, EGF, wortmannin (catalog no. W1628), and phorbol 12-myristate 13-acetate were from Sigma, and bisindolylmaleimide (catalog no. sc-24004) and the general Akt inhibitor (catalog no. sc-221226) were from Santa Cruz Biotechnology.
Cell Culture and Induction of Apoptosis
HeLa cells were used in this study because they are a well established system to study apoptosis. HT1080 cells (a gift of Dr. Avraham Raz, Wayne State University), which can be induced to undergo apoptosis without the addition of CHX were used in some protein degradation studies. Both cell lines were cultured in DMEM containing 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, in 5% CO2 at 37 °C. NIH3T3 cells were cultured in the same medium except that 10% bovine calf serum replaced the fetal bovine serum. For the induction of apoptosis, HeLa cells were incubated with 10 ng/ml TNFα and 10 μg/ml CHX for the indicated periods of time at 37 °C, 5% CO2. Alternatively, HeLa cells were induced into apoptosis by incubation with 1 μm STS for the indicated time. HT1080 cells were induced by incubation with 1 μg/ml mouse monoclonal anti-human CD95 antibody (catalog no. AHS9552, Invitrogen) with or without 10 μm MG132. The progression through apoptosis was monitored (25) using an annexin V-FITC/phosphatidylinositol staining kit (Invitrogen) according to the manufacturer's protocol. The samples were analyzed using a Becton-Dickinson FACScan cytofluorometer at the Wayne State University Karmanos Cancer Institute, Flow Cytometry Facility.
FAM129B Knockdown
HeLa cells were grown in six-well plates to 20–30% confluence. Cells were transfected with siRNA directed against the FAM129B mRNA and, as a negative control, with a scrambled siRNA, using RNAi Lipofectamine RNAiMAX (Invitrogen), according to the manufacturer's protocol. The oligonucleotides used for these studies were commercially available: 1) SC-92820-A, CUGUGGGAUGAUCUCAGAU (Santa Cruz Biotechnology); 2) FAM129B Stealth RNAiTM siRNA HSS127849 (Invitrogen), UCACGGACAUGAACCUGAACGUCAU; 3) SC-92820-B, CAUCAUCCCUCCCUGAUAU (Santa Cruz Biotechnology); and 4) FAM129B Stealth RNAiTM siRNA HSS185602 (Invitrogen), CAGUAUGGCGUGGCUCUCUUCAACA. To assess the extent to which the expression of FAM129B was suppressed, cell extracts were isolated at various times following transfection, and the cell lysate was analyzed by Western blotting. Equivalent amounts of total protein were analyzed as determined by the Lowry method using BSA as a standard. Immunoblotting of β-tubulin was used to verify that equal amounts of total protein had been loaded on the gel.
Western Blot Analysis
Whole-cell extracts were prepared in lysis buffer containing 20 mm Tris-HCl, pH 7.5, 137 mm NaCl, 1% Triton X-100, 10% glycerol, 0.2 mm PMSF supplemented with a 1× mixture of phosphatase and protease inhibitors (Sigma). Protein samples were heated at 95 °C for 10 min and analyzed by SDS-PAGE on a 4–12% gradient gel. Western blots were developed using the Western Lighting Plus-ECL reagent (NEL104001EA, PerkinElmer Life Sciences). The resulting immunoblots were scanned with an HP Scanjet 4c, and the software UNSCAN-IT (Silk Scientific) was used to quantify the signal intensities.
Cell Fractionation
Cytoplasmic and nuclear fractions were prepared using the Qproteome Nuclear Protein kit (Qiagen) according to the manufacturer's protocol. The purity of the fractions was assessed by Western blotting of nuclear PARP and cytoplasmic (β-tubulin) marker proteins.
Caspase Assays
The intracellular levels and activation of caspase-8 and caspase-3 were followed by Western blotting using antibodies specific for the proenzymes and activated species. Caspase-3 activity was measured using the EnzChek Caspase-3 Assay Kit II (Molecular Probes, Invitrogen). Briefly, 50 μl of the supernatant was added to an individual well of a 96-well microfluorescent plate and incubated with or without 1 μl of Ac-DEVD-CHO inhibitor for 10 min at room temperature. After incubation, 50 μl of the 2× working substrate (5 μm Z-DEVD-R110) were added to each well and further incubated for 30 min. Fluorescence was measured at 485 nm excitation and 538 nm emission using a Germini XS spectrofluorometer (Molecular Devices, Union City, CA). Caspase-3 activity was expressed as arbitrary units of fluorescence.
Immunofluorescence Microscopy
HeLa cells grown on coverslips in six-well plates were fixed with 3.7% formaldehyde for 10 min at room temperature and then permeabilized with 0.2% Triton X-100 in PBS for 15 min at room temperature. The permeabilized cells were blocked using 3% bovine serum albumin in PBS for 1 h at room temperature. Cells were incubated with the primary antibody, rabbit anti-FAM129B, and/or mouse anti-β-catenin monoclonal antibody, overnight at 4 °C. The cells were then incubated with chicken anti-rabbit IgG antibody conjugated with Alexa Fluor 594 (1:2000) and a chicken anti-mouse IgG antibody conjugated with Alexa Fluor 488 (1:2000) (Molecular Probes, Invitrogen) as secondary antibodies for 1 h at room temperature. After extensively washing with PBS, the cells were counterstained with Hoechst 33342, mounted, and visualized using a Leica TCS SP5 laser-scanning confocal microscope (Microscopy, Imaging and Cytometry Resources Core Facility at Wayne State University). The images were analyzed using the Leica LAS AF Imaging software.
Cloning of FAM129B
The complete coding region of human FAM129B was amplified from the pOBT7 clone (Invitrogen, catalog no. 5456246). This cDNA clone included two introns that were excised using PCR-based deletion. The PCR product was then amplified using primers that included an Xho1 site (5′-CAC CCT CGA GGG GGA CGT GCT GTC CAC GCA CCT GGA CG-3′) and a Kpn1 site (5′GGC GGT ACC CTA GAA CTC AGT CTG CAC CCC TGC ACT G-3′). The digested product was then ligated into the Xho1 and Kpn1 sites of the pEGFP-C3 vector (Clontech) to generate the recombinant FAM129B expression plasmid with GFP-fused to the amino end. The fidelity of the construct was verified by nucleotide sequencing. Cotransfection of siRNA and the FAM129B construct was carried out using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
RESULTS
Because cancer progression and invasion depend on the suppression of apoptosis, the potential role of FAM129B in programmed cell death was investigated.
Intracellular Localization of FAM129B
HeLa cells were fractionated into nuclear and cytoplasmic fractions and analyzed by immunoblotting (Fig. 2). In exponentially growing HeLa cells, FAM129B was localized exclusively in the cytoplasmic compartment (Fig. 2A) along with the cytoplasmic marker protein β-tubulin. FAM129B was not detected in the nuclear fraction, although the nuclear marker PARP was found only in this fraction. Similar results were observed for the mouse embryonic fibroblast cell line NIH3T3 (Fig. 2B).
FIGURE 2.

Intracellular localization of FAM129B in exponential and confluent cell cultures. A, HeLa cells (5 × 106) were harvested during the late exponential growth phase, and the cell extracts were fractionated into cytoplasmic (Cyt) and nuclear (Nuc) fractions (as described under “Experimental Procedures”). The fractions were analyzed by immunoblotting using antibodies directed against FAM129B, PARP, p53, CDK6, and CDK2. B, the same procedure was used to analyze NIH3T3 cell fractions. C, immunofluorescence microscopy of exponentially growing HeLa cells stained with a Hoechst 33342 DNA stain and with antibodies directed against rabbit FAM129B and the secondary antibody, chicken anti-rabbit IgG Alexa Fluor 594. The same protocol was followed for the cells in confluent HeLa cells (D) and an exponentially growing HeLa culture showing some mitotic cells (E). F, immunofluorescence co-localization (as described under “Experimental Procedures”) of FAM129B and β-catenin. The confluent cells were co-stained with rabbit antibodies directed against FAM129B and mouse antibodies directed against β-catenin. The secondary antibodies were Alexa Fluor 594-conjugated anti-rabbit IgG and an Alexa Fluor 488-conjugated anti-mouse IgG antibody. The cells were also stained with Hoechst 33342.
These results were confirmed by immunofluorescence microscopy. FAM129B was found to be dispersed throughout the cytoplasm and was completely excluded from the nucleus in exponentially growing HeLa cells where there was little cell-cell contact (Fig. 2C). In confluent HeLa cell cultures, FAM129B migrated to the plasma membrane and appeared to be localized at the cell junctions (Fig. 2D). Interestingly, localization at the cell membrane was observed during telophase (Fig. 2E). The localization of FAM129B at the cell-cell junction was confirmed by fluorescence microscopy colocalization of FAM129B and β-catenin, a protein that is part of the adherens junction, the complex present at the cell-cell contacts (23). The yellow pixels concentrated at the cell membrane in the merged image (Fig. 2F) indicate that FAM129B (red fluorescence) and β-catenin (green fluorescence) partially colocalize.
Several inhibitors and activators of signaling molecules were tested in an attempt to alter the intracellular location of FAM129B. UO126 (10 μm), EGF (100 ng/ml), the Erk1/2 pathway inhibitor and activator, respectively, a general Akt inhibitor (10 μm), the PKC inhibitor, bisindolylmaleimide (4 μm), the PKC activator phorbol 12-myristate 13-acetate (100 nm), and the PI3K inhibitor wortmannin (1 μm). Cells were incubated for 4 h with each of these compounds, but none promoted either the membrane association or dissociation of FAM129B.
Knockdown of FAM129B in HeLa Cells
The expression of the FAM129B gene was silenced in HeLa cells using siRNA technology. Four oligonucleotides corresponding to several coding and noncoding regions of the human FAM129B gene (Fig. 1) were tested. All four oligonucleotides effectively knocked down FAM129B gene expression, whereas no effect was observed with control oligonucleotides with a scrambled sequence (Fig. 3). Oligonucleotide SC-92820-A, targeted to the 3′-noncoding sequence, which suppressed the expression of the gene by >95% within 72 h, was selected for these studies (Fig. 3, A and C). Similarly, immunofluorescence microscopy confirmed that FAM129B was depleted from the transfected cells (Fig. 3B).
FIGURE 3.

siRNA silencing of FAM129B expression. A, FAM129B knockdown with two different siRNAs, SC-92820-A (siRNA-1) and HSS127849 (siRNA-2) (see Fig. 1 and “Experimental Procedures”). The controls (Cont) for each siRNA had a similar length, and nucleotide composition but the sequence was scrambled. Extracts of the siRNA transfected cells were analyzed by immunoblotting using FAM129B antibodies, and β-tubulin antibodies were used as a loading control. The same blot was developed for a total of 1 and 5 min. B, indirect immunofluorescence confirmed the specific localization of endogenous FAM129B at the cell junction in confluent HeLa cells. HeLa cells were transfected with either a control scrambled siRNA (left, Control) or a specific siRNA directed against FAM129B (right). Cells were fixed 72 h post-transfection, blocked, and incubated with a 1:100 dilution of rabbit anti-FAM129B antibody at 4 °C overnight. Cells were then washed three times and incubated at room temperature for 1 h with a 1/2000 dilution of chicken anti-rabbit IgG antibody coupled with Alexa Fluor 594. The cell nucleus was stained with Hoechst 33342. C, the time course for FAM129B knockdown using siRNA (SC-92820-A). The cells were transfected with FAM129B siRNA (SC-92820-A) or with the control siRNA. β-Tubulin served as a loading control.
FAM129B Knockdown Accelerated Onset of Apoptosis
Silencing the expression of FAM129B neither induced apoptosis nor significantly affected cell growth. However, when apoptosis was induced by TNFα, a ligand that activates the extrinsic apoptotic pathway, and CHX (24), the cells entered apoptosis more rapidly than control cells that were transfected with a scrambled siRNA. The initiation and progression of apoptosis was monitored by an annexin V assay (Fig. 4A). Annexin V, in the presence of calcium ions selectively binds to phosphatidylserine, a lipid displayed on the cell surface during the early stages of apoptosis (24). FACS analysis (Fig. 4, B and C) indicated that after 4 h of exposure to TNFα/CHX, the percentage of apoptotic cells was 3-fold higher in cells in which FAM129B had been depleted. Immunofluorescence microscopy showed extensive fragmentation of nuclear DNA in cells transfected with FAM129B siRNA after induction by TNFα/CHX (Fig. 4D). In contrast, the DNA appeared intact in untransfected cells in the same population.
FIGURE 4.

Effect of FAM129B knockdown on apoptotic progression. A, HeLa cells transfected with control siRNA or with siRNA directed against FAM129B were exposed to TNFα/CHX following the standard protocol (25) (as described under “Experimental Procedures”). Fluorescence microscopy of cells stained with Hoechst 33342 and FITC-conjugated annexin V was carried out immediately after the addition of TNFα/CHX (0 h) and at 4 h post-exposure. The green FITC fluorescence bound to the cell surface provides an indication of the progression through apoptosis. B, an annexin V apoptosis assay (25) (as described under “Experimental Procedures”) of cells transfected with the scrambled siRNA control (siControl) or with FAM129B siRNA (siFAM129B). Samples were analyzed by FACS at the indicated times after induction of apoptosis by exposure to TNFα/CHX. FACS data are presented in a two-dimensional dot display showing the fluorescence intensity of cells with bound annexin V (AnnV; x axis) plotted versus the fluorescence intensity of cells dyed with the nonvital stain, propidium iodide (y axis). The lower right quadrant represents the early apoptotic cells expressed as a percentage of the total cell population, whereas the upper right quadrant represents cells in late apoptosis or necrosis. C, the percentage of apoptotic cells transfected with scrambled siRNA (○) or FAM129B siRNA (●) plotted against the time of exposure to TNFα/CHX. D, HeLa cells in which FAM129B expression was silenced were incubated with TNFα/CHX for 2 h, fixed, and then immunostained with anti-FAM129B antibodies (red) and Hoechst 33342 (blue). The field shown in the figure was selected because it gives a comparison between untransfected cells and cells in which FAM129B was silenced. Those cells in which FAM129B had been effectively knocked down as indicated by the lack of red staining exhibited extensive degradation of nuclear DNA (arrows), whereas the DNA appeared intact in the untransfected cells that still expressed FAM129B.
Effect of FAM129B Knockdown Was Mediated by a Caspase-dependent Pathway
In the extrinsic pathway, the binding of TNFα to its receptor results initially in the activation of caspase-8. Activation is a consequence of the cleavage of procaspase-8 into 41-kDa and 43-kDa proteolytic fragments and subsequently to 18-kDa and 10-kDa fragments that associate to form the heterotetrameric, active caspase-8. In HeLa cells transfected with the control siRNA (Fig. 5A), the 41/43-kDa intermediates began to accumulate after 4 h of exposure to TNFα/CHX. The active 18-kDa species was not yet detectable. Note that the 10-kDa subunit is not recognized by this antibody. In contrast, significant amounts of the intermediates were visible after 2 h in cells transfected with FAM129B siRNA. After a 4-h exposure, significant amounts of the 18-kDa subunit of the active species had accumulated.
FIGURE 5.

Activation of caspases upon TNFα/CHX exposure. Apoptosis was induced by TNFα/CHX for the indicated times (0, 2, and 4 h) in HeLa cells in which FAM129B was knocked down by exposure for 72 h with siRNA (SC-92820-A) and in control cultures transfected with scrambled siRNA (control). A, the cell extracts were analyzed by Western blotting using antibodies directed against FAM129B and the mouse monoclonal antibody 1C12 that recognizes full-length human procaspase-8, the 41 and 43-kDa proteolytic intermediates and the 18-kDa subunit of the activated caspase. B, Western blotting of cell extracts using an antibody directed against intact and cleaved PARP and an antibody recognizing the 17 and 12-kDa subunits of activated caspase-3. The cell extracts in A and B were also immunoblotted with β-tubulin antibodies.
The next step in the extrinsic pathway is the activation of the executioner caspase-3, the proteolytic enzyme primarily responsible for degradation of the proteins that occurs during apoptosis (26). Caspase-3, a 35-kDa protein, is cleaved into 17-kDa and 12-kDa fragments that associate forming a heterotetramer. Following exposure to TNFα/CHX for 2 h, HeLa cells transfected with control siRNA, had barely detectable levels of active caspase-3 (Fig. 5B). However, caspase-3 was rapidly activated in FAM129B knockdown cells. In agreement with this result, immunofluorescence microscopy (Fig. 6A) showed that the extent of caspase-3 activation was much greater following TNFα/CHX induction of FAM129B-depleted cells. This result was confirmed by directly assaying the caspase-3 activity in control and knockdown cells (Fig. 6B). Caspase-3 activity was 4.4-fold higher in cells in which the FAM129B gene had been silenced.
FIGURE 6.

Caspase-3 activity following FAM129B knockdown. A, HeLa cells were treated with either FAM129B siRNA or with the scrambled siRNA control for 72 h. The cells were then induced to undergo apoptosis by exposure to TNFα (10 ng/ml) and CHX (10 μg/ml) for 2 h. The fixed cells were incubated for 2 h at room temperature with rabbit antibodies recognizing only activated caspase-3. The secondary antibody was Alexa Fluor 594-conjugated anti-rabbit IgG. B, HeLa cells (1 × 106) were either treated with FAM129B siRNA or with the scrambled siRNA control for 72 h. The cells were then induced to undergo apoptosis as described in A for the indicated times. The cells were then harvested, lysed, and assayed for caspase-3 activity using a synthetic substrate Z-DEVD-R110 (as described under “Experimental Procedures”). The unfilled and gray shaded bars correspond to the control and FAM129B knockdown cells, respectively.
A major caspase-3 target is PARP, an antiapoptotic protein involved in DNA repair (27, 28). PARP is inactivated during apoptosis by cleavage of the 116-kDa active enzyme, rendering it unable to repair the massive DNA damage. Again, PARP cleavage occurred much more rapidly following TNFα/CHX stimulation of FAM129B-depleted cells.
FAM129B Was Degraded during Apoptosis
The intracellular levels of FAM129B decreased as TNFα/CHX induced apoptosis proceeded (Fig. 7A). However, the interpretation of this experiment was complicated because CHX inhibits protein synthesis. The inhibitor promotes efficient TNFα induction of apoptosis by blocking the synthesis of antiapoptotic proteins (24). Thus, the intracellular levels of FAM129B were also monitored in HeLa cells induced to apoptosis by STS, which activates the intrinsic apoptotic pathway (29). Under the conditions used in these experiments (Fig. 7B), STS-induced apoptosis proceeded more rapidly than that observed for TNFα/CHX induction (Fig. 7A). The decrease in the intracellular level of FAM129B, β-catenin, PARP, and Akt and the extent of proteolytic activation of caspase-8 and caspase-9 were monitored as a function of time following exposure to STS (Fig. 7, B and C). After a lag time of ∼1 h, during which time the caspases began to be activated, PARP disappeared rapidly. The intracellular level of PARP was reduced by 50% after ∼3 h. The rate of degradation of FAM129B and β-catenin was approximately the same with a half-time of 4.1 h. Half of the Akt was degraded after 5 h. All four proteins were barely detected 12 h after induction of apoptosis.
FIGURE 7.

FAM129B levels as apoptosis progresses. A, HeLa cells (1 × 106) were exposed to TNFα/CHX, and the intracellular levels of endogenous FAM129B and PARP were assessed by immunoblotting with the corresponding antibodies at the indicated times. β-Tubulin served as a loading control. B, apoptosis was induced with STS as described in under “Experimental Procedures”. Cells were harvested at the indicated times, and the relative levels of PARP, β-catenin, FAM129B, and Akt, as well as caspase-8 and caspase-9 and their cleavage products were assessed by immunoblotting. C, the relative levels of the proteins, FAM129B (●), PARP (○), Akt (□), and β-catenin (△) was determined by scanning the Western blot in B, and the results were expressed as a percentage of the level at the time apoptosis was induced.
The potent proteasome inhibitor, MG132 strongly potentiates apoptosis induced by antibodies directed against CD95 (30). In HT1080 cells, increased apoptosis in the presence of the inhibitor (Fig. 8) was indicated by significantly higher levels of cleaved PARP and activated caspase-8 and caspase-3 (compare lanes 2 and 4). The degradation of FAM129B was also accelerated by MG132, indicating that its reduced intracellular level was not a consequence of proteolysis mediated by the proteasome. However, a broad spectrum caspase inhibitor blocked the activation of caspase-8 and caspase-3 and the degradation of both FAM129B and PARP (lane 5).
FIGURE 8.
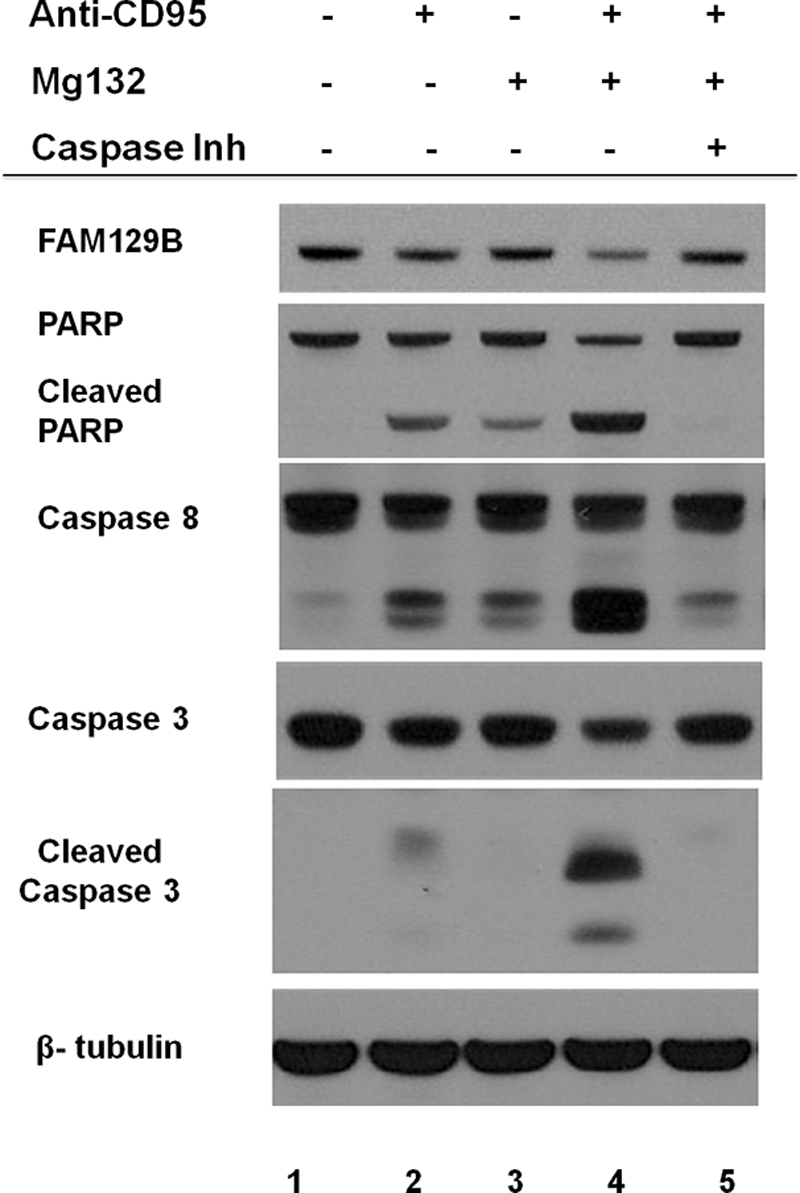
Effect of Mg132 and a general caspase inhibitor on FAM129B degradation. HT1080 cells were incubated in the presence (+) or absence (-) of anti-CD95 and Mg132 for 4 h. The relative intracellular levels of FAM129B and full-length and cleaved PARP, caspase-8, and caspase-3 were determined by immunoblotting. β-Tubulin served as a loading control. The effect of a general caspase inhibitor (Caspase Inh), Z-VAD-fmk, that targets all caspases, on the level of these proteins was also tested following induction of apoptosis with both anti-CD95 and Mg132.
Recombinant FAM129B Reversed Effect of Endogenous FAM129B Knockdown
The results thus far indicated that FAM129B knockdown accelerates the onset of apoptosis. To confirm that the result was specifically due to the depletion of intracellular FAM129B, the coding region was cloned and expressed in HeLa cells as a fusion protein with EGFP appended to the amino end of the polypeptide (see “Experimental Procedures”). The full-length protein was expressed at moderately high levels in HeLa cells. Immunoblotting (Fig. 9) using anti-GFP antibody showed that the recombinant protein had the expected molecular mass of 110 kDa. Fluorescence microscopy (data not shown) demonstrated that the intracellular localization of the recombinant protein mimicked the behavior of endogenous FAM129B. The siRNA used to suppress the endogenous FAM129B (Fig. 1, SC-92820-A) was targeted to the 3′-untranslated region and therefore cannot silence the expression of the recombinant FAM129B gene. To determine whether recombinant FAM129B could rescue the siRNA knockdown effect, HeLa cells were co-transfected with siRNA (SC-92820-A) and EGFP-FAM129B or the EGFP vector for 48 h, followed by TNFα/CHX treatment for an additional 4 h. HeLa cells co-transfected with the EGFP vector, and the siRNA entered apoptosis more rapidly as indicated by increased cleavage of caspase-3 and PARP (Fig. 9, lane 3). However, these effects were partially reversed in cells that were co-transfected with EGFP-FAM129B as indicated by the suppression of PARP and caspase-3 cleavage (Fig. 9, lane 4).
FIGURE 9.

Recombinant FAM129B restores the resistance to apoptosis. HeLa cells were co-transfected with FAM129B siRNA, the EGFP vector or recombinant EGFP-FAM129B for 48 h using Lipofectamine 2000. Apoptosis was then induced by incubation with TNFα/CHX for an additional 4 h. Immunoblotting using EGFP-specific antibodies showed that recombinant EGFP-FAM129B (lanes 2 and 4) and as a control, the EGFP protein (EGFP vector) (lanes 1 and 3) were both highly expressed in cells treated with TNFα/CHX and in untreated cells. Expression of the recombinant protein had no effect on the level of PARP or caspase-3 in the untreated controls (lane 2) compared with cells transfected with only the vector (lane 1). Following induction of apoptosis, the cells transfected with EGFP vector (lane 3) exhibited increased PARP cleavage and caspase-3 activation, whereas these effects were diminished in cells transfected with recombinant FAM129B (lane 4). β-Tubulin served as a loading control.
DISCUSSION
MINERVA or FAM129B was shown recently by Old and colleagues (5) to be implicated in cell invasion in human melanoma cells in a process controlled by the Erk1/2 signaling cascade. Cancer progression and metastasis requires the suppression of apoptosis to allow the aberrant cells to survive and proliferate in blood and distal tissues.
Silencing FAM129B gene expression did not induce apoptosis or affect the growth rate of HeLa cells. Rather, FAM129B knockdown accelerated the response to apoptotic signals. The annexin V assay indicated that apoptosis induced by TNFα/CHX proceeds 3–4-fold more rapidly in the FAM129B knockdown cells. This result is significant because if FAM129B knockdown simply induced apoptosis, it would be difficult to sort out whether FAM129B directly affects the apoptotic pathways as opposed to some other crucial function that leads to apoptosis as a secondary consequence. There are several additional observations that support the notion that FAM129B directly affects apoptosis: 1) knockdown with three different oligonucleotides gave an identical phenotype making off target effects unlikely; 2) FAM129B knockdown accelerated apoptosis induced by three different stimuli, TNFα/CHX and anti-CD95, which activate the extrinsic pathway and STS, an intrinsic pathway activator, as indicated by an increased rate of PARP cleavage and caspase activation (only TNFα/CHX data shown); and 3) rescue experiments showing that the elevated rate of apoptosis can be partially reversed in cells in which the endogenous protein has been depleted when transiently transfected with recombinant FAM129B.
The adherens junction must be dismantled with the onset of apoptosis (22). The cytoplasmic domain of cadherin is cleaved by caspase-3, whereas the extracellular domain that links the cells together is cleaved by a metalloprotease that is also activated during apoptosis (31). In addition, there is a caspase-3-dependent proteolytic cleavage of β-catenin (16). Once cleaved, β-catenin and cadherin are no longer linked, and the junction is lost. The actin cytoskeleton retracts and the cell shrinks and eventually disintegrates.
In agreement with the studies of Old et al. (5), immunofluorescence microscopy indicated that FAM129B can be localized in the cytosol or associated with the cell junctions. In melanoma cells, FAM129B was found dispersed throughout the cytoplasm when the MAPK (Erk1/2) cascade was active. However, exposure to the MAPKK inhibitor, UO126, which effectively shuts down the cascade, resulted in the migration of FAM129B to the cell junctions. We also found FAM129B to be cytosolic in exponentially growing HeLa cells (Fig. 2C). However, as the cells became confluent, FAM129B was exclusively localized to the cell junctions (Fig. 2D). As observed previously (5) in melanoma cells, FAM129B at the junctions of HeLa cells co-localized with β-catenin (Fig. 2F), indicating that it is near the adherens junctions.
In rapidly growing cells, MAPK activity is generally elevated so that the cytosolic location of FAM129B observed in exponentially growing HeLa cells is in accord with the previous study. However, migration to the cell junctions may be dependent on factors other than MAP kinase phosphorylation in HeLa cells since incubation of exponentially growing cultures with the MAPKK inhibitor, UO126, did not result in the relocation of FAM129B to the cell membrane (data not shown). Similarly, the activation of the MAP kinase cascade by EGF did not alter the FAM129B intracellular location. Several other inhibitors and activators of signaling pathways also had no effect on the association of FAM129B with the cell junction. Rather, FAM129B was invariably observed at the cell-cell junctions whenever two cells were in contact in both confluent and preconfluent HeLa cell cultures.
The intracellular dynamics and distribution of FAM129B appeared to closely parallel that of β-catenin. FAM129B and β-catenin were localized together at cell-cell contacts, both disappeared as the junction disassembles and both were degraded at approximately the same rate. In this sense, FAM129B behaves as a normal component of the adherens junction. The only exception is that FAM129B, unlike β-catenin, which also acts as a transcriptional regulator, was not observed in the nucleus. The Wnt signaling pathway controls the fate of β-catenin once it is released from the adherens junction (23). Phosphorylation of β-catenin by glycogen synthase kinase 3, a key component of the pathway (32), prevents it from entering the nucleus to activate the expression of Wnt pathway target genes. Instead β-catenin undergoes proteolytic degradation. Akt phosphorylation inhibits glycogen synthase kinase 3 (32), so Akt inhibitors would be expected to increase glycogen synthase kinase 3 phosphorylation of β-catenin and promote its degradation. In contrast, the Akt inhibitor had no effect on FAM129B, although more extensive studies are required to conclusively rule out the role of the Wnt pathway in FAM129B turnover. However, there is no evidence thus far that FAM129B has a role in transcriptional regulation.
We also observed that FAM129B was localized in the plasma cell membrane during mitosis. The localization was not confined to the area of contact between the nascent daughter cells but rather was dispersed throughout the entire plasma membrane (Fig. 2E). This unexpected result suggests that FAM129B may play a role in mitosis or cytokinesis, perhaps in the reorganization of the cytoskeleton.
The signal that triggers the relocation of FAM129B to the cell junction in HeLa cells is unknown. One possibility is that FAM129B has an intrinsic affinity for the adherens junction where it binds once the junction is assembled. Perhaps more likely, there may be specific signals that trigger its relocation. In addition to the four serines that are phosphorylated by MAPK, two other phosphoserines have been found in the proline-rich region near the carboxyl end of FAM129B (5). Experiments are planned to identify the kinase responsible for these modifications and to determine whether their phosphorylation alters the intracellular localization of FAM129B.
Studies are also underway to determine whether FAM129B is physically associated with cadherin or any of the catenins to confirm that it is localized in the adherens junction and to establish whether its location there is related to its antiapoptotic function. One attractive hypothesis is that the FAM129B suppresses apoptosis by stabilizing the junction perhaps by protecting the components of the complex from proteolytic degradation. Experiments are underway to investigate this phenomenon.
FAM129B was degraded during apoptosis without the accumulation of smaller proteolytic fragments. Perhaps the epitope, a short sequence near the carboxyl end of the peptide, was present on a small fragment that was not visible in the blot and that the larger fragments thus eluded detection. Alternatively, following an initial cleavage, the protein may be rapidly degraded to small fragments. Anti-apoptotic proteins and signaling molecules (33) are among the early, specific caspase targets. One of the proteins first cleaved is PARP which abolishes the ability of the enzyme to repair the DNA damage occurring in apoptotic cells (34). The phosphatidylinositol 3-phosphate-regulated protein kinase, Akt, that has been shown to antagonize apoptosis (35), was degraded some what more slowly by caspases than PARP in accord with previous reports (33). The rate of degradation of FAM129B and β-catenin was intermediate between PARP and Akt. It is likely that FAM129B degradation is also mediated by caspases. Proteolysis by the proteasome can be ruled out since degradation proceeded rapidly in the presence of the proteasome inhibitor, Mg132. FAM129B degradation was blocked by a broad spectrum caspase inhibitor, although we cannot be sure whether this effect was due to inhibition of the protease or simply the arrest of the apoptotic program in general.
There is evidence that the cytoplasmic domain of cadherin also suppresses apoptosis (31). The rapid cleavage of FAM129B and cadherin that occurred during apoptosis may be a mechanism that ensures that the antiapoptotic function of these proteins is eliminated once programmed cell death is initiated.
One crucial facet of the mechanism through which FAM129B promotes cancer cell invasion is likely to be the suppression of apoptosis. The protein has some of the hallmarks of a signaling molecule. The PH domain near the amino end of the FAM129B polypeptide is found in many signaling molecules. One of the functions of the PH domain is to dock proteins to the plasma membrane. This domain is also found in FAM129C, the other family member that binds to membrane surfaces. The proline-rich region at the carboxyl terminus is a potential target of SH3 domains, a module found in proteins involved in many signaling cascades. The detailed mechanism through which FAM129B exerts its effects on apoptosis and cell invasion is currently under investigation.
Metastasis is the result of the disruption of the precise balance between proliferation and apoptosis (36). It is thought that a large fraction of metastatic tumor cells are lost by apoptosis (9, 37). Thus, chemotherapeutic approaches that can tip the balance in favor of apoptosis would be expected to be effective in combating proliferative disorders.
Acknowledgments
We thank Dr. Avraham Raz for the generous gift of HT1080 cells and Drs. Quanwen Li and Zhiwei Wang for reagents and helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant GM/CA60371. This work was also supported by internal funds from the Office of the Provost (Wayne State University). The Microscopy, Imaging and Cytometry Resource Core is supported in part by NIH Center Grant P30CA22453 to the Karmanos Cancer Institute, Wayne State University, and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University.
- PARP
- poly(ADP-ribose)polymerase
- CHX
- cycloheximide
- PH domain
- pleckstrin homology domain
- STS
- staurosporine
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- EGFP
- enhanced GFP.
REFERENCES
- 1. Majima S., Kajino K., Fukuda T., Otsuka F., Hino O. (2000) Jpn. J. Cancer Res. 91, 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adachi H., Majima S., Kon S., Kobayashi T., Kajino K., Mitani H., Hirayama Y., Shiina H., Igawa M., Hino O. (2004) Oncogene 23, 3495–3500 [DOI] [PubMed] [Google Scholar]
- 3. Matsumoto F., Fujii H., Abe M., Kajino K., Kobayashi T., Matsumoto T., Ikeda K., Hino O. (2006) Hum. Pathol. 37, 1592–1600 [DOI] [PubMed] [Google Scholar]
- 4. Boyd R. S., Adam P. J., Patel S., Loader J. A., Berry J., Redpath N. T., Poyser H. R., Fletcher G. C., Burgess N. A., Stamps A. C., Hudson L., Smith P., Griffiths M., Willis T. G., Karran E. L., Oscier D. G., Catovsky D., Terrett J. A., Dyer M. J. (2003) Leukemia 17, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 5. Old W. M., Shabb J. B., Houel S., Wang H., Couts K. L., Yen C. Y., Litman E. S., Croy C. H., Meyer-Arendt K., Miranda J. G., Brown R. A., Witze E. S., Schweppe R. E., Resing K. A., Ahn N. G. (2009) Mol. Cell 34, 115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smalley K. S., Haass N. K., Brafford P. A., Lioni M., Flaherty K. T., Herlyn M. (2006) Mol. Cancer Ther. 5, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 7. Lowe S. W., Lin A. W. (2000) Carcinogenesis 21, 485–495 [DOI] [PubMed] [Google Scholar]
- 8. Townson J. L., Naumov G. N., Chambers A. F. (2003) Curr. Mol. Med. 3, 631–642 [DOI] [PubMed] [Google Scholar]
- 9. Mehlen P., Puisieux A. (2006) Nat. Rev. Cancer 6, 449–458 [DOI] [PubMed] [Google Scholar]
- 10. Thompson C. B. (1995) Science 267, 1456–1462 [DOI] [PubMed] [Google Scholar]
- 11. McDonnell T. J., Troncoso P., Brisbay S. M., Logothetis C., Chung L. W., Hsieh J. T., Tu S. M., Campbell M. L. (1992) Cancer Res. 52, 6940–6944 [PubMed] [Google Scholar]
- 12. Hague A., Moorghen M., Hicks D., Chapman M., Paraskeva C. (1994) Oncogene 9, 3367–3370 [PubMed] [Google Scholar]
- 13. Kerr J. F., Winterford C. M., Harmon B. V. (1994) Cancer 73, 2013–2026 [DOI] [PubMed] [Google Scholar]
- 14. Schmitt C. A., Lowe S. W. (1999) J. Pathol. 187, 127–137 [DOI] [PubMed] [Google Scholar]
- 15. Grossman D., Altieri D. C. (2001) Cancer Metastasis Rev. 20, 3–11 [DOI] [PubMed] [Google Scholar]
- 16. Brancolini C., Lazarevic D., Rodriguez J., Schneider C. (1997) J. Cell Biol. 139, 759–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takeichi M. (1995) Curr. Opin. Cell Biol. 7, 619–627 [DOI] [PubMed] [Google Scholar]
- 18. Gumbiner B. M. (2005) Nat. Rev. Mol. Cell Biol. 6, 622–634 [DOI] [PubMed] [Google Scholar]
- 19. Drees F., Pokutta S., Yamada S., Nelson W. J., Weis W. I. (2005) Cell 123, 903–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pokutta S., Drees F., Yamada S., Nelson W. J., Weis W. I. (2008) Biochem. Soc. Trans. 36, 141–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Green K. J., Getsios S., Troyanovsky S., Godsel L. M. (2010) Cold Spring Harb. Perspect Biol. 2, a000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzanne M., Steller H. (2009) J. Biol. 8, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nelson W. J., Nusse R. (2004) Science 303, 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aggarwal B. B. (2003) Nat. Rev. Immunol. 3, 745–756 [DOI] [PubMed] [Google Scholar]
- 25. Bossy-Wetzel E., Green D. R. (2000) Methods Enzymol. 322, 15–18 [DOI] [PubMed] [Google Scholar]
- 26. Wajant H. (2002) Science 296, 1635–1636 [DOI] [PubMed] [Google Scholar]
- 27. Satoh M. S., Lindahl T. (1992) Nature 356, 356–358 [DOI] [PubMed] [Google Scholar]
- 28. Simbulan-Rosenthal C. M., Rosenthal D. S., Iyer S., Boulares H., Smulson M. E. (1999) Mol. Cell Biochem. 193, 137–148 [PubMed] [Google Scholar]
- 29. Antonsson A., Persson J. L. (2009) Anticancer Res. 29, 2893–2898 [PubMed] [Google Scholar]
- 30. Kim H. A., Kim Y. H., Song Y. W. (2003) J. Rheumatol. 30, 550–558 [PubMed] [Google Scholar]
- 31. Steinhusen U., Weiske J., Badock V., Tauber R., Bommert K., Huber O. (2001) J. Biol. Chem. 276, 4972–4980 [DOI] [PubMed] [Google Scholar]
- 32. Cohen P., Frame S. (2001) Nat. Rev. Mol. Cell Biol. 2, 769–776 [DOI] [PubMed] [Google Scholar]
- 33. Widmann C., Gibson S., Johnson G. L. (1998) J. Biol. Chem. 273, 7141–7147 [DOI] [PubMed] [Google Scholar]
- 34. Nicholson D. W., Thornberry N. A. (1997) Trends Biochem. Sci. 22, 299–306 [DOI] [PubMed] [Google Scholar]
- 35. Franke T. F., Kaplan D. R., Cantley L. C. (1997) Cell 88, 435–437 [DOI] [PubMed] [Google Scholar]
- 36. Hoffman B., Liebermann D. A. (1994) Oncogene 9, 1807–1812 [PubMed] [Google Scholar]
- 37. Wyllie A. H., Kerr J. F., Currie A. R. (1980) Int. Rev. Cytol. 68, 251–306 [DOI] [PubMed] [Google Scholar]