

Abstract
α-Conotoxins are peptides from cone snails that target the nicotinic acetylcholine receptor (nAChR). RgIA and Vc1.1 have analgesic activity in animal pain models. Both peptides target the α9α10 nAChR and inhibit N-type calcium channels via GABAB receptor activation, but the mechanism of action of analgesic activity is unknown. PeIA has previously been shown to inhibit the α9α10 and α3β2 nAChRs. In this study, we have determined the structure of PeIA and shown that it is also a potent inhibitor of N-type calcium channels via GABAB receptor activation. The characteristic α-conotoxin fold is present in PeIA, but it has a different distribution of surface-exposed hydrophobic and charged residues compared with Vc1.1. Thus, the surface residue distribution, rather than the overall fold, appears to be responsible for the 50-fold increase in selectivity at the α3β2 nAChR by PeIA relative to Vc1.1. In contrast to their difference in potency at the nAChR, the equipotent activity of PeIA and Vc1.1 at the GABAB receptor suggests that the GABAB receptor is more tolerant to changes in surface residues than is the nAChR. The conserved Asp-Pro-Arg motif of Vc1.1 and RgIA, which is crucial for potency at the α9α10 nAChR, is not required for activity at GABAB receptor/N-type calcium channels because PeIA has a His-Pro-Ala motif in the equivalent position. This study shows that different structure-activity relationships are associated with the targeting of the GABAB receptor versus nAChRs. Furthermore, there is probably a much more diverse range of conotoxins that target the GABAB receptor than currently realized.
Keywords: GABA Receptors, Nicotinic Acetylcholine Receptors, Peptide Conformation, Peptides, Toxins
Introduction
α-Conotoxins are peptides from cone snail venoms that antagonize nicotinic acetylcholine receptors (nAChRs)5 (1–3). They typically comprise 12–19 amino acids and are characterized by two disulfide bonds that have a CysI-CysIII and CysII-CysIV connectivity. Their three-dimensional structures incorporate an α-helix centered around CysIII (4), and they are regarded as rigid molecular frameworks based on their well defined secondary structure and disulfide bracing (5). α-Conotoxins have a range of potential therapeutic applications, including the treatment of pain and disease states such as Parkinson disease (6, 7).
nAChRs comprise five subunits and a range of different receptor subtypes exist depending on the subunit composition of the pentamer (8, 9). Specific subtypes have different expression patterns and pharmacological and biophysical profiles. Targeting individual subtypes can potentially result in drug leads with minimal side effects (10, 11). For example, the α9α10 nAChR subtype has been proposed as a potential target for pain relief (12, 13). So far, the only α-conotoxins that have been reported to be potent antagonists of the α9α10 nAChR subtype are PeIA (14), RgIA (15), and Vc1.1 (16). PeIA and Vc1.1 belong to the same structural subclass of α-conotoxins in that they both contain four and seven residues, respectively, in their two loops. In contrast, RgIA has four and three residues, respectively, in the two loops. The sequences of the three peptides are shown in Fig. 1, which highlights that RgIA and Vc1.1 have identical sequences in loop 1 but very different sequences in loop 2. PeIA and Vc1.1 have similar sequences in loop 2 but significantly different charge distributions in loop 1.
FIGURE 1.
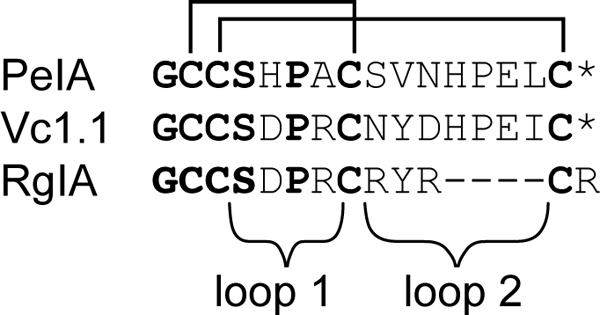
Sequences of α-conotoxins that target the α9α10 nAChR. The cysteine residues are connected in the native disulfide connectivity as indicated. The asterisk represents an amidated C terminus.
RgIA and Vc1.1 exhibit analgesic activity in animal pain models when delivered via injection (16–18), and a cyclic form of Vc1.1 has been shown recently to be orally active, an exciting finding that augers well for its development as a therapeutic agent (19). However, the mechanism by which the analgesic activity of these peptides is exerted is unclear. Electrophysiological studies have shown that RgIA and Vc1.1 inhibit N-type Ca2+ channel currents in dorsal root ganglion (DRG) neurons and Cav2.2 channels expressed in Xenopus oocytes via activation of the G protein-coupled GABAB receptor (13, 20). The fact that the post-translationally modified version of Vc1.1 (Vc1a), which has Pro-6 hydroxylated and Glu-14 in loop 2 converted to a γ-carboxyglutamic acid, is a potent inhibitor of the α9α10 nAChR subtype but does not have analgesic activity (7, 21) suggests that GABAB activation is responsible for the analgesic activity. Furthermore, Vc1.1 reversal of mechanical allodynia is antagonized by pretreatment with the orally active GABAB receptor antagonist SCH50911 (22). However, RgIA and Vc1.1 do not potently displace [3H]CGP54626 binding in HEK293T cells transiently transfected with GABAB1b and GABAB2 subunits or activate human GABAB receptors coupled to G protein-activated inwardly rectifying K+ (GIRK1/4) channels expressed in Xenopus oocytes (12). Thus, further studies are required to resolve the precise mechanism of action of RgIA and Vc1.1 that underlies their analgesic activity.
Analysis of the structure-activity relationships of new peptides that target α9α10 or GABAB receptors might shed light on the mechanism of action of analgesic α-conotoxins. In this study, we have determined the activity of PeIA on GABAB receptor modulation of N-type Ca2+ channels for comparison with its activity at the α9α10 nAChR. We also determined the three-dimensional structure of PeIA. This study reveals that an α-conotoxin with a distinctive loop 1 compared with Vc1.1 and RgIA is also a potent inhibitor of N-type Ca2+ channels via GABAB receptor activation.
EXPERIMENTAL PROCEDURES
Peptide Synthesis
PeIA was synthesized using methods described previously for Vc1.1 (23). Oxidation of the disulfide bonds was achieved by stirring overnight in 0.1 m ammonium bicarbonate buffer.
NMR Spectroscopy
Spectra were recorded at 600 MHz (Bruker AVANCE NMR spectrometer) on a sample containing 1 mm PeIA in 10% D2O and 90% H2O. The two-dimensional spectra were recorded and three-dimensional structures were calculated as described previously (24). Distance restraints were obtained from a NOESY spectrum recorded with a 200-ms mixing time at 290 K.
Oocyte Electrophysiology
RNA preparation, oocyte preparation, and expression of nAChR subunits in oocytes obtained from Xenopus laevis were carried out as described previously (21). Plasmids with cDNA encoding the rat α9 and α10 nAChR subunits were kindly provided by Dr. A. B. Elgoyhen (Universidad de Buenos Aires, Buenos Aires, Argentina), and all other nAChR subunit clones were kindly provided by Dr. J. Boulter (UCLA, Los Angeles, CA). All oocytes were injected with 5 ng of cRNA and then kept at 18 °C in ND96 buffer (96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 5 mm HEPES, pH 7.4) supplemented with 50 mg/liter gentamycin, 5 mm pyruvic acid, and 5% horse serum 2–5 days before recording.
Membrane currents were recorded from Xenopus oocytes using a two-electrode voltage clamp amplifier (OC-725C oocyte clamp, Warner Instruments, Hamden, CT). Recording electrodes were pulled from borosilicate glass (GC150T-15, Harvard Apparatus Ltd., Edenbridge, UK) and had resistances of 0.3–1.5 megohms when filled with 3 m KCl. All recordings were conducted at room temperature (21–23 °C) using a bath solution of ND96 buffer as described above. During recordings, the oocytes were voltage-clamped in a small recording chamber (≤40 μl) at a holding potential of −80 mV and superfused continuously with ND96 buffer via gravity-fed tubes at 0.1–0.2 ml/min, with 5-min incubation times for the bath-applied conotoxins. Acetylcholine (ACh) was applied via gravity-fed tubes until peak current amplitude was obtained (1–3 s), with 1–2-min washout periods between applications. Data were sampled at 500 Hz and filtered at 200 Hz. Peak current amplitude was measured before and following incubation of the peptide.
All data were pooled (n = 3–7 for each data point) and represent arithmetic means ± S.E. Concentration-response curves for antagonists were fitted by unweighted nonlinear regression to the logistic equation Ex = EmaxXn/(Xn + IC50n), where Ex is the response, Emax is the maximal response, X is the antagonist concentration, n is the slope factor, and IC50 is the antagonist concentration producing half-maximal inhibition of the agonist response. Computation was done using SigmaPlot 8.0 (Jandel Corp., San Rafael, CA).
Electrophysiological Recording from Rat DRG Neurons
DRG neurons were enzymatically dissociated from ganglia of 7–14-day-old Wistar rats according to standard protocols. Briefly, rats were killed by cervical dislocation in accordance with the University of Queensland and RMIT University Animal Ethics Committees, the spinal column was hemisegmented, and the spinal cord was removed. Ganglia were removed, rinsed in cold Hanks' balanced salt solution (Invitrogen), minced, and incubated in 1 mg/ml collagenase (type 2, 405 units/mg; Worthington) in Hanks' balanced salt solution at 37 °C for ∼30 min. Following incubation, ganglia were rinsed three times with warm (37 °C) DMEM (Invitrogen) supplemented with 10% fetal calf serum and 1% penicillin/streptomycin and gently triturated with a fire-polished Pasteur pipette. Cells were plated on glass coverslips, incubated at 37 °C in 95% O2 and 5% CO2, and used within 4–36 h.
The external recording solution for rat DRG neurons contained 150 mm tetraethylammonium chloride, 2 mm BaCl2, 10 mm d-glucose, and 10 mm HEPES, pH 7.3–7.4. Recording electrodes were filled with an internal solution containing 140 mm CsCl, 1 mm MgCl2, 5 mm MgATP, 0.1 mm NaGTP, 5 mm 1,2-bis(O-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetracesium salt, and 10 mm HEPES, pH 7.3, with CsOH and had resistances of 1.0–2.5 megohms. Membrane currents were recorded using the whole-cell configuration of the patch-clamp technique with an Axopatch 200B amplifier (Molecular Devices Corp., Sunnyvale, CA). A voltage protocol using step depolarizations from −80 to −10 mV was used when examining high voltage-activated (HVA) Ca2+ channel currents. Test potentials of 150-ms duration were applied every 20 s. CGP55845 hydrochloride was obtained from Tocris Bioscience (Bristol, UK), and ω-conotoxin CVID was prepared as described previously (25). Drugs were applied via perfusion (∼1 ml/min) in the bath solution. Leak and capacitive currents were subtracted using a −P/4 pulse protocol. Currents were generated by a computer using pCLAMP 9.2 software (Molecular Devices Corp.), filtered at 2 kHz, and sampled at 8 kHz with a Digidata 1322A digitizer (Molecular Devices Corp.). Sampled data were stored digitally on a computer for further analysis.
RESULTS
PeIA was synthesized using solid-phase chemistry, and one major disulfide isomer was formed during oxidation. We confirmed that this isomer contained the native disulfide connectivity using an analysis of NMR chemical shifts compared with Vc1.1, as shown in Fig. 2A. The three-dimensional structure of PeIA was determined using simulated annealing based on NMR data, and the ensemble of the 20 lowest energy structures is shown in Fig. 2B. The major element of secondary structure is an α-helix spanning residues 6–10. In addition, type I β-turns are present at residues 1–4, 2–5, and 9–12. The secondary structure present in PeIA is similar to that determined for Vc1.1 (24).
FIGURE 2.

NMR chemical shifts and structure of PeIA. A, the secondary shifts of PeIA (red) and Vc1.1 (blue) were calculated by subtracting random-coil α-H shifts (33) from the experimental shifts. The negative secondary shifts for residues 6–11 are indicative of helical structure. The similarities in secondary shifts between the two peptides indicate that the three-dimensional structures are similar. B, superposition of the 20 lowest energy structures of PeIA (red) and Vc1.1 (blue). Despite the majority of secondary shifts being negative, only residues 6–10 are formally recognized as an α-helix. Similarly, in the structures calculated previously for Vc1.1, only residues 6–12 are recognized as an α-helix despite the negative secondary shifts at the N and C termini (30). It appears that the turn regions at the N and C termini of PeIA and Vc1.1 result in the negative secondary shifts present in these regions. C, surface representations of PeIA (left) and Vc1.1 (right). Hydrophobic residues are shown in green, cysteines in yellow, polar in cyan, positively charged in blue, and negatively charged residues in red.
The activity of the synthesized α-conotoxin PeIA was assessed on ACh-evoked currents in Xenopus oocytes expressing different nAChR subunit combinations. Concentration-response curves revealed an inhibition of α9α10 and α3β2 nAChR currents with IC50 values and Hill slopes of 54.9 ± 9.0 nm (n ≥ 4) and nH = 0.6 and 97.5 ± 10.9 nm (n ≥ 4) and nH = 0.8, respectively (Fig. 3, A and B). In contrast, no significant inhibition of ACh-evoked currents was observed at α4β2, human α7, and muscle αβγδ nAChRs in the presence of 1 μm PeIA.
FIGURE 3.

Effect of α-conotoxin PeIA on nAChR subtypes expressed in Xenopus oocytes and GABAB-mediated inhibition of N-type Ca2+ channels in rat DRG neurons. A, representative superimposed currents evoked by ACh in α9α10, α3β2, and α7 nAChRs expressed in oocytes in the absence (Control) and presence of 100 nm and 1 μm PeIA. Rat α9α10 and α3β2 nAChRs were activated by 30 μm and 100 μm ACh, respectively, whereas human α7 nAChRs were activated by 200 μm ACh. B, concentration-response relationships obtained for the inhibition of ACh-evoked current amplitudes following 5 min of incubation of α9α10 (●) and α3β2 (○) nAChRs with PeIA giving IC50 values of 54.9 and 97.5 nm, respectively. No significant inhibition was observed with concentrations up to 1 μm PeIA at α4β2 (▾), human α7 (△) and muscle αβγδ (■) nAChRs. Concentration-response data (means ± S.E., n = 4–7 for each data point) were fitted using the logistic equation (see “Experimental Procedures”).
Depolarization-activated whole-cell Ba2+ currents elicited by voltage steps from a holding potential of −80 to −10 mV were inhibited by PeIA in a concentration-dependent manner. The concentration-response relationship obtained for inhibition of HVA Ca2+ channel currents in rat DRG neurons by PeIA gave an IC50 of 1.1 nm (Fig. 4, A and B). In the presence of the selective N-type Ca2+ channel inhibitor ω-conotoxin CVID, 100 nm PeIA failed to further inhibit the inward Ba2+ current. CVID (200 nm) alone reduced the Ca2+ channel current amplitude to 57.2 ± 10.8% of the control, and in the presence of CVID + PeIA, the current amplitude was 53.6 ± 12.7% of the control (n = 4) (Fig. 4C). The expression of the GABAB1 and GABAB2 receptor subunits in rat DRG neurons has been confirmed at both the mRNA and protein levels (26). Furthermore, inhibition of N-type Ca2+ channel currents in rat DRG neurons by the GABAB agonist baclofen has been reported previously (27–29). The inhibition of HVA Ca2+ channel currents by PeIA (100 nm) was antagonized in the presence of the selective GABAB receptor antagonist CGP55845A (1 μm) to 87.9 ± 6.1% of the control (n = 8) (Fig. 4D). Lower concentrations of CGP55845 (100 and 300 nm) also antagonized the effects of PeIA (n = 3) (Fig. 4B). Bath application of 1 μm CGP55845A alone did not affect HVA Ca2+ channel current amplitude (96.4 ± 5.1% of the control). In preliminary experiments, PeIA (100 nm) inhibited the peak amplitude of depolarization-activated Ca2+ channel currents to 63.3 ± 4.4% (n = 3) of the control in baclofen-sensitive Xenopus oocytes expressing recombinant Cav2.2 (α1B-b + α2δ + β3) (13). Taken together, these data suggest that PeIA inhibits ω-conotoxin-sensitive N-type Ca2+ channels via activation of the G protein-coupled GABAB receptor in DRG neurons.
FIGURE 4.

Effect of α-conotoxin PeIA on GABAB-mediated inhibition of N-type Ca2+ channels in rat DRG neurons. A, superimposed depolarization-activated whole-cell Ba2+ currents elicited by voltage steps from a holding potential of −80 to −10 mV in the absence (Control) and presence of 10 nm PeIA. B, concentration-response relationship obtained for inhibition of HVA Ca2+ channel currents in DRG neurons by PeIA (n = 4–13 cells for each data point) giving an IC50 of 1.1 nm. The effect of PeIA was antagonized in the presence of 100 and 300 nm CGP55845 (n = 3). Data points represent means ± S.E. of normalized peak current amplitude. C, representative time course of inhibition of peak Ba2+ current amplitude in the presence of the selective N-type Ca2+ channel inhibitor ω-conotoxin CVID, followed by application of PeIA (100 nm). pF, picofarads. D, bar graph of the relative inhibition of HVA Ca2+ channel currents by 100 nm PeIA alone, in the presence of 1 μm CGP55845 alone, and after application of 100 nm PeIA in the presence of CGP55845. Numbers in parentheses reflect the number of cells. PeIA significantly reduced the HVA Ca2+ channel currents compared with CGP55845 and CGP55845 + PeIA in an unpaired t test (p < 0.0001). Cells treated with CGP55845 alone and PeIA + CGP55845 were not significantly different in a paired t test (p = 0.312).
DISCUSSION
We report here, for the first time, that PeIA is a new member of the select group of conotoxins known to target G protein-coupled GABAB receptors as well as the α9α10 nAChR. The inhibition of N-type Ca2+ channels via GABAB receptor modulation in DRG neurons by PeIA is similar to that reported for Vc1.1 (1.1 nm for PeIA compared with 1.7 nm for Vc1.1) (13). Our observations allow some deductions regarding structure-activity relationships to be made. As well as their shared Cys framework, PeIA and Vc1.1 share two residues in common in loop 1 (Ser-4 and Pro-6) and four residues in loop 2 (residues 12–14 are identical, and residue 15 involves a conservative Leu-to-Ile substitution). Consequently, it appears that the residues that differ, i.e. residues 5 and 7 and residues 9–11, are not critically important for activity at the GABAB receptor based on the similar levels of activity for the two peptides. In particular, our results clearly show that there is not an absolute requirement for a negatively charged residue at position 5 and a positively charged residue at position 7 for GABAB receptor activity, as might have been supposed from the conservation of these residues in Vc1.1 and RgIA. The results further suggest that there is probably a much more diverse range of conotoxins that target the GABAB receptor than is currently realized based on the substantial differences seen in loop 1 between PeIA on the one hand and Vc1.1 and RgIA on the other.
The activities of PeIA and Vc1.1 at the α9α10 nAChR are also similar, but there are differences observed for other nAChR subtypes. For instance, PeIA is a potent antagonist of the α3β2 subtype, whereas Vc1.1 is >50-fold less active (14, 30). A comparison of the structures of PeIA and Vc1.1 is shown in Fig. 2 (B and C). The overall fold of PeIA is similar to that of Vc1.1, but there are differences in the surface characteristics of PeIA in terms of the distribution of charged and hydrophobic residues. These surface differences presumably account for the differences in activity at specific nAChR subtypes.
Mutagenesis studies on Vc1.1 (23) and RgIA (31) have shown that the Asp-Pro-Arg motif in loop 1 is crucial for activity at the α9α10 nAChR. However, in PeIA, the equivalent residues are His-Pro-Ala, yet it remains a potent antagonist of the α9α10 nAChR. Interestingly, mutation of Arg-7 in Vc1.1 to alanine, the equivalent residue in PeIA, significantly decreases activity at the α9α10 receptor. Our observations reported here suggest that the loss of the positive charge at position 7 in Vc1.1 can be compensated for by the introduction of a positive charge at position 5. It is likely that the proline residue in PeIA will also be critical for activity, but this probably derives from a structural, rather than functional, role of this residue. Previous studies have shown that when this proline is mutated to alanine in Vc1.1, significant perturbations in structure are observed (23). Furthermore, we recently showed that a naturally occurring α-conotoxin, LtIa, which does not contain this conserved proline, does not have a well defined structure in solution (32).
PeIA, Vc1.1, and RgIA all have different pI values, varying from 4.5 to 9, and this distinction is reflected in the charge distribution on the surfaces of these molecules. These differences appear more critical at the nAChR based on the different potencies observed at different subtypes. By contrast, the three peptides have similar potency at the GABAB/N-type Ca2+ channel, indicating that electrostatic interactions are not crucial for activity at this receptor. A more complete understanding of the structure-activity relationships of α-conotoxins will require further mutational studies and the testing of naturally occurring α-conotoxins at a range of membrane receptors, but the studies reported here provide a valuable starting point.
In summary, we have shown that PeIA has a well defined three-dimensional structure in solution and report for the first time that it is a potent inhibitor of N-type Ca2+ channels coupled to GABAB receptors. The information reported here on the structure-activity relationships of α-conotoxins that have applications as analgesic agents should potentially be useful in the design of novel drug leads for the treatment of pain.
This work was supported by Australian Research Council Grant DP0986281 and National Health and Medical Research Council Grant APP456074.
- nAChR
- nicotinic acetylcholine receptor
- DRG
- dorsal root ganglion
- ACh
- acetylcholine
- HVA
- high voltage-activated.
REFERENCES
- 1. McIntosh J. M., Gardner S., Luo S., Garrett J. E., Yoshikami D. (2000) Eur. J. Pharmacol. 393, 205–208 [DOI] [PubMed] [Google Scholar]
- 2. Dutton J. L., Craik D. J. (2001) Curr. Med. Chem. 8, 327–344 [DOI] [PubMed] [Google Scholar]
- 3. Terlau H., Olivera B. M. (2004) Physiol. Rev. 84, 41–68 [DOI] [PubMed] [Google Scholar]
- 4. Millard E. L., Daly N. L., Craik D. J. (2004) Eur. J. Biochem. 271, 2320–2326 [DOI] [PubMed] [Google Scholar]
- 5. Hu S. H., Gehrmann J., Alewood P. F., Craik D. J., Martin J. L. (1997) Biochemistry 36, 11323–11330 [DOI] [PubMed] [Google Scholar]
- 6. Olivera B. M., Quik M., Vincler M., McIntosh J. M. (2008) Channels 2, 143–152 [DOI] [PubMed] [Google Scholar]
- 7. Livett B. G., Sandall D. W., Keays D., Down J., Gayler K. R., Satkunanathan N., Khalil Z. (2006) Toxicon 48, 810–829 [DOI] [PubMed] [Google Scholar]
- 8. Dani J. A., Bertrand D. (2007) Annu. Rev. Pharmacol. Toxicol. 47, 699–729 [DOI] [PubMed] [Google Scholar]
- 9. Albuquerque E. X., Pereira E. F., Alkondon M., Rogers S. W. (2009) Physiol. Rev. 89, 73–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gotti C., Riganti L., Vailati S., Clementi F. (2006) Curr. Pharm. Des. 12, 407–428 [DOI] [PubMed] [Google Scholar]
- 11. Dwoskin L. P., Bardo M. T. (2009) Neuropsychopharmacology 34, 244–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McIntosh J. M., Absalom N., Chebib M., Elgoyhen A. B., Vincler M. (2009) Biochem. Pharmacol. 78, 693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Callaghan B., Haythornthwaite A., Berecki G., Clark R. J., Craik D. J., Adams D. J. (2008) J. Neurosci. 28, 10943–10951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McIntosh J. M., Plazas P. V., Watkins M., Gomez-Casati M. E., Olivera B. M., Elgoyhen A. B. (2005) J. Biol. Chem. 280, 30107–30112 [DOI] [PubMed] [Google Scholar]
- 15. Ellison M., Haberlandt C., Gomez-Casati M. E., Watkins M., Elgoyhen A. B., McIntosh J. M., Olivera B. M. (2006) Biochemistry 45, 1511–1517 [DOI] [PubMed] [Google Scholar]
- 16. Vincler M., Wittenauer S., Parker R., Ellison M., Olivera B. M., McIntosh J. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 17880–17884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vincler M., McIntosh J. M. (2007) Expert Opin. Ther. Targets 11, 891–897 [DOI] [PubMed] [Google Scholar]
- 18. Satkunanathan N., Livett B., Gayler K., Sandall D., Down J., Khalil Z. (2005) Brain Res. 1059, 149–158 [DOI] [PubMed] [Google Scholar]
- 19. Clark R. J., Jensen J., Nevin S. T., Callaghan B. P., Adams D. J., Craik D. J. (2010) Angew. Chem. Int. Ed. Engl. 49, 6545–6548 [DOI] [PubMed] [Google Scholar]
- 20. Callaghan B., Adams D. J. (2010) Channels 4, 51–54 [DOI] [PubMed] [Google Scholar]
- 21. Nevin S. T., Clark R. J., Klimis H., Christie M. J., Craik D. J., Adams D. J. (2007) Mol. Pharmacol. 72, 1406–1410 [DOI] [PubMed] [Google Scholar]
- 22. Klimis H., Adams D. J., Callaghan B., Nevin S., Alewood P. F., Vaughan C. W., Mozar C. A., Christie M. J. (2011) Pain 152, 259–266 [DOI] [PubMed] [Google Scholar]
- 23. Halai R., Clark R. J., Nevin S. T., Jensen J. E., Adams D. J., Craik D. J. (2009) J. Biol. Chem. 284, 20275–20284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Millard E. L., Nevin S. T., Loughnan M. L., Nicke A., Clark R. J., Alewood P. F., Lewis R. J., Adams D. J., Craik D. J., Daly N. L. (2009) J. Biol. Chem. 284, 4944–4951 [DOI] [PubMed] [Google Scholar]
- 25. Lewis R. J., Nielsen K. J., Craik D. J., Loughnan M. L., Adams D. A., Sharpe I. A., Luchian T., Adams D. J., Bond T., Thomas L., Jones A., Matheson J. L., Drinkwater R., Andrews P. R., Alewood P. F. (2000) J. Biol. Chem. 275, 35335–35344 [DOI] [PubMed] [Google Scholar]
- 26. Towers S., Princivalle A., Billinton A., Edmunds M., Bettler B., Urban L., Castro-Lopes J., Bowery N. G. (2000) Eur. J. Neurosci. 12, 3201–3210 [DOI] [PubMed] [Google Scholar]
- 27. Dolphin A. C., Scott R. H. (1987) J. Physiol. 386, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tatebayashi H., Ogata N. (1992) J. Physiol. 447, 391–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Campbell V., Berrow N., Dolphin A. C. (1993) J. Physiol. 470, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clark R. J., Fischer H., Nevin S. T., Adams D. J., Craik D. J. (2006) J. Biol. Chem. 281, 23254–23263 [DOI] [PubMed] [Google Scholar]
- 31. Ellison M., Feng Z. P., Park A. J., Zhang X., Olivera B. M., McIntosh J. M., Norton R. S. (2008) J. Mol. Biol. 377, 1216–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luo S., Akondi K. B., Zhangsun D., Wu Y., Zhu X., Hu Y., Christensen S., Dowell C., Daly N. L., Craik D. J., Wang C. I., Lewis R. J., Alewood P. F., McIntosh J. M. (2010) J. Biol. Chem. 285, 12355–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wishart D. S., Bigam C. G., Holm A., Hodges R. S., Sykes B. D. (1995) J. Biomol. NMR 5, 67–81 [DOI] [PubMed] [Google Scholar]