

Abstract
It is well documented that protein kinase A (PKA) acts as a negative regulator of M phase promoting factor (MPF) by phosphorylating cell division cycle 25 homolog B (Cdc25B) in mammals. However, the molecular mechanism remains unclear. In this study, we identified PKA phosphorylation sites in vitro by LC-MS/MS analysis, including Ser149, Ser229, and Ser321 of Cdc25B, and explored the role of Ser149 in G2/M transition of fertilized mouse eggs. The results showed that the overexpressed Cdc25B-S149A mutant initiated efficient MPF activation by direct dephosphorylation of Cdc2-Tyr15, resulting in triggering mitosis prior to Cdc25B-WT. Conversely, overexpression of the phosphomimic Cdc25B-S149D mutant showed no significant difference in comparison with the control groups. Furthermore, we found that Cdc25B-Ser149 was phosphorylated at G1 and S phases, whereas dephosphorylated at G2 and M phases, and the phosphorylation of Cdc25B-Ser149 was modulated by PKA in vivo. In addition, we examined endogenous and exogenous Cdc25B, which were expressed mostly in the cytoplasm at the G1 and S phases and translocated to the nucleus at the G2 phase. Collectively, our findings provide evidence that Ser149 may be another potential PKA phosphorylation target of Cdc25B in G2/M transition of fertilized mouse eggs and Cdc25B as a direct downstream substrate of PKA in mammals, which plays important roles in the regulation of early development of mouse embryos.
Keywords: Cyclin-dependent Kinase, Cell Cycle, MS, Protein Kinase A (PKA), Protein Phosphorylation, CDC25B, M Phase-promoting Factor (MPF), Fertilized Mouse Eggs, In Vitro Phosphorylation
Introduction
Progression through the cell division cycle and transitions between its various phases are regulated by the activation of specific cyclin-dependent kinase complexes, which are activated by Cdc25 phosphatases (1–5). Cdc25 proteins are dual-specificity phosphatases that uniquely function to dephosphorylate specific tyrosine/threonine residues on cyclin-dependent kinases. Three isoforms have been identified in mammalian cells: Cdc25A, Cdc25B, and Cdc25C (6, 7). Although all three Cdc25 family members are associated with mitosis and meiosis, recent evidence has demonstrated that Cdc25B plays an essential role during the G2/M transition, and in mitosis, Cdc25B is a more potent activator of MPF2 than Cdc25A or Cdc25C (8–12).
PKA is a cAMP-dependent serine/threonine kinase composed of two regulatory subunits and two catalytic subunits. Thus, it plays a pivotal role in cell growth, differentiation, and proliferation as well as in cell cycle control (13–15). In Xenopus embryonic extracts, oscillations in cAMP concentration and PKA activity accompany cell cycle progression (16, 17); they are low at mitosis, increase during the M/G1 transition, and remain high until the next mitosis. High PKA activity decreases MPF activity and vice versa. This effect occurs through a mechanism involving Cdc25 phosphorylation (16) and cyclin B degradation (17). Studies in Xenopus oocytes have shown that Cdc25C is phosphorylated on Ser287 by PKA, whereas the heat-stable inhibitor of PKA induces dephosphorylation of Ser287, indicating that Cdc25C functions downstream of PKA (18). However, in mice, studies suggest that Cdc25B is the essential phosphatase for meiotic resumption and therefore the likely target of PKA (12, 19).
The fertilized mouse egg is the most simple and natural cell cycle model in a vertebrate closely related to humans. Little is known about the mechanism of early development of fertilized mouse eggs, especially that of the G2/M transition. Our previous research showed that PKA negatively regulates cell cycle progression of fertilized mouse eggs by inhibiting MPF (20). However, the link between PKA activity and Cdc25B inhibition remains unclear. To explore the effects of PKA on Cdc25B in mammalian cells, Cdc25B has been predicted as a potential PKA substrate in mice with the Scansite software, and three potential PKA phosphorylation sites, including Ser149, Ser229, and Ser321, were predicted. Acting as a candidate target of PKA, in our previous study, it has been proven that the Ser321 of Cdc25B (corresponding to Ser323 of the human protein) plays a critical regulatory role not only in meiotic resumption of mouse oocytes but also in the development of mouse embryos at the one-cell stage by modification of phosphorylation and dephosphorylation, whereas the Ser229 of Cdc25B has no effects on the development of fertilized mouse eggs (19, 21). However, the role of Ser149 in the mitotic cell cycle of fertilized mouse eggs remains unknown. In this study, to further explore the role of the Ser149 site of Cdc25B (equivalent to Ser151 in the human protein) in regulating the development of fertilized mouse eggs, we identified PKA phosphorylation sites in vitro by LC-MS/MS analysis, including Ser149, Ser229, and Ser321 of Cdc25B, which correspond to the phosphorylation sites predicted by Scansite software. Our findings show that residue Ser149 of Cdc25B, as well as Ser321 in mammalian cells, is likely a potential target of PKA involved in the mitosis of fertilized mouse eggs and plays an important role in G2/M transition of fertilized mouse eggs and in the subcellular localization of Cdc25B. Our results further prove that Cdc25B is the direct downstream substrate of PKA and PKA regulates the early development of fertilized mouse eggs by phosphorylation of the Ser149 and Ser321 residues of Cdc25B.
EXPERIMENTAL PROCEDURES
Kunming genealogy-specific pathogen-free mice (females at 4 weeks and 18 g; males at 8 weeks and 30 g) were obtained from the Department of Laboratory Animals, China Medical University. All experiments were performed at China Medical University in accordance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals. Reagents, unless otherwise specified, were from Sigma.
Prokaryotic Expression and Purification of Recombinant Cdc25B Protein
The cDNA of mouse Cdc25B, pBSK-Cdc25B-WT, was a kind gift from Dr. Tony Hunter (The Salk Institute). A 603-bp fragment encoding 201 amino acids (130–330) was amplified by PCR with pBSK-Cdc25B-WT as a template, and PCR was performed with 30 cycles (primers: 5′-CGCGGATCCATTCAGGCAGCCAGTCGGGT-3′ and 5′-CCGCTCGAGTCAGATGGGTCGGATCACACTGC-3′). Each PCR cycle consisted of 50 s at 94 °C, 50 s at 64 °C, and 60 s at 72 °C. The PCR product was then subcloned into the pGEX-4T-2 vector. The resulting recombinant plasmid, pGEX-4T-2-Cdc25B201, was sequenced to verify the correct gene insertion.
The method of protein expression was a modification of the procedures described in Refs. 22 and 23. GST-Cdc25B201 was expressed for 3 h at 27 °C in Escherichia coli BL21(DE3) (Takara) transformed with pGEX-4T-2-Cdc25B201 in the presence of 0.1 mm isopropyl-β-d-thiogalactopyranoside (Takara). The proteins were purified by the glutathione-Sepharose 4B protein chromatography purification kit directions (GE Healthcare). Total protein induced by isopropyl-β-d-thiogalactopyranoside, vector-expressed protein, and purified protein were identified by 10% SDS-PAGE and Western blotting.
Phosphorylation of GST-Cdc25B201 Fusion Protein in Vitro and LC-MS/MS Analysis of PKA Phosphorylation Site
Purified GST-Cdc25B201 protein (400 μg) was incubated with 6 μl of PKA (New England Biolabs) and 10 μl of ATP (New England Biolabs) at 30 °C for 45 min. After separation by 10% SDS-PAGE, the band of phosphorylated Cdc25B201 protein was excised from the gel, and in-gel digestion was performed (24). To obtain maximal sequence coverage of protein, the digested products were collected at 1, 2, 4, and 12 h. The sample was combined and analyzed with LC-linear trap quadrupole-MS/MS (Thermo Fisher Scientific, Waltham, MA) with MS/MS (MS2) and MS/MS/MS (MS3) strategy (25). MS data were searched with SEQUEST (26), and the spectra of all identified phosphopeptides were manually verified.
Collection and Culture of One-cell Stage Mouse Embryos
Mouse embryos at the one-cell stage were collected and cultured according to the method described by our previous report (21) on the basis of the method of Hogan and Costantini (27).
Plasmid Construction and Site-directed Mutagenesis
The pBluescript II/SK-Cdc25B-S149A (pBSK-Cdc25BS149A) construct was prepared by mutating Ser149 of Cdc25B to alanine with pBSK-Cdc25B-WT as a template and a site-directed mutagenesis kit (Stratagene). The primers were FWD1 (5′-CCATAAAACGCTTCCGAGCCTTACCAGTGAG-3′) and REV2 (5′-GGTAAGGCTCGGAAGCGTTTTATGGTAAACTG-3′). The pBSK-Cdc25B-S149A/S321A construct was prepared by mutating Ser321 of Cdc25B to alanine with pBSK-Cdc25B-S149A as a template. The primers were FWD3 (5′-GCTCTTCCGCTCCCCAGCCATGCCATGCA-3′) and REV4 (5′-CTGGGGAGCGGAAGAGCCTCTGGCACTTG-3′). Mutants mimicking phosphorylation were also constructed. pBSK-Cdc25B-S149D and pBSK-Cdc25B-S321D mutants in which residues 149 and 321 were replaced by aspartic acid were generated with pBSK-Cdc25B-S149A and pBSK-Cdc25B-S321A (kept by our laboratory (21)) as templates. The primers in pBSK-Cdc25B-S149D were FWD5 (5′-CCATAAAACGCTTCCGAGACTTACCAGTGAG-3′) and REV6 (5′-GGTAAGTCTCGGAAGCGTTTTATGGTAAACTG-3′). The primers for pBSK-Cdc25B-S321D were FWD7 (5′-CTCTTCCGCTCCCCAGACATGCCATGCAGTG-3′) and REV8 (5′-CATGTCTGGGGAGCGGAAGAGCCTCTGGCAC-3′). pBSK-Cdc25B-S149D/S321D was prepared with pBSK-Cdc25B-S149A/S321A as a template and the primer pairs FWD5 and REV6 and FWD7 and REV8. All of the above recombinant plasmids were sequenced to verify the correct gene insertion and successful mutation.
The three types of plasmids (pBSK-Cdc25B-WT, pBSK-Cdc25B-S149A, and pBSK-Cdc25B-S149D) were subcloned into pEGFP-C3 vector. The recombinant plasmids were named pEGFP-Cdc25B-WT, pEGFP-Cdc25B-S149A, and pEGFP-Cdc25B-S149D, respectively.
In Vitro Transcription
According to our previous report (21), all of the pBluescript II/SK constructs were linearized with XbaI and transcribed in vitro into 5′-capped mRNA for microinjection by the mMESSAGE mMACHINE kit (Ambion).
Microinjection and Morphological Analysis
Various Cdc25B-mRNAs or plasmids were microinjected into the cytoplasm or nucleus of one-cell embryos at the S or G1 phase according to our previous report (21), The typical injection volumes were 5% (10 pl, cytoplasm) and 1% (2 pl, nuclear) of the total cell volume per egg. mRNAs were diluted to various concentrations in nuclease-free TE buffer (5 mmol/liter Tris-HCL and 0.5 mmol/liter EDTA, pH 7.4). Eggs in the control groups were microinjected with or without TE buffer.
To observe the effects of PKA on Cdc25B, the fertilized eggs at the S phase (22 h after human chorionic gonadotropin (hCG) injection) were cultured in M16 medium with dibutyryl cAMP (dbcAMP, Sigma) and H-89 (Santa Cruz Biotechnology) for 1 h. Subsequently, the embryos microinjected with various Cdc25B-mRNAs were put back to M16 medium. Mitotic stages (G1, S, G2, and M phases) were defined as described previously (28). The rate of cleavage, namely, the number of two-cell embryos resulting from the division of a one-cell embryo, was counted in three independent experiments under a phase contrast microscope at 31, 34, or 30 h after hCG injection in the absence or presence of dbcAMP and/or H-89. Morphological analysis was performed, and the subcellular location of Cdc25B in embryos of the plasmid microinjection groups was detected with fluorescence microscopy.
Assay of MPF Activity
MPF kinase activity was measured by a histone H1 kinase assay (29). The kinase assay was performed according to a similar procedure to that described in our previous report (21).
Western Blotting
Embryos at the indicated times were lysed, subjected to SDS-PAGE (10%), transferred onto nitrocellulose membranes, and immunoblotted with antibodies to Tyr(P)15 of Cdc2 (1:500; Santa Cruz Biotechnology), Cdc25B (1:200; Santa Cruz Biotechnology), phospho-Cdc25B-pSer149 and nonphospho-Cdc25B-Ser149 (1:200; Signalway Antibody), and GST and β-actin (1:500; Beyotime Institute of Biotechnology). Proteins were visualized by the enhanced chemiluminescence (ECL) detection system (Pierce Biotechnology).
The phospho-Cdc25B-pSer149 and nonphospho-Cdc25B-Ser149 antibodies were raised in New Zealand White Rabbits against the keyhole limpet hemocyanin-conjugated phosphopeptide IKRFRpSLPVRLLC or the nonphosphopeptide IKRFRSLPVRLLC.
Immunofluorescence
Embryos were fixed with 2% paraformaldehyde for 40 min, permeabilized with 0.1% Triton X-100 for 10 min, and then blocked with 3% BSA in PBS for 40 min. Fertilized eggs were stained with monoclonal goat anti-mouse Cdc25B antibody (1:200, Santa Cruz Biotechnology) overnight at 4 °C. After three washes with PBS, embryos were incubated with FITC rabbit anti-goat IgG (1:200, Beijing Zhongshan Biotechnology) at 37 °C for 30 min. The DNA was stained with 25 μg/ml Hoechst 33258 for 3 min at 37 °C. Signals of subcellular localization of endogenous Cdc25B and DNA staining were detected by fluorescence microscope at 488 and 260 nm, respectively.
Statistical Analysis
All experiments were performed independently at least three times. A Student's t test or x2 test was used to evaluate the differences between multiple experimental groups with SSPS software (version 13.0). A probability level of 0.05 was considered significant.
RESULTS
Ser149, Ser229, and Ser321 Sites of Cdc25B Protein Are Phosphorylated by PKA in Vitro
The protein expression results showed that a clear band of GST-Cdc25B201 fusion protein was observed in 50 kDa of the expected size, and vector-derived GST produced a 26-kDa band, whereas the band of phosphorylated GST-Cdc25B201 protein was seen in ∼55 kDa in 10% SDS-PAGE (see supplemental Fig. S1). The results of LC-MS/MS analysis of phosphorylated GST-Cdc25B201 were shown in Fig. 1, A–F. Three PKA phosphorylation sites, Ser149, Ser229, and Ser321, were identified. The coverage rate for a given sample is above 97% (Fig. 1F), and the covered sequence is shown in black letters.
FIGURE 1.

Cdc25B201 LC-MS/MS analysis identified three PKA phosphorylation sites, including Ser149, Ser229, and Ser321. A, MS2 spectrum of the phosphorylated peptide, 147FRS*LPVR153, containing Ser149; S* indicates that serine residue 149 is phosphorylated by PKA. B, MS3 spectrum of the phosphorylated peptide 147FRS#LPVR153, containing Ser149, S# indicates neutral loss of H3PO4 from sequence ions. C, MS2 spectrum of the phosphorylated peptide 319SPS*MPCSVIRPI330 containing Ser321; S* indicates that serine residue 321 is phosphorylated by PKA. D, MS3 spectrum of the phosphorylated peptide 319SPS#MPCSVIRPI330 containing Ser321; S# indicates neutral loss of H3PO4 from sequence ions. E, MS2 spectrum of the phosphorylated peptide 220RQAFTQRPSS*APDLMCLTTEWK241, containing Ser229; S* indicates that serine residue 229 is phosphorylated by PKA. F, amino acid sequence of the Cdc25B201 protein. The LC-MS/MS analysis coverage rate of the given sample is >97% (the covered sequence is shown in boldface letters). The underlined fragments indicate the identified phosphorylated peptide; pS represents phosphorylated serine, including Ser(P)149, Ser(P)229, and Ser(P)321.
Overexpression of Cdc25B-WT and Cdc25B-S/A Mutants Promotes Onset of Mitosis of Fertilized Mouse Eggs
Western blotting showed that the Cdc25B protein accumulated to higher levels in the various mRNA microinjection groups than in the control groups at 4 h after microinjection (Fig. 2A), indicating that various exogenous Cdc25B-mRNAs could be translated efficiently in fertilized mouse eggs. Furthermore, there was no difference among the microinjection groups.
FIGURE 2.

Western blot analysis of Cdc25B protein expression and phosphorylation status of Cdc2-Tyr15, cleavage rate, and MPF activity after mRNAs encoding Cdc25B-WT, Cdc25B-S149A, Cdc25B-S321A, Cdc25B-S149A/S321A, Cdc25B-S149D, Cdc25B-S321D, or Cdc25B-S149D/S321D were microinjected into fertilized mouse eggs in the absence of dbcAMP and H-89 in M16 medium. A, Western blot analysis of Cdc25B protein expression at 4 h after microinjection of Cdc25B mRNAs. B, the cleavage rate in cultured mouse embryos of various Cdc25B mRNAs injection at 31 h after hCG injection. The cleavage rates were calculated with data from three independent experiments. C, Western blot analysis of the phosphorylation status of Cdc2-Tyr15 in the various Cdc25B mRNA injection groups. The eggs were collected at 26.5, 27, 27.5, 28, 28.5, and 29 h after hCG injection. D, MPF activity in the control groups, including no injection and TE buffer groups. E, MPF activity in eggs injected with Cdc25B-WT-mRNA. F, MPF activity in eggs injected with Cdc25B-S149A-mRNA. G, MPF activity in eggs injected with Cdc25B-S321A-mRNA. H, MPF activity in eggs injected with Cdc25B-S149A/S321A-mRNA. I, MPF activity in eggs injected with Cdc25B-S149D-mRNA. J, MPF activity in eggs injected with Cdc25B-S321D-mRNA. K, MPF activity in eggs injected with Cdc25B-S149D/S321D-mRNA. For each point, five eggs were collected, and MPF activity was examined by scintillation counting and autoradiography. Each value was expressed as the mean ± S.D. from three independent experiments. A total of ∼200 eggs were loaded onto each lane in Western blot.
The cleavage of fertilized mouse eggs was observed at 26–31 h after hCG injection, and the cleavage rate were calculated at 31 h. In the two control groups, cleavage of the embryos had begun at 28.5–29 h after hCG injection and 58.2% (no injection) and 59.3% (TE injection) of embryos had reached the two-cell stage at 31 h (Fig. 2B). In contrast, embryos injected with Cdc25B-WT-mRNA entered the M phase at 27.5–28 h after hCG injection, and the cleavage rate was as high as 76.4% at 31 h, markedly higher than that of the control groups (p < 0.01). Embryos microinjected with mRNAs of either Cdc25B-S149A or Cdc25B-S321A entered the M phase at 26.5–27 h after hCG injection, and almost 90.5 and 91.2% of embryos, respectively, had reached the two-cell stage at 31 h (Fig. 2B) A similar pattern was seen in the Cdc25B-S149A/S321A-injected eggs, but the cleavage rate was as high as 98.3% at 31 h after hCG injection and markedly higher than that of the Cdc25B-S149A and Cdc25B-S321A groups (p < 0.05). However, embryos microinjected with Cdc25B-S/D-mRNAs entered M phase at 28.5–29 h after hCG injection, and the cleavage rates were ∼60% at 31 h (Fig. 2B), similar to the control groups (p > 0.05).
Overexpression of Cdc25B-WT and Cdc25B-S/A Mutants Activates MPF by Direct Dephosphorylation of Cdc2-pTyr15
To illustrate the effects of microinjection of various Cdc25B-mRNAs on MPF activity in fertilized eggs, we measured the MPF activity at 26 h after hCG injection every 30 min. In the control groups, MPF activity was consistently low at 26–28 h after hCG injection, increasing gradually at 28 h, reaching its maximal level at 29 h and then decreasing slowly (Fig. 2D). However, MPF activity in Cdc25B-WT-mRNA-injected eggs reached its highest level at 28 h and then began to slowly decline (Fig. 2E). MPF activity increased sharply at 26.5 h, peaked at 27 h, and then decreased slowly in Cdc25B-S149A, Cdc25B-S321A, and Cdc25B-S149A/S321A-mRNA-injected eggs (Fig. 2, F–H), but the peak of MPF activity in Cdc25B-S/D-mRNA-injected eggs appeared at 29 h (Fig. 2, I–K). Furthermore, to identify the relationship between the phosphorylation status of Cdc2-Tyr15 and MPF activity, Western blotting was carried out at 26.5, 27, 27.5, 28, 28.5, and 29 h after hCG injection in the control and microinjection groups (Fig. 2C). In the control groups, there were strong singals of Cdc2-Tyr15 phosphorylation at 26.5–28 h, a reduced phosphorylation level at 28.5 h, and no signal at 29 h after hCG injection, which demonstrated that MPF was fully activated. Meanwhile, in the Cdc25B-WT group, there was a weak Cdc2-Tyr15 phosphorylation signal at 27.5 h and no signal at 28–29 h after hCG injection. In the Cdc25B-S/A mutant groups, the strong signals of Cdc2-Tyr15 phosphorylation were detected at 26.5 h, but no signal was found at 27–29 h after hCG injection. Conversely, the inhibitory phosphorylation of Cdc2-Tyr15 was still observed at 28–28.5 h, but no signal was detected at 29 h in the Cdc25B-S/D mutant groups.
Microinjection of Cdc25B-S/A Mutants Overcomes G2 Arrest Induced by dbcAMP
It has been reported that cAMP analogs, such as dbcAMP, which can activate PKA, play an important role in the regulation of meiosis arrest in mouse oocytes (30, 31). On the basis of our previous experimental research (21), 2 mm dbcAMP led to maximal G2 arrest, and none of the mouse eggs was able to enter the M phase of mitosis, suggesting inhibition of the G2/M transition. Therefore, to further explore whether PKA could phosphorylate the Ser149 site of Cdc25B, various Cdc25B mRNAs were each microinjected into embryos at the S phase. The injected eggs were pretreated with 2 mm dbcAMP in M16 medium for about 1 h. Cdc25B protein was highly expressed at 4 h after microinjection in the microinjection groups compared with the control groups (Fig. 3A), and there was no significant difference among the microinjection groups, indicating that the various exogenous Cdc25B mRNAs could be translated efficiently in fertilized mouse eggs.
FIGURE 3.

Western blot analysis of Cdc25B protein expression and phosphorylation status of Cdc2-Tyr15, cleavage rate, and MPF activity after mRNAs encoding Cdc25B-WT, Cdc25B-S149A, Cdc25B-S321A, Cdc25B-S149A/S321A, Cdc25B-S149D, Cdc25B-S321D, or Cdc25B-S149D/S321D were microinjected into fertilized mouse eggs in the presence of 2 mmol/liter dbcAMP in M16 medium. A, Western blot analysis of Cdc25B protein expression at 4 h after various Cdc25B mRNA microinjection. B, the cleavage rate in cultured mouse embryos of various Cdc25B mRNA injections at 34 h after hCG injection. The cleavage rates were calculated with data from three independent experiments. C, Western blot analysis of the phosphorylation status of Cdc2-Tyr15 in the various mRNA-injected eggs. The eggs were collected at 29, 30, 31, 32, and 33 h after hCG injection. D, MPF activity of fertilized mouse eggs in the control groups, including no injection and TE buffer groups. E, MPF activity in eggs injected with Cdc25B-WT-mRNA. F, MPF activity in eggs injected with Cdc25B-S149A-mRNA. G, MPF activity in eggs injected with Cdc25B-S321A-mRNA. H, MPF activity in eggs injected with Cdc25B-S149A/S321A-mRNA. I, MPF activity in eggs injected with Cdc25B-S149D-mRNA. J, MPF activity in eggs injected with Cdc25B-S321D-mRNA. K, MPF activity in eggs injected with Cdc25B-S149D/S321D-mRNA. For each point, five eggs were collected, and MPF activity was examined by scintillation counting and autoradiography. Each value was expressed as the mean ± S.D. from three independent experiments. A total of ∼200 eggs were loaded onto each lane in Western blot.
In the presence of 2 mm dbcAMP, the cleavage of fertilized mouse eggs was observed at 29–34 h after hCG injection, and the cleavage rate was calculated at 34 h. As shown in Fig. 3B, the cleavage rate of embryos induced by dbcAMP in each group decreased obviously, and hardly any eggs reached the two-cell stage at 34 h after hCG injection in the control and Cdc25B-S/D groups. Meanwhile, parts of embryos microinjected with Cdc25B-WT-mRNA entered M phase at 32 h, and the cleavage rate was as high as 21.5% at 34 h, significantly higher than that of the control groups (p < 0.01). In contrast, embryos microinjected with either Cdc25B-S149A or Cdc25B-S321A-mRNA entered M phase at 29.5–30 h after hCG injection, and nearly 89.5 and 90.1% of embryos, respectively, had developed to the two-cell stage at 34 h. Eggs injected with Cdc25B-S149A/S321A-mRNA resumed mitosis at 29–29.5 h after hCG injection, and nearly 97.8% of eggs had entered the M phase at 34 h, which was markedly higher than that in the Cdc25B-S149A and Cdc25B-S321A groups (p < 0.05).
Overexpression of Cdc25B-S/A Mutants Can Activate MPF Efficiently in Presence of dbcAMP
To illustrate the effects of PKA activity on MPF activity in fertilized eggs in the presence of 2 mm dbcAMP, MPF activity was measured beginning at 29 h after hCG injection at 30-min intervals. As shown in Fig. 3, MPF activity was stably low at 29–34 h in the control groups, which indicated that MPF activity was inhibited by PKA activation (Fig. 3D). Meanwhile, MPF activity in Cdc25B-WT-mRNA-injected eggs increased weakly at 32 h after hCG injection and then decreased gradually (Fig. 3E). However, MPF activity had a sharp increase at 29.5 h, peaked at 30 h, and then decreased slowly in Cdc25B-S149A and Cdc25B-S321A groups (Fig. 3, F and G). In the Cdc25B-S149A/S321A group, MPF activity reached its maximal level at 29.5 h after hCG injection, remained at this level for 30 min, and then decreased slowly (Fig. 3H). In contrast, MPF activity in eggs injected with the Cdc25B-S/D mutants stayed at a low level for 29–34 h after hCG injection, which is similar to the MPF activity pattern observed in the control groups (Fig. 3, I–K).
Meanwhile, the phosphorylation status of Cdc2-Tyr15 was detected by Western blotting at 29, 30, 31, 32, and 33 h after hCG injection to identify the relationship between MPF activity and the phosphorylation of Cdc2-Tyr15 in the fertilized mouse eggs induced by dbcAMP. In the control groups, strong bands of phosphorylated Cdc2-Tyr15 were identified at 29–33 h after hCG injection, demonstrating that MPF activity was inhibited (Fig. 3C). In the Cdc25B-WT group, Cdc2-Tyr15 phosphorylation was weakly detected at 31–33 h after hCG injection, which was coincident with the MPF activity. However, in the Cdc25B-S/A mutant groups, there was almost no signal at 30–33 h after hCG injection, which implied that MPF was completely activated. In contrast, the inhibitory phosphorylation signals of Cdc2-Tyr15 were still observed at 33 h after hCG injection in the Cdc25B-S/D mutant groups, as in the control groups.
Microinjection of Cdc25B-WT and Cdc25B-S/A Mutants Accelerates Mitotic Entry of Fertilized Mouse Eggs with H-89
It has been well documented that H-89 inhibits the PKA catalytic subunit and easily diffuses through the cell membrane (32, 33). To further test whether PKA can affect the mitotic cell cycle, one-cell stage mouse embryos at S phase were incubated in M16 medium containing various concentrations of H-89. With increasing concentrations of H-89 (0–50 μm), the G2 phase of eggs was decreased, and the cleavage rate was accelerated. A concentration of 40 μm H-89 induced all of the mouse eggs entering the M phase of mitosis at 31 h after hCG injection (Fig. 4A). Therefore, after the eggs had been pretreated with 40 μm H-89 in M16 medium for ∼1 h, various Cdc25B-mRNAs were injected into fertilized mouse eggs at S phase. The Cdc25B protein was more highly expressed at 4 h after microinjection in the microinjection groups than in the control groups (Fig. 4B), and there was no significant difference among the microinjection groups, indicating that the various exogenous Cdc25B mRNAs could be translated efficiently in fertilized mouse eggs.
FIGURE 4.

Western blot analysis of Cdc25B protein expression and phosphorylation status of Cdc2-Tyr15, cleavage rate, and MPF activity after mRNAs encoding Cdc25B-WT, Cdc25B-S149A, Cdc25B-S321A, Cdc25B-S149A/S321A, Cdc25B-S149D, Cdc25B-S321D, or Cdc25B-S149D/S321D were microinjected into fertilized mouse eggs in the presence of 40 μmol/liter H-89 in M16 medium. A, bar graph analysis of cleavage rate changes with different concentration of H-89 at 31 h after hCG injection. B, Western blot analysis of Cdc25B protein expression at 4 h after microinjection of various Cdc25B mRNAs. C, the cleavage rate in cultured mouse embryos injected with various Cdc25B mRNA at 30 h after hCG injection. The cleavage rates were calculated with data from three independent experiments. D, Western blotting detection of the phosphorylation status of Cdc2-Tyr15 in the various mRNA-injected eggs. The eggs were collected at 26, 26.5, 27, 27.5, and 28 h after hCG injection. E, MPF activity of fertilized mouse eggs in the control groups, including no injection and TE buffer groups. F, MPF activity in eggs injected with Cdc25B-WT-mRNA. G, MPF activity in eggs injected with Cdc25B-S149A-mRNA. H, MPF activity in eggs injected with Cdc25B-S321A-mRNA. I, MPF activity in eggs injected with Cdc25B-S149A/S321A-mRNA. J, MPF activity in eggs injected with Cdc25B-S149D-mRNA. K, MPF activity in eggs injected with Cdc25B-S321D-mRNA. L, MPF activity in eggs injected with Cdc25B-S149D/S321D-mRNA. For each point, five eggs were collected, and MPF activity was examined by scintillation counting and autoradiography. Each value was expressed as the mean ± S.D. from three independent experiments. A total of ∼200 eggs were loaded onto each lane in Western blot.
In the presence of 40 μm H-89, the cleavage of fertilized mouse eggs was observed at 26–30 h after hCG injection, and the cleavage rate was calculated at 30 h. Fig. 4C shows that cleavage rate of embryos induced by H-89 in each group increased markedly, and the control embryos entered M phase at 27.5–28 h after hCG injection, nearly 78.2% (no injection) and 79.4% (TE injection) of embryos had developed to the two-cell stage at 30 h after hCG injection. However, eggs injected with Cdc25B-WT and Cdc25B-S/A-mRNAs resumed mitosis at 26–26.5 h after hCG injection, and >90% of eggs had developed to the two-cell stage at 30 h, markedly higher than that in the control groups (p < 0.05). The eggs injected with Cdc25B-S/D mutants entered M phase at 27.5–28 h, and ∼80% of eggs had reached the two-cell stage at 30 h after hCG injection, which was similar to the control groups.
Overexpression of Cdc25B-WT and Cdc25B-S/A Mutants Can Reinforce MPF Activity Early in Presence of H-89
In the presence of H-89, MPF activity was detected beginning at 26 h after hCG injection at 30-min intervals. As shown in Fig. 4, in the control groups, there was a striking increase in MPF activity at 27.5 h after hCG injection, then MPF activity peaked at 28 h, indicating that inhibition of PKA may activate MPF early (Fig. 4E). MPF activity in Cdc25B-WT and Cdc25B-S/A mutants groups reached its maximal level at 26.5 h after hCG injection and then decreased gradually (Fig. 4, F–I). However, MPF activity in eggs injected with Cdc25B-S/D mutants peaked at 28 h after hCG injection, as in the control groups (Fig. 4, J–L).
To identify the relationship between MPF activity and the phosphorylation of Cdc2-Tyr15 in the fertilized mouse eggs with H-89, we carried out Western blotting analysis at 26, 26.5, 27, 27.5, and 28 h after hCG injection. The inhibitory phosphorylation signals of Cdc2-Tyr15 were detected at 26–27.5 h, and no phosphorylation was found at 28 h after hCG injection in the control groups (Fig. 4D). However, in the Cdc25B-WT and Cdc25B-S/A mutants groups, there were strong bands of Cdc2-Tyr15 phosphorylation at 26 h, and no signal was detected at 26.5–28 h after hCG injection. On the contrary, the inhibitory phosphorylation bands of Cdc2-Tyr15 were observed at 26–27.5 h in the Cdc25B-S/D groups, and there still was a weak Cdc2-Tyr15 phosphorylation signal at 28 h after hCG injection.
Expression of Cdc25B Protein and Phosphorylation Status of Cdc25B-Ser149
We used a goat monoclonal antibody against the Cdc25B N terminus to detect Cdc25B expression at different phases in fertilized mouse eggs. As shown in Fig. 5A, Cdc25B protein band observed in both the G1 and S phases had a slightly different apparent molecular weight from that in seen in the G2 and M phases, as we reported previously (21). To identify whether Cdc25B-Ser149 was phosphorylated in vivo, we collected fertilized mouse eggs at different phases in the absence or presence of dbcAMP and/or H-89 and then measured the phosphorylation status of Cdc25B-Ser149 by the phosphospecific antibody, and nonphosphorylation status by the nonphosphospecific antibody. Fig. 5, B and D, showed that a phosphorylated Cdc25B-Ser149 band was observed at G1 and S phases, whereas no phosphorylation of Cdc25B-Ser149 was observed at G2 and M phases in eggs with or without H-89. However, a strong phosphorylated Cdc25B-Ser149 band was found not only in the G1 and S phases, but also in the G2 and M phases in the presence of dbcAMP (Fig. 5C). Conversely, a nonphosphorylated Cdc25B-Ser149 band was detected mainly during the G2 and M phases (Fig. 5, B–D), and it is the strongest in the presence of H-89 (Fig. 5D). Taken together, these results demonstrated that Cdc25B-Ser149 is phosphorylated at G1 and S and dephosphorylated at G2 and M in vivo and is responsible for the G2/M transition.
FIGURE 5.
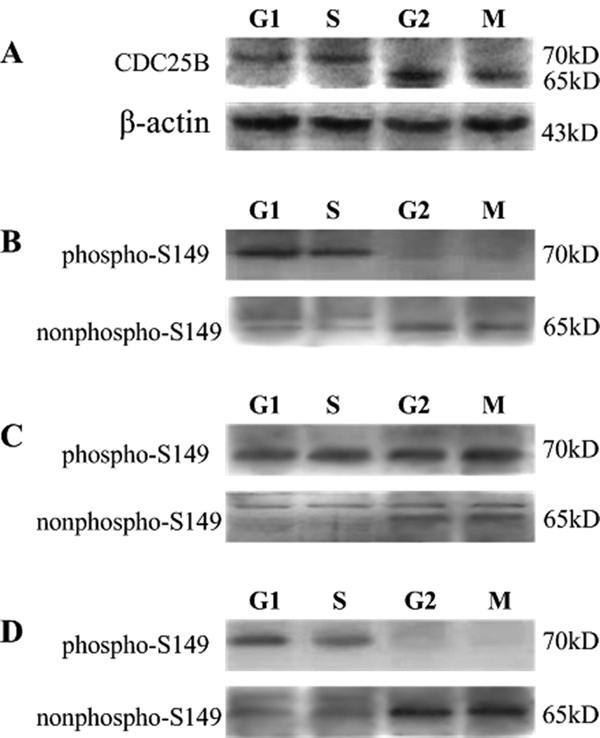
Endogenous Cdc25B protein expression and detection of the phosphorylated and unphosphorylated status of Cdc25B-Ser149 in fertilized mouse eggs at different phases of the cell cycle by Western blotting. A, endogenous Cdc25B protein expression at different phases of the cell cycle was detected with goat polyclonal antibody against the Cdc25B N terminus in the absence of dbcAMP and H-89 in M16 medium. The control for loading (β-actin) is shown. B, Western blot analysis of the phosphorylation and nonphosphorylation status of Cdc25B-Ser149 in fertilized mouse eggs at the G1, S, G2, and M phases with a phosphospecific and a non-phosphospecific Cdc25B-Ser149 antibody, respectively, in the absence of dbcAMP and H-89. C, the phosphorylated and unphosphorylated status of Cdc25B-Ser149 at the G1, S, G2, and M phases was measured in the presence of 2 mmol/liter dbcAMP. D, the phosphorylated and unphosphorylated form of Cdc25B-Ser149 at the G1, S, G2, and M phases was detected in the presence of 40 μmol/liter H-89. The molecular mass of proteins is indicated on the right side of the figure. A total of ∼200 eggs were loaded onto each lane in Western blot.
Subcellular Localization of Endogenous Cdc25B in Fertilized Mouse Eggs
We observed the subcellular localization of endogenous Cdc25B at every phase of cell cycle in fertilized mouse eggs with indirect immunofluorescence. As shown in Fig. 6, green fluorescent Cdc25B signals were detected mainly in the cytoplasm of G1 phase eggs (Fig. 6A), whereas in S phase eggs, the fluorescent signals were observed throughout the whole cell (Fig. 6B). The green fluorescent signals were translocated to the nucleus of eggs at the G2 phase, and the signal in the nucleus was stronger than that in the cytoplasm (Fig. 6C). However, the fluorescent signals became distributed in the cytosol again and decreased in the nucleus during the transition from the M phase to the two-cell stage (Fig. 6D).
FIGURE 6.

The distribution of endogenous Cdc25B in fertilized mouse eggs. A, the green fluorescent signals were mainly detected in the cytoplasm, particularly obviously in the cytoplasmic cortex (arrow) at the G1 phase. B, the green fluorescent signals were observed in the whole cell at the S phase. C, the green fluorescent signals were seen in the whole cell, but nuclear accumulation of signals (arrow) was detected at the G2 phase. D, the green fluorescent signals were mainly detected in the cytoplasmic cortex at late M stage.
Subcellular Localization of Exogenously Expressed Cdc25B
To observe the subcellular localization of exogenous Cdc25B, the pEGFP-Cdc25B-WT, pEGFP-Cdc25B-S149A, pEGFP-Cdc25B-S149D, and pEGFP-C3 plasmids were microinjected into fertilized mouse eggs, respectively, at the G1 phase (20 h after hCG injection), and then the microinjected eggs were transferred into M16 medium. When mouse embryos injected with pEGFP-Cdc25B-WT or pEGFP-Cdc25B-S149A entered S phase, a distinct GFP signal was detected throughout the whole cell. Then, the green fluorescent signals in the nucleus were much stronger than those in the cytosol during the G2 phase. In contrast, the green fluorescent signals in the nucleus apparently weakened and became distributed again mainly in the cytoplasm in the two-cell stage (Fig. 7, A and B). However, the emergence of pEGFP-Cdc25B-S149D and pEGFP-C3 fluorescent signals lagged ∼1 h compared with the pEGFP-Cdc25B-WT, and green fluorescence signals were detected in the whole cells, but mainly in the cytoplasm in the pEGFP-Cdc25B-S149D-injected eggs (Fig. 7C), and diffused throughout the whole cells in the pEGFP-C3-injected eggs (Fig. 7D).
FIGURE 7.

Expression and localization of pEGFP-Cdc25B in the one-cell stage of development of fertilized mouse eggs. A, fertilized mouse eggs injected with pEGFP-Cdc25B-WT. B, fertilized mouse eggs injected with pEGFP-Cdc25B-S149A. In these two groups, the green fluorescent signals were observed in the whole cell at the S stage, and much stronger signals were detected in the nucleus at the G2 phase (double arrow). However, in the two-cell stage, green fluorescent signals were mainly detected in the cytoplasm (arrow). C, fertilized mouse eggs injected with pEGFP-Cdc25B-S149D. The green fluorescent signals were detected in the whole cell but were much more obvious in the cytoplasm (arrow). D, eggs injected with pEGFP-C3. The green fluorescent signals diffused evenly throughout the whole cell, and there was no difference among the various phases of the cell cycle. Scale bar, 20 μm.
DISCUSSION
PKA plays a critical role in regulating the cell cycle progression of eukaryotic organisms. Cdc25B has been predicted to be a potential PKA substrate in mice (19, 21, 34, 35); however, there has been no proof of its phosphorylation in vitro until now. Further investigation was needed to explore how PKA phosphorylates Cdc25B in mammalians. Our results showed that the PKA phosphorylation sites of Cdc25B are Ser149, Ser229, and Ser321, corresponding to the phosphorylation sites predicted by the Scansite software; therefore, Cdc25B is the direct downstream substrate of PKA in mammalian cells.
We further explored the role of Ser149 of Cdc25B in regulating the development of fertilized mouse eggs. Our findings showed that overexpressed Cdc25B-WT, Cdc25B-S149A, Cdc25B-S321A, and Cdc25B-S149A/S321A in fertilized mouse eggs, not only accelerated the mitotic G2/M transition but also stimulated an increase in the cleavage rate in agreement with our previous results (21). Interestingly, Cdc25B-S149A performed similarly to Cdc25B-S321A, but much less efficiently than the Cdc25B-S149A/S321A mutant, which indicated that residue Ser149 of Cdc25B as well as Ser321 is likely direct target of PKA in mammals and cooperates in the regulation of early development of fertilized mouse eggs.
To test directly whether inhibition of fertilized mouse eggs by Ser149 of Cdc25B was indeed due to the endogenous PKA, we attempted to activate or inhibit specifically endogenous PKA activity and examine the function of Cdc25B. The cleavage rate of embryos induced by dbcAMP in each group decreased obviously, but a great number of embryos microinjected with mRNAs of either Cdc25B-S149A, Cdc25B-S321A, or Cdc25B-S149A/S321A entered the M phase at 34 h after hCG injection. However, most eggs injected with Cdc25B-WT-mRNA hardly entered the M phase. In contrast, in the presence of H-89, fertilized mouse eggs injected with Cdc25B-WT and Cdc25B-S/A-mRNAs resumed mitosis quickly. Meanwhile, overexpression of phosphomimic Cdc25B-S/D mutants cannot promote cell cycle progression and has no significant difference from the control groups. Our findings further identify that Ser149 of Cdc25B may be another important phosphorylation target of PKA in G2/M transition of fertilized mouse eggs.
It is well documented that Cdc25 proteins are dual-specificity phosphatases that not only uniquely function to dephosphorylate specific tyrosine/threonine residues on Cdc2 during the G2/M transition but also are key regulators of the meiotic resumption of mouse oocytes (35–38). Our previous research fully indicated that PKA acts as a negative regulator of MPF by phosphorylating Cdc25B-Ser321 (21). In this study, we further elucidated the effects of the Ser149 residue of Cdc25B on MPF activity and Cdc2-Tyr15 phosphorylation status. The results showed that MPF activity without dbcAMP and H-89 peaked in Cdc25B-S/A mutant groups earlier than in the Cdc25B-WT and control groups. However, in the presence of dbcAMP, MPF activity remained at a relatively low level at 29–34 h after hCG injection in the control groups, and the peak of MPF activity appeared in the Cdc25B-S/A-mutants groups, ∼2.5–3 h later than in the Cdc25B-S/A mutants groups without dbcAMP and H-89, suggesting that PKA activation is responsible for the inhibition of MPF activity. Interestingly, MPF activity reached its maximum level in the Cdc25B-S149A/S321A group 30 min earlier than in Cdc25B-S149A and Cdc25B-S321A groups, which indicates that the function of Cdc25B-S149A/S321A is stronger than that of Cdc25B-S149A and Cdc25B-S321A. Overall, overexpression of Cdc25B-S/A mutants can overcome G2 arrest induced by dbcAMP. In contrast, in the presence of H-89, MPF activity reached its maximum level in Cdc25B-WT and Cdc25B-S/A-mutants groups 30 min earlier than in the Cdc25B-S/A mutants groups without H-89 and dbcAMP, which suggests that PKA inhibition is responsible for the activation of MPF activity. Meanwhile, Western blotting showed that the phosphorylation status of Cdc2-Tyr15 at different time in each group was coincident with the MPF activity, whether dbcAMP or H-89 is added or not. These data suggest that the unphosphorylated Cdc25B can activate MPF by the direct dephosphorylation of Cdc2-pTyr15 in the mitotic cell cycle of fertilized mouse eggs.
With the purpose of probing relationship between Cdc25B-Ser149 phosphorylation and G2/M transition, we measured the phosphorylation status of Cdc25B-Ser149 in eggs at G1, S, G2 and M phases, respectively, by Western blotting with phosphospecific and non-phosphospecific antibodies in the absence or presence of dbcAMP and/or H-89. The results suggest that Cdc25B-Ser149 is phosphorylated at the G1 and S phases, whereas Cdc25B-Ser149 is dephosphorylated at the G2 and M phases in vivo, corresponding to our previous reports about the Ser321 site of Cdc25B (21). Taken together, our findings prove that PKA regulates the early development of fertilized mouse eggs by phosphorylation of Ser149 and Ser321 of Cdc25B, and the residue Ser149 of Cdc25B cooperates with Ser321. When these two sites were simultaneously mutated to the unphosphorylated residue Ala, the Cdc25B mutants promoted the onset of mitosis prior to a single point mutant, whether dbcAMP was added or not. If PKA phosphorylated Ser149, Ser321, or both sites of Cdc25B during the G1 and S phases, Cdc25B phosphatase was inhibited to some extent, resulting in retardation of mitosis progression. On the contrary, dephosphorylation of Ser149 or Ser321 in the G2 phase induced the activation of Cdc25B, which can activate MPF efficiently and resume mitosis by the direct dephosphorylation of Cdc2-Tyr15.
In addition to protein levels and phosphorylation status, the subcellular distribution of the Cdc25 family may be an important regulator of G2/M transition. Using CHO cells, Woo et al. (39) observed that Cdc25B partitioned primarily in the cytoplasm during G1 and progressively migrated to the nucleus as cells transitioned from the S to the G2/M phase. It is reported that 14-3-3 binding to the Ser323 site of Cdc25B directly inhibits the activity of Cdc25B in HeLa cells, thereby blocking the access of substrate, the catalytic site of MPF, and controlling the progression of mitosis (40). In contrast, mutation of the Ser323 site, resulting in the complete loss of 14-3-3 binding, increases the access of the catalytic site of MPF and the nuclear localization sequence (41). Therefore, it is necessary to trigger mitosis by Cdc25 shuttling in and out of the nucleus of fertilized eggs and vice versa. In this study, we investigated the effects of endogenous and exogenous Cdc25B subcellular location on the development of fertilized mouse eggs at the one-cell stage by immunofluorescence and microinjection of the plasmids of pEGFP-Cdc25B-WT, pEGFP-Cdc25B-S149A, and pEGFP-Cdc25B-S149D, respectively. Changes in the endogenous and exogenous fluorescent signals suggest that Cdc25B transferred from cytoplasm to nucleus at G2 phase, which may be in connection with phosphorylation/dephosphorylation of Cdc25B.
Collectively, the aforementioned findings can be accommodated in the following working model. Cdc25B may exist primarily in the cytoplasm for PKA phosphorylation of the Ser149 and Ser321 residues of Cdc25B, resulting in a direct interaction between phosphorylated Cdc25B and the 14-3-3 protein, which prevents Cdc25B from entering the nucleus, and then retards cell cycle progression. Phosphorylated Cdc25B is dephosphorylated by an unknown protein phosphatase, activated, and then transferred from the cytoplasm to the nucleus to stimulate MPF to trigger mitosis. Afterward, Cdc25B may be transferred from the nucleus to the cytoplasm again or may be degraded in the nucleus when Cdc25B completes its mission in late M phase. Our findings suggest that Ser321 and/or Ser149 of Cdc25B may be phosphorylated and bind to the 14-3-3 protein, which selectively reduces the rate of Cdc25B nuclear import, resulting in more efficient exclusion of Cdc25B from the nucleus, thus negatively controlling the G2/M transition. When the Ser149 and Ser321 of Cdc25B were mutated to unphosphorylated Ala, the 14-3-3 binding would be abrogated, and homeostasis of nucleus export and import would be restored. It is noteworthy that the various changes in Cdc25B-S/D mutant-injected eggs correspond to those of eggs in the control groups whether they were pretreated with dbcAMP or H-89 or not, indicating that Cdc25B-S/D mutants is different from Cdc25B-S/A mutants, possessing the phosphomimic function.
In the future, it is of great importance to probe the molecular mechanisms modulating the association of Cdc25B with 14-3-3 proteins, dephosphorylation of Cdc25B and MPF activity by the shuttling of Cdc25B in and out of the nucleus of fertilized mouse eggs.
Acknowledgment
We thank Dr. Tony Hunter (Laboratory of Molecular Biology, The Salk Institute) for the kind gift of the full-length mouse Cdc25B cDNA clone.
This work was supported by National Nature Science Foundation of China Grant 30900514.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Figs. S1–S3.
- MPF
- M phase/maturation promoting factor
- PKA
- protein kinase A
- dbcAMP
- dibutyryl cAMP
- hCG
- human chorionic gonadotropin
- FWD
- forward
- REV
- reverse.
REFERENCES
- 1. Strausfeld U., Labbé J. C., Fesquet D., Cavadore J. C., Picard A., Sadhu K., Russell P., Dorée M. (1991) Nature 351, 242–245 [DOI] [PubMed] [Google Scholar]
- 2. Morgan D. O. (1995) Nature 374, 131–134 [DOI] [PubMed] [Google Scholar]
- 3. Nilsson I., Hoffmann I. (2000) Prog. Cell Cycle Res. 4, 107–114 [DOI] [PubMed] [Google Scholar]
- 4. Alonso A., Sasin J., Bottini N., Friedberg I., Friedberg I., Osterman A., Godzik A., Hunter T., Dixon J., Mustelin T. (2004) Cell 117, 699–711 [DOI] [PubMed] [Google Scholar]
- 5. Han S. J., Conti M. (2006) Cell Cycle 5, 227–231 [DOI] [PubMed] [Google Scholar]
- 6. Boutros R., Dozier C., Ducommun B. (2006) Curr. Opin. Cell Biol. 18, 185–191 [DOI] [PubMed] [Google Scholar]
- 7. Rudolph J. (2007) Biochemistry 46, 3595–3604 [DOI] [PubMed] [Google Scholar]
- 8. Karlsson C., Katich S., Hagting A., Hoffmann I., Pines J. (1999) J. Cell Biol. 146, 573–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lincoln A. J., Wickramasinghe D., Stein P., Schultz R. M., Palko M. E., De Miguel M. P., Tessarollo L., Donovan P. J. (2002) Nat. Genet. 30, 446–449 [DOI] [PubMed] [Google Scholar]
- 10. Donzelli M., Draetta G. F. (2003) EMBO Rep. 4, 671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bugler B., Quaranta M., Aressy B., Brezak M. C., Prevost G., Ducommun B. (2006) Mol. Cancer Ther. 5, 1446–1451 [DOI] [PubMed] [Google Scholar]
- 12. Boutros R., Lobjois V., Ducommun B. (2007) Nat. Rev. Cancer 7, 495–507 [DOI] [PubMed] [Google Scholar]
- 13. Eyers P. A., Liu J., Hayashi N. R., Lewellyn A. L., Gautier J., Maller J. L. (2005) J. Biol. Chem. 280, 24339–24346 [DOI] [PubMed] [Google Scholar]
- 14. Duncan F. E., Moss S. B., Williams C. J. (2006) Dev. Dyn. 235, 2961–2968 [DOI] [PubMed] [Google Scholar]
- 15. Kovo M., Kandli-Cohen M., Ben-Haim M., Galiani D., Carr D. W., Dekel N. (2006) Reproduction 132, 33–43 [DOI] [PubMed] [Google Scholar]
- 16. Grieco D., Avvedimento E. V., Gottesman M. E. (1994) Proc. Natl. Acad. Sci. U.S.A. 91, 9896–9900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grieco D., Porcellini A., Avvedimento E. V., Gottesman M. E. (1996) Science 271, 1718–1723 [DOI] [PubMed] [Google Scholar]
- 18. Duckworth B. C., Weaver J. S., Ruderman J. V. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16794–16799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y., Zhang Z., Xu X. Y., Li X. S., Yu M., Yu A. M., Zong Z. H., Yu B. Z. (2008) Dev. Dyn. 237, 3777–3786 [DOI] [PubMed] [Google Scholar]
- 20. Yu B., Wang Y., Liu Y., Liu Y., Li X., Wu D., Zong Z., Zhang J., Yu D. (2005) Dev. Dyn. 232, 98–105 [DOI] [PubMed] [Google Scholar]
- 21. Cui C., Zhao H., Zhang Z., Zong Z., Feng C., Zhang Y., Deng X., Xu X., Yu B. (2008) Biol. Reprod. 79, 991–998 [DOI] [PubMed] [Google Scholar]
- 22. Studier F. W. (2005) Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 23. Bouché J. P., Froment C., Dozier C., Esmenjaud-Mailhat C., Lemaire M., Monsarrat B., Burlet-Schiltz O., Ducommun B. (2008) J. Proteome Res. 7, 1264–1273 [DOI] [PubMed] [Google Scholar]
- 24. Li X., Gerber S. A., Rudner A. D., Beausoleil S. A., Haas W., Villén J., Elias J. E., Gygi S. P. (2007) J. Proteome Res. 6, 1190–1197 [DOI] [PubMed] [Google Scholar]
- 25. Jiang X., Han G., Feng S., Jiang X., Ye M., Yao X., Zou H. (2008) J. Proteome Res. 7, 1640–1649 [DOI] [PubMed] [Google Scholar]
- 26. Eng J. K., McCormack A. L., Yates J. R., 3rd (1994) J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 27. Hogan B., Costantini F., Lacy E. (1986) Manipulating the Mouse Embryo: A Laboratory Manual, pp. 249–256, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 28. Zhang Z., Su W. H., Feng C., Yu D. H., Cui C., Xu X. Y., Yu B. Z. (2007) Mol. Reprod. Dev. 74, 1247–1254 [DOI] [PubMed] [Google Scholar]
- 29. Gallicano G. I., McGaughey R. W., Capco D. G. (1997) Mol. Reprod. Dev. 46, 587–601 [DOI] [PubMed] [Google Scholar]
- 30. Schultz R. M., Montgomery R. R., Belanoff J. R. (1983) Dev. Biol. 97, 264–273 [DOI] [PubMed] [Google Scholar]
- 31. Chen J., Hudson E., Chi M. M., Chang A. S., Moley K. H., Hardie D. G., Downs S. M. (2006) Dev. Biol. 291, 227–238 [DOI] [PubMed] [Google Scholar]
- 32. Vitolo O. V., Sant'Angelo A., Costanzo V., Battaglia F., Arancio O., Shelanski M. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 13217–13221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamada K., Inoue H., Kida S., Masushige S., Nishiyama T., Mishima K., Saito I. (2006) Pathobiology 73, 1–7 [DOI] [PubMed] [Google Scholar]
- 34. Schultz R. (2009) Cell Cycle 8, 516–517 [PubMed] [Google Scholar]
- 35. Pirino G., Wescott M. P., Donovan P. J. (2009) Cell Cycle 8, 665–670 [DOI] [PubMed] [Google Scholar]
- 36. Lammer C., Wagerer S., Saffrich R., Mertens D., Ansorge W., Hoffmann I. (1998) J. Cell Sci. 111, 2445–2453 [DOI] [PubMed] [Google Scholar]
- 37. Lobjois V., Jullien D., Bouché J. P., Ducommun B. (2009) Biochim. Biophys. Acta 1793, 462–468 [DOI] [PubMed] [Google Scholar]
- 38. Solc P., Saskova A., Baran V., Kubelka M., Schultz R. M., Motlik J. (2008) Dev. Biol. 317, 260–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Woo E. S., Rice R. L., Lazo J. S. (1999) Oncogene 18, 2770–2776 [DOI] [PubMed] [Google Scholar]
- 40. Forrest A., Gabrielli B. (2001) Oncogene 20, 4393–4401 [DOI] [PubMed] [Google Scholar]
- 41. Giles N., Forrest A., Gabrielli B. (2003) J. Biol. Chem. 278, 28580–28587 [DOI] [PubMed] [Google Scholar]