

Abstract
The protein CrV2 is encoded by a polydnavirus integrated into the genome of the endoparasitoid Cotesia rubecula (Hymenoptera:Braconidae:Microgastrinae) and is expressed in host larvae with other gene products of the polydnavirus to allow successful development of the parasitoid. CrV2 expression has previously been associated with immune suppression, although the molecular basis for this was not known. Here, we have used time-resolved Förster resonance energy transfer (TR-FRET) to demonstrate high affinity binding of CrV2 to Gα subunits (but not the Gβγ dimer) of heterotrimeric G-proteins. Signals up to 5-fold above background were generated, and an apparent dissociation constant of 6.2 nm was calculated. Protease treatment abolished the TR-FRET signal, and the presence of unlabeled CrV2 or Gα proteins also reduced the TR-FRET signal. The activation state of the Gα subunit was altered with aluminum fluoride, and this decreased the affinity of the interaction with CrV2. It was also demonstrated that CrV2 preferentially bound to Drosophila Gαo compared with rat Gαi1. In addition, three CrV2 homologs were detected in sequences derived from polydnaviruses from Cotesia plutellae and Cotesia congregata (including the immune-related early expressed transcript, EP2). These data suggest a potential mode-of-action of immune suppressors not previously reported, which in addition to furthering our understanding of insect immunity may have practical benefits such as facilitating development of novel controls for pest insect species.
Keywords: G-proteins, Immunosuppressor, Innate Immunity, Insect, Viral Immunology, Cotesia rubecula Bracovirus, Immune Signaling, Insect Immunity, Pireis rapae, Polydnaviruses
Introduction
Polydnaviruses (PDVs)2 are endogenous particles that are produced by some parasitoid wasps and injected, along with the wasp egg(s), into the hemocoel of host insects causing a range of developmental and immune effects that allow the parasitoid to successfully develop (1–4). Members of the Polydnaviridae are divided into two paraphyletic groups as follows: bracoviruses (BVs; genus Bracovirus) and ichnoviruses (genus Ichnovirus), which occur in some members of the wasp families, Braconidae and Ichneumonidae, respectively (5). Recent evidence suggests that these genera should perhaps be placed into separate virus families, as there is good evidence that Bracovirus and Ichnovirus derived from different ancestral viruses (6, 7). PDVs are replicated only in the ovaries of female wasps and are not known to affect the wasp (8). PDVs are highly unusual in that individual particles only contain a subset of the expressed genes and do not contain any genes for virus structure/replication. Therefore, the virus is only able to be transmitted vertically (between wasp generations) through the integration of the PDV genome into wasp chromosomes (8–10). Indeed, it was the analysis of nonpackaged PDV genes (such as capsid proteins) that allowed the different ancestral viruses to be differentiated. Prior to this, a range of encapsidated PDV genes and/or gene families was postulated as being immune-suppressive, targeting cellular and humoral (cell-free) aspects of the innate insect immune system (4). Proteins encoded by these genes include protein-tyrosine phosphatases (11), viral ankyrins (vankyrins) (12, 13), host translation inhibitory factors (14, 15), EGF-like proteins (16), CrV1 homologs (17–19), EP1-like proteins (20), the H4 histone (21), and C-type lectins (22–25). Other PDV proteins are also thought to target insect immunity, but as yet their class and potential mode-of-action are yet to be elucidated. CrV2, expressed by the Cotesia rubecula bracovirus (CrBV), is an example of such a protein. CrV2 is expressed by CrBV only in the larvae of two closely related butterflies (Pieris spp.) and is secreted into the hemolymph. It was thought to be temporally associated with short term, subtle immune dysfunction in hemocyte cells that take up the secreted protein, although the molecular basis for its effects is not yet understood (26). The protein does not cause toxic effects on the cells that recover their immune function once significant expression of CrV2 (and other early expressed proteins) is reduced. Here, we present data that infer a specific, high affinity interaction between CrV2 and invertebrate/mammalian Gα subunits of heterotrimeric G-proteins, which are important cell-signaling proteins, indirectly implicated in vertebrate and invertebrate immune function (see under “Discussion” and Refs. 27–32).
Heterotrimeric G-proteins consist of three subunits, the Gα subunit and a dimer of Gβ and Gγ (Gβγ). There are a variety of subtypes for each G-protein class. The Gα subunit binds guanine nucleotides and exchanges GDP for GTP upon activation of the G-protein-coupled receptor (GPCR) with which the G-protein is usually associated. Nucleotide exchange causes conformation changes such that the Gα subunit and the Gβγ dimer interact with downstream effectors, including various enzymes and ion channels that alter cellular metabolism (33). We previously developed a TR-FRET (time-resolved Förster resonance energy transfer) assay utilizing a terbium-chelate donor and Alexa546 acceptor to demonstrate interactions of the Gα subunit with the Gβγ dimer and the regulator of G-protein signaling 4 (RGS4) (34). The assay was recently adapted by others to monitor the effect of allosteric modulators of RGS4 in altering RGS4:Gα affinity and further developed into a high throughput screen for other such modulators (35). Generally, TR-FRET uses a lanthanide donor fluorophore (such as terbium or europium) in combination with an appropriate acceptor (with an excitation spectrum overlapping the donor emission spectrum). When the fluorophores are in close proximity (<100 Å), energy transfer occurs between them, and acceptor emission is measured to determine binding kinetics (36). Using a lanthanide donor increases the signal:noise ratio when the long lived luminescence (characteristic of lanthanides) is exploited with time-gated measurements that eliminate short lived background signals. Lanthanides also exhibit other favorable properties, including multiple emission peaks, a large Stokes shift, and nonpolarized emission (37, 38).
As part of validating the original TR-FRET assay, we determined that the CrV2 protein appeared to specifically interact with mammalian Gα subunits. Here, we adapt our TR-FRET assay to demonstrate a high affinity (nanomolar) interaction of acceptor-labeled CrV2 with mammalian and insect donor-labeled Gα. We also demonstrate that recombinant CrV2 is taken up by a specific hemocyte morphotype from larval Pieris rapae, which are key immune cells, where it could potentially interact with cellular proteins such as Gα. These results are intriguing as they suggest that insect hemocyte immune function could be regulated through G-protein signaling pathways and that some PDVs subvert host immunity by producing proteins that interact with Gα to alter immune signaling. This mode-of-action for immune suppression has not previously been reported, and its elucidation has the potential to facilitate the development of novel insect controls.
EXPERIMENTAL PROCEDURES
All chemicals and reagents were of analytical grade and purchased from Sigma unless otherwise stated. All buffers were made in Milli-Q water.
Production and Purification of Recombinant CrV2 and G-protein Subunits
To produce CrV2 used in cell and far Western blot experiments, PCR was used to generate full CrV2 constructs containing the N-terminal signal peptide with a His tag incorporated at the 3′ end and NotI and KpnI restriction enzyme sites at the 5′ and 3′ ends, respectively. The forward primer sequence was 5′-gcg gcc gca tgt tgt cta caa agc-3′, and the reverse primer sequence was 5′-ggt acc tta gtg atg gtg atg gtg atg ggg atg atc tcg agc cct-3′. PCR products were produced with proofreading polymerase and cloned into pCR®-Blunt (Invitrogen) to allow subsequent restriction of the cloned products for insertion into pFastBac1 (Invitrogen). Recombinant baculovirus was then generated using the Bac-to-Bac system (Invitrogen) as per the manufacturer's instructions. Sf9 cells at 2 × 106 cells/ml were infected with amplified virus at a multiplicity of infection of ∼2 for 48–72 h in suspension at 27 °C with shaking. Cells were harvested by centrifugation, and expression was confirmed by Western blot. The media containing secreted CrV2 were stored at −20 °C or −80 °C for longer term storage.
To produce purified recombinant CrV2 for TR-FRET experiments, the coding sequence without the first 21 N-terminal amino acids, which contain the secretory signal peptide, was cloned into pQE30 (Qiagen) and transformed into M15[pREP4] Escherichia coli as per Glatz et al. (26). Expression of recombinant CrV2 was induced in 400-ml bacterial cultures in YT broth (8 g/liter tryptone, 5 g/liter yeast extract, 2.5 g/liter NaCl, pH 7.0) as per the manufacturer's instructions. Induced bacterial cells were pelleted by centrifugation and stored at −80 °C. Cells were later resuspended in 10 ml of TBP buffer (50 mm Tris, pH 8, 10 mm β-mercaptoethanol, 0.02 mg/ml phenylmethanesulfonyl fluoride, 0.03 mg/ml benzamidine). Lysozyme was added to a final concentration of 0.2 mg/ml and gently mixed at 4 °C for 30 min. MgCl2 was then added to a final concentration of 5 mm followed by DNase I to a concentration of 0.01 mg/ml. Mixing was continued for a further 30 min. Twenty percent (w/v) cholate solution (50 mm NaHEPES, pH 8.0, 3 mm MgCl2, 50 mm NaCl, and 200 g/liter cholic acid (Na+)) was added to give a final cholate concentration of 1% (v/v). The preparation was then gently stirred (1 h at 4 °C) before ultracentrifugation in a Beckman Coulter OptimaTM LE-80K at 100,000 × g for 40 min. Ni-NTA-agarose beads (800 μl; Qiagen) in TBP (50%) were added to the supernatant and incubated on ice for 30 min with occasional stirring. Supernatants were then applied to gravity-fed columns. Columns were then washed with 20 ml of TBP containing 100 mm NaCl followed by washing with 5 ml of TBP containing 100 mm NaCl and 10 mm imidazole. All washing procedures were carried out at 4 °C. His-tagged CrV2 was eluted from the column in 400-μl fractions using TBP containing 100 mm NaCl and 250 mm imidazole. Elution fractions were run on a polyacrylamide gel and stained using Coomassie Blue. Elutions containing CrV2 were identified and fractions pooled. Protein concentration was determined according to Bradford (39) or by laser densitometry, before aliquoting and storage at −80 °C.
G-protein subunits were produced from 1 to 2 liters of Sf9 cells infected with the desired recombinant baculovirus. Recombinant full-length Drosophila Gαo (isoform I)-expressing baculovirus was produced using the Bac-to-Bac expression system as described by the manufacturer (Invitrogen). Cell culture, virus amplifications, and infections, as well as the purification of G-protein subunits using Ni-NTA-agarose beads and fluorescent labeling of G-proteins or CrV2 with cysteine-reactive Alexa Fluor 546 C5-maleimide (Invitrogen) or CS124-DTPA-EMCH-Tb (Invitrogen), were performed as described by Leifert et al. (34).
Antibody Labeling of CrV2 in Hemocytes in Vitro
Larval P. rapae were surface-sterilized in ice-cold 70% ethanol for a few minutes and then kept on ice until they were bled from a severed proleg directly into an ice-chilled 1.5-ml microcentrifuge tube containing ∼200 μl of Sf900II medium (Invitrogen) saturated with phenylthiourea (which inhibits melanization reactions). Hemocytes were aliquoted into wells of a printed glass slide and incubated at room temperature until cells attached, and spreading was observed. Obviously, spread cells were considered to be a plasmatocyte morphotype and attached cells that retained a rounded, apparently unspread, conformation were considered as a granulocyte morphotype. Insect cell culture medium containing secreted baculovirus-expressed CrV2 was applied to the hemocyte-containing slides and incubated for the desired time. Medium was then removed, and cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 4 min, and washed in TBST (8.8 g/liter NaCl, 0.2 g/liter KCl, 3 g/liter Trizma (Tris base), 500 μl/liter Tween 20 detergent). Anti-CrV2 polyclonal rabbit antibodies (26) in TBST (1:500) were added to the glass slide and incubated for 1.5–2 h at room temperature. Cells were washed with TBST, and fluorphore-conjugated anti-rabbit antibody (1:200–250) with 1:50 dilution of FITC-phalloidin (0.1 mg/ml) in TBST were applied to slides for 1 h at room temperature in the dark. Cells were then washed with TBST and stained with 1:10,000 dilution (1 μg/ml) of DAPI for 5 min. Cells were then washed with TBST and PBS; a coverslip was applied, and the coverslip sealed with nail varnish.
TR-FRET Assays
The interaction between Alexa546 (Alexa)- and CS124-DTPA-EMCH-Tb (Tb)-labeled proteins was measured using TR-FRET as described in Leifert et al. (34). Briefly, these experiments were conducted in black 96-well plates. 20× working solutions of proteins were made in TMN buffer (50 mm Tris, pH 7.6, 100 mm NaCl, 10 mm MgCl2). Five microliters of each was then applied to opposite sides of the well such that mixing did not occur. Other indicated components such as proteinase K could also be added in this manner where required. TMN buffer was then added to give a final assay volume of 100 μl, and the reaction was initiated by mixing of all components. TR-FRET was then measured using a Victor3 multilabel plate reader (PerkinElmer Life Sciences) using an excitation wavelength of 340 nm and a delay of 50 μs, before measuring the emission at 572 nm for 900 μs. Where appropriate, measurements were ceased so that unlabeled proteins or buffer could be added and then measurements resumed.
[35S]GTPγS Binding Assay
40 nm purified Gα subunit or CrV2 was mixed with 1 nm [35S]GTPγS in a final volume of 100 μl of TMN buffer and incubated in a shaking water bath for 90 min at 27 °C. Twenty five microliters in triplicate were then filtered through glass microfiber 1-μm filter papers (GFCs; Filtech), and unbound [35S]GTPγS was removed by washing with three times with 4 ml of TMN buffer. The filters were then dried, and the amount of bound [35S]GTPγS was measured by scintillation counting for 60 s in Pico Pro VialsTM with 4 ml of Ultima GoldTM scintillation mixture (PerkinElmer Life Sciences) using a Wallac 1410 liquid scintillation counter.
Far Western Assay
Purified Drosophila Gαo and bovine serum albumin were subjected to SDS-PAGE on an “any kDa” TGX gel (Bio-Rad) using a nonreducing sample buffer. Proteins were transferred onto a Hybond-C nitrocellulose membrane (GE Healthcare) at 100 V for 1 h. Membranes were blocked with 5% skim milk powder in TBS buffer (8.8 g/liter NaCl, 0.2g/liter KCl, 3 g/liter Trizma) for >2 h. Membranes were then probed with CrV2 in Sf900II medium or conditioned medium overnight. Membranes were then washed with TBST buffer (see above) and probed with rabbit anti-CrV2 (1:10,000) in blocking solution for 2 h. Following washing with TBST, membranes were probed with alkaline phosphatase-conjugated goat anti-rabbit IgG (1:20,000) in blocking solution for 2 h. Membranes were washed with TBST and developed using 5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt and nitro blue tetrazolium chloride to detect any CrV2 bound to Gαo.
TR-FRET Data Analyses
Data were analyzed using PrismTM 4.00 (GraphPad software Inc., San Diego). Data are presented as mean ± S.E., where n is equal or greater than 3. Where n = 2, data are presented as the mean, and error bars represent the range of the duplicates. If error bars are not visible, they are small and therefore hidden by the symbols.
Amino Acid Sequence Analyses
The CrV2 amino acid sequence was subjected to protein Blast (blast.ncbi.nlm.nih.gov) to detect any similar protein sequences. Three similar sequences were detected. ClustalW2 software was then used to align the amino acid sequences of the four similar proteins and to produce a simple phylogenetic tree of the four protein sequences. Protein BLAST was also used to determine the amino acid identity between the two experimental Gα subunits used here, i.e. rat Gαi1 (GenBankTM accession number NP_037277) and Drosophila melanogaster Gαo isoform I (GenBankTM accession number AAS64873).
RESULTS
Identification of CrV2 Homologs
CrV2 amino acid sequence was subjected to protein Blast, resulting in detection of three similar amino acid sequences, which are putative homologs (Fig. 1). One sequence (EPL-7) is a hypothetical protein determined from the genome sequence of Cotesia plutellae bracovirus (CpBV). The other two proteins are derived from the Cotesia congregata bracovirus (CcBV); One of these encodes the early expressed immune suppression-related transcript, EP2 (40), and the second is a hypothetical protein CcBV_31.9 derived from the total CcBV genome sequence. ClustalW2 software was used to align the amino acid sequences of the four proteins.
FIGURE 1.

A CrV2 gene family exists in Cotesia-associated bracoviruses. ClustalW2 software was used to align CrV2 against three putative homologs detected via protein Blast. Amino acid numbers are presented at the end of each line of sequence. Proteins are as follows: EPL-7, hypothetical protein from CpBV genome sequence (GenBankTM accession number ABK63352), CrV2, CrV2 expressed by CrBV (GenBankTM accession number AY631272); EP2, EP2 expressed by CcBV (GenBankTM accession number AJ632305), CcBV_31.9, putative protein from CcBV genome sequence (GenBankTM accession number YP184881). The alignment is presented in order of increasing size of the first deletion common to three genes but is absent from hypothetical EPL-7 protein. There is another conserved deletion downstream of the first (relative to hypothetical EPL-7 protein) in the central region of the protein. The high level of conservation indicates that the four genes are homologous. Consensus symbols follow convention of ClustalW2 software: * = complete conservation; : = conserved substitution; . = semi-conserved substitutions.
Uptake of CrV2 by Hemocytes
Application of hemocytes to the wells of a glass slide results in adherent cells attaching to the slide surface. A subpopulation of cells was then observed to spread, although other cells remain more rounded. These cells are considered as plasmatocytes and granulocytes, respectively. CrV2 was only detected in association with the unspread, rounded cells following incubation of all hemocytes with CrV2 (Fig. 2). These results suggest that CrV2 is likely to modulate some function of granulocytes while not directly affecting plasmatocytes. However, apart from the uptake of CrV2, the effect on granulocytes produced no other phenotype observable under our conditions.
FIGURE 2.

Uptake of CrV2 by specific P. rapae hemocytes. Pieris larvae were bled into media saturated with phenylthiourea; the hemocytes were applied to the wells of a glass slide and allowed to adhere and spread. CrV2 in media was then applied to the cells and incubated for 45 min. A, merged image of bright field and red fluorescence from TRITC secondary antibody bound to anti-CrV2 shows CrV2 associated with the small round hemocytes. B, merged blue and red fluorescence images showing the nucleus (blue) stained with DAPI and CrV2 (red). C, merged blue, red, and green fluorescent image showing the nucleus (blue), CrV2 (red), and the cytoskeleton stained with FITC-conjugated phalloidin (green).
Interaction of CrV2 with Gαi1 Measured by TR-FRET
Recombinant bacterially expressed CrV2 was successfully purified using Ni-NTA chromatography, and the protein was labeled with the fluor Alexa546. G-protein subunits were expressed in Sf9 cells and also purified using Ni-NTA chromatography. These terbium-labeled (or unlabeled) G-proteins were functional in receiving signals from appropriate GPCRs in a reconstituted system as shown by Leifert et al. (34). Purified CrV2 labeled with Alexa546 (CrV2-Alexa) as the acceptor for TR-FRET, was mixed with purified Gαi1 labeled with CS124-DTPA-EMCH-Tb (Gαi1-Tb) as the fluorescent donor exhibiting long lived luminescence. Upon mixing of the two labeled proteins and excitation of terbium at 340 nm, an increase in gated emission of Alexa546 (acceptor) fluorescence at 572 nm occurred with time. Signals up to 5-fold greater than the background could be achieved and the signal increased upon addition of increasing amounts of CrV2-Alexa (Fig. 3A). Background fluorescence was generated by a low amount of Gαi1-Tb emission in the 572 nm channel, whereas background contributions from Alexa546 and other buffer components were negligible because typical autofluorescence had decayed during the 50-μs delay (after excitation) before the emission signal was measured. When increasing concentrations of CrV2-Alexa were added to 10 nm Gαi1-Tb, saturation was achieved at ∼ 25 nm CrV2-Alexa, and an apparent dissociation constant (Kd) of 6.2 nm was produced (Fig. 3B). This indicated that a high affinity interaction was occurring between CrV2-Alexa and Gαi1-Tb, which was comparable with the affinity between the Gα subunit and the Gβγ dimer (2 nm) measured using the same technique (34). To show the TR-FRET signal was attributable to a specific protein/protein interaction, proteinase K, a broad spectrum serine protease that cleaves peptide bonds at the carboxylic sides of aliphatic, aromatic, or hydrophobic amino acids, was used to digest the proteins. This reduced the TR-FRET signal (Fig. 3C) and suggested that generation of the TR-FRET signal was due to a specific interaction between complete CrV2 and Gαi1 proteins, which were destroyed by protease digestion.
FIGURE 3.

CrV2-Alexa association with Gαi1-Tb increases TR-FRET to saturation, and treatment with proteinase K abolishes TR-FRET. TR-FRET measurements were taken using a Victor3 plate reader set for time-resolved fluorescence with the following parameters: λex 340 nm, λem 572 nm, 50-μs delay. and 900-μs counting duration. A, 10 nm Gαi1-Tb (■) was mixed with 20 nm (▴), 40 nm (●), or 100 nm (♦) CrV2-Alexa. Data shown are mean (n = 2). B, 10 nm Gαi1-Tb was mixed with increasing concentrations (0–40 nm) of CrV2-Alexa, and TR-FRET was measured following 10 min of incubation. The background from 10 nm Gαi1-Tb has been deducted, and an apparent dissociation constant (Kd) of 6.2 nm was calculated. Data shown are mean ± S.E. (n = 3). C, 10 nm Gαi1-Tb was mixed with 20 nm CrV2-Alexa ± 0.1 mg/ml proteinase K. After an incubation period of 30 min at 37 °C, TR-FRET measurements were taken. Data shown are mean ± S.E. (n = 3) of the ratio of acceptor emission: donor emission.
The addition of an excess of unlabeled binding partners, including Gαi1 or CrV2 (Fig. 4, A and B, respectively), rapidly decreased the TR-FRET signal, although with the addition of buffer, the signal remained steady. This further showed that a specific interaction between the proteins was occurring (i.e. reduced TR-FRET was not the result of increased assay volume) resulting in a significant TR-FRET signal because unlabeled CrV2 or Gα appeared to compete for binding to labeled binding partners. Gβ4γ2 could also inhibit the association of Gα-Tb with CrV2-Alexa (Fig. 4C), and this was particularly interesting because assuming that CrV2 does not bind to Gβγ, this could suggest that Gβγ and CrV2 have overlapping binding sites on the Gαi1 subunit and/or disrupts the binding of the other. To gain an insight as to whether CrV2 could also be associating with the Gβγ dimer, CrV2-Alexa was mixed with Gβ4γ2 labeled with terbium (Gβγ-Tb). However, this failed to produce a substantial TR-FRET signal compared with that generated by CrV2 and Gαi1 or Gαi1 and Gβγ (Fig. 5). Although the labeling efficiency of the Gβγ subunits with terbium may differ from Gα, the similar level of background terbium luminescence suggested that the amount of terbium present was not significantly less and did not result in the lack of TR-FRET signal. This was further established by the strong TR-FRET signal generated from the interaction of Gβγ-Tb with Gαi1-Alexa. The activation state of the Gα subunit could also modulate the interaction with CrV2. The addition of excess GDP (or GTPγS; data not shown) produced similar association curves over time, whereas the addition of aluminum fluoride (AlF3) by adding 10 mm NaF and 30 μm AlCl3 appeared to decrease the maximum fluorescence achieved (Fig. 6).
FIGURE 4.

Unlabeled binding partners compete with Gαi1-Tb for binding to CrV2-Alexa. TR-FRET measurements were taken with the following parameters: λex 340 nm, λem 572 nm, 50-μs delay, and 900-μs counting duration. A, unlabeled Gαi1 competes for binding to CrV2-Alexa reducing the TR-FRET signal. B, unlabeled CrV2 competes for binding to Gαi1-Tb reducing the TR-FRET signal. A and B, 10 nm Gαi1-Tb was mixed with 20 nm CrV2-Alexa in 100 μl of TMN buffer. At 5 min, 2 μm of unlabeled Gαi1 or 70 nm of unlabeled CrV2 (■) or an equivalent volume of TMN buffer (▴) was added and TR-FRET measurements over the time period shown. Backgrounds of 10 nm Gαi1-Tb with buffer added at 5 min (●) and 10 nm Gαi1-Tb + the indicated unlabeled protein (♦) are shown. Data shown are mean (n = 2). C, Gβ4γ2 inhibits CrV2-Alexa association with Gαi1-Tb. 10 nm Gαi1-Tb was mixed with 20 nm CrV2-Alexa with or without 240 nm Gβ4γ2 in 100 μl of TMN buffer. Backgrounds of Gαi1-Tb and Gαi1-Tb + Gβ4γ2 have been deducted as appropriate. Data shown are mean (n = 2). a.u., arbitrary units.
FIGURE 5.
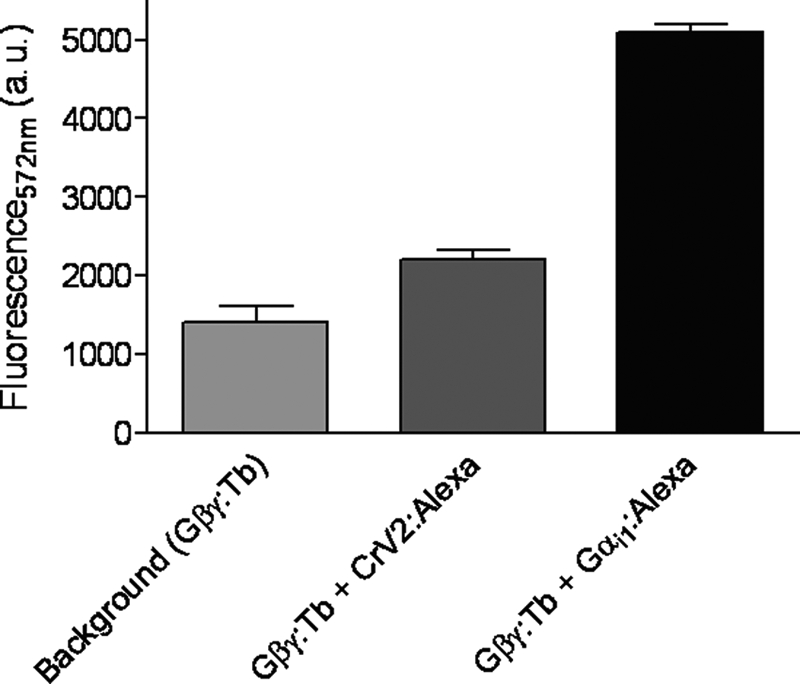
CrV2-Alexa interacts only minimally with Gβγ-Tb. 10 nm Gβγ-Tb was mixed with 20 nm CrV2-Alexa or 10 nm Gαi1-Alexa to a final volume of 100 μl with TMN buffer. TR-FRET measurements were taken with the following parameters: λex 340 nm, λem 572 nm, 50-μs delay, and 900-μs counting duration over the time period shown. Data shown are mean (n = 2).
FIGURE 6.

Alteration of the activation state of Gαi1 using aluminum fluoride decreases the interaction with CrV2. 10 nm Gαi1-Tb was mixed with 20 nm CrV2-Alexa with excess amounts of GDP (2.5 μm) or aluminum fluoride produced by the addition of NaF (10 mm) and AlCl3 (30 μm). TR-FRET measurements were immediately taken with the following parameters: λex 340 nm, λem 572 nm, 50-μs delay, and 900-μs counting duration. Background of Gαi1-Tb with GDP or aluminum fluoride has been deducted. Data shown are mean ± S.E. (n = 3). a.u., arbitrary units.
Because insect Gα subunits would likely be the target for CrV2 in vivo and show relatively little variation between different organisms and Gα subclasses, we used the TR-FRET assay to compare the CrV2/Gα interaction using Drosophila Gαo. Protein Blast showed that there is 69% amino acid identity between the two experimental Gα proteins, rat Gαi1 and Drosophila Gαo isoform I. Recombinant baculovirus-expressed Drosophila Gαo was purified (Fig. 7A) and shown to be similarly functional in binding [35S]GTPγS as rat Gαi1, whereas CrV2 did not bind significant amounts of [35S]GTPγS (Fig. 7B). This was a good indication that a functional Drosophila Gαo subunit had been expressed and purified from Sf9 cells. Fig. 7C shows that when increasing concentrations of purified unlabeled Gα subunits were added to the TR-FRET assay of CrV2/Gαi1 interactions, Drosophila Gαo competed for binding to CrV2-Alexa at lower concentrations than unlabeled mammalian Gαi1, with an IC50 of 41 nm compared with 241 nm. This indicated that Drosophila Gαo had a higher affinity for CrV2 than mammalian Gαi1 and was the preferred binding partner of the two tested subunits.
FIGURE 7.

Drosophila Gαo purified from Sf9 cells binds to [35S]GTPγS and competes for binding to CrV2. A, polyacrylamide gel showing eluted fractions of Drosophila Gαo from 1.7 liters of infected cells purified using Ni-NTA chromatography. B, 40 nm Gα or CrV2 was mixed with 1 nm [35S]GTPγS in a final volume of 100 μl of TMN buffer and incubated in a shaking water bath for 90 min at 27 °C. 25 μl was then filtered through GFC filters, and unbound [35S]GTPγS was removed by washing with TMN buffer. The amount of bound [35S]GTPγS was then measured by scintillation counting. Data shown are mean ± S.E. (n = 6) of filter triplicates of two experiments. C, CrV2-Alexa binds preferentially to Drosophila Gαo when 20 nm CrV2-Alexa was mixed with 20 nm mammalian Gαi1-Tb and doses (0–900 nm) of unlabeled Drosophila Gαo (■) or mammalian Gαi1 (▾) were added to compete with labeled proteins. TR-FRET measurements were taken with the following parameters: λex 340 nm, λem 572 nm, 50-μs delay, and 900-μs counting duration over the shown time period. Data shown are mean ± S.E. (n = 3). a.u., arbitrary units.
Interaction of CrV2 with Gαo Confirmed by Far Western Blot
To confirm the specific interaction observed by TR-FRET, Gαo was blotted onto a nitrocellulose membrane and was labeled by probing with recombinant CrV2, and then bound CrV2 was detected with anti-CrV2 (Fig. 8). A range of results producing no detection indicated the blotted Gαo was only detectable due to prior CrV2 binding, viz. Gα was not detected when CrV2 was absent or when anti-CrV2 was not applied. Furthermore, CrV2 did not bind to bovine serum albumin under the same conditions.
FIGURE 8.
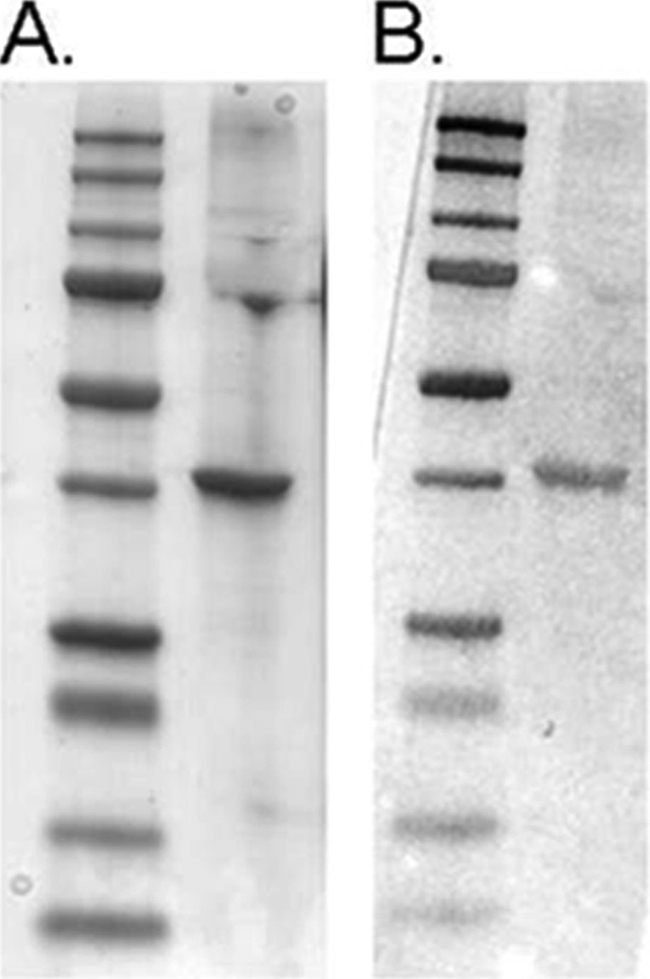
Far Western blot confirms Gαo interaction with CrV2. A, Coomassie-stained gel of Gαo (right lane) run by SDS-PAGE with molecular weight marker (Bio-Rad precision plus dual color protein standard) in left lane. B, duplicate samples of A transferred to nitrocellulose and probed with CrV2. CrV2 binding was detected using rabbit anti-CrV2 and alkaline phosphatase-conjugated secondary antibody. CrV2 bound to the band of Gαo but did not bind to BSA under the same conditions (data not shown). Gαo was not detected when probe CrV2 or anti-CrV2 was absent (data not shown).
DISCUSSION
CrV2 Homologs
The detection of CrV2 homologs in the genomic sequences of CcBV and CpBV indicates that there is a family of CrV2-related proteins that are expressed by Cotesia-associated PDVs (Fig. 1). Two of the proteins (EPL-7 and CcBV_31.9) are hypothetical, and their sequences were directly translated from total virus genome sequences. The third is the confirmed transcript EP2, which like CrV2 transcripts was detected soon after parasitization and is thought to be immune-suppressive (26, 40). Relative to the hypothetical EPL-7 protein, the other homologs have two conserved deletions occurring within the first 100 amino acids of the respective proteins.
Each of the four homologs have identifiable, highly conserved secretion signals (containing a high proportion of hydrophobic amino acids) at their N termini and are predicted to be glycosylated; secreted CrV2 is also detected in large amounts in the hemolymph of parasitized larvae (26, 40). It is therefore likely that like CrV2, all of the homologs would be secreted from infected cells. CrV2 protein was also detectable at 24 h post-parasitization (by antibody staining of hemocytes and Western analyses of tissues and serum from parasitized larvae). This is in contrast to the CrV2 transcript, which had been reduced so as to be undetectable by Northern hybridization as early as 12 h post-parasitization (26, 41). The large amount of hemocyte-related CrV2 protein at a time where high level expression is not detected and corresponding (transient) loss of hemocyte immune function, both suggested that these key immune cells are the target for CrV2 (26).
CrV2 was also shown to exist as a trimer in the hemolymph of infected larvae, and a C-terminal coiled-coil region (starting approximately at amino acid 270) was predicted to be responsible for the trimeric structure (26). This region is again well conserved, with a large percentage of “redundant” amino acid substitutions occurring between the homologs in this region, indicating that each protein forms polymeric structures in the hemolymph. Therefore, the key functional region is likely to lie within the highly conserved central parts of the amino acid sequence. This could be assessed by generating mutants with targeted amino acid substitutions in that region.
Specific Uptake of CrV2 by Granulocytes
Application of CrV2 to hemocytes showed that CrV2 was taken up from surrounding medium. Moreover, the recombinant protein was only associated with a specific type of hemocyte that once attached to the slide surface displayed a smaller, more rounded morphology, typical of granulocytes. This suggests that CrV2 targets a particular immune function carried out by granulocytes, which may not occur in plasmatocytes. Infection with PDVs has previously been observed to have effects on a single hemocyte type. Interestingly, Strand and Pech (42) reported that Microplitis demolitor bracovirus induced apoptosis specifically in granulocytes. However, to date our studies have not shown that the presence of CrV2 results in apoptosis, and apoptosis has not been observed in the Cotesia-Pieris system.
Specific in Vitro Interaction between CrV2 and Gα
In this study, we used an established in vitro TR-FRET assay (34, 35) to identify a novel interaction between the G-protein subunit Gα, and CrV2. We demonstrated that CrV2 binds to Gα subunits from rat and Drosophila, with nanomolar affinity (Kd = 6.2 nm). Far Western analysis also confirmed the presence of a specific interaction with Drosophila Gαo. There are relatively few proven/proposed Gα-binding proteins, and these proteins generally have affinities in the nanomolar range. For example, the TR-FRET assay used here was previously applied to calculate Kd values of 2.4 and 14.6 nm for the interaction of Gαi1 with Gβγ and RGS4, respectively (34). Other assays have produced comparable results, the mammalian Gβγ/Gαi1 interaction was shown to have a Kd of 3 nm using flow cytometry (43). A regulator of G-protein signaling, RGS4, was shown to interact with GTP-activated Gq protein with a Kd of <1 nm (44). Ozaki et al. (31) used surface plasmon resonance on immobilized bovine G-protein to calculate Kd values for the invertebrate proteins tachyplesin and mastoparan (both discussed below) of 880 and 220 nm, respectively. Therefore, the high affinity of the CrV2/Gα interaction (Kd = 6.2 nm for rat Gαi1) is highly unlikely to occur through chance alone and suggests that the interaction between Gα and CrV2 could also occur in vivo as part of immune suppression by CrBV.
A specific interaction is also supported by the finding that the CrV2/Gα interaction can be modulated by AlF3, which changes the conformation of the Gα subunit to mimic the “transition state” (45, 46). This is similar to the association of Gα with Gβγ, whereby AlF3 causes dissociation of the subunits and a decrease in TR-FRET signal and is in contrast to the result seen with RGS4 where an increase in TR-FRET signal with Gα was achieved in the presence of AlF3 (34). The decrease in signal generated by the interaction of Gα with CrV2, upon the addition of AlF3, could be a result of the inability of CrV2 to bind to Gα subunits in the transition state or, alternatively, a conformational change in the bound CrV2/Gα complex resulting in the donor and acceptor labels being moved further apart and decreasing FRET efficiency (resulting in a lower signal). The fact that AlF3 does not return the signal to background level could indicate that conformational change or a decrease in affinity is more likely than a complete loss of interaction. The physiological importance of these data is unclear because although commonly employed for this purpose within published literature, the presence of AlF3 “mimics” an in vivo Gα activation state; actual AlF3-activated Gα proteins do not occur in vivo.
It also appears that the binding site of CrV2 could overlap with that of Gβγ because Gβγ can compete with CrV2 for binding to Gα. This may also in part explain the effect of AlF3 in decreasing the interaction between CrV2 and Gα because AlF3 changes the conformation of Gα in switch regions known to be important for Gβγ binding (45, 46). The fact that CrV2 binds to Gα subunits from rat and Drosophila is not particularly surprising as the two experimental proteins showed a high level of amino acid identity (69%). Analyses of partial Gα amino acid sequences from lepidopteran cell lines have shown that Gαq and Gαi subtypes identical between the tested lepidopteran species were between 88 and 98% identical to other invertebrates and 88 and 90% identical to known mammalian subunits (47). Similarly to CrV2, invertebrate proteins mastoparan (from Vespa spp. wasps) and tachyplesin (from the horseshoe crab, Tachypleus tridentatus) are also known to bind to mammalian G-proteins even though they interact with homologous invertebrate proteins in vivo (31, 48). CrV2 had a higher affinity for the Drosophila protein (Fig. 6C), which could be due to an evolved preference for Gαo rather than Gαi1 or the invertebrate origin of the protein rather than the mammalian. Regardless of the reasons for CrV2 interaction with mammalian Gα and the preference for the tested Drosophila subunit, these data nevertheless suggest that there is physiological importance in the CrV2 interaction with Gα subunits in insects. However, the specific metabolic consequences of the interaction between CrV2 and Gαi1 require further investigation.
G-protein Relationship with Invertebrate Immunity
Why would CrV2 target Gα? Most studies on G-protein-coupled receptors, G-proteins, and their associated signal transduction pathways have been conducted in mammalian organisms. In mammals, G-proteins regulate various effectors, including enzymes and ion channels to mediate processes in most bodily systems, including the nervous, circulatory, and immune systems (49). G-proteins are also known to be important signaling molecules in invertebrates, but as yet, the specific immune-associated biochemical pathways and protein interactions have not been elucidated. However, there is a range of evidence that has indirectly linked invertebrate immunity and G-protein signaling. For example, in the mollusc Mytilus galloprovinicialis, corticotrophin-releasing hormone GPCR subtypes are involved with mediating cell shape changes in immunocytes through G-protein pathways (50). Exocytotic responses that release defense-related molecules from the intracellular stores of hemocytes are an important part of the immune response of invertebrates in detection of pathogens. The invertebrate Styela plicata induces the release of such molecules through a G-protein pathway where pharmacological reagents, known to inhibit G-proteins and tubulin microtubule assembly, decreased the release of C3-like proteins (51). Exocytosis has also been shown to be regulated by G-protein subunits, including Gαi, Gαo, and Gβγ subunits (52, 53), and immune pathways are induced by activating adenylate cyclases to produce cyclic AMP (or phospholipases to produce inositol 1,4,5-trisphosphate and diacylglycerol) and cause release of divalent cations to activate protein kinases (27, 32), all of which are classical downstream effects of G-proteins. A specific example is data showing that cAMP levels are related to the immune responses of hemocytes in larval lepidopteran insects (29).
There are also other examples of proteins from invertebrates that interact with G-proteins to apparently modulate immune responses. Tachyplesin (discussed above) is a major granular component of hemocytes of the horseshoe crab and is an antimicrobial peptide with broad spectrum activity against both Gram-positive and -negative bacteria (30). Moreover, tachyplesin has been found to induce hemocyte exocytosis in a positive feedback mechanism to amplify the immune response to an infection through a G-protein signaling pathway and was also demonstrated to interact directly with a bovine G-protein with a Kd of 0.88 μm (31, 54), which is relatively low in affinity compared with the CrV2 interactions we propose here. A similar process is likely to occur in other invertebrates, but as yet, tachyplesin homologs have not been discovered. Tachyplesin shares a number of structural similarities with the wasp venom protein, mastoparan (see above), that has also been found to regulate cellular responses by inducing exocytosis of substances from mammalian cells such as histamine from rat mast cells, serotonin from platelets, catecholamines from chromaffin cells and prolactin from the anterior pituitary (48). As mentioned previously, mastoparan was shown to directly interact with bovine G-proteins (31) and also to increase the GTPase activity and rate of nucleotide exchange to purified bovine Gαo independently of a GPCR (48). Interestingly, mastoparan was also associated with G-protein dysfunction leading to decreased expression of genes associated with LPS-induced (i.e. bacteria-induced) responses of mammalian endothelial and immune cells (28). This is interesting as it represents another example of a wasp-derived protein that interacts with G-proteins from a wide variety of organisms and is associated indirectly with immune regulation.
Concluding Remarks
Here, we have provided evidence that CrV2 accumulates in a specific type of hemocyte where it would be expected to modulate immune functions. The existence of CrV2 homologs also suggests that CrV2 has an important role in modulating host physiology that is conserved among a number of braconid species. We have also shown an in vitro interaction at physiologically relevant concentrations of CrV2 with Gα subunits. Although there is indirect evidence that G-proteins are involved in cellular immune cascades in invertebrates, there have not been reports of potentially immune-suppressive proteins that interact with G-proteins or their signaling cascades. We therefore propose that CrV2 provides further evidence of the involvement of Gα in insect immune signaling and that the interaction between CrV2 and Gα could represent a novel mode-of-action for immune suppression by PDVs, which are known to express a range of immune-suppressive proteins in parasitized insects (1, 3).
Given this combined evidence for Gα involvement in the immune response of invertebrates, and the data we present here showing nanomolar affinity between the proteins, we hypothesize that CrV2 targets Gα subunits in specific immune cells (granulocytes) to disrupt Gα-associated immune signaling by these cells. This would likely produce an abrogation of normal immune processes such as exocytosis. Further work establishing the interaction in vivo and elucidation of the novel CrV2 mode-of-action will likely be useful in understanding the molecular signaling mechanisms in invertebrate immunology. Such knowledge will be useful for designing nontoxic, immune-related compounds for control of pest insects.
Acknowledgments
We thank Prof. Rick Neubig (University of Michigan) for the provision of His-Gαi1, His-Gγ2, Gαi1, and Gγ2 baculoviruses and Prof. James Garrison (University of Virginia) for the provision of Gβ4/Gβ1 baculovirus stocks.
This work was supported by the OzNano2Life research initiative of the Australian Department of Education, Science, and Technology, CSIRO Emerging Science Area for Nanotechnology funding scheme, Australian Research Council Grant F09943013, a University of Queensland grant (to S. A.), and a Horticulture Australia Ltd. Grant VG08048 (to R. G.).
- PDV
- polydnavirus
- TR-FRET
- time-resolved FRET
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- BV
- bracovirus
- Ni-NTA
- nickel-nitrilotriacetic acid
- GPCR
- G-protein-coupled receptor
- TRITC
- tetramethylrhodamine isothiocyanate.
REFERENCES
- 1. Webb B. A., Strand M. R. (2005) In Comprehensive Molecular Insect Science (Gilbert L. I., Latrou K., Gill S. S. eds) pp. 260–323, Elsevier, San Diego [Google Scholar]
- 2. Beckage N. E., Gelman D. B. (2004) Annu. Rev. Entomol. 49, 299–330 [DOI] [PubMed] [Google Scholar]
- 3. Kroemer J. A., Webb B. A. (2004) Annu. Rev. Entomol. 49, 431–456 [DOI] [PubMed] [Google Scholar]
- 4. Glatz R. V., Asgari S., Schmidt O. (2004) Trends Microbiol. 12, 545–554 [DOI] [PubMed] [Google Scholar]
- 5. Webb B. A., Beckage N. E., Hayakawa Y., Lanzrein B., Stoltz D. B., Strand M. R., Summers M. D. (2005) in 8th Report of the International Committee on Taxonomy of Viruses (Fauquet C. M., Mayo M. A., Maniloff J., Desselberger U., Ball L. A. eds) pp. 255–267, Elsevier/Academic Press, San Diego [Google Scholar]
- 6. Bézier A., Annaheim M., Herbinière J., Wetterwald C., Gyapay G., Bernard-Samain S., Wincker P., Roditi I., Heller M., Belghazi M., Pfister-Wilhem R., Periquet G., Dupuy C., Huguet E., Volkoff A. N., Lanzrein B., Drezen J. M. (2009) Science 323, 926–930 [DOI] [PubMed] [Google Scholar]
- 7. Volkoff A. N., Jouan V., Urbach S., Samain S., Bergoin M., Wincker P., Demettre E., Cousserans F., Provost B., Coulibaly F., Legeai F., Béliveau C., Cusson M., Gyapay G., Drezen J. M. (2010) PLoS Pathog. 6, e1000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beckage N. E. (1997) Bioscience 48, 305–311 [Google Scholar]
- 9. Stoltz D. B. (1990) J. Gen. Virol. 71, 1051–1056 [DOI] [PubMed] [Google Scholar]
- 10. Stoltz D. B. (1993) in Parasites and Pathogens of Insects (Beckage N. E., Thompson S. N., Federici B. A. eds) pp. 80–101, Academic Press, New York [Google Scholar]
- 11. Ibrahim A. M., Kim Y. (2008) Naturwissenschaften 95, 25–32 [DOI] [PubMed] [Google Scholar]
- 12. Kroemer J. A., Webb B. A. (2006) J. Virol. 80, 12219–12228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi M., Chen Y. F., Huang F., Liu P. C., Zhou X. P., Chen X. X. (2008) Virology 375, 374–382 [DOI] [PubMed] [Google Scholar]
- 14. Barandoc K. P., Kim Y. (2009) Comp. Biochem. Physiol. Part D Genomics Proteomics 4, 218–226 [DOI] [PubMed] [Google Scholar]
- 15. Nalini M., Kim Y. (2007) J. Insect Physiol. 53, 1283–1292 [DOI] [PubMed] [Google Scholar]
- 16. Beck M. H., Strand M. R. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 19267–19272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Asgari S., Schmidt O., Theopold U. (1997) J. Gen. Virol. 78, 3061–3070 [DOI] [PubMed] [Google Scholar]
- 18. Gitau C. W., Gundersen-Rindal D., Pedroni M., Mbugi P. J., Dupas S. (2007) J. Insect Physiol. 53, 676–684 [DOI] [PubMed] [Google Scholar]
- 19. Labropoulou V., Douris V., Stefanou D., Magrioti C., Swevers L., Iatrou K. (2008) Cell. Microbiol. 10, 2118–2128 [DOI] [PubMed] [Google Scholar]
- 20. Kwon B., Kim Y. (2008) Dev. Comp. Immunol. 32, 932–942 [DOI] [PubMed] [Google Scholar]
- 21. Gad W., Kim Y. (2008) J. Gen. Virol. 89, 931–938 [DOI] [PubMed] [Google Scholar]
- 22. Glatz R., Schmidt O., Asgari S. (2003) J. Biol. Chem. 278, 19743–19750 [DOI] [PubMed] [Google Scholar]
- 23. Lee S., Nalini M., Kim Y. (2008) Comp. Biochem. Physiol. A Mol. Integr. Physiol. 149, 351–361 [DOI] [PubMed] [Google Scholar]
- 24. Nalini M., Choi J. Y., Je Y. H., Hwang I., Kim Y. (2008) J. Insect Physiol. 54, 1125–1131 [DOI] [PubMed] [Google Scholar]
- 25. Teramato T., Tanaka T. (2003) J. Insect Physiol. 49, 463–471 [DOI] [PubMed] [Google Scholar]
- 26. Glatz R., Schmidt O., Asgari S. (2004) J. Gen. Virol. 85, 2873–2882 [DOI] [PubMed] [Google Scholar]
- 27. Cytryñska M., Zdybicka-Barabas A., Jakubowicz T. (2006) J. Insect Physiol. 52, 744–753 [DOI] [PubMed] [Google Scholar]
- 28. Lentschat A., Karahashi H., Michelsen K. S., Thomas L. S., Zhang W., Vogel S. N., Arditi M. (2005) J. Immunol. 174, 4252–4261 [DOI] [PubMed] [Google Scholar]
- 29. Marin D., Dunphy G. B., Mandato C. A. (2005) J. Insect Physiol. 51, 575–586 [DOI] [PubMed] [Google Scholar]
- 30. Nakamura T., Furunaka H., Miyata T., Tokunaga F., Muta T., Iwanaga S., Niwa M., Takao T., Shimonishi Y. (1988) J. Biol. Chem. 263, 16709–16713 [PubMed] [Google Scholar]
- 31. Ozaki A., Ariki S., Kawabata S. (2005) FEBS J. 272, 3863–3871 [DOI] [PubMed] [Google Scholar]
- 32. Solon E., Gupta A. P., Gaugler R. (1996) Dev. Comp. Immunol. 20, 307–321 [DOI] [PubMed] [Google Scholar]
- 33. Oldham W. M., Hamm H. E. (2008) Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 34. Leifert W. R., Bailey K., Cooper T. H., Aloia A. L., Glatz R. V., McMurchie E. J. (2006) Anal. Biochem. 355, 201–212 [DOI] [PubMed] [Google Scholar]
- 35. Blazer L. L., Roman D. L., Chung A., Larsen M. J., Greedy B. M., Husbands S. M., Neubig R. R. (2010) Mol. Pharmacol. 78, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Selvin P. R. (2002) Annu. Rev. Biophys. Biomol. Struct. 31, 275–302 [DOI] [PubMed] [Google Scholar]
- 37. Nishioka T., Yuan J., Matsumoto K. (2007) in BioMEMS and Biomedical Nanotechnology (Ferrari M., Ozkan M., Heller M. J. eds) Vol. 3, pp. 437–446, Springer-Verlag Inc., New York [Google Scholar]
- 38. Roda A., Guardigli M., Michelini E., Mirasoli M. (2009) Anal. Bioanal. Chem. 393, 109–123 [DOI] [PubMed] [Google Scholar]
- 39. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 40. Harwood S. H., Beckage N. E. (1994) Insect Biochem. Mol. Biol. 24, 685–698 [Google Scholar]
- 41. Asgari S., Hellers M., Schmidt O. (1996) J. Gen. Virol. 77, 2653–2662 [DOI] [PubMed] [Google Scholar]
- 42. Strand M. R., Pech L. L. (1995) J. Gen. Virol. 76, 283–291 [DOI] [PubMed] [Google Scholar]
- 43. Sarvazyan N. A., Remmers A. E., Neubig R. R. (1998) J. Biol. Chem. 273, 7934–7940 [DOI] [PubMed] [Google Scholar]
- 44. Dowal L., Elliott J., Popov S., Wilkie T. M., Scarlata S. (2001) Biochemistry 40, 414–421 [DOI] [PubMed] [Google Scholar]
- 45. Wall M. A., Posner B. A., Sprang S. R. (1998) Structure 6, 1169–1183 [DOI] [PubMed] [Google Scholar]
- 46. Sondek J., Lambright D. G., Noel J. P., Hamm H. E., Sigler P. B. (1994) Nature 372, 276–279 [DOI] [PubMed] [Google Scholar]
- 47. Knight P. J., Grigliatti T. A. (2004) Arch. Insect Biochem. Physiol. 57, 142–150 [DOI] [PubMed] [Google Scholar]
- 48. Higashijima T., Uzu S., Nakajima T., Ross E. M. (1988) J. Biol. Chem. 263, 6491–6494 [PubMed] [Google Scholar]
- 49. Offermanns S. (2003) Prog. Biophys. Mol. Biol. 83, 101–130 [DOI] [PubMed] [Google Scholar]
- 50. Malagoli D., Franchini A., Ottaviani E. (2000) Peptides 21, 175–182 [DOI] [PubMed] [Google Scholar]
- 51. Raftos D. A., Fabbro M., Nair S. V. (2004) Dev. Comp. Immunol. 28, 181–190 [DOI] [PubMed] [Google Scholar]
- 52. Lang J., Nishimoto I., Okamoto T., Regazzi R., Kiraly C., Weller U., Wollheim C. B. (1995) EMBO J. 14, 3635–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blackmer T., Larsen E. C., Bartleson C., Kowalchyk J. A., Yoon E. J., Preininger A. M., Alford S., Hamm H. E., Martin T. F. (2005) Nat. Neurosci. 8, 421–425 [DOI] [PubMed] [Google Scholar]
- 54. Kurata S., Ariki S., Kawabata S. (2006) Immunobiology 211, 237–249 [DOI] [PubMed] [Google Scholar]