

Abstract
We previously showed that thrombin induces interleukin (IL)-8/CXCL8 expression via the protein kinase C (PKC)α/c-Src-dependent IκB kinase α/β (IKKα/β)/NF-κB signaling pathway in human lung epithelial cells. In this study, we further investigated the roles of Rac1, phosphoinositide 3-kinase (PI3K), and Akt in thrombin-induced NF-κB activation and IL-8/CXCL8 expression. Thrombin-induced IL-8/CXCL8 release and IL-8/CXCL8-luciferase activity were attenuated by a PI3K inhibitor (LY294002), an Akt inhibitor (1-l-6-hydroxymethyl-chiro-inositol-2-((R)-2-O-methyl-3-O-octadecylcarbonate)), and the dominant negative mutants of Rac1 (RacN17) and Akt (AktDN). Treatment of cells with thrombin caused activation of Rac and Akt. The thrombin-induced increase in Akt activation was inhibited by RacN17 and LY294002. Stimulation of cells with thrombin resulted in increases in IKKα/β activation and κB-luciferase activity; these effects were inhibited by RacN17, LY294002, an Akt inhibitor, and AktDN. Treatment of cells with thrombin induced Gβγ, p85α, and Rac1 complex formation in a time-dependent manner. These results imply that thrombin activates the Rac1/PI3K/Akt pathway through formation of the Gβγ, Rac1, and p85α complex to induce IKKα/β activation, NF-κB transactivation, and IL-8/CXCL8 expression in human lung epithelial cells.
Keywords: Chemokines, Epithelial Cell, G Protein-coupled Receptors (GPCR), Inflammation, Lung, NF-kappa B, Phosphatidylinositol 3-Kinase, Signal Transduction
Introduction
Thrombin is a procoagulant serine protease, is released during initiation of intravascular coagulation by tissue injury, and is well known for its pivotal role in the coagulation cascade, but it also promotes a wide range of cellular responses including inflammatory and immune responses (1). Accumulating evidence reveals that thrombin exerts its action through proteolytic activation of its receptors, named protease-activated receptors (PARs).2 To date, four PARs have been identified, and each receptor was shown to modulate a variety of pathophysiological processes such as chemokine release (2, 3). PAR1, PAR3, and PAR4 are activated by thrombin, whereas PAR2 is activated by trypsin and cofactor Xa (4). Previous reports showed that thrombin plays important roles in lung inflammation, such as inducing the accumulation of neutrophils in the airway and elevating tumor necrosis factor (TNF)-α, MIP-2, and keratinocyte-derived chemokine levels in bronchoalveolar lavage fluid of thrombin-treated mice (2, 5). Moreover, thrombin, acting through PAR1 and PAR4, can induce the release of proinflammatory mediators such as prostaglandin E2, interleukin (IL)-6, and IL-8/CXCL8 in lung epithelial cells (2, 6). Thus, thrombin appears to be a common feature of a variety of inflammatory lung diseases and plays pivotal roles in lung inflammation.
The CXC chemokine IL-8/CXCL8 is a potent chemoattractant and controls neutrophil chemotaxis toward sites of infection and induced airway inflammation (7, 8). IL-8/CXCL8 is produced by a wide array of cell types, including lung epithelial cells in response to various stimuli (2, 6). IL-8/CXCL8 is abundant in sputum from patients with chronic obstructive pulmonary disease, chronic bronchitis, bronchiectasis, and cystic fibrosis and in bronchoalveolar lavage fluid from patients with diffuse panbronchiolitis (8–10), and it is related to the neutrophil chemoattractant activity exhibited by these diseases. IL-8/CXCL8 expression is primarily regulated by activation of activator protein (AP)-1, nuclear factor (NF)-IL-6, and NF-κB transcription factors (11). Interestingly, NF-κB appears to be essential for the induction of IL-8/CXCL8 transcription (12), whereas AP-1 and NF-IL-6 are required for maximal gene expression (13). It was reported that thrombin coupling of PAR1 and PAR4 in different cell types is required for NF-κB activation and gene expression (2, 4, 14). A previous study demonstrated that thrombin acting through PAR1 induces NF-κB activation and intercellular adhesion molecule (ICAM)-1 expression by engaging the Gαq/protein kinase C (PKC)δ/Akt and Gβγ/phosphoinositide 3-kinase (PI3K)/Akt signaling pathways (14). Moreover, several studies demonstrated that thrombin induces NF-κB activation and protein expression through multiple signaling pathways such as PKCα/c-Src, Rac1, extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), c-Jun N-terminal kinase (JNK), IκB kinases α/β (IKKα/β), and PI3K/Akt (2, 14–16). Our previous study showed that thrombin-induced NF-κB activation and IL-8/CXCL8 expression are mediated through the PAR1- and PAR4-dependent phosphoinositide (PI)-phospholipase C (PLC)/PKCα/c-Src and IKKα/β signaling pathways (2), but little information is available about the roles of Rac1 and PI3K/Akt in regulating NF-κB activation and IL-8/CXCL8 expression following thrombin stimulation in human lung epithelial cells.
Rac, a member of the Rho family of small GTPases, is divided into three major classes: Rac1, Rac2, and Rac3 (17). Rac1 acts as a molecular switch to mediate multiple cellular mechanisms such as actin cytoskeletal organization and transcriptional regulation of genes (18). A previous report showed that Rac can be activated by Gβγ subunits of the G protein (19). Moreover, Rac was reported to induce activation of NF-κB by stimulation with NF-κB-inducing kinase (NIK), IKKβ, and phosphorylation of the IκB signal pathway, resulting in transcription of multiple genes (20–22). More recently, Diebold et al. (23) reported that thrombin mediates pulmonary artery smooth muscle cell proliferation and plasminogen activator inhibitor-1 expression through activation of Rac1-dependent NF-κB activation signaling pathways.
PI3K is a family of lipid kinases involved in mitogenic signal transduction and gene expression (24). PI3K can be activated by either receptor tyrosine kinase or a G protein-coupled receptor. Once activated, PI3K generates phosphatidyinositol-3,4,5-triphosphate, leading to the recruitment and activation of Akt (25). Akt, a serine/threonine kinase, acts as a direct downstream effector of PI3Ks and is activated by multiple intracellular signaling pathways (26, 27). The PI3K/Akt pathway plays a critical role in Ras-mediated cellular transformation, adhesion, and survival (28). Moreover, Akt can stimulate signaling pathways that mediate NF-κB activity in Jurkat T-cells (29). Akt also activates NF-κB through phosphorylation of IKKs, which, in turn, phosphorylate IκB, allowing NF-κB to be translocated to the nucleus and induce gene expression (30). It was reported that activation of PI3K and Akt mediated NF-κB activation and gene expression in human lung epithelial cells (31). We previously reported that thrombin induced NF-κB activation and IL-8/CXCL8 expression via PAR1- and PAR4-dependent PI-PLC/PKCα/c-Src/IKKα/β signaling pathways (2). In this connection, we propose another signaling pathway of Rac1-dependent PI3K/Akt, which is associated with NF-κB activation and IL-8/CXCL8 expression. In this study, we found that thrombin might activate the Rac1/PI3K/Akt pathway through formation of a Rac1, Gβγ, and p85α complex to induce IKKα/β activation and NF-κB transactivation, ultimately causing IL-8/CXCL8 expression and release in human lung epithelial cells.
EXPERIMENTAL PROCEDURES
Materials
Thrombin (from bovine plasma) and enolase were purchased from Sigma. SFLLRN-NH2 (a PAR1 agonist peptide), TFRGAP-NH2 (a PAR3 agonist peptide), and GYPGQV-NH2 (a PAR4 agonist peptide) were purchased from Bachem (Merseyside, UK). LY294002, PD98059, SB203580, and Ro-32-0432 were obtained from Calbiochem. The Akt inhibitor (1-l-6-hydroxymethyl-chiro-inositol-2-((R)-2-O-methyl-3-O-octadecylcarbonate)) was purchased from Enzo Life Sciences (Lausen, Switzerland). The pure histone H2B from calf thymus was purchased from Roche Applied Science (Mannheim, Germany). SP600125 was purchased from Tocris Bioscience (Bristol, UK). Lung A549 epithelial cells were obtained from American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle's medium (DMEM)/Ham's F-12, fetal calf serum (FCS), and penicillin/streptomycin were purchased from Invitrogen. An antibody specific for α-tubulin was purchased from BD Transduction Laboratories. The IL-8/CXCL8 enzyme-linked immunosorbent assay (ELISA) kit was obtained from R&D Systems (Minneapolis, MN). Protein A/G beads, antibodies specific for IKKα/β, Akt, Rac1, Gβ, Gγ, and p85α, as well as anti-mouse and anti-rabbit IgG-conjugated horseradish peroxidase (HRP) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies specific for phospho-Akt (Ser-473) and phospho-IKKα (Ser-180)/IKKβ (Ser-181) were purchased from New England Biolabs (Beverly, MA). Anti-mouse and anti-rabbit immunoglobulin G (IgG)-conjugated alkaline phosphatases were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA). IL-8/CXCL8 wild-type (-133)-luciferase (IL-8/CXCL8 WT-Luc) was kindly provided by Dr. Naofumi Mukaida (Kanazawa University, Kanazawa, Japan). The pcDNA was kindly provided by Dr. M.-C. Chen (Taipei Medical University, Taipei, Taiwan). pGL2-ELAM-Luc (which is under the control of an NF-κB-binding site) and pBK-CMV-LacZ were kindly provided by Dr. W.-W. Lin (National Taiwan University, Taipei, Taiwan). Rac1 expression construct sequences carrying the T17N (dominant negative, RacN17) mutation and a Rac activity assay kit were purchased from Upstate Biotech Millipore (Lake Placid, NY). [γ-32P]ATP (6,000 Ci/mmol) was purchased from Amersham Biosciences (Buckinghamshire, UK). The Myc-His-tagged expression construct for the dominant negative Akt1-K179M mutant (AktDN) was a kind gift from Prof. C.-M. Teng (National Taiwan University, Taipei, Taiwan). All materials for SDS-PAGE were purchased from Bio-Rad. All other chemicals were obtained from Sigma.
Cell Culture
A549 cells were cultured in DMEM/Ham's F-12 nutrient mixture containing 10% FCS, 100 units/ml penicillin G, and 100 μg/ml streptomycin in a humidified 37 °C incubator. After reaching confluence, cells were seeded onto 6-cm dishes for immunoblotting or the kinase assays, onto 12-well plates for cell transfection and the κB-luciferase and IL-8/CXCL8-luciferase assays, and onto 24-well plates for the IL-8/CXCL8 assays.
IL-8/CXCL8 Measurement
A549 cells (1 × 105 cells/well) were seeded onto 24-well plates in DMEM/Ham's F-12 with 10% FCS overnight. The next day, the growth medium was removed and replaced with 0.5 ml of basal medium devoid of FCS before drug treatment. IL-8/CXCL8 released into the culture medium after thrombin (10 units/ml) treatment or after being pretreated with specific inhibitors as indicated followed by thrombin stimulation was assayed using the IL-8/CXCL8 ELISA kit following the manufacturer's instructions. After finishing, cells were detached, and cell numbers were determined by a trypan blue assay.
Transfection and Luciferase Assays
A549 cells (2 × 105 cells/well) were seeded onto 12-well plates, and cells were transfected the following day using Lipofectamine PlusTM with either 0.5 μg of pGL2-ELAM-Luc or 0.2 μg of IL-8/CXCL8 WT-Luc and 0.5 μg of pBK-CMV-LacZ. After 24 h, the medium was aspirated and replaced with fresh DMEM/Ham's F12 containing 10% FBS and then stimulated with thrombin (10 units/ml) for another 24 h before being harvested. To assess the effects of the indicated inhibitors, drugs were added to cells 20 min before thrombin addition. To assay the effect of RacN17, cells were cotransfected with RacN17, pGL2-ELAM-Luc, IL-8/CXCL8-WT-Luc, and pBK-CMV-LacZ. Luciferase activity was determined with a luciferase assay system (Promega) and was normalized on the basis of LacZ expression. The level of induction of luciferase activity was compared as the ratio of cells with and without stimulation. To determine the transfection efficiency, cells were transfected with 1 μg of pEGFP, a green fluorescent protein (GFP) expression vector, for 24 h. After treatment, the medium was aspirated and replaced with fresh DMEM/Ham's F12 containing 10% FBS for another 24 h. Cells were observed under inverted laser scanning confocal microscopy (Olympus). The transfection efficacy was defined as the percentage of cells expressing GFP. The transfection rate of GFP was about 33% (data not shown).
Western Blot Analysis
To determine the protein levels of IKKα/β phosphorylated at Ser-180 (IKKα) or at Ser-181 (IKKβ), IKKα/β, phospho-Akt (Ser-473), Akt, and α-tubulin in A549 cells, proteins were extracted, and a Western blot analysis was performed as described previously (2). Briefly, A549 cells were cultured in 6-cm dishes. After reaching confluence, cells were treated with the vehicle and thrombin or pretreated with specific inhibitors as indicated followed by thrombin. After incubation, cells were washed twice in ice-cold phosphate-buffered saline (PBS) and solubilized in extraction buffer containing 10 mm Tris (pH 7.0), 140 mm NaCl, 2 mm phenylmethylsulfonyl fluoride (PMSF), 5 mm dithiothreitol, 0.5% Nonidet P-40, 0.05 mm pepstatin A, and 0.2 mm leupeptin. Samples of equal amounts of protein (80 μg) were subjected to SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane, which was then incubated in TBST buffer (150 mm NaCl, 20 mm Tris-HCl, and 0.02% Tween 20; pH 7.4) containing 5% nonfat milk. Proteins were visualized by specific primary antibodies and then incubated with HRP-conjugated secondary antibodies. The immunoreactivity was detected using enhanced chemiluminescence (ECL) following the manufacturer's instructions. Quantitative data were obtained using a densitometer with Image-Pro Plus image analysis software systems (Eastman Kodak Co.).
Immunoprecipitation and Protein Kinase Assays
A549 cells were grown in 6-cm dishes. After reaching confluence, cells were treated with 10 units/ml thrombin for the indicated time intervals or pretreated with specific inhibitors as indicated followed by thrombin. After incubation, cells were washed twice with ice-cold PBS, lysed in 1 ml of lysis buffer containing 20 mm Tris-HCl (pH 7.5), 1 mm MgCl2, 125 mm NaCl, 1% Triton X-100, 1 mm PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 25 mm β-glycerophosphate, 50 mm NaF, and 100 μm sodium orthovanadate, and centrifuged at 14,000 × g for 30 min. The supernatant was then immunoprecipitated with polyclonal antibodies against Akt, IKKα, or IKKβ in the presence of A/G-agarose beads overnight. The beads were washed three times with lysis buffer and two times with kinase buffer containing 20 mm HEPES (pH 7.4), 20 mm MgCl2, and 2 mm dithiothreitol. The kinase reactions were performed by incubating immunoprecipitated beads with 20 μl of kinase buffer supplemented with 20 μm ATP and 3 μCi of [γ-32P]ATP at 30 °C for 30 min. To assess the Akt and IKKα/β activities, 50 μg/ml histone H2B and 0.5 μg of GST-IκBα protein (amino acids 1–317) were respectively added as the substrates. The reaction mixtures were analyzed by 15% (Akt kinase activity) and 12% (IKKα/β kinase activity) SDS-PAGE followed by autoradiography.
Rac Activity Assay
Rac activity was measured using a Rac activity assay kit. The assay was performed according to the manufacturer's instructions. Briefly, cells were washed twice with ice-cold PBS, lysed in 1 ml of magnesium lysis buffer (25 mm HEPES (pH 7.5), 150 mm NaCl, 5% IGEPAL CA-630, 10 mm MgCl2, 5 mm EDTA, 10% glycerol, 10 μg/ml aprotinin, and 10 μg/ml leupeptin), and centrifuged at 14,000 × g for 30 min. The lysate (0.8 ml) was incubated with 5 μg of PAK1 p21-binding domain (PBD)-agarose at 4 °C overnight. The beads were washed three times with magnesium lysis buffer and centrifuged at 8,000 × g for 5 min. Bound Rac proteins were then solubilized in 20 μl of 2× Laemmli sample buffer and quantitatively detected by Western blotting (12% SDS-PAGE) using a mouse monoclonal anti-Rac antibody with the ECL system.
Co-immunoprecipitation
A549 cells were grown in 6-cm dishes. After reaching confluence, cells were treated with 10 units/ml thrombin for the indicated time intervals. Cells were harvested, lysed in 1 ml of pulldown buffer (40 mm Tris-HCl (pH 8.0), 500 mm NaCl, 0.1% Nonidet P-40, 6 mm EGTA, 10 mm β-glycerophosphate, 10 mm NaF, 300 μm sodium orthovanadate, 2 mm PMSF, 10 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm dithiothreitol), and centrifuged at 14,000 × g for 30 min. The supernatant was then immunoprecipitated with 1 μg of specific antibodies against Gβ, Gγ, Rac1, or p85α in the presence of protein A/G beads at 4 °C overnight. The immunoprecipitated beads were washed three times with pulldown buffer and centrifuged at 8,000 × g for 5 min. Samples were fractionated on 12% (for Rac1), 8% (for p85α), or 15% (for Gβ) SDS-PAGE, transferred to a PVDF membrane, and subjected to immunoblot analysis using a 1:1,000 antibody dilution specific for Rac1, p85α, or Gβ.
Statistical Analysis
Results are presented as the mean ± S.E. of at least three independent experiments. One-way analysis of variance followed by, when appropriate, Bonferroni's multiple range test was used to determine the statistical significance of the difference between means. Values of p < 0.05 were considered statistically significant.
RESULTS
Involvement of Rac1 in Thrombin-induced IL-8/CXCL8 Expression and Release
Our previous study found that thrombin (0.3–10 units/ml) induced IL-8/CXCL8 release in a concentration-dependent manner, with a maximal effect at 10 units/ml in A549 cells and primary normal human bronchial epithelial cells (2). Thrombin-induced IL-8/CXCL8 release by A549 cells being similar to the normal human bronchial epithelial cell response supports the use of A549 cells in this study. Small GTPases such as Rac1 were shown to participate in signaling pathways leading to the induction of IL-8/CXCL8 expression by various stimuli (32). To explore whether Rac1 might mediate thrombin-induced IL-8/CXCL8 expression and release, the Rac1 expression construct for the dominant negative Rac1-T17N mutant (RacN17) was used. As shown in Fig. 1A, transfection of A549 cells with RacN17 (0.5 and 1 μg) markedly inhibited thrombin-induced IL-8/CXCL8 release. When cells were treated with 1 μg of RacN17, the basal levels of IL-8/CXCL8 and thrombin-induced IL-8/CXCL8 release were inhibited by 28 ± 18 and 55 ± 13%, respectively (n = 3). (Fig. 1A). Furthermore, we found that RacN17 inhibited the thrombin-induced increase in IL-8/CXCL8-luciferase activity, with 1 μg of RacN17 reducing the basal IL-8/CXCL8-luciferase activity and thrombin response by 67 ± 9 and 92 ± 3% (n = 3), respectively (Fig. 1B). Next, we directly measured Rac activity in response to thrombin. Fig. 1C shows that treatment of A549 cells with 10 units/ml thrombin induced an increase in Rac activity in a time-dependent manner, as assessed by immunoblot samples for Rac1 immunoprecipitated from lysates using PAK1 PBD-agarose. The response began at 1 min and was sustained to 30 min of treatment (Fig. 1C). Taken together, these results imply that Rac1 activation is involved in thrombin-induced IL-8/CXCL8 expression and release.
FIGURE 1.

Rac1 is involved in thrombin-induced IL-8/CXCL8 release and IL-8/CXCL8-luciferase activity in A549 cells. A, A549 cells were transiently transfected with either 1 μg of pcDNA (mock) or 0.5 or 1 μg of RacN17 for 24 h. Cells were stimulated with an equivant vehicle control (double-distilled H2O (−)) or 10 units/ml thrombin for another 24 h, and then IL-8/CXCL8 levels were determined. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with thrombin treatment. B, A549 cells were either transiently transfected with 0.2 μg of IL-8/CXCL8 WT-Luc and 0.5 μg of pBK-CMV-LacZ or cotransfected with 1 μg of pcDNA (mock) or with 0.5 or 1 μg of RacN17 for 24 h and then stimulated with 10 units/ml thrombin for another 24 h. Cells were harvested for the luciferase assay as described under “Experimental Procedures.” Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with thrombin treatment. C, for Rac activity, cells were incubated with vehicle or 10 units/ml thrombin for the indicated time intervals, and cell lysates were then immunoprecipitated with PAK-1 PBD-agarose. The Rac activity assay is described under “Experimental Procedures.” Rac1 and α-tubulin protein levels were determined in total lysates by a Western blot analysis as the loading control. Traces represent results from three independent experiments with similar results.
Involvement of PI3K/Akt in Thrombin-induced IL-8/CXCL8 Expression and Release
Rac1 might activate a number of signal pathways including PI3K/Akt (33). Next, to determine whether PI3K and its downstream protein kinase, Akt, are involved in the signal transduction pathway leading to IL-8/CXCL8 expression and release caused by thrombin, a PI3K inhibitor (LY294002), an Akt inhibitor (1-l-6-hydroxymethyl-chiro-inositol-2-((R)-2-O-methyl-3-O-octadecylcarbonate)), and AktDN were used. Pretreatment of cells with LY294002 (10 μm) and the Akt inhibitor (1 μm) significantly attenuated the thrombin-induced IL-8/CXCL8 release by 43 ± 12 and 47 ± 10%, respectively (Fig. 2A). Thrombin-induced IL-8/CXCL8 release was completely inhibited in cells transfected with AktDN (0.5 μg) (n = 3) (Fig. 2A). In parallel with IL-8/CXCL8 release, thrombin-induced IL-8/CXCL8-luciferase activity was also inhibited by LY294002 (10 μm) by 68 ± 12% (Fig. 2B). Transfection of A549 cells with AktDN (0.5 μg) almost completely abolished thrombin-induced IL-8/CXCL8 luciferase activity. These results showed that PI3K and Akt are involved in thrombin-induced IL-8/CXCL8 release and expression.
FIGURE 2.

PI3K/Akt and MAPKs are involved in thrombin-mediated IL-8/CXCL8 release and IL-8/CXCL8-luciferase activity in A549 cells. A, cells were transfected with 0.5 μg of pcDNA (mock) or with 0.5 μg of AktDN for 24 h or were pretreated with an equivalent vehicle control (DMSO), 10 μm LY294002, or 1 μm of the Akt inhibitor (Akt inh) for 30 min and then incubated with 10 units/ml thrombin for another 24 h. IL-8/CXCL8 levels were determined. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with thrombin treatment. B, A549 cells were transfected with 0.2 μg of IL-8/CXCL8 WT-Luc and 0.5 μg of pBK-CMV-LacZ or cotransfected with 0.5 μg of pcDNA (mock) or with 0.5 μg of AktDN for 24 h or were pretreated with an equivalent vehicle control (DMSO) or 10 μm LY294002 for 30 min and then incubated with 10 units/ml thrombin for another 24 h. Cells were harvested for the luciferase assay as described in the legend for Fig. 1. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with thrombin treatment. C, cells were pretreated with an equivalent vehicle control (DMSO), 10 μm PD98059, 3 μm SB203580, or 10 μm SP600125 for 30 min and then incubated with an equivant vehicle control (double-distilled H2O (−)) or 10 units/ml thrombin for another 24 h. IL-8/CXCL8 levels were determined. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with thrombin treatment.
Involvement of MAPKs in Thrombin-induced IL-8/CXCL8 Release
Several reports demonstrated that MAPKs, including ERK, p38 MAPK, and JNK, are associated with the secretion of IL-8/CXCL8 by thrombin stimulation in vascular smooth muscle cells, macrophages, and lung epithelial cells (34–36). Therefore, we further investigated whether MAPKs play a role in thrombin-induced CXCL8 release in human lung epithelial cells. We found that pretreatment of A549 cells with 30 μm PD98059 (a MAPK kinase (MEK) inhibitor) and 10 μm SP600125 (a JNK inhibitor) both inhibited thrombin-induced IL-8/CXCL8 release by 34 ± 6 and 42 ± 3%, respectively (Fig. 2C). However, 3 μm SB203580 (a p38 MAPK inhibitor) did not affect thrombin-induced IL-8/CXCL8 release (Fig. 2C). These results show that ERK and JNK, but not p38 MAPK, are also involved in thrombin-induced IL-8/CXCL8 release and expression. In this study, we focused on molecular mechanisms of the Rac1-dependent PI3K/Akt signal pathway in thrombin-induced IL-8/CXCL8 expression.
Thrombin Causes Increases in Akt Ser-473 Phosphorylation and Kinase Activity
Because serine phosphorylation of residue 473 in Akt causes enzymatic activation (37), an antibody specific against phosphorylated Akt (Ser-473) was used to examine Akt phosphorylation. When cells were treated with 10 units/ml thrombin for various time intervals, Akt Ser-473 phosphorylation increased at 5 min, peaked at 10 min, and declined after 30 min of treatment (Fig. 3A, upper panel). The protein level of Akt1/2 was not affected by thrombin treatment (Fig. 3A, lower panel). Using the histone H2B as an Akt substrate, treatment of cells with 10 units/ml thrombin increased the Akt activity in a time-dependent manner. Maximal activation was detected at 10 min after stimulation, and the response declined after 20 min of treatment (Fig. 3B). Moreover, both 300 μm SFLLRN-NH2 (a PAR1 agonist peptide) and 300 μm GYPGQV-NH2 (a PAR4 agonist peptide) also induced increases in Akt Ser-473 phosphorylation (Fig. 3C). In contrast, 300 μm TFRGAP-NH2 (a PAR3 agonist peptide) did not induce Akt Ser-473 phosphorylation (Fig. 3C). These results suggest that PAR1 and PAR4 may be involved in thrombin-induced Akt Ser-473 phosphorylation.
FIGURE 3.

Akt activation is caused by thrombin, a PAR1 agonist, and a PAR4 agonist in A549 cells. A, cells were incubated with 10 units/ml thrombin for the indicated time intervals. Cell lysates were prepared, and then immunoblotted with antibodies for phospho-Akt, Akt1/2, or α-tubulin. Ser473-p, phosphorylated Ser-473. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with the control (0-min time point) group. B, A549 cells were incubated with 10 units/ml thrombin for 0–60 min, and cell lysates were then immunoprecipitated with antibodies specific for Akt1/2. One set of immunoprecipitates was subjected to the kinase assay (KA) described under “Experimental Procedures” using the histone H2B as a substrate. The other set of immunoprecipitates was subjected to 10% SDS-PAGE and analyzed by immunoblotting (IB) with the anti-Akt1/2 antibody. Equal amounts of the immunoprecipitated kinase complex present in each kinase assay were confirmed by immunoblotting for Akt1/2. α-Tubulin protein in total cell lysates served as the loading control. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with the control (0 min time point) group. C, cells were incubated with an equivalent vehicle control (double-distilled H2O), 10 units/ml thrombin, 300 μm SFLLRN-NH2, 300 μm TFRGAP-NH2, and 300 μm GYPGQV-NH2 for 30 min, and then Akt phosphorylation, Akt1/2, and α-tubulin protein levels were determined. Data are presented as the mean ± S.E. of three experiments performed in duplicate. *, p < 0.05, as compared with the vehicle group.
Rac1 and PI3K Mediate Thrombin-induced Akt Activation
Next, we investigated the roles of Rac1 and PI3K in thrombin-mediated Akt Ser-473 phosphorylation and kinase activity. As shown in Fig. 4, transfection of A549 cells with RacN17 (1 μg) markedly attenuated thrombin-induced Akt Ser-473 phosphorylation (Fig. 4A). Thrombin-induced Akt Ser-473 phosphorylation was inhibited in cells transfected with RacN17 (1 μg) by 98 ± 3% (Fig. 4A). In addition, 1 μg of RacN17 partially inhibited the basal level of Akt Ser-473 phosphorylation by 53 ± 10% (Fig. 4A). In parallel with Akt Ser-473 phosphorylation, thrombin-induced Akt kinase activity was also inhibited by RacN17 (Fig. 4B). None of these treatments had any effect on Akt expression (Fig. 4, A and B). We further examined whether PI3K mediates Akt Ser-473 phosphorylation and Akt kinase activity. We found that thrombin-induced Akt Ser-473 phosphorylation was inhibited by 10 μm LY294002 by 64 ± 10% (Fig. 5A). Similarly, thrombin-induced Akt kinase activity was also inhibited by 10 μm LY294002 (Fig. 5B). Based on these results, we suggest that activation of Rac1 and PI3K occurs upstream of Akt in the thrombin-induced signaling pathway. A previous study demonstrated that Akt lies downstream of both PKCδ and PI3K in mediating thrombin-induced NF-κB activation and ICAM-1 expression in human umbilical vein endothelial cells (14). Our previous report showed that PKCα mediated thrombin-induced NF-κB activation and IL-8/CXCL8 expression (2). In addition to PI3K, there is an upstream molecule in Akt activation by thrombin stimulation. To also address the possibility that PKCα contributes to Akt Ser-473 phosphorylation induced by thrombin stimulation, a PKCα inhibitor, Ro-32-0432, was used. Treatment with 10 μm Ro-32-0432 did not affect thrombin-induced Akt Ser-473 phosphorylation (Fig. 5C). This result suggests that PKCα is not involved in thrombin-induced Akt phosphorylation.
FIGURE 4.
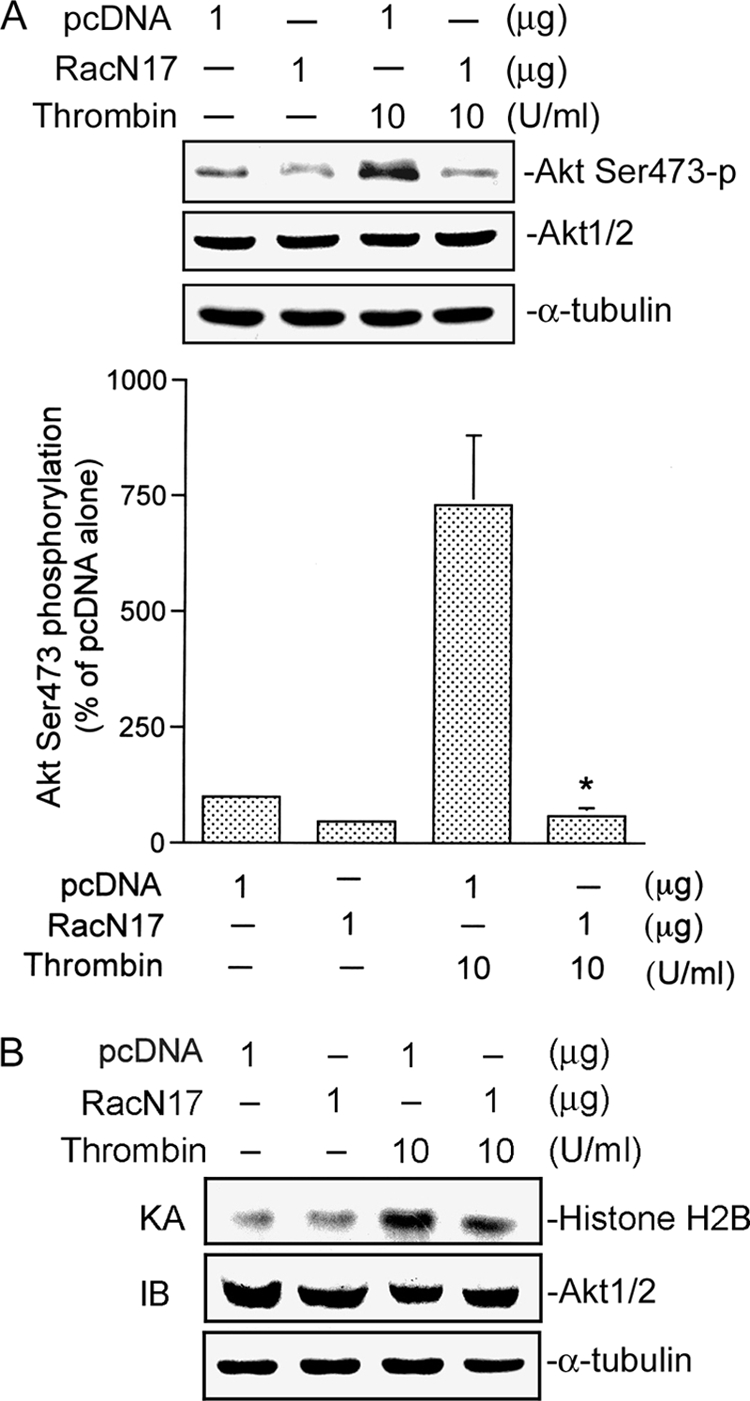
Involvement of Rac1 in thrombin-mediated Akt Ser-473 phosphorylation and kinase activity in A549 cells. A, cells were transfected with 1 μg of pcDNA (mock) or 1 μg of RacN17 for 24 h and then incubated with the vehicle or 10 units/ml thrombin for another 30 min. Akt Ser-473 phosphorylation was shown by immunoblotting with an antibody specific for phosphorylated Akt Ser-473. Equal loading in each lane is shown by the similar intensities of Akt1/2 or α-tubulin. Typical traces represent three experiments with similar results. Results are expressed as the mean ± S.E. (n = 3). *, p < 0.05, as compared with the thrombin-treated group. B, cells were transfected with 1 μg of pcDNA (mock) or 1 μg of RacN17 for 24 h and then incubated with vehicle or 10 units/ml thrombin for another 30 min. Cell lysates were then immunoprecipitated with antibodies specific for Akt1/2. One set of immunoprecipitates was subjected to the kinase assay (KA) described under “Experimental Procedures” using the histone H2B as a substrate). The other set of immunoprecipitates was subjected to 10% SDS-PAGE and analyzed by immunoblotting (IB) with the anti-Akt1/2 antibody. Equal amounts of the immunoprecipitated kinase complex present in each kinase assay were confirmed by immunoblotting for Akt1/2. α-Tubulin protein in total cell lysates served as the loading control. Typical traces represent two experiments with similar results.
FIGURE 5.
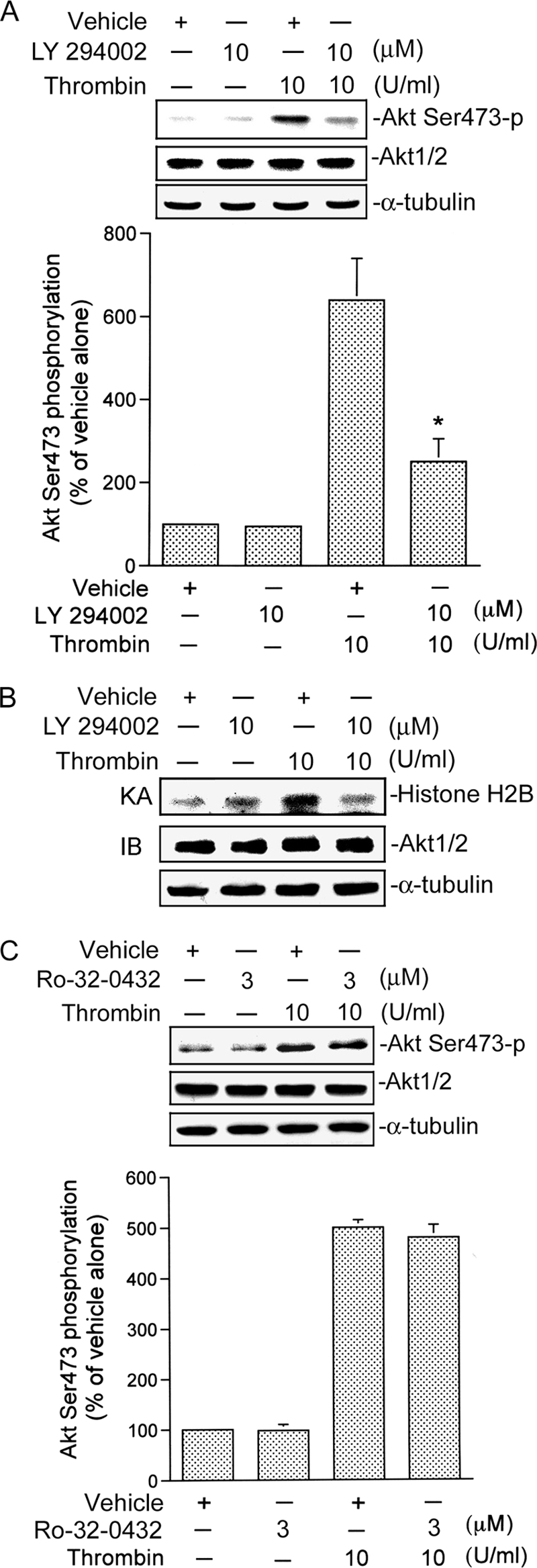
Involvement of PI3K in thrombin-induced Akt Ser-473 phosphorylation and kinase activity in A549 cells. A and C, cells were pretreated with an equivalent vehicle control (DMSO), 10 μm LY294002 (A), or 3 μm Ro-32-0432 (C) for 30 min and then incubated with the vehicle or 10 units/ml thrombin for another 30 min. Akt Ser-473 phosphorylation was shown by immunoblotting with an antibody specific for phosphorylated Akt Ser-473. Equal loading in each lane is shown by the similar intensities of Akt1/2 or α-tubulin. Typical traces represent three experiments with similar results. Results are expressed as the mean ± S.E. (n = 3). *, p < 0.05, as compared with the thrombin-treated group. B, cells were pretreated with an equivalent vehicle control (DMSO) or 10 μm LY294002 for 30 min and then incubated with vehicle or 10 units/ml thrombin for another 30 min. Cell lysates were then immunoprecipitated with antibodies specific for Akt1/2. One set of immunoprecipitates was subjected to the kinase assay (KA) described as in the legend for Fig. 4. The other set of immunoprecipitates was subjected to 10% SDS-PAGE and analyzed by immunoblotting (IB) with the anti-Akt1/2 antibody. Equal amounts of the immunoprecipitated kinase complex present in each kinase assay were confirmed by immunoblotting for Akt1/2. α-Tubulin protein in total cell lysates served as the loading control. Typical traces represent two experiments with similar results.
Rac1, PI3K, and Akt Mediate Thrombin-induced IKKα/β Activation
We further examined whether IKKα/β activation occurred through the Rac1/PI3K/Akt signaling pathway. Transfection of A549 cells with 1 μg of RacN17 for 24 h or pretreatment of cells with 10 μm LY294002 and 10 μm of an Akt inhibitor for 30 min markedly attenuated thrombin-induced IKKα/β phosphorylation by 80 ± 5, 94 ± 6, and 89 ± 3%, respectively (Fig. 6, A and B). None of these treatments had any effect on IKKα/β expression (Fig. 6, A and B). In addition, thrombin-induced IKKα/β kinase activity was also inhibited by 1 μg of RacN17, 10 μm LY294002, and 0.5 μg of AktDN (Fig. 7, A–C). These results suggest that Rac1, PI3K, and Akt play important roles in thrombin-induced IKKα/β activation.
FIGURE 6.

Involvement of Rac1, PI3K, and Akt in thrombin-induced IKKα/β phosphorylation in A549 cells. A and B, cells were transfected with 1 μg of pcDNA (mock) or 1 μg of RacN17 for 24 h (A) or pretreated with an equivalent vehicle control (DMSO), 10 μm LY294002, or 10 μm of an Akt inhibitor (Akt inh) for 30 min (B) and then incubated with 10 units/ml thrombin for another 30 min. IKKα/β phosphorylation was shown by immunoblotting with an antibody specific for phosphorylated IKKα/β. Equal loading in each lane is shown by the similar intensities of IKKα/β or α-tubulin. Typical traces represent three experiments with similar results. Results are expressed as the mean ± S.E. (n = 3). *, p < 0.05, as compared with the thrombin-treated group.
FIGURE 7.
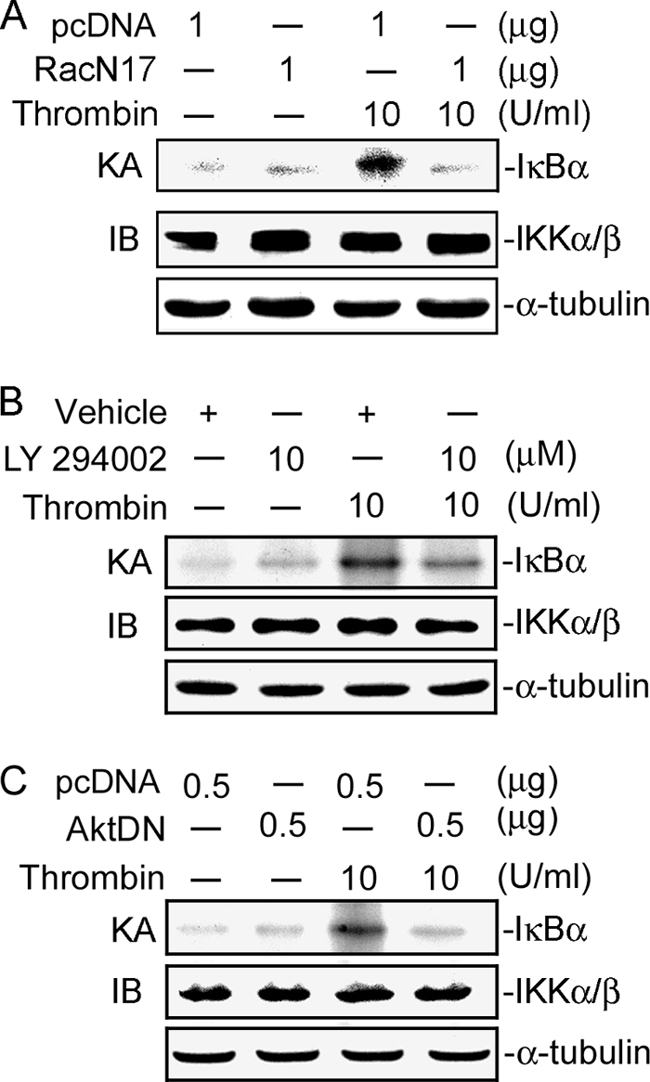
Involvement of Rac1, PI3K, and Akt in thrombin-induced IKKα/β kinase activity in A549 cells. A–C, cells were transfected with1 μg of pcDNA (mock), 1 μg of RacN17 (A), or 0.5 μg of AktDN (C) for 24 h or were pretreated with an equivalent vehicle control (DMSO) or 10 μm LY294002 for 30 min (B) and then incubated with vehicle or 10 units/ml thrombin for another 30 min. Whole-cell lysates were then immunoprecipitated with antibodies specific for IKKα and IKKβ. One set of immunoprecipitates was subjected to the kinase assay (KA) described under “Experimental Procedures” using the IκBα-GST fusion protein as a substrate. The other set of immunoprecipitates was subjected to 10% SDS-PAGE and analyzed by immunoblotting (IB) with the anti-IKKα/β antibody. Equal amounts of the immunoprecipitated kinase complex present in each kinase assay were confirmed by immunoblotting for IKKα/β. α-Tubulin protein in total cell lysates served as the loading control. Typical traces represent two experiments with similar results.
Rac1, PI3K, and Akt Mediate Thrombin-induced NF-κB Activation
We further examined whether the activation of NF-κB occurs through the Rac1/PI3K/Akt signaling pathway. Using transient transfection with pGL2-ELAM-κB-luciferase as an indicator of NF-κB activity, we found that the thrombin-induced increase in κB-luciferase activity was inhibited by transfection of cells for 24 h with 1 μg of RacN17 or 0.5 μg of AktDN or by pretreating cells for 30 min with 10 μm LY294002 by 72 ± 5, 66 ± 6, and 75 ± 8%, respectively (Fig. 8). Taken together, these data suggest that activation of the Rac1/PI3K/Akt pathway is required for thrombin-induced NF-κB activation in A549 epithelial cells.
FIGURE 8.
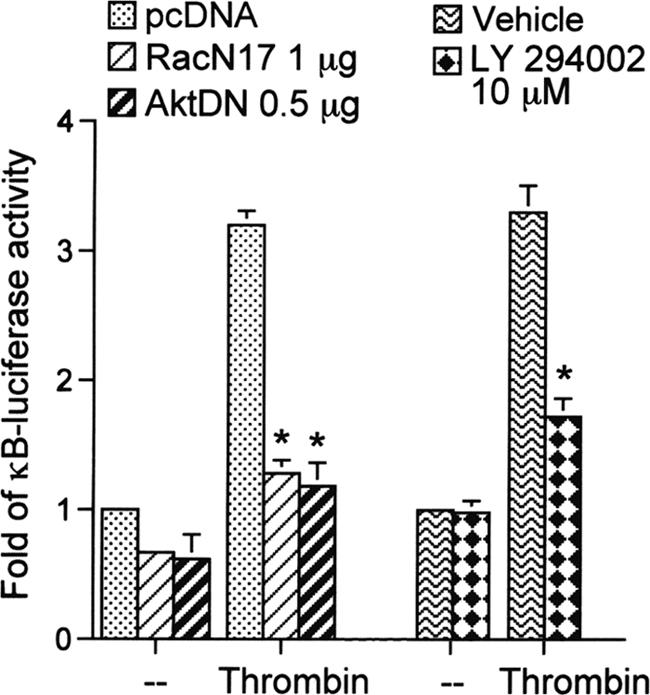
Involvement of Rac1, PI3K, and Akt in thrombin-induced NF-κB activation in A549 cells. A549 cells were transiently transfected with 0.5 μg of pGL2-ELAM-Luc and 0.5 μg of pBK-CMV-LacZ for 24 h, cotransfected with 1 μg of pcDNA (mock), 1 μg of RacN17, or 0.5 μg of AktDN for 24 h, or pretreated with an equivalent vehicle control (DMSO) or 10 μm LY294002 for 30 min and then stimulated with vehicle or 10 units/ml thrombin for another 24 h. Luciferase activities were determined as described under “Experimental Procedures.” Results are expressed as the mean ± S.E. (n = 3). *, p < 0.05, as compared with the thrombin-treated group.
Rac1 Is Associated with p85α and Gβγ upon Thrombin Stimulation
We next investigated whether thrombin can induce interactions among Rac1, p85α, and Gβγ. As shown in Fig. 9A, treatment of A549 cells with 10 units/ml thrombin caused the association of Rac1 and p85α, as detected by immunoblotting using the antibody to Rac1 after immunoprecipitation of p85α. The association of Rac1 and p85α occurred at 1 min, was sustained to 10 min, and then declined after 20 min (Fig. 9A). We further found that the association of Gβγ and p85α occurred at 0.5–3 min as detected by immunoblotting using an antibody to p85α after the immunoprecipitation of Gβγ (Fig. 9B). Treatment of A549 cells with thrombin induced the association of Rac and Gβ. This response peaked at 1 min, was sustained to 3 min, and declined after 5 min of treatment (Fig. 9C). These results suggest that thrombin induces Rac1 activation by interacting with Gβγ and p85α in A549 lung epithelial cells.
FIGURE 9.
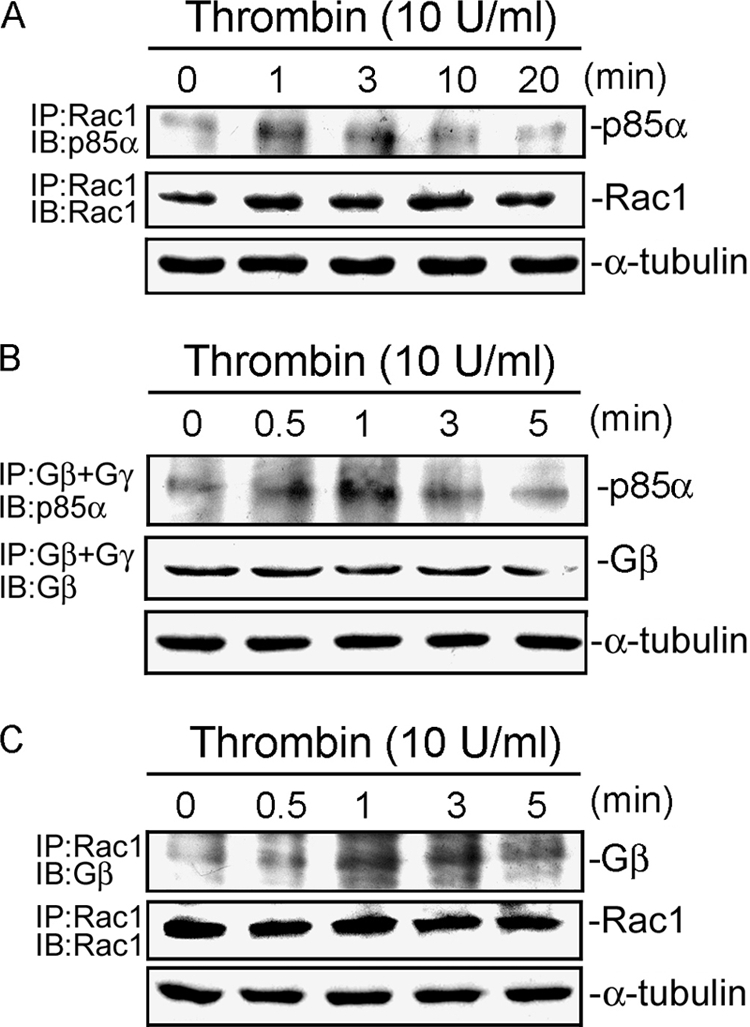
Rac1 is associated with Gβγ and p85α upon thrombin stimulation in A549 cells. A, cells were stimulated with 10 units/ml thrombin for 0–20 min and were then lysed. Cell extracts were immunoprecipitated with an anti-Rac1 antibody, and then immunoblotted with an anti-p85α or anti-Rac1 antibody. IP, immunoprecipitation; IB, immunoblotting. B, A549 cells were stimulated with 10 units/ml thrombin for 0–5 min and then lysed. Cells extracts were immunoprecipitated with anti-Gβ and anti-Gγ antibodies and then immunoblotted with an anti-p85α or anti-Gβ antibody. C, cells were treated with thrombin for the indicated time intervals and then immunoprecipitated with the anti-Rac1 antibody and immunoblotted with the anti-Gβ or anti-Rac1 antibody. α-Tubulin protein in total cell lysates served as the loading control. Typical traces are representative of three experiments with similar results.
DISCUSSION
It is known that increased thrombin levels occur in most inflammatory pulmonary diseases and that thrombin plays a critical role in various pathological processes. For example, thrombin modulates lung inflammation by stimulating the secretion of proinflammatory mediators (2, 6). A large body of evidence has accumulated to suggest that IL-8/CXCL8 plays a pivotal role in respiratory diseases such as chronic obstructive pulmonary diseases, chronic bronchitis, bronchiectasis, and asthma (8, 10, 11, 38). In the present study, we found that thrombin induced IL-8/CXCL8 expression and release in human lung epithelial cells, suggesting that thrombin and IL-8/CXCL8 expression and release contribute to the pathogenesis of lung diseases. Our previous study showed that thrombin activates the PI-PLC/PKCα/c-Src pathway, which in turn initiates IKKα/β and NF-κB activation and ultimately induces IL-8/CXLC8 expression and release in human lung epithelial cells (2). In the present study, we provide the first description of a second pathway linking the small GTP-binding protein, Rac1, to thrombin-stimulated PI3K/Akt activation, IKKα/β activation, NF-κB transcriptional activation, and subsequent IL-8/CXCL8 expression in human lung epithelial cells.
Activation of Rac by cell surface receptors including G protein-coupled receptors occurs through incompletely characterized effector routes. A previous study showed that in A549 cells, thrombin leads to activation of Rac through the PI-PLC pathway (39). However, the molecular mechanisms by which Rac becomes activated by thrombin in lung epithelial cells are still unclear. In other cell models, various Gi and Gq/G11 G protein-coupled receptors, including P2Y12, PAR1, and M1 muscarinic receptors, were shown to activate Rac (19, 40, 41). In many cases, however, Rac activation is critically dependent on PI3K activity. For example, lysophosphatidic acid-induced Rac activation in neuroblastoma cells is dependent on PI3K activation (42). In addition, the intracellular calcium-dependent PKC signal pathway mediates thrombin-induced Rac activation in human prostate carcinoma cells (43). Moreover, Soulet et al. (19) demonstrated that P2Y12-induced Rac activation is mediated by Gβγ subunits in human platelets. In this study, we found that thrombin induced an increase in Rac activity. Furthermore, we found that thrombin can induce an association of Rac1 with Gβγ after 1 min of treatment. Our data also revealed that Gβγ mediated thrombin-induced Rac activity in human lung epithelial cells. PI3Ks are divided into three classes according to their substrate specificity and molecular structure (25). Class IA and IB PI3Ks are heterodimeric enzymes composed of an adaptor subunit coupled to a catalytic subunit (25). Previous studies showed that the class IB PI3K p110γ catalytic subunit is associated with the p101 adaptor subunit and is activated by Gβγ (24, 25). The class IA PI3K p85α adaptor subunit is associated with the p110α catalytic subunit and is activated by tyrosine kinase, Ras, and Rac1 (24, 25, 44). In this study, we found that thrombin can induce the association of p85α and Gβγ. Furthermore, we also found that thrombin can induce Rac1 and p85α complex formation. In support of our findings, Bokoch et al. (44) demonstrated that the active GTP-bound form of Rac1 can directly bind to p85 and increase PI3K activity. Therefore, p85α may function as an adaptor for Gβγ and Rac1. Taken together, these results suggest that the rapid signal complex assembly involved Gβγ, Rac1, and p85α in human lung epithelial cells stimulated by thrombin.
Rac was shown to be required for the activation of the NF-κB pathway and mediates protein expression by various stimuli (33, 45). Mainiero et al. (45) reported that activation of Rac1 is required for IL-8/CXCL8 release caused by β1 integrin. Moreover, Rac was shown to be activated by TNF-α and to mediate IL-6 and IL-8/CXCL8 release in HeLa cells (32). In this study, we found that treatment of A549 cells with thrombin caused activation of Rac1, and a Rac1 dominant negative mutant (RacN17) inhibited thrombin-induced NF-κB activation and IL-8/CXCL8 expression. These results indicate that Rac1 is required for NF-κB activation and IL-8/CXCL8 expression caused by thrombin in human lung epithelial cells. Furthermore, a large body of evidence has implicated the PI3K/Akt pathway involvement in gene expression by various stimuli (29, 31). However, it is not clear whether thrombin stimulates the Rac1-dependent PI3K/Akt pathway in lung epithelial cells and whether activation of the PI3K/Akt pathway is involved in thrombin-induced IL-8/CXCL8 expression. The results of this study showed that the Rac1 and PI3K pathway is essential for Akt activation and IL-8/CXCL8 expression stimulated by thrombin. This is based on the fact that RacN17 and LY294002, a PI3K-specific inhibitor, blocked Akt activation and IL-8/CXCL8 expression and release. Moreover, AktDN and an Akt inhibitor suppressed thrombin-induced IL-8/CXCL8 expression and release. This is therefore consistent with reports that Akt plays an important role in mediating the effects of PI3K on lung epithelial cell IL-8/CXCL8 expression with human rhinovirus infection (46). Many chemokines including IL-8/CXCL8 are dependent upon MAPK pathways by thrombin stimulation in various cell types (34–36). Chung et al. (34) reported that in vascular smooth muscle cells, MAPKs mediate thrombin-induced IL-8/CXCL8 release. Another report showed that thrombin-induced IL-8/CXCL8 release depends on ERK and JNK in human lung epithelial cells (A549) (36). In this study, we present data that confirmed the role of ERK and JNK in thrombin-induced IL-8/CXCL8 release in human lung epithelial cells. We found that a MEK inhibitor (PD98059) and a JNK inhibitor (SP600125) both significantly inhibited thrombin-induced IL-8/CXCL8 release. However, we found that a p38 MAPK inhibitor (SB203580) did not affect thrombin-induced IL-8/CXCL8 release. These results indicate that ERK and JNK, but not p38 MAPK, also participate in thrombin-induced CXCL8 release.
Thrombin-induced cellular effects in different kinds of cells including human lung epithelial cells are primarily mediated by G protein-coupled receptors known as PARs. Upon thrombin stimulation, activated PARs couple with the G protein and activate downstream signaling molecules to mediate IL-8/CXCL8 expression in lung epithelial cells (2, 6). Human lung epithelial cells express PAR1–4 (6). PAR1 and PAR4 are considered essential for the release of lung epithelial cell inflammatory mediators (2). A previous study showed that in human platelets, stimulation of PAR1 and PAR4 induced Akt phosphorylation (47). In this study, our results also revealed that thrombin, a PAR1 agonist peptide (SFLLRN-NH2), and a PAR4 agonist peptide (GYPGQV-NH2), but not a PAR3 agonist peptide (TFRGAP-NH2), induced increases in Akt Ser-473 phosphorylation. Thus, we suggest that thrombin can induce Akt Ser-473 activation in A549 cells through PAR1 and PAR4. This is consistent with our previous report that thrombin activates PAR1 and PAR4 to induce IL-8/CXCL8 expression in A549 epithelial cells (2).
Recent investigations clarified multiple signaling intermediates involved in the regulation of NF-κB, which regulates the expressions of many genes (2, 48). Our previous study showed that NF-κB activation contributes to thrombin-induced IL-8/CXCL8 expression in human lung epithelial cells (2). We also found that thrombin induces IKKα/β activation, IκBα phosphorylation, and IκBα degradation, as well as an increase in NF-κB activity. Moreover, Rho family GTPases were previously shown to induce the transcriptional activity of NF-κB (49). A previous report showed that in A549 cells, a Rac1-dependent reactive oxygen species signal pathway mediates TNF-α-induced NF-κB activation (50). Moreover, Sanlioglu et al. (51) showed that Rac1 leads to activation of NF-κB through the IKKα/β complex in RAW 264.7 macrophages. Given that PI3K/Akt plays a critical role in NF-κB activation (31), we speculated that PI3K/Akt might be upstream of NF-κB and investigated this possibility. In this study, we found that a Rac1 dominant negative mutant (RacN17), a PI3K inhibitor (LY294002), an Akt inhibitor, and an Akt dominant negative mutant (AktDN) blocked thrombin-induced IKKα/β phosphorylation, kinase activity, and NF-κB reporter activity, suggesting that Rac1, PI3K, and Akt are involved in thrombin-mediated NF-κB activation through an increase in IKKα/β activity.
A previous report demonstrated that thrombin induced NF-κB activation and ICAM-1 expression by engaging parallel Gαq/PKCδ/Akt and Gβγ/PI3K/Akt pathways that converge at Akt (14). In this study, we found that thrombin induced NF-κB activation and IL-8/CXCL8 expression by engaging parallel PI-PLC/PKCα/c-Src and Rac1/PI3K/Akt signaling pathways that converge at IKKα/β in human lung epithelial cells. There is evidence of that IKKα/β activation by one or more upstream protein kinases such as NF-κB-inducing kinase, Akt, c-Src, and PKC (14, 52–54). Our previous study (2) found that inhibition of PKCα and c-Src prevented IKKα/β phosphorylation and kinase activity. Moreover, thrombin induced PKCα, c-Src, and IKKα/β complex formation (2). Lallena et al. (54) also showed that PKCα directly binds IKKβ to mediate phorbol 12-myristate 13-acetate-generated signals to the IKK complex (54). Another report showed that c-Src is involved in IKKα/β phosphorylation in NCI-H292 epithelial cells (53). Furthermore, in this study, we found that A549 cells transfected with a dominant negative mutant of Rac1 (RacN17) or treatment with a PI3K inhibitor (LY294002) both inhibited Akt Ser-473 phosphorylation and kinase activity. However, Ro-32-0432 (a selective PKCα inhibitor) did not affect thrombin-induced Akt phosphorylation. These results suggest that Rac1 and PI3K, but not PKCα, are upstream kinases of Akt in A549 cells. This suggestion was further supported by a previous study, which showed that IL-8/CXCL8-induced Akt phosphorylation is mediated by PI3K but not PKCα/βII in human peripheral blood mononuclear cells (55). Another study also showed that hemoglobin-induced Akt phosphorylation is inhibited by a PI3K inhibitor (LY294002), but not by a PKC inhibitor (Ro-31-8220), in dendritic cells (56). Taken together, these results indicate that thrombin-induced NF-κB activation and IL-8/CXCL8 expression are, however, engaging parallel PI-PLC/PKCα/c-Src and Rac1/PI3K/Akt pathways that converge at IKKα/β by thrombin stimulation in human lung epithelial cells.
In conclusion, the present study together with our previous report (2) indicates that treatment of human lung epithelial cells with thrombin causes activation of IKKα/β and NF-κB and IL-8/CXCL8 expression and release through two separate pathways: the PI-PLC/PKCα/c-Src and Gβγ/Rac1/PI3K/Akt pathways. This is the first study to show that thrombin-induced Rac1 activation can occur through recruitment of Gβγ and p85α in human lung epithelial cells. Fig. 10 is a schematic representation of the signaling pathways of thrombin-induced IL-8/CXCL8 transcription in human lung epithelial cells. Discovery of the signaling pathways of thrombin in IL-8/CXCL8 expression and release may help elucidate the roles of thrombin in lung inflammation and diseases.
FIGURE 10.
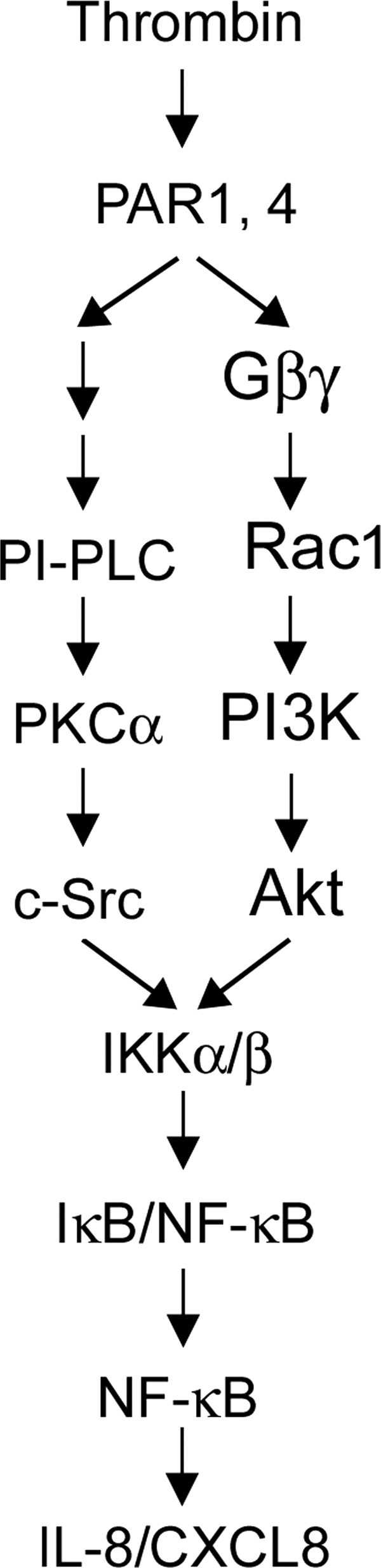
Schematic summary of how signal transduction by thrombin induces IL-8/CXCL8 expression in human lung epithelial cells. Thrombin-induced Rac1 activation may occur through the Gβγ subunit in A549 cells. Thrombin-induced activation of the Rac1/PI3K/Akt and PI-PLC/PKCα/c-Src cascades results in an increase in IKKα/β activation, which in turn induces NF-κB activation and ultimately causes IL-8/CXCL8 expression and release in A549 lung epithelial cells.
This work was supported by Grants NSC92-2320-B-038-022 and NSC93-2320-B-038-045 from the National Science Council of Taiwan.
- PAR
- protease-activated receptor
- DN
- dominant negative mutant
- ICAM-1
- intercellular adhesion molecule-1
- IKKα/β
- IκB kinases α/β
- PBD
- p21-binding domain
- PI
- phosphoinositide
- PLC
- phospholipase C
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Steinhoff M., Buddenkotte J., Shpacovitch V., Rattenholl A., Moormann C., Vergnolle N., Luger T. A., Hollenberg M. D. (2005) Endocr. Rev. 26, 1–43 [DOI] [PubMed] [Google Scholar]
- 2. Lin C. H., Cheng H. W., Hsu M. J., Chen M. C., Lin C. C., Chen B. C. (2006) J. Immunol. 177, 3427–3438 [DOI] [PubMed] [Google Scholar]
- 3. Sokolova E., Reiser G. (2007) Pharmacol. Ther. 115, 70–83 [DOI] [PubMed] [Google Scholar]
- 4. Coughlin S. R. (2000) Nature 407, 258–264 [DOI] [PubMed] [Google Scholar]
- 5. Moffatt J. D., Lever R., Page C. P. (2004) Br. J. Pharmacol. 143, 269–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Asokananthan N., Graham P. T., Fink J., Knight D. A., Bakker A. J., McWilliam A. S., Thompson P. J., Stewart G. A. (2002) J. Immunol. 168, 3577–3585 [DOI] [PubMed] [Google Scholar]
- 7. Kunkel S. L., Standiford T., Kasahara K., Strieter R. M. (1991) Exp. Lung. Res. 17, 17–23 [DOI] [PubMed] [Google Scholar]
- 8. Richman-Eisenstat J. B., Jorens P. G., Hébert C. A., Ueki I., Nadel J. A. (1993) Am. J. Physiol. 264, L413–L418 [DOI] [PubMed] [Google Scholar]
- 9. Chung K. F. (2005) Curr. Drug Targets Inflamm. Allergy 4, 619–625 [DOI] [PubMed] [Google Scholar]
- 10. Sakito O., Kadota J., Kohno S., Abe K., Shirai R., Hara K. (1996) Respiration 63, 42–48 [DOI] [PubMed] [Google Scholar]
- 11. Mukaida N., Shiroo M., Matsushima K. (1989) J. Immunol. 143, 1366–1371 [PubMed] [Google Scholar]
- 12. Mukaida N., Morita M., Ishikawa Y., Rice N., Okamoto S., Kasahara T., Matsushima K. (1994) J. Biol. Chem. 269, 13289–13295 [PubMed] [Google Scholar]
- 13. Hoffmann E., Dittrich-Breiholz O., Holtmann H., Kracht M. (2002) J. Leukoc. Biol. 72, 847–855 [PubMed] [Google Scholar]
- 14. Rahman A., True A. L., Anwar K. N., Ye R. D., Voyno-Yasenetskaya T. A., Malik A. B. (2002) Circ. Res. 91, 398–405 [DOI] [PubMed] [Google Scholar]
- 15. Rahman A., Anwar K. N., Uddin S., Xu N., Ye R. D., Platanias L. C., Malik A. B. (2001) Mol. Cell. Biol. 21, 5554–5565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryu J., Pyo H., Jou I., Joe E. (2000) J. Biol. Chem. 275, 29955–29959 [DOI] [PubMed] [Google Scholar]
- 17. Bokoch G. M. (2005) Trends Cell Biol. 15, 163–171 [DOI] [PubMed] [Google Scholar]
- 18. Van Aelst L., D'Souza-Schorey C. (1997) Genes Dev. 11, 2295–2322 [DOI] [PubMed] [Google Scholar]
- 19. Soulet C., Hechler B., Gratacap M. P., Plantavid M., Offermanns S., Gachet C., Payrastre B. (2005) J. Thromb. Haemost. 3, 2296–2306 [DOI] [PubMed] [Google Scholar]
- 20. Mohan S., Koyoma K., Thangasamy A., Nakano H., Glickman R. D., Mohan N. (2007) Am. J. Physiol. Cell Physiol. 292, C362–C371 [DOI] [PubMed] [Google Scholar]
- 21. Perona R., Montaner S., Saniger L., Sánchez-Pérez I., Bravo R., Lacal J. C. (1997) Genes Dev. 11, 463–475 [DOI] [PubMed] [Google Scholar]
- 22. Sulciner D. J., Irani K., Yu Z. X., Ferrans V. J., Goldschmidt-Clermont P., Finkel T. (1996) Mol. Cell. Biol. 16, 7115–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Diebold I., Djordjevic T., Hess J., Görlach A. (2008) Thromb. Haemost. 100, 1021–1028 [PubMed] [Google Scholar]
- 24. Cantley L. C. (2002) Science 296, 1655–1657 [DOI] [PubMed] [Google Scholar]
- 25. Oudit G. Y., Sun H., Kerfant B. G., Crackower M. A., Penninger J. M., Backx P. H. (2004) J. Mol. Cell Cardiol. 37, 449–471 [DOI] [PubMed] [Google Scholar]
- 26. Franke T. F., Kaplan D. R., Cantley L. C. (1997) Cell 88, 435–437 [DOI] [PubMed] [Google Scholar]
- 27. Franke T. F., Kaplan D. R., Cantley L. C., Toker A. (1997) Science 275, 665–668 [DOI] [PubMed] [Google Scholar]
- 28. Osada M., Tolkacheva T., Li W., Chan T. O., Tsichlis P. N., Saez R., Kimmelman A. C., Chan A. M. (1999) Mol. Cell. Biol. 19, 6333–6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kane L. P., Shapiro V. S., Stokoe D., Weiss A. (1999) Curr. Biol. 9, 601–604 [DOI] [PubMed] [Google Scholar]
- 30. Romashkova J. A., Makarov S. S. (1999) Nature 401, 86–90 [DOI] [PubMed] [Google Scholar]
- 31. Chang M. S., Lee W. S., Chen B. C., Sheu J. R., Lin C. H. (2004) Mol. Pharmacol. 66, 561–571 [DOI] [PubMed] [Google Scholar]
- 32. Williams L. M., Lali F., Willetts K., Balague C., Godessart N., Brennan F., Feldmann M., Foxwell B. M. (2008) Mol. Immunol. 45, 2446–2454 [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez E., Kou R., Michel T. (2006) J. Biol. Chem. 281, 3210–3216 [DOI] [PubMed] [Google Scholar]
- 34. Chung S. W., Park J. W., Lee S. A., Eo S. K., Kim K. (2010) Biochem. Biophys. Res. Commun. 396, 748–754 [DOI] [PubMed] [Google Scholar]
- 35. Zheng L., Martins-Green M. (2007) J. Leukoc. Biol. 82, 619–629 [DOI] [PubMed] [Google Scholar]
- 36. Ostrowska E., Reiser G. (2008) Biochem. Biophys. Res. Commun. 366, 1030–1035 [DOI] [PubMed] [Google Scholar]
- 37. Alessi D. R., Andjelkovic M., Caudwell B., Cron P., Morrice N., Cohen P., Hemmings B. A. (1996) EMBO J. 15, 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- 38. John A. E., Zhu Y. M., Brightling C. E., Pang L., Knox A. J. (2009) J. Immunol. 183, 4682–4692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kawkitinarong K., Linz-McGillem L., Birukov K. G., Garcia J. G. (2004) Am. J. Respir. Cell Mol. Biol. 31, 517–527 [DOI] [PubMed] [Google Scholar]
- 40. Azim A. C., Barkalow K., Chou J., Hartwig J. H. (2000) Blood 95, 959–964 [PubMed] [Google Scholar]
- 41. Street M., Marsh S. J., Stabach P. R., Morrow J. S., Brown D. A., Buckley N. J. (2006) J. Cell Sci. 119, 1528–1536 [DOI] [PubMed] [Google Scholar]
- 42. Van Leeuwen F. N., Olivo C., Grivell S., Giepmans B. N., Collard J. G., Moolenaar W. H. (2003) J. Biol. Chem. 278, 400–406 [DOI] [PubMed] [Google Scholar]
- 43. Price L. S., Langeslag M., ten Klooster J. P., Hordijk P. L., Jalink K., Collard J. G. (2003) J. Biol. Chem. 278, 39413–39421 [DOI] [PubMed] [Google Scholar]
- 44. Bokoch G. M., Vlahos C. J., Wang Y., Knaus U. G., Traynor-Kaplan A. E. (1996) Biochem. J. 315, 775–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mainiero F., Soriani A., Strippoli R., Jacobelli J., Gismondi A., Piccoli M., Frati L., Santoni A. (2000) Immunity 12, 7–16 [DOI] [PubMed] [Google Scholar]
- 46. Newcomb D. C., Sajjan U., Nanua S., Jia Y., Goldsmith A. M., Bentley J. K., Hershenson M. B. (2005) J. Biol. Chem. 280, 36952–36961 [DOI] [PubMed] [Google Scholar]
- 47. Reséndiz J. C., Kroll M. H., Lassila R. (2007) J. Thromb. Haemost. 5, 2484–2493 [DOI] [PubMed] [Google Scholar]
- 48. Chen B. C., Liao C. C., Hsu M. J., Liao Y. T., Lin C. C., Sheu J. R., Lin C. H. (2006) J. Immunol. 177, 681–693 [DOI] [PubMed] [Google Scholar]
- 49. Montaner S., Perona R., Saniger L., Lacal J. C. (1998) J. Biol. Chem. 273, 12779–12785 [DOI] [PubMed] [Google Scholar]
- 50. Kim H., Hwang J. S., Woo C. H., Kim E. Y., Kim T. H., Cho K. J., Kim J. H., Seo J. M., Lee S. S. (2008) Exp. Mol. Med. 40, 167–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sanlioglu S., Williams C. M., Samavati L., Butler N. S., Wang G., McCray P. B., Jr., Ritchie T. C., Hunninghake G. W., Zandi E., Engelhardt J. F. (2001) J. Biol. Chem. 276, 30188–30198 [DOI] [PubMed] [Google Scholar]
- 52. Hatada E. N., Krappmann D., Scheidereit C. (2000) Curr. Opin. Immunol. 12, 52–58 [DOI] [PubMed] [Google Scholar]
- 53. Huang W. C., Chen J. J., Inoue H., Chen C. C. (2003) J. Immunol. 170, 4767–4775 [DOI] [PubMed] [Google Scholar]
- 54. Lallena M. J., Diaz-Meco M. T., Bren G., Payá C. V., Moscat J. (1999) Mol. Cell. Biol. 19, 2180–2188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hsu Y. L., Hung J. Y., Ko Y. C., Hung C. H., Huang M. S., Kuo P. L. (2010) Carcinogenesis 31, 587–596 [DOI] [PubMed] [Google Scholar]
- 56. Ogasawara N., Oguro T., Sakabe T., Matsushima M., Takikawa O., Isobe K., Nagase F. (2009) J. Cell. Biochem. 108, 716–725 [DOI] [PubMed] [Google Scholar]