

Abstract
His(E7) to Trp replacements in HbA lead to markedly biphasic bimolecular CO rebinding after laser photolysis. For isolated mutant subunits, the fraction of fast phase increases with increasing [CO], suggesting a competition between binding to an open conformation with an empty E7 channel and relaxation to blocked or closed, slowly reacting states. The rate of conformational relaxation of the open state is ∼18,000 s−1 in α subunits and ∼10-fold faster in β subunits, ∼175,000 s−1. Crystal structures were determined for tetrameric α(WT)β(Trp-63) HbCO, α(Trp-58)β(WT) deoxyHb, and Trp-64 deoxy- and CO-Mb as controls. In Trp-63(E7) βCO, the indole side chain is located in the solvent interface, blocking entry into the E7 channel. Similar blocked Trp-64(E7) conformations are observed in the mutant Mb crystal structures. In Trp-58(E7) deoxy-α subunits, the indole side chain fills both the channel and the distal pocket, forming a completely closed state. The bimolecular rate constant for CO binding, k′CO, to the open conformations of both mutant Hb subunits is ∼80–90 μm−1 s−1, whereas k′CO for the completely closed states is 1000-fold slower, ∼0.08 μm−1 s−1. A transient intermediate with k′CO ≈ 0.7 μm−1 s−1 is observed after photolysis of Trp-63(E7) βCO subunits and indicates that the indole ring blocks the entrance to the E7 channel, as observed in the crystal structures of Trp(E7) deoxyMb and βCO subunits. Thus, either blocking or completely filling the E7 channel dramatically slows bimolecular binding, providing strong evidence that the E7 channel is the major pathway (≥90%) for ligand entry in human hemoglobin.
Keywords: Hemoglobin Myoglobin, Ligand-binding Protein, Molecular Dynamics, Oxygen Binding, Protein Conformation, Protein Structure
Introduction
Hemoglobins represent a very diverse protein family with members occurring in all six kingdoms of life (1). The functions of these proteins differ significantly, ranging from oxygen storage and transport, NO dioxygenation, and nitrite reduction to sensing intracellular levels of diatomic gases for transcriptional regulation and chemotaxis (2). The central chemical events for these functions are movement of ligands into the distal portion of the heme pocket, internal bond formation with the heme iron, and electrostatic stabilization or steric hindrance of the bound ligand by surrounding active site amino acids. Although the quantum mechanical details of bond formation remain to be resolved, there is general agreement on the biophysical mechanisms governing steric hindrance and hydrogen bonding between amino acid side chains and the bound ligand (3–10). In contrast, the pathways for ligands movement from solvent through the protein and into the active site are still under debate.
As recently reviewed by Elber (11), molecular dynamics simulations and other computational approaches have almost uniformly suggested that there are multiple routes for ligand entry into and escape from the active sites of hemoglobins and myoglobins (12–27). Many of these pathways are coincident with apolar cavities, which have been identified as xenon docking sites (16, 17, 28–30). However, as also pointed out by Elber (11), there is little or no direct experimental evidence in support of multiple pathways (31, 32). In the cases of mammalian myoglobins, human hemoglobin, and HbI from Scapharca inaequivalvis, almost all of the experimental evidence suggests that ≥75% of ligands enter and exit the distal pocket through a transient channel between the heme propionates, which is produced by outward rotation of the distal histidine (His(E7)).2 Although ligands do migrate into internal cavities immediately after photodissociation, they appear to return to the distal pocket and escape through the E7 gate (33–41).
The structures of the active sites and the E7 channels in Mb and the subunits of human HbA are very similar, and thus analogous mechanisms for ligand binding are inferred. Until very recently, there were little experimental and computational data regarding the pathways of ligand migration into HbA. Mouawad et al. (15) observed the formation of transient cavities in the α and β subunits of human HbA during simulations of the T to R conformational change and postulated these cavities could allow ligands to diffuse through the globin matrix. Sottini et al. (16, 17) used molecular modeling approaches to find potential xenon binding cavities in human HbA and suggested that ligands could use these apolar voids as pathways to enter or escape the active site. Savino et al. (30) reported crystal structures of Tyr(B10)/Gln(E7) deoxyHbA with xenon atoms partially occupying the sites identified in molecular modeling experiments. Thus, if only the theoretical and structural literature is examined, multiple internal pathways appear to occur for ligand entry into all globins, including HbA, and the heme pocket appears to be accessible from virtually any direction within the globin matrices.
In a recent publication (35), we demonstrated that the association rate constants for O2 and NO binding to distal histidine mutants of isolated α and β subunits depend markedly on the size of the amino acid at the E7 helical position, decreasing from roughly 150 μm−1 s−1 for the Gly or Ala mutants to 5.0–0.5 μm−1 s−1 for the equilibrium deoxygenated forms of the Trp(E7) mutants. However, the most remarkable result was that, when the isolated Trp(E7) αCO and βCO subunits were examined by flash photolysis, multiple bimolecular rebinding phases were observed. One phase was very fast and similar to that seen for Gly(E7) and Ala(E7) mutants, and one was very slow and similar to that seen when the equilibrium deoxy forms of the mutants were mixed with CO in a stopped flow apparatus.
These observations suggest that, after photolysis, the E7 gate is transiently open, with the Trp(E7) side chain located in the solvent, and bimolecular rebinding competes with rotation of the indole side chain back into the E7 channel to close it. This conclusion is supported by geminate recombination studies with E7 mutants of both HbA subunits (35). The fraction of geminate CO recombination in wild type His(E7) and Phe(E7) α and β subunits is small, 0.1–0.2, because the aromatic side chains of these amino acids rapidly relax to closed conformations that partially block access to the iron atom, reducing the rate of internal bond formation. In contrast, the fractions of geminate recombination in Gly(E7), Ala(E7), and Leu(E7) are 4–5-fold larger due to greater accessibility of the iron atom and large rates of internal bond formation. Similar increases in both the rate of internal bond formation and the fraction of geminate rebinding were observed with the α- and βTrp(E7) mutants, indicating that the indole rings are present in open conformations.
In this paper, we have investigated quantitatively the kinetics of CO binding to Trp-58(E7) α and Trp-63(E7) β subunits of human HbA after laser photolysis, when transient unliganded intermediates are generated, and in simple rapid mixing experiments, when the properties of the equilibrium deoxy forms are being measured. Detailed mechanisms were derived to fit the data and assign rate constants for the interconversion of the open and closed conformers and ligand binding to the open and closed states. The stereochemical features of the open and closed forms were examined by determining crystal structures of α(Trp-58(E7))β(WT) deoxyHb and α(WT)β(Trp-63(E7)). The structures of the CO and deoxy forms of Trp-64(E7) Mb were determined as controls.
EXPERIMENTAL PROCEDURES
Protein Expression, Purification, and Preparation of Isolated Subunits
Recombinant hemoglobins Hb α(Trp-58)β(WT) and Hb α(WT)β(Trp-63) were expressed and purified as described previously (35, 42, 43). The sperm whale myoglobin variant Mb Trp-64 was prepared according to the protocol originally designed by Springer and Sligar (44) and modified by Carver et al. (45). Preparation of the isolated Hb subunits was described by Birukou et al. (35). Separation of the α and β chains was assessed by cellulose-acetate strip electrophoresis (Helena Laboratories, Beaumont, TX).
Kinetic Methods
CO binding to equilibrium deoxygenated forms of monomeric αTrp-58, βTrp-63, and Mb Trp-64 was measured in a stopped flow rapid mixing apparatus. CO rebinding to the unliganded intermediates transiently formed after ligand photodissociation was followed after a 0.5-μs excitation pulse from a Phase-R model 2100B dye laser, as described in previous work (35, 46, 47).
Crystallization, Data Collection, and Structure Solution
Recombinant HbCO α(WT)β(Trp-63) was crystallized using the batch method initially designed by Perutz (48). Concentrated HbCO solution (in 10 mm sodium phosphate buffer, pH 7) was mixed with 3.4 m sodium/potassium phosphate, pH 6.7 (precipitant), to yield ∼20 mg/ml HbCO in 2.3 m sodium/potassium phosphate, pH 6.7. 100 μl of the mixture was placed in a glass tube, sealed, and purged with CO, and a drop of toluene was added to the batch. A small aliquot of dilute seeding solution, prepared by finely grinding previously grown WT HbCO crystals in mother liquor, was added to the mutant HbCO mixture to initiate crystal formation. Crystals grew over 2–3 days at room temperature.
Crystals of MbCO Trp-64 were obtained using the method reported by Quillin et al. (49). 10 μl of 60 mg/ml Mb solution was added to 3.2 m ammonium sulfate in 20 mm Tris-HCl, 1 mm EDTA, pH 9.0, to a final [(NH4)2SO4] equal to 2.4–2.6 m. The mixture was placed in a glass tube, seeded as above (using a small amount of a sperm whale WT MbCO seeding suspension), sealed, and purged with CO.
DeoxyHb α(Trp-58)β(WT) was obtained from CO-bound protein by successive oxidation and reduction. First, the met form of the protein was generated by incubating concentrated HbCO with excess potassium ferricyanide in a 5-ml round bottom flask on ice under constant illumination with strong light to promote CO photodissociation. The gas space above the sample was continuously purged with N2. After prolonged incubation, an aliquot of the protein mixture was passed through a Sephadex G-25 column to remove excess ferricyanide and then tested for the residual HbCO in flash photolysis experiments. The whole procedure was repeated until no detectable CO photolysis and rebinding was observed. Fully oxidized protein was buffer-exchanged against 10 mm ammonium phosphate, pH 7, concentrated to ∼60 mg/ml, and degassed. The protein samples were reduced in an N2-filled anaerobic glovebox (Vacuum Atmospheres, Hawthorne, CA) by the addition of a 10-fold excess of sodium dithionite.
Crystallization of recombinant deoxyHb was performed according to the procedure described by Brucker (50), which was adapted from Perutz (48). The precipitant solution was prepared by mixing 0.5 ml of 2 m monobasic ammonium phosphate, 1.5 ml of 2 m dibasic ammonium phosphate and 8 ml of 4 m ammonium sulfate and then degassed. Then all solutions were brought inside the glove box, and precipitant was mixed with concentrated deoxyHb solution to give 10 mg/ml Hb and 2.26–2.80 m ammonium phosphate/sulfate. 120 μl of the resultant mixture was placed in a glass tube and sealed. Large crystals were formed in 2–4 weeks.
Just before data collection, crystals were bathed in a mounting solution containing 10–15% glycerol in the appropriate mother liquor. For CO complexes, the mounting solution was saturated with 1 atm of pure carbon monoxide; for deoxyHb, the mounting solution was saturated with 1 atm of ultrapure N2, and a few grains of sodium dithionite were added to remove any residual oxygen contamination that occurred during transfer.
A cryo-cooling N2 system was used to maintain low temperature (100 K) in the environment of the mounted crystals to reduce radiation damage. The continuous flush with ultracold N2 gas also sustains anaerobic conditions around crystal to prevent O2 binding to deoxyHb and CO replacement by O2 in HbCO and MbCO crystals. Complete x-ray diffraction data sets for each of the globins were obtained from single crystals using copper Kα radiation (λ = 1.5418 Å) from a Rigaku RUH3R rotating anode x-ray generator operated at 50 kV and 90 mA and a Rigaku R-AXIS IV++ image plate detector (Rigaku Americas Co.). Data were collected, scaled, and reduced using d*TREK software (51).
The structure of Trp-64 MbCO was solved by molecular replacement using CNS (52) and the structure of recombinant wild type sperm whale metMb (Protein Data Bank code 2MBW (53)) as a starting model with the solvent molecules omitted. The best solution using data from 15.0 to 4.0 Å yielded a correlation coefficient of 0.65 and an R-factor of 0.35 after rigid body refinement. This model was further refined by conjugate gradient minimization and then simulated annealing, interspersed with manual fitting. The slow cool protocol was also used to calculate unbiased OMIT maps to confirm the correct placement of Trp-64. At this stage, crystallographic refinement was pursued in PHENIX (54). After an initial round of simulated annealing refinement, several macrocycles that included bulk solvent correction and anisotropic scaling of the data, individual coordinate refinement with minimization, and individual isotropic ADP refinement were carried out with maximum likelihood as the target. The CO ligand was modeled into the electron density maps at this point, and refinement was continued with restraints imposed on the expected iron-CO geometry. Solvent molecules were then included, and combined TLS and individual ADP refinements were carried out in the final stages.
The structures of HbCO α(WT)β(Trp-63), deoxyHb α(Trp-58)β(WT), and deoxyMb Trp-64 were solved in CNS by direct Fourier synthesis using the structures of wild type human HbCO (Protein Data Bank code 2DN3 (55)), deoxyHb (Protein Data Bank code 2DN2 (55)), and sperm whale Trp-64 MbCO, respectively, as starting models. The final refinements were carried out in PHENIX as described above. Map fitting and other manipulations with molecular models were performed using the graphic software COOT (56). The accession codes for the models, crystal parameters, and statistics of x-ray data collection and refinement are provided in Table 1. Figs. 4–6 were prepared using the PyMOL Molecular Graphics System, version 1.2r3pre (Schrödinger, LLC).
TABLE 1.
Crystallization and structure determination parameters of Hb and Mb Trp(E7) mutants
| Protein | DeoxyHb α(Trp-58(E7))β(WT) | HbCO α(WT)β(Trp-63(E7)) | MbCO Trp-64(E7) | DeoxyMb Trp-64(E7) |
|---|---|---|---|---|
| Protein Data Bank entry code | 3NMM | 3NL7 | 3NML | 3OGB |
| Crystal data | ||||
| Resolution range (Å) | 52.67–1.60 | 51.27–1.80 | 41.59–1.68 | 23.57–1.60 |
| Space group | P 21 | P 41212 | P 212121 | P 212121 |
| Unit cell parameters (Å, degrees) | a = 62.55, b = 81.83, c = 53.44, β = 99.76 | a = b = 53.21, c = 191.78 | a = 38.93, b = 46.96, c = 89.50 | a = 38.83, b = 47.13, c = 88.93 |
| Reflections (measured/unique) | 176,967/62,570 | 94,990/25,409 | 71,043/19,145 | 78,267/21,466 |
| Completeness (%) | 89.5 (72.3)a | 95.5 (98.8) | 98.9 (94.7) | 96.6 (91.0) |
| 〈I〉/σ〈I〉 | 15.9 (4.2) | 8.8 (3.5) | 19.7 (7.0) | 15.5 (4.1) |
| Redundancy | 2.83 (2.64) | 3.74 (3.46) | 3.71 (3.20) | 3.65 (3.66) |
| Rmerge (%) | 3.8 (18.7) | 7.4 (26.0) | 4.1 (13.4) | 4.3 (26.3) |
| Refinement | ||||
| Resolution range (Å) | 28.82–1.60 | 31.11–1.80 | 22.71–1.68 | 23.57–1.60 |
| R-Factor (%) | 17.3 | 18.6 | 16.6 | 18.0 |
| Rfree (%) | 19.8 | 23.3 | 19.6 | 20.3 |
| Root mean square deviations from ideal values | ||||
| Bond length (Å) | 0.013 | 0.028 | 0.019 | 0.006 |
| Bond angles (degrees) | 1.571 | 1.281 | 1.242 | 1.088 |
| Ramachandran plot | ||||
| Residues in most favorable region (%) | 94.4 | 93.2 | 92.8 | 92.0 |
| Residues in additional allowed region (%) | 5.6 | 6.8 | 7.2 | 8.0 |
a Parameters in parentheses are for the outer resolution shell.
FIGURE 4.

Side views of wild type and Trp(E7) mutants of HbA and Mb. The electron density was contoured at 1.3 σ and is shown as a blue net. The color code for atoms is as follows: green, carbon; blue, nitrogen; red, oxygen; orange, iron. The key residues are shown clockwise: Trp(E7) or His(E7), His(F8), Val(E11), and Leu(B10). The hemoglobin subunits are part of a tetrameric structure with wild type or native subunit partners. A, top panels, native deoxy-α (55) (Protein Data Bank code 2DN2) with the distal pocket water removed and Trp-58(E7) deoxy-α (Protein Data Bank code 3NMM); bottom panels, native CO-β (55) (Protein Data Bank code 2DN3) and Trp-63(E7) βCO (Protein Data Bank code 3NL7). B, top panels, wild type deoxyMb (49) (Protein Data Bank code 2MGL) with the distal pocket water removed and Trp-64(E7) deoxyMb (Protein Data Bank code 3OGB); bottom panels, wild type MbCO (49) (Protein Data Bank code 2MGK) and Trp-64(E7) MbCO (Protein Data Bank code 3NML).
FIGURE 5.

Front view of Trp-58(E7) α subunit in hybrid deoxyHb. Different conformers of α His-45(CE3) are observed in the α1 and α2 subunits of deoxygenated α(Trp-58(E7))β(WT) tetramers (Protein Data Bank code 3NMM). The stick models of the key amino acids are colored as follows: white, carbon; blue, nitrogen; red, oxygen. The atoms of the other amino acids are represented as space-filling yellow spheres. The orange sphere represents the heme iron. The blue dotted line in the α2 subunit represents a hydrogen bond between His-45(CE3) and the heme 6-propionate with an interatomic distance of 3.0 Å.
FIGURE 6.

Models of open, closed, and blocked kinetic states. A, model for one of the blocked states based on the crystal structure of Trp-64(E7) deoxyMb (Protein Data Bank code 3OGB). B, hypothetical model of an open state based on the Trp(E7) βCO structure shown in D, in which the E7 indole side chain has moved further away from the porphyrin ring by rotation about the Cα–Cβ bond to an angle similar to that for the indole side chain in Trp(E7) deoxyMb (A). C, closed conformation observed in the structure of deoxy-α(Trp-58(E7))β(WT) HbA (Protein Data Bank code 3NMM). D, blocked conformer based on the observed structure of α(WT)β(Trp-63(E7)) HbCO with the ligand removed (Protein Data Bank code 3NL7) and blue sphere representing the water molecule interconnecting the indole nitrogen atom with the heme propionates. The color scheme is identical to that in Fig. 5.
RESULTS
Bimolecular CO Rebinding to Mutant/Wild Type Hybrid HbA Tetramers
Dramatic differences are observed for bimolecular CO rebinding to hemoglobin tetramers containing α- versus βTrp(E7) mutants with wild type partner subunits (Fig. 1A). In our dye laser experiments, the observed time courses represent bimolecular rebinding to the various, transiently generated conformational states of unliganded hemoglobin. In the case of wild type HbA at 1000 μm CO, the observed time course is markedly biphasic with a large rapid phase due to ligand binding to the R quaternary state (∼40%) and a slow, accelerating phase due to binding to T state tetramers (∼60%) (Fig. 1A, middle time course). After complete ligand photodissociation, the initial HbCO complex undergoes the R → T state transition at a rate of ∼104 to 105 s−1 (46). Thus, at high [CO], there is competition between rapid CO binding to the R state and the quaternary conformational change that leads to the slowly reacting hemoglobin conformation.
FIGURE 1.

CO binding to Trp(E7) mutants of the α and β subunits of human HbA. A, bimolecular rebinding to ∼100 μm native HbA, α(Trp-58(E7))β(WT), and α(WT)β(Trp-63(E7)) tetramers at [CO] = 1000 μm, after complete photolysis with a 0.5-μs excitation pulse from an Phase-R model 2100B dye laser, which “pumps” all of the ligands into the solvent phase. B, slow phases for bimolecular CO rebinding to ∼50 μm isolated Trp(E7) α and β subunits at 1000 μm CO after complete photolysis; the inset shows the rapid bimolecular binding phases, which occur on a microsecond time scale. C, stopped flow, rapid mixing experiments for CO binding to equilibrium deoxygenated forms of ∼5 μm isolated Trp(E7) α and β subunits at [CO] = 200 μm after mixing. The light gray lines in B and C represent fits to single or multiple exponential expressions. Conditions were 100 mm sodium phosphate, pH 7, 20 °C.
Replacing His(E7) with Trp in one pair of subunits dramatically alters the time course for bimolecular CO rebinding to the resulting hybrid Hb tetramer after laser photolysis. The α(Trp-58)β(WT) HbCO mutant shows almost 80% rapid binding, whereas the time course for CO binding to the α(WT)β(Trp-63) mutant is almost 100% slow. The fast phase for α(Trp-58) β(WT) is itself biphasic with apparent bimolecular R-state rate constants of ∼60 μm−1 s−1 (∼35%) and 5 μm−1 s−1 (∼45%). The latter rate constant is similar to that expected for R-state wild type β subunits (k′CO ≈ 7 μm−1 s−1, (35)). The slow phase for the α(Trp-58)β(WT) mutant suggests a bimolecular rate constant of ∼0.2 μm−1 s−1, which is similar to CO rebinding to the T-state HbA (57, 58). Thus, it appears that the Trp(E7) mutation in α subunits enhances rapid CO rebinding after laser photolysis by speeding up the intrinsic rate of binding to the mutated R-state subunit, which in turn generates partially liganded intermediates that prevent the R to T transition from occurring. This result was unexpected because we thought that the large indole side chain of Trp(E7) would inhibit ligand binding markedly.
The Trp(E7) mutation does significantly decrease the rate of CO binding when it is made in β subunits. As shown in Fig. 1A (upper curve), bimolecular CO rebinding to α(WT)β(Trp-63) is virtually all slow, with little or no rapid phase, and the apparent bimolecular rate, ∼0.1 μm−1 s−1, is roughly 2-fold smaller than that observed for the slow phase of CO rebinding to wild type HbA. Thus, as expected, the Trp(E7) mutation slows CO binding to R-state β subunits, which in turn allows more time for the R → T transition to take place and results in more slowly reacting conformations.
Bimolecular CO Binding to Isolated α and βTrp(E7) Mutants
In order to reduce the complexity of the results and to avoid problems in partial photolysis experiments due to the significant differences in quantum yields of the native versus mutant subunits (35), we isolated the individual α- and βTrp(E7) subunits and examined them separately in both laser photolysis and stopped flow rapid mixing experiments (Fig. 1, B and C) (35). Under these conditions, the subunits exist in high affinity, R-state-like conformations with none of the quaternary constraints found in the tetramer. However, CO binding to isolated αTrp(E7) subunits is still biphasic, with one extremely fast phase (∼90 μm−1 s−1) comprising 79% of the total absorbance change at 1000 μm CO and a small slow phase (0.1 μm−1 s−1). Analogous experiments with βTrp(E7) subunits show a small fast phase, 33%, with an apparent rate constant for CO binding equal to ∼80 μm−1 s−1, and two slow processes, with apparent rate constants equal to 0.7 μm−1 s−1 (49%) and 0.06 μm−1 s−1 (18%). As described by Birukou et al. (35), we attributed the fast phases to bimolecular CO binding to open conformers of the α- and βTrp(E7) mutants in which the Trp(E7) side chain has rotated out into solvent and does not obstruct the E7 channel. The bimolecular rate constants for the fast phases of both subunits approach the diffusion limit and are similar to or larger than those for Gly or Ala(E7) mutants where the E7 tunnel is completely open to the solvent (35). The slowly reacting components were assigned to species in which the pathway for ligand movement into the distal pocket is severely hindered or completely closed by the indole ring of Trp(E7). However, in the previous work, we did not examine in detail the kinetic mechanism for the competition between rapid bimolecular ligand binding to the open conformers and relaxation of the indole side chain to the closed, slowly reacting conformations.
In order to define the kinetic mechanism, we measured the ligand concentration dependence of the observed time courses for bimolecular CO rebinding to the isolated mutant subunits after laser photolysis (Fig. 2). In the case of the αTrp(E7) mutant, the extent of rapid bimolecular rebinding diminishes from 79% at 1000 μm CO to 25% at 50 μm. These results demonstrate that the two phases are not due to heterogeneity in the sample but, instead, to open and closed conformers, which interconvert on the same time scale as ligand binding. We assume that the conformational transition represents rotation of the Trp(E7) side chain into the E7 channel, markedly inhibiting ligand binding from solvent. Thus, at low [CO], where the pseudo-first order rate of ligand binding is relatively small, conformational relaxation from the open to the closed forms occurs almost completely before CO can rebind from solvent. As a result, the observed time course shows primarily a slow phase (Fig. 2A, top trace). At high [CO], the opposite situation occurs; rapid bimolecular rebinding happens before the Trp(E7) rotation can take place, and the time course shows a dominant fast phase (Fig. 2A, bottom trace). When the amplitudes of the fast and slow phases are equal, the rate of the conformational change must be roughly equal to the pseudo-first order rate of fast bimolecular rebinding, which for αTrp(E7) is roughly 18,000 s−1 at ∼250 μm CO, pH 7, 20 °C.
FIGURE 2.

Rapid bimolecular CO rebinding to Trp(E7) α and β subunits after laser photolysis. Concentrations of CO are listed beside each curve, and the lines represent fits to either a single exponential process with an offset or a two-exponential process with a second very slow phase. Experiments were conducted in 100 mm sodium phosphate, pH 7, 20 °C. A, time courses for ∼50 μm isolated Trp-58(E7) αCO. B, time courses for ∼50 μm isolated Trp-63(E7) βCO. Complete photolysis was achieved with a 0.5-μs excitation pulse from a Phase-R model 2100B dye laser.
In contrast to the α mutant, bimolecular CO rebinding to isolated βTrp(E7) subunits is mostly a slow process, and only at very high [CO] is there a significant fraction of the fast phase, 33% at 1000 μm (Fig. 2B). In addition, the rate of the fast phase does not show a large dependence on [CO], increasing from ∼190,000 to 260,000 s−1 over a 20-fold increase in ligand concentration. The observed rate of the fast phase at [CO] → 0 represents the rate of interconversion between open and closed conformations and equals ∼175,000 s−1, which is roughly 10-fold greater than the estimated rate for the same conformational relaxation in αTrp(E7) subunits.
Rapid Mixing Experiments with Unliganded Trp(E7) Subunits
To prove that the closed, slowly reacting forms represent the equilibrium, deoxygenated conformation, we mixed deoxygenated solutions of α- and βTrp(E7) subunits with various [CO] in stopped flow experiments. As shown in Fig. 1C, only slow phases are seen in these mixing experiments, even at very low ligand concentrations, where fast bimolecular binding could be observed. Thus, the slowly reacting closed conformations are completely favored at equilibrium. The open, rapidly reacting unliganded conformers are transient and only observed immediately after laser photolysis.
In the rapid mixing experiments with isolated αTrp(E7) subunits, two slow phases are observed (Fig. 1C). The initial slow phase corresponds to the slow bimolecular phase for CO rebinding observed after photolysis. The second, smaller phase is 5-fold slower. This second slow phase represents a conformer of deoxy-αTrp(E7), which is even more restricted to ligand binding than the initial closed state, forms much more slowly, and is not observed in the laser experiments. Trent et al. (59) observed similar discrepancies between the slow phases observed in laser photolysis versus rapid mixing experiments with the nonsymbiotic hexacoordinate hemoglobin from rice, nsHb1. They proposed that, at equilibrium, deoxygenated rice nsHb1 exists in two structurally distinct, slowly interconverting unliganded forms (59, 60).
In the case of βTrp(E7), two significantly different slow CO binding phases are observed after laser photolysis, suggesting transitions to at least two distinct slowly reacting conformers. In rapid mixing experiments with isolated βTrp(E7) subunits, only the very slow phase seen in the laser experiments is observed (Fig. 1C). The dominant middle phase from the flash photolysis time courses is absent in the stopped flow experiments, implying that at equilibrium there is primarily just one closed βTrp(E7) conformer that reacts very slowly with CO.
Ligand Binding to Mb Trp(E7)
As a control, we re-examined ligand binding to Trp-64(E7) sperm whale Mb to look more carefully for slow interconversions between open and closed states. Both O2 and CO association time courses show no heterogeneity after laser photolysis at either low or high ligand concentrations (Fig. 3A), and the observed bimolecular rate constants are identical to those observed in stopped flow experiments at low [CO] (Fig. 3B) and to those reported previously (61). The absolute value of k′CO for the Trp(E7) Mb is roughly the same as that observed for wild type Mb, ∼0.7 μm−1 s−1 and 10–20-fold smaller than that observed for Gly(E7) and Ala(E7) mutants. Thus, either the Trp(E7) side chain cannot enter and completely close the E7 gate in myoglobin or, if multiple conformations exist, they interconvert very rapidly, resulting in population averaging, with the observed rate of ligand binding being proportional to the equilibrium fraction of open conformers.
FIGURE 3.

Bimolecular CO binding to Trp-64(E7) sperm whale Mb. A, time courses for bimolecular CO rebinding to ∼50 μm Trp-64(E7) Mb after complete photolysis by the 0.5-μs dye laser at [CO] = 1000 and ∼50 μm. The fitted and estimated observed pseudo-first order rate constants were ∼700 s−1 and ∼35 s−1, respectively. B, dependence of the pseudo-first order rate constant on [CO] for CO binding to the equilibrium deoxygenated form of Trp-64(E7) Mb measured in stopped flow, rapid mixing experiments at low ligand concentrations. The slope of the fitted line gives an association rate constant equal to 0.69 μm−1 s−1, which is identical to the value computed from the laser photolysis data in A.
Crystal Structures of Trp(E7) Mutants
To examine the structures of the open and closed conformations of the Trp(E7) side chains, we attempted to crystallize the deoxygenated and CO forms of Hb α(WT)β(Trp-63(E7)), Hb α(Trp-58(E7))β(WT), and sperm whale Mb Trp-64(E7). Despite extensive efforts, high quality crystals of the deoxy form of α(WT)β(Trp-63(E7)) and the CO form of α(Trp-58(E7))β(WT) were not obtained.
Fortunately, the high resolution crystal structures of deoxyHb α(Trp-58(E7))β(WT) (Protein Data Bank code 3NMM), HbCO α(WT)β(Trp-63(E7)) (Protein Data Bank code 3NL7), MbCO Trp-64(E7) (Protein Data Bank code 3NML), and deoxyMb Trp-64(E7) (Protein Data Bank code 3OGB) provide plausible structural models for the closed, blocked, and open conformers of the mutants defined in the kinetic mechanisms proposed under “Discussion.” The crystal properties and statistical parameters for x-ray diffraction data collection and model refinement of all four proteins are given in Table 1, and electron density maps of the active sites are shown in Fig. 4.
Remarkably, the Trp(E7) mutations do not cause any significant global perturbations of the structures of either HbA or Mb. Alignment of deoxy-α(Trp-58(E7))β(WT) with native deoxy-HbA gave average root mean square deviation values of 0.26 Å for the Cα atoms in α chains, and 0.17 Å for the Cα atoms in β subunits. In the case of the CO forms of α(WT)β(Trp-63(E7)) and HbA, the root mean square deviation of the Cα atoms of the β subunits is 0.18 Å, whereas the value for the native versus wild type α chains is 0.10 Å. Thus, the functional changes resulting from the large indole side chain appear to be localized to the E7 channel and active site, which we interpret in terms of greater blocking of ligand entry into the active site and less accessibility of the iron atom.
Deoxy-α(Trp-58(E7))β(WT), a Model for the Closed Conformer
The upper panels in Fig. 4A compare the structures of the ligand binding sites in native versus Trp-58(E7) α subunits in deoxyHbA tetramers. As described by Park et al. (55), the distal histidine side chain moves inward in deoxy-α and -β subunits compared with its position in the O2 or CO complexes, as shown for the native βCO subunit in Fig. 4A (bottom left). In the native deoxy-α structure, an internal water molecule is also present directly above the iron atom and hydrogen bonded to the Nϵ atom of the E7 imidazole side chain (not shown in Fig. 4A (top left)). In the structure of the deoxygenated αTrp-58(E7) subunit, the large indole ring has rotated into the distal pocket, completely closes the E7 channel for ligand entry, and hinders access to the iron atom from any direction. CO binding to this conformation is impossible without outward rotation of the Trp(E7) side chain, and as a result, we have assigned this structure as a model for the closed, very slowly reacting states observed in our kinetic mechanisms.
There are two other major changes in the structure of the distal pocket of deoxy-αTrp-58(E7) compared with that of the native subunit. First, the side chain of Leu(B10) has rotated clockwise, ∼90° around the Cα–Cβ bond, causing an expansion of the distal pocket to make room for the large indole side chain. Second, multiple conformations are observed for the α His-45(CE3) side chain (Fig. 5). In α subunits of native deoxyHbA, the CE3 imidazole is pointing toward the carboxyl oxygen atoms of the heme 6-propionate, forming a strong electrostatic interaction that stabilizes bound heme (55, 62). In the α1 subunit of the α(Trp-58(E7))β(WT) deoxytetramer, the CE3 imidazole has rotated out into solvent and is far away from the heme propionate (Fig. 5, α1 conformer). The space between His(CE3) and the heme propionate is filled with several high occupancy water molecules (positions not shown). However, in the α2 subunit of the mutant tetramer, the position of the His(CE3) is the same as in the native subunit (Fig. 5, α2 conformer). These results imply that the His-45(CE3) interaction with the heme 6-propionate must be disrupted before the Trp(E7) side chain can enter the distal pocket.
In contrast, there is no interaction between the heme 6-propionate and the spatially equivalent but much smaller Ser-44(CD3) side chain in β subunits (55). This lack of stabilization of the heme 6-propionate in β subunits explains the higher rates of hemin dissociation from β subunits in native HbA (62). The lack of electrostatic interaction between the CD3 side chain and the heme 6-propionate may also facilitate inward rotation of the large Trp(E7), accounting in part for the more rapid conformational transitions between the open and closed states in β subunits.
CO Form of α(WT))β(Trp-63(E7)), a Model for a Blocked Conformer
The distal histidine remains in the distal pocket of the α and β subunits of native HbA and sperm whale Mb when ligands are bound (see Fig. 4). The Nϵ atom of His(E7) donates a hydrogen bond to bound ligands in all three globins, which is manifested as decreased rates of ligand dissociation and shifts of the ligand stretching frequencies (3). When His(E7) is replaced by Trp in αCO and βCO subunits, the stretching frequency of the bound ligand, νC-O, increases from ∼1950 to ∼1970 cm−1, indicating the loss of the positive electrostatic field donated by the native distal histidine (35). This result suggests that there is no interaction with either the edge of the aromatic indole ring or the pyrrole N-H atoms and that the side chain is not in the distal pocket near bound CO. This prediction is verified by the structure of the Trp-63(E7) βCO subunit in which the large indole ring is precluded from the active site by the bound ligand (Fig. 4A, bottom right).
The indole side chain of Trp(E7) βCO is located outside of the distal pocket with most of the indole ring exposed to solvent. The nitrogen atom of the indole ring is hydrogen-bonded to a water molecule, which, in turn, is donating hydrogen atoms to the oxygen atoms of both heme propionates. These electrostatic interactions appear to be quite strong and cause a downward displacement of the Cα atom of Trp(E7) by 0.67 Å with respect to the position of the Cα of His(E7) in native βCO subunits. Although not in the distal pocket and interacting with the bound ligand, the βTrp(E7) side chain does physically block the entrance to the E7 channel in the crystal structure, and ligand entry through the E7 pathway requires outward rotation of the indole ring.
Trp-64(E7) MbCO and deoxyMb, Another Model for a Blocked Conformer
A comparison of the crystal structures of wild type and Trp-64(E7) MbCO and deoxyMb is shown in Fig. 4B. In both Trp(E7) Mb structures, the indole ring has moved further away from the heme ring, by additional rotation about the Cα–Cβ bond when compared to Trp(E7) βCO subunits. The entire side chain is in the solvent phase. The aromatic ring has rotated 180° about the Cβ–Cδ bond, causing the phenyl portion of the indole ring to be closer to the heme than the pyrrole. This indole ring orientation is similar to that seen in the closed Trp(E7) deoxy-α structure shown in Fig. 4A (upper right) but is the opposite of that seen for Trp(E7) βCO. The E7 channel is significantly but not completely blocked in the Trp(E7) Mb structures, and small rotations about the Cβ–Cδ bond would open up the channel completely.
The solution FTIR spectrum for Trp-64(E7) MbCO shows two νC-O bands, one at 1969 cm−1 (60%) and another at 1942 cm−1 (40%) (61). The high frequency band probably reflects the properties of the conformers shown in Fig. 4B, whereas the low frequency band implies hydrogen bonding from internal water molecules that may have penetrated into distal pocket and interacted with the bound CO when the Trp(E7) side chain populates an open conformation. However, the spectrum of Trp(E7) metMb(FeIII) has a broad Soret peak at 395 nm, which is indicative of a five-coordinate hemin complex and no coordinated water (49). Thus, the active site of Trp(E7) Mb appears to be primarily apolar with ligand entry partially blocked.
DISCUSSION
Direct measurements of the rates of opening and closing of the distal histidine (E7) gate in mammalian myoglobins and hemoglobin subunits have proved to be difficult and rare. In time-resolved x-ray analyses of photolyzed MbCO, the ligand is observed to move between the distal pocket and the Xe4 and Xe1 cavities on nanosecond time scales, but no rotation of the His(E7) side chain out into solvent is observed as the total ligand electron density disappears to generate a transient unliganded state on nanosecond time scales. Similarly, no observable open conformer is observed as bound ligand electron density reappears after several hundred ms due to bimolecular rebinding from the mother liquor (41, 63, 64). The imidazole side chain of His(E7) does rotate completely out into solvent in low pH wild type and neutral pH F46V MbCO crystal structures, and these open conformers correlate with large bimolecular rates of ligand entry (34, 65–68).
Tian et al. (65, 66) estimated that the rate of opening and closing of the His(E7) gate in Mb is on the order of 1–10 μs−1, which competes with internal geminate rebinding of CO and water entry but not with bimolecular binding from solvent (69, 70). Thus, CO rebinding to WT MbCO after laser photolysis is a simple one-step process on microsecond to millisecond time scales. The unliganded intermediate generated after a 0.5-μs photolysis pulse has spectral and kinetic properties identical to the equilibrium deoxygenated state examined in rapid mixing experiments.
Much less is known about opening and closing of the E7 channel in the subunits of human hemoglobin. Recently, Jenkins et al. (71) reported a crystal structure of a new quaternary substate of high affinity HbA, in which β His-63(E7) rotates into solvent, creating a direct channel to the active site. However, as with Mb, only single bimolecular rebinding phases are observed after laser photolysis of the O2, CO, and NO complexes of isolated wild type and native α and β subunits (35). Thus, any movements of the His(E7) side chain and water entry are very rapid and, like those in Mb, occur on submicrosecond time scales (72).
However, replacing His(E7) with Trp in Hb subunits causes the appearance of complex bimolecular rebinding time courses on microsecond to millisecond time scales (Figs. 1 and 2). The open and closed states of the indole side chain interconvert on microsecond time scales and compete with bimolecular ligand rebinding from solvent (Figs. 1 and 2). In contrast, water movement into the distal pocket is much faster than the rotations of the E7 side chains (72) and probably has little effect on the observed kinetics, particularly in the case of the closed conformers, where the large indole ring excludes solvent from the active site (Figs. 4 and 6).
The kinetic results with the Trp(E7) mutants also provide evidence that the E7 gate regulates bimolecular ligand binding to the α and β subunits. The crystal structures in Figs. 4–6 are the first structural models for these transitions in human HbA. In effect, the large indole ring of Trp(E7) exaggerates the degree of opening and closing caused by the small, native His(E7) imidazole ring, which is why we were able to obtain the blocked and closed crystal structures under physiological conditions.
Simple Linear Conformational Relaxation Mechanisms
The simplest mechanism for explaining the rapid and slow bimolecular binding phases observed after laser photolysis of Trp(E7) αCO is shown in Scheme 1. It is analogous to the mechanism proposed 14 years ago for ligand binding to non-symbiotic plant hemoglobins (73–76), in which the fast process represents competition between His(E7) coordination and ligand binding to an open conformation, whereas the slow phase reflects the displacement of the coordinated distal histidine by ligands incoming from solvent.
SCHEME 1.

Simple linear scheme for ligand binding to Hb subunits.
In αTrp(E7) subunits, bound CO displaces the large indole side chain, pushing it out into solvent. Thus, after photodissociation, the initial conformer is open (Hbopen in Scheme 1, modeled in Fig. 6B), and there is a competition between rapid bimolecular binding and conformational relaxation of the indole side chain back into the distal pocket to form the closed, slowly reacting deoxy-α conformer (Hbclosed in Scheme 1, modeled in Fig. 6C).
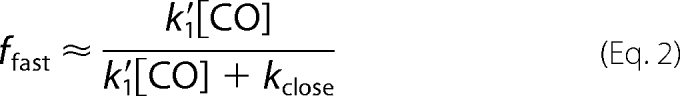 |
 |
Scheme 1, without allowing binding to Hbclosed by alternative routes (k′alt), successfully describes the dependence of the observed fast rate and the fraction of the fast phase on [CO] for isolated Trp(E7) α subunits but cannot reproduce the almost linear dependence of kslow on [CO] (Fig. 7). The hyperbolic dependence of the slow phase rate, kslow, on [CO] (dashed line in Fig. 7B) was obtained by simultaneously fitting the dependences of kfast, ffast, and kslow on [CO] to the expressions in Equations 1–3. The best fit values of k′1, kclose, and kopen are ∼95 μm−1 s−1, ∼15,000 s−1, and ∼120 s−1, respectively. In Scheme 1, a linear dependence of kslow on [CO] can only be obtained when kclose ≫ k′1[CO], but then ffast would be ∼0.0, and the time courses would show completely slow bimolecular rebinding. Thus, Scheme 1 cannot quantitatively describe bimolecular CO rebinding to Hb αTrp(E7) subunits, although the basic idea of a competition between closing of the E7 gate and ligand binding to an open state is qualitatively correct.
FIGURE 7.

Dependences of the observed fast and slow rates for bimolecular CO binding and fraction of the fast phase on [CO] for both laser photolysis and rapid mixing experiments with isolated Trp-58(E7) α subunits. A, dependence of kfast (open circles) and ffast (open squares) on [CO]. B, dependence of kslow (light gray symbols) and kultraslow (dark symbols) on [CO]. The diamonds represent rates determined in stopped flow, rapid mixing experiments with the equilibrium deoxy forms of the Trp(E7) α subunits. The light gray circles for the slow rates represent data from laser photolysis experiments. Black lines represent fits to the model described in Scheme 2, and the fitted parameters are listed in the text after Equation 6. The rates for the ultraslow phase were fit using the model described in the supplementary material (Section A3 (Scheme 2c)). The fitted rate constants for the interconversion of the slow α subunit forms are as follows: ks = 610 s−1, k−s = 200 s−1. The dashed line represents a fit to Scheme 1, which requires a hyperbolic dependence of kslow on [CO]. The experimental conditions were 100 mm sodium phosphate, pH 7, 20 °C.
The linear dependence of kslow on [CO] can be explained by direct ligand binding to the closed αTrp(E7) subunits as indicated in Scheme 1 by the gray dashed arrow and question mark. In this interpretation, ligand binding does not require a transition to an open E7 channel state, and the closed conformer can bind ligands that migrate to the active site via alternative pathways. If the rate of outward movement of Trp(E7) were very small, the observed slow bimolecular rate for CO binding would reflect the speed of recombination via non-E7 gate routes.
A major argument against using Scheme 1 with alternative binding to the Hbclosed state is that the structure of the deoxy-αTrp-58(E7) distal pocket (Figs. 4A, upper right, 5, and 6C) indicates that access to the iron atom is prohibited by the bulky indole side chain, which not only blocks the E7 channel but also lies directly above the iron atom. Thus, any binding event requires outward rotation of the αTrp(E7) side chain to allow access to the iron atom.
Detailed Mechanism for Ligand Binding to Isolated αTrp-58(E7) Subunits
Analysis of the molecular models of open and closed states of αTrp(E7) subunits demonstrates that it is impossible to position the Trp side chain into the E7 channel and the distal pocket by simple rotation about Cα–Cβ (transition from Fig. 6A to Fig. 6C) or Cβ–Cγ (transition from Fig. 6D to Fig. 6C) bonds. Severe steric clashes between the edge of the indole ring and the surface of the heme group occur regardless of whether the pyrrole or phenyl portion of the indole ring is pointing toward the protein interior. Thus, there must be transient expansion of the heme pocket in the open state (conformer Hb*open) to increase the size of the E7 channel and allow inward movement of the large Trp side chain.
A mechanism that can quantitatively describe the kinetics of CO binding to α mutant subunits and take into account transient expansion of the distal pocket is shown in Scheme 2. Here, both open conformers (Hbopen and Hb*open) react rapidly with CO at pseudo-first order rates, k1 = k′1[CO], but only the expanded Hb*open allows the Trp side chain to immediately move into the distal pocket, close the E7 channel, and form the slowly reacting Hbclosed conformer. The linear dependence of the slow phase rate on [CO] requires that the rate of closing of the E7 gate in Hb*open is very rapid compared with the interconversion of open states (i.e. k3 ≫ k2 and k−2).
SCHEME 2.

Mechanism for ligand binding to αTrp-58(E7).
The observation of only slow phases in rapid mixing experiments demonstrates that closing is highly favored and almost complete at equilibrium (k3 ≫ k−3). For these conditions, a steady state assumption can be made for [Hb*open] during both the fast and slow phases after laser photolysis, and k−3 can be neglected during the fast phase. An analytical solution for the formation of HbCO as a function of time during the fast phase can be derived (see supplemental Equations S1–S9) and provides the following expressions for kfast and ffast after laser photolysis of Trp(E7) αCO.
Both expressions show a complex quadratic dependence on [CO]. However, if k3 ≫ k′1[CO], k2, and k−2, only the k3 terms in the numerators and denominators need to be considered. Then kfast ≈ k′1[CO] + k2, and ffast ≈ k′1[CO]/(k′1[CO] + k2), which are analogous to the simple expressions for Scheme 1 in Equations 1–3.
The binding of CO to Hbclosed, which occurs during the slow phase after laser photolysis and in rapid mixing experiments, can be analyzed by assuming a steady state for both open conformers and taking into account the rate of the Hbclosed to Hb*open transition, k−3. A complete derivation for kslow is given in supplemental Equations S11–S21, and the final expression is as follows.
 |
This expression also implies a complex quadratic dependence on [CO], but if k3 ≫ k′1[CO], k−3, k2, and k−2, it simplifies to kslow ≈ k′1[CO]k−3/k3 = k′1[CO]/K3 at high [CO], which prescribes a linear dependence on [CO] and an inverse dependence on the equilibrium constant for the Hb*open to Hbclosed transition, K3 (see supplemental Equations S20 and S21). Thus, the apparent bimolecular rate constant for the slow phase can be interpreted as the rate constant for binding to the open form times the equilibrium fraction of Hb*open, 1/(1 + K3), which equals 1/K3 when K3 is large.
The full expressions in Equations 4–6 were used to successfully fit plots of kfast, kslow, and ffast values versus [CO] for bimolecular CO rebinding to Trp(E7) α subunits after laser photolysis (Fig. 7). The fitted value of k′1 for bimolecular CO binding to the open α forms is 92 μm−1 s−1. The fitted values of k2 and k−2 for the interconversion of open conformers are ∼18,000 and ∼21,000 s−1 and on the same order as the pseudo-first order rates of CO binding to Hbopen. The fitted rate of the Hb*open to Hbclosed transition was 100 times larger, k3 = 2,010,000 s−1, whereas k−3, the rate for opening the closed E7 channel, is much smaller, 2900 s−1. As a result, K3 (k3/k−3) is equal to 690, and the equilibrium fraction of deoxy-α subunits in the open state is 1/(1 + K3) ≈ 1/K3 = 0.0015, which accounts for the 1000-fold smaller bimolecular rate constant for CO binding to the closed conformer, k′slow ≈ 0.1 μm−1 s−1.
The fitting analysis indicates that the absolute values of k3 and k−3 are poorly defined, but k3 must be ≥∼1,000,000 s−1. Thus, the expansion of the distal pocket and inward movement of the indole side chain are concerted processes, with the rate-limiting step being the increase in size of the active site (i.e. k2 ≤ k3 in Scheme 2). In contrast, the ratio of k3/k−3 is well determined by the observed dependence of the slow phase rate on [CO] and must be ∼700. In addition, the detailed shape of the plot of kslow versus [CO] (Fig. 7B, gray symbols) does exhibit curvature at low [CO], reflecting the quadratic expressions for [CO] in Equation 6.
In this interpretation, the slow bimolecular rates observed in rapid mixing and flash photolysis experiments reflect the very small fraction (≤0.001) of open states at equilibrium in the unliganded form instead of reflecting alternative routes with very high kinetic barriers. Unfortunately, distinguishing between ligand binding by alternative pathways and the E7 gate cannot be achieved unambiguously by kinetic data alone. We favor Scheme 2 because the crystal structure suggests that outward rotation of Trp(E7) is needed for any ligand to bind, and the more complex quadratic expression in Equation 6 does emulate the observed curvature in plots of kslow versus [CO] (Fig. 7B, solid line).
Ultraslow Rates of Binding to the Equilibrium Forms of αTrp-58(E7) Subunits
The first slow phase observed for CO binding to equilibrium deoxy-αTrp(E7) subunits in stopped flow experiments matches exactly the slow phase observed in the laser photolysis experiments (Fig. 7B, see gray diamonds in the upper curve). The second ultraslow phase is unique to the mixing experiments. Similar ultraslowly reacting deoxy forms are seen in stopped flow rapid mixing experiments with non-symbiotic, hexacoordinate plant hemoglobins (59) and the heme protein transcription factor CooA (77) with CO. These slowly reacting forms can be modeled by an additional linear transition to an even more hindered, less accessible form of Hbclosed. Thus, it appears that, after the initial closed state is formed, the Trp side chain induces further expansion of the distal pocket to bury itself even deeper in the protein interior, giving rise to the observed ultraslow phase (a more detailed mechanistic description is provided in supplemental Equations S22–S28).
Mechanisms for Ligand Binding to Isolated βTrp-63(E7) Subunits
As shown in Fig. 1B, three phases are observed for CO rebinding to Trp(E7) β subunits after laser photolysis. A small, very fast phase is followed by a dominant, moderately slow middle phase and then a smaller phase with a rate similar to that of the slow phase observed after laser photolysis of Trp(E7) αCO subunits. The rate of the fast phase, kfast, shows a linear dependence on [CO] with a large y intercept, reflecting the rate of conformational relaxation to the slowly reacting species (blocked and closed) (Fig. 8B). The competition between conformational relaxation and rapid ligand rebinding to the open conformer of βTrp(E7) is demonstrated by the increases in ffast and the decreases in the fractions of the middle, fmiddle, and slow, fslow, phases with increasing [CO] (Fig. 8A). The middle and slow phase rates, kmiddle and kslow, both show a linear dependence on [CO]. Thus, in the case of β subunits, two slowly reacting conformers are formed from the unliganded open state immediately after photolysis.
FIGURE 8.

Dependences of the observed rates and the fractions of the fast and slow phases on [CO] for CO binding to isolated βTrp-63(E7) subunits. A, squares represent the observed fractions of the fast (open), middle (gray), and slow (dark) phases (ffast, fmiddle, fslow, respectively) observed after laser photolysis of Trp(E7) βCO subunits (see Figs. 2 and 3) B, open circles represent the observed rate for the fast phase, kfast. C, light gray circles and dark circles represent the rates for the middle, kmiddle, and slow, kslow, phases, respectively, measured after laser photolysis. The light gray diamonds represent the slow phase measured in stopped flow rapid mixing experiments with the equilibrium forms of Trp(E7) deoxy-β subunits. The black lines represent fits to the model described in Scheme 3 for the blocked and closed states, and the fitted parameters are listed in the text after Equation 13. The experiments were conducted in 100 mm sodium phosphate, pH 7, 20 °C.
We have assigned the slowest process to a closed conformation of Trp(E7), similar to Hbclosed for α subunits, in which the indole ring fills both the E7 channel and the distal pocket, restricting ligand access to the iron (see Fig. 6C). The middle phase is assigned to bimolecular ligand rebinding to Hbblocked, a deoxygenated blocked conformer resembling the structure shown in Fig. 6D for the mutant subunit in α(WT)β(Trp-63(E7)) HbCO, in which the indole side chain is located outside the distal pocket, being held in place by a water molecule that interconnects the indole nitrogen atom and two heme propionates. In contrast to the closed state, in Hbblocked, the Trp side chain blocks only the entrance to the E7 gate but not access to the iron from within the heme pocket. Thus, the blocked conformer can directly bind ligands migrating via alternative routes.
The blocked-to-closed transition cannot be achieved by a simple one-step rotation of the indole side chain about the Cα–Cβ bond due to a severe clash with the heme and other residues lining the inner surface of the distal pocket. Instead, the indole side chain in the blocked state shown in Fig. 6D first rotates outward about the Cα-Cβ bond, forming a transient open state similar to that proposed in Fig. 6B and then “flips” its orientation about the Cβ–Cγ bond to generate a conformer analogous to that in Fig. 6A and finally swings back into the distal pocket by rotation about the Cα–Cβ bond to form a closed state similar to that seen for deoxy-αTrp(E7) subunits in Fig. 6C. Thus, Hbclosed cannot be formed directly from the blocked β conformer without first returning to an open conformation (i.e. from Fig. 6D to 6B to 6A to 6C).
A mechanism for ligand binding to βTrp-63(E7), which accommodates the structural interpretations described above, is shown in Scheme 3. The open conformation of the protein is generated immediately after CO photodissociation, and then either CO rebinds rapidly to Hbopen or Hbopen interconverts to Hbblocked and Hbclosed. As in the case of αTrp-58(E7) subunit, the transition from Hbopen to Hbclosed involves the expansion of the initial open state (Hbopen → Hb*open) to accommodate the indole side chain orientation shown in Fig. 6C. In contrast, relaxation of Hbopen to the blocked state (see Fig. 6, B and D) does not require any significant structural perturbations at the active site and can be described as a single-step process.
SCHEME 3.
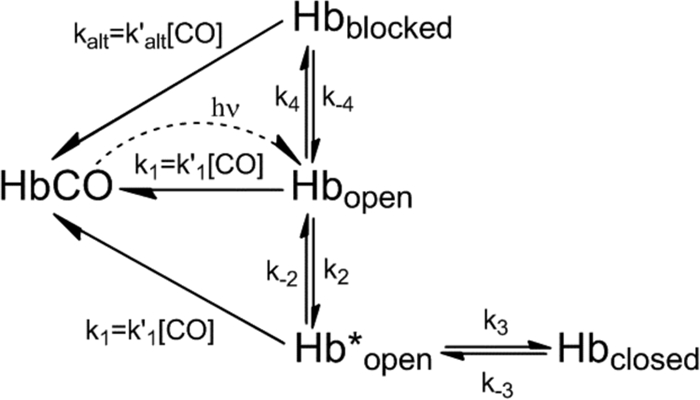
Mechanism for ligand binding to βTrp-63(E7).
In this mechanism, we have modeled CO binding to the blocked state in terms of ligand movement into the active site by alternative routes without requiring opening of the blocked state, and thus, the middle phase rates in Figs. 1B and 8C are postulated to represent k′alt[CO]. Alternatively, these rates could represent the fraction of open states derived from Hbblocked (i.e. 1/(1 + K4), where K4 = k4/k−4). We cannot discriminate between these interpretations of the middle phase and have chosen to use Scheme 3 as a compromise that allows the alternative route interpretation for analyzing βTrp(E7) subunits.
We assumed a steady-state approximation for Hb*open to derive expressions for the dependences of kfast, ffast, fmiddle, and fslow on [CO] and neglected the rates k′alt[CO], k−3, and k−4, because CO binding to the blocked and closed species is significantly slower than the fast phase (see supplemental Equations S29–S44).
When k3 ≫ k1, k2, and k−2, simple expressions for the observed photolysis parameters can be obtained.
 |
 |
 |
Equations 7–10 are analogous to the expressions for the linear mechanism in Scheme 1 and the simplified equations for Scheme 2.
An expression for the rate of CO binding to the Hbblocked state was derived by applying steady-state approximations for both open intermediates (Hbopen and Hb*open) and ignoring CO binding to Hbclosed because kmiddle is observed to be ≥10-fold faster than kslow. The detailed derivation can be found in supplemental Equations S45–S50.
Thus, the middle phase is a sum of two binding processes: 1) ligand binding via alternative routes directly to Hbblocked (expressed as k′alt[CO]) and 2) CO recombination to open conformers originating from Hbblocked (expressed as the k−4(1 −fmiddle) term). Although Equation 11 predicts a complex dependence on [CO] because fmiddle is inversely proportional to [CO], but if k−4 is small, kmiddle is effectively equal to k′alt[CO] at high ligand concentration.
An analytical solution for the slow phase after laser photolysis of Trp(E7) βCO subunits was derived assuming a steady state for Hbopen, Hb*open, and Hbblocked in Scheme 3 (see supplemental Equations S51–S58). When k3 ≫ k1, k−2, k2, and k−3, the expression for kslow can be simplified to the following.
 |
The 1/K3 term represents the fraction of Hb*open formed from Hbclosed when K3 ≫ 1. If k−4 ≪ k′alt[CO] so that k-4 + k′alt[CO] ≈ k′alt[CO], the ratio in parentheses reduces to (k′1[CO] + k4)/(k′1[CO] + k2 + k4), which is ∼(1 − fslow). As a result, the final form of Equation 12 becomes Equation 13.
 |
This is analogous to the rate expression for the middle phase (Equation 11). Thus, the rate for slow CO recombination to Hbclosed is the sum of all possible binding processes: 1) CO binding to Hb*open (Hbclosed → Hb*open → HbCO); 2) CO binding to Hbopen (Hbclosed → Hb*open → Hbopen → HbCO); and 3) CO binding to Hbblocked (Hbclosed → Hb*open → Hbopen → Hbblocked → HbCO).
The dependences of fractions and rates of fast, middle, and slow phases on [CO] were fitted to full expressions for ffast, fmiddle, fslow, kfast, kmiddle, and kslow. The solid lines in Fig. 8 represent best fits with parameter values k′1 = 81 μm−1 s−1, k2 = 51,000 s−1, k−2 = 14,000 s−1, k3 = 3,500,000 s−1, k−3 = 2150 s−1, k4 = 125,000 s−1, k−4 = 100 s−1, and k′alt = 0.61 μm−1 s−1.
Generally, most of these values are similar to the analogous parameters for αTrp(E7) subunit (except for k4, k−4, and k′alt, which are unique to βTrp(E7) subunits). The values of k3 and k−3 are undetermined; however, k3 must be at least 10-fold larger than k4. At the same time, the equilibrium constant for the closed conformer, K3 = k3/k−3, is defined and equals ∼1600. This large value of K3 determines the very small fraction of open conformers compared with Hbclosed and explains the markedly slow bimolecular rate of CO binding to the closed species.
Only slow phases are seen when isolated deoxy-βTrp(E7) subunits are rapidly mixed with CO in stopped flow experiments (Figs. 2C and 8C). Thus, at equilibrium, the closed Trp(E7) conformer is also favored in β subunits, whereas the blocked conformer is a transient state, which is primarily seen after photolysis. This conclusion is supported quantitatively by equilibrium analysis of Scheme 3 (supplemental Equations S59–S62), which indicates that the fraction of closed state for deoxy-βTrp(E7) subunits is ∼0.8–0.9.
Comparison of the Mechanisms for α and β Subunits
In both subunits, ligand binding to the open state is fast (∼90–80 μm−1 s−1) and comparable with that for Gly and Ala(E7) mutants (∼90–50 μm−1 s−1) (35). The rate of the decay of the initial open state (Hbopen) to the closed or blocked states is 10 times faster in β subunits than in α subunits (175,000 s−1 versus 18,000 s−1, respectively). The multiple orientations of His-45(CE3) in the crystal structure of deoxy-α(Trp-58)β(WT) (Fig. 5) provide a possible explanation for these differences. In the wild type structures of either deoxy-α or αCO subunits, His(CE3) forms a salt bridge with the heme 6-propionate, which would have to be disrupted for either outward or inward movement of the distal tryptophan. In β subunits, the equivalent CD3 amino acid is Ser-44. This Ser residue is located too far away to interact strongly with the heme 6-propionate, which could explain the more rapid formation of the expanded open state (Hb*open) in β subunits and account for the higher rates of inward movement of the βTrp(E7) side chain.
This idea is supported by the lack of a closed conformation in Trp(E7) Mb. In wild type Mb, Arg(CD3) forms strong hydrogen bonds with the heme 6-propionate and water molecules at the entrance to the E7 channel, and the heme-7-propionate is fixed in position by strong electrostatic interactions with His-97(FG3), both of which would inhibit expansion of the E7 channel. These interactions reduce the rate of hemin loss markedly in metMb compared with Hb subunits in R state methemoglobin (62) and will also inhibit expansion of the E7 channel, preventing inward movement of the Trp(E7) side chain in Mb.
The lifetime of Hbopen in β subunits is also shortened by the appearance of the unique blocked conformer of βTrp-63(E7). The formation of the blocked conformer in β subunits appears to depend on the orientation of the heme propionates, with the carboxyl atoms lying below the heme plane and the presence of a water molecule between them (Figs. 4A, bottom right). In α subunits and Mb, the existence of this blocked conformer is unlikely, because the heme 6-propionate is involved in strong electrostatic interactions with either His-45(CD3) (α subunits) or Arg-45(CD3) (Mb), both of which keep the 6-propionate “arm” above the heme plane (Fig. 4). In the blocked conformer of Trp(E7) Mb (Figs. 4B and 6A), the indole ring has rotated 180° about the Cβ–Cγ bond, causing the pyrrole nitrogen atom to be far away from the heme propionates.
In general, there appears to be more conformational flexibility in the β subunits. This idea is supported by the observations that β subunits bind and release all of the diatomic gases ∼2 times more rapidly and react much more rapidly with larger ligands than the α subunits (78). The large rates of formation of the blocked and closed states in β subunits add further support to this conclusion.
Opening and Closing of the E7 Gate in Wild Type and Mutant Mb and Hb
Roughly 15 years ago Champion and co-workers (65, 66) examined the interconversion of the open and closed states of the His(E7) side chain in sperm whale MbCO. Using double-pulse flash photolysis methods, they were able to kinetically select and then measure the fraction of open Mb molecules after the initial excitation pulse. The interconversion rate between open and closed forms was estimated to be 1,400,000 s−1 in aqueous solutions of Mb at neutral pH. We assume that a similar and probably greater rate occurs between the open and closed forms of His(E7) in hemoglobin subunits, which are, in general, less rigid, with fewer electrostatic interactions between His(E7), surrounding amino acid side chains, and the heme propionates. Thus, the smaller, naturally occurring His(E7) side chain can move rapidly between open and closed conformational states on submicrosecond time scales, probably without significant expansion of the distal portion of the heme pocket. In contrast, inward movement of the larger Trp(E7) side chain, which requires expansion of the distal pocket, occurs at rates equal to ∼18,000 s−1 and ∼51,000 s−1 in the α and β subunits, respectively, and competes with bimolecular ligand rebinding after laser photolysis.
Morikis et al. (39) and Tian et al. (65, 66) estimated that in Mb, the His gate is open 3–5% of the time at neutral pH. In HbA, similar fractions of open His(E7) states (∼0.07) are obtained from comparisons of CO binding to the wild type Hb subunits (k′CO ≈ 6 μm−1s−1) and to the open forms (∼80–90 μm−1 s−1). The slow phase rate constants, k′CO ≈ 0.1 μm−1 s−1, observed in rapid mixing experiments indicate that the equilibrium fraction of open Trp(E7) conformers is 50-fold smaller (∼0.001) than that observed for His(E7). The larger more apolar Trp side chain prefers the interior, closed position and can be accommodated at equilibrium in the distal pockets of both the α and β subunits.
In contrast to the Hb subunits, the indole side chain in Trp(E7) Mb does not appear to form a closed conformer but remains in a blocked conformation in both the liganded and unliganded states (Fig. 4B, right panels). Rotation about the Cα–Cβ Trp(E7) bond to bring the indole ring into a closed, internal conformation is restricted by close contacts with the heme ring and by significant steric clashes with Val-68(E11). In addition, the cluster of charged amino acids (Arg-45, Asp-60, and Lys-66) and ordered water molecules located in the vicinity of the pyrrole portion of the indole ring may electrostatically stabilize the observed Trp-64(E7) conformation. This structural interpretation is supported by the observation of simple kinetics after laser photolysis of Trp(E7) MbCO (Fig. 3A). The observed bimolecular rate for CO binding to the Mb mutant is close to that for the wild type protein (Table 2), indicating that there is no dramatic increase in hindrance at the iron atom due to filling of the distal pocket with the indole side chain.
TABLE 2.
Comparison of wild type, Gly(E7), and Trp(E7) association rate constants for CO and O2 binding to isolated HbA subunits and Mb
| Globin and conformer | k′CO | Open → closed, blocked | k′O2 | k′entry |
|---|---|---|---|---|
| μm−1s−1 | s−1 | μm−1s−1 | μm−1s−1 | |
| His(E7) α | 5.5a | ≥1,400,000b | 40a | 36a |
| Gly(E7) α | 100a | NAc | 250a | 150a |
| Trp(E7) α | ||||
| Open | 92 | 210a,d | 160a | |
| Closed | 0.12 | 18,000 | ∼0.5a | 0.14a |
| His(E7) β | 7.2a | ≥1,400,000b | 52a | 42a |
| Gly(E7) β | 40a | NA | 160a | 89a |
| Trp(E7) β | ||||
| Open | 80 | 130d | 250a | |
| Blocked | 0.7 | 130,000 | 1.7a | 1.2a |
| Closed | 0.06 | 50,000 | 0.13a | |
| His(E7) Mb | 0.51e | 1,400,000b | 17e | 34f |
| Gly(E7) Mb | 5.8e | NA | 140e | 540f |
| Trp(E7) Mb | 0.70 | NA | 6.2e | 8.6f |
a Rate parameter taken from Birukou et al. (35).
b The interconversion rate for Mb was taken from Tian et al. (65, 66); corresponding parameters for Hb subunits were assumed to be equal to or larger than that for Mb (see “Discussion”).
c NA, not applicable.
d Computed as the slope of the linear dependence of the observed rate on [ligand].
e Rate parameter taken from Springer et al. (81).
f Rate parameter taken from Scott et al. (34).
The bimolecular rate of CO binding to Mb is limited by internal iron-ligand bond formation and is not affected significantly by the bimolecular rate of ligand entry (79, 80). Because the Trp(E7) side chain can only block the entrance to the E7 channel and does not influence iron reactivity, it has little effect on the overall association rate constant for CO binding. In contrast, the bimolecular rate constant for O2 binding to Mb is governed more strongly by the rate of ligand entry. As a result, k′O2 (the oxygen association rate constant) for Trp(E7) Mb is ∼3-fold less than for wild type Mb due to blocking of the entrance to the E7 channel (Table 2).
Scott et al. (34) estimated that the specific bimolecular rate constants for ligand entry into the distal pockets of wild type and Trp(E7) Mb are 34 and 8.7 μm−1 s−1, respectively. In contrast, the slow phases for O2 binding to the Trp(E7) Hb subunits exhibit bimolecular rate constants on the order of 2–5 μm−1 s−1, which is significantly smaller than for Trp(E7) Mb (Table 2) and suggests that the side chain of Trp(E7) can enter the distal pockets of the α and β subunits of human hemoglobin but not in Mb.
CONCLUSIONS
We have demonstrated opening and closing of the E7 gate in HbA on microsecond time scales. Replacing the distal His with the much larger Trp decreases the rates of interconversion between open and blocked or closed conformations ∼10–100-fold in comparison with estimated values for wild type α and β subunits (Table 2). In both Hb subunits, the large indole side chain can enter the distal pocket and form a closed conformer, which obstructs the E7 channel, restricts access to the iron atom, and lowers the bimolecular rate of ligand binding 50–100-fold compared with wild type human HbA. In β subunits, an additional conformation is formed in which the Trp side chain only blocks the entrance to the E7 channel but not access to the iron atom within the distal pocket. Bimolecular ligand binding to this blocked conformer is still 10-fold slower than to wild type protein, suggesting that, if alternative ligand migration pathways occur, those routes are taken ≤10% of the time in β subunits containing His(E7).
Much smaller effects are observed for sperm whale Trp(E7) Mb, where k′entry (the bimolecular rate constant of ligand entry into the distal pocket) is only ∼3–4-fold less than the value for the wild type protein, implying that more ligands could take alternative routes in Mb. Alternatively, this smaller effect can be explained by a more flexible indole ring in the blocked conformer of Trp(E7) Mb in comparison with that in the blocked βTrp(E7) state (see Fig. 6, A and D). In Mb, the Trp(E7) side chain appears to have more rotational freedom about the Cα–Cβ and Cβ–Cγ bonds.
These structural and kinetic data for the Trp(E7) mutants argue strongly that the E7 channel is the major route for ligand entry and escape, particularly for β subunits of human hemoglobin. If the alternative pathways based on xenon binding and computational predictions (30) were major routes for ligand migration in Hb, it would be hard to understand how the blocked conformer of the βTrp(E7) mutant subunit (Fig. 6) could have such large effects on the time courses for bimolecular ligand rebinding after laser photolysis.
Acknowledgments
We acknowledge Anastasiya Birukova for preparing some of the recombinant Hb samples and Mallory Salter for making suggestions to improve the manuscript.
Footnotes
This work was supported, in whole or in part, by National Institutes of Health Grants GM035649 and HL047020. This work was also supported by Robert A. Welch Foundation Grant C0612 (to J. S. O.) and a Welch Foundation predoctoral fellowship (to I. B.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Methods and Derivations.
The atomic coordinates and structure factors (codes 3NMM, 3NML, 3NL7, and 3OGB) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
The helical nomenclature refers to the position of the amino acid along each helix of the globin fold. The helices are designated A–H starting from the N terminus of each subunit, and the positions are numbered from the N- to C-terminal amino acids of each helix. For example, His(E7) refers to the histidine at the seventh position along the E-helix, which, in the α and β subunits, is His-58 and His-63, respectively.
REFERENCES
- 1. Vinogradov S. N., Hoogewijs D., Bailly X., Arredondo-Peter R., Gough J., Dewilde S., Moens L., Vanfleteren J. R. (2006) BMC Evol. Biol. 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vinogradov S. N., Moens L. (2008) J. Biol. Chem. 283, 8773–8777 [DOI] [PubMed] [Google Scholar]
- 3. Phillips G. N., Teodoro M. L., Li T. S., Smith B., Olson J. S. (1999) J. Phys. Chem. B 103, 8817–8829 [Google Scholar]
- 4. Franzen S. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16754–16759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Franzen S. (2002) J. Am. Chem. Soc. 124, 13271–13281 [DOI] [PubMed] [Google Scholar]
- 6. Olson J. S., Phillips G. N. (1997) J. Biol. Inorg. Chem. 2, 544–552 [Google Scholar]
- 7. Kozlowski P. M., Vogel K. M., Zgierski M. Z., Spiro T. G. (2001) J. Porphyrins Phthalocyanines 5, 312–322 [Google Scholar]
- 8. Spiro T. G., Kozlowski P. M. (2001) Acc. Chem. Res. 34, 137–144 [DOI] [PubMed] [Google Scholar]
- 9. Zhang Z., Benabbas A., Ye X., Yu A., Champion P. M. (2009) J. Phys. Chem. B. 113, 10923–10933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ionascu D., Gruia F., Ye X., Yu A., Rosca F., Beck C., Demidov A., Olson J. S., Champion P. M. (2005) J. Am. Chem. Soc. 127, 16921–16934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Elber R. (2010) Curr. Opin. Struct. Biol. 20, 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Elber R., Karplus M. (1990) J. Am. Chem. Soc. 112, 9161–9175 [Google Scholar]
- 13. Elber R., Gibson Q. H. (2008) J. Phys. Chem. B. 112, 6147–6154 [DOI] [PubMed] [Google Scholar]
- 14. Golden S. D., Olsen K. W. (2008) Methods Enzymol. 437, 417–437 [DOI] [PubMed] [Google Scholar]
- 15. Mouawad L., Maréchal J. D., Perahia D. (2005) Biochim. Biophys. Acta 1724, 385–393 [DOI] [PubMed] [Google Scholar]
- 16. Sottini S., Abbruzzetti S., Spyrakis F., Bettati S., Ronda L., Mozzarelli A., Viappiani C. (2005) J. Am. Chem. Soc. 127, 17427–17432 [DOI] [PubMed] [Google Scholar]
- 17. Sottini S., Abbruzzetti S., Viappiani C., Bettati S., Ronda L., Mozzarelli A. (2005) J. Phys. Chem. B 109, 11411–11413 [DOI] [PubMed] [Google Scholar]
- 18. Ruscio J. Z., Kumar D., Shukla M., Prisant M. G., Murali T. M., Onufriev A. V. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 9204–9209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brunori M., Bourgeois D., Vallone B. (2004) J. Struct. Biol. 147, 223–234 [DOI] [PubMed] [Google Scholar]
- 20. Martí M. A., Capece L., Bikiel D. E., Falcone B., Estrin D. A. (2007) Proteins 68, 480–487 [DOI] [PubMed] [Google Scholar]
- 21. Bidon-Chanal A., Martí M. A., Estrin D. A., Luque F. J. (2007) J. Am. Chem. Soc. 129, 6782–6788 [DOI] [PubMed] [Google Scholar]
- 22. Boechi L., Martí M. A., Milani M., Bolognesi M., Luque F. J., Estrin D. A. (2008) Proteins 73, 372–379 [DOI] [PubMed] [Google Scholar]
- 23. Bossa C., Amadei A., Daidone I., Anselmi M., Vallone B., Brunori M., Di Nola A. (2005) Biophys. J. 89, 465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bossa C., Anselmi M., Roccatano D., Amadei A., Vallone B., Brunori M., Di Nola A. (2004) Biophys. J. 86, 3855–3862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cohen J., Schulten K. (2007) Biophys. J. 93, 3591–3600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cohen J., Olsen K. W., Schulten K. (2008) Methods Enzymol. 437, 439–457 [DOI] [PubMed] [Google Scholar]
- 27. Tetreau C., Blouquit Y., Novikov E., Quiniou E., Lavalette D. (2004) Biophys. J. 86, 435–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tilton R. F., Jr., Kuntz I. D., Jr., Petsko G. A. (1984) Biochemistry 23, 2849–2857 [DOI] [PubMed] [Google Scholar]
- 29. Schoenborn B. P., Watson H. C., Kendrew J. C. (1965) Nature 207, 28–30 [DOI] [PubMed] [Google Scholar]
- 30. Savino C., Miele A. E., Draghi F., Johnson K. A., Sciara G., Brunori M., Vallone B. (2009) Biopolymers 91, 1097–1107 [DOI] [PubMed] [Google Scholar]
- 31. Huang X., Boxer S. G. (1994) Nat. Struct. Biol. 1, 226–229 [DOI] [PubMed] [Google Scholar]
- 32. Tomita A., Sato T., Ichiyanagi K., Nozawa S., Ichikawa H., Chollet M., Kawai F., Park S. Y., Tsuduki T., Yamato T., Koshihara S. Y., Adachi S. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 2612–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scott E. E., Gibson Q. H. (1997) Biochemistry 36, 11909–11917 [DOI] [PubMed] [Google Scholar]
- 34. Scott E. E., Gibson Q. H., Olson J. S. (2001) J. Biol. Chem. 276, 5177–5188 [DOI] [PubMed] [Google Scholar]
- 35. Birukou I., Schweers R. L., Olson J. S. (2010) J. Biol. Chem. 285, 8840–8854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blouin G. C., Schweers R. L., Olson J. S. (2010) Biochemistry 49, 4987–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Knapp J. E., Pahl R., Cohen J., Nichols J. C., Schulten K., Gibson Q. H., Srajer V., Royer W. E., Jr. (2009) Structure 17, 1494–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nienhaus K., Knapp J. E., Palladino P., Royer W. E., Jr., Nienhaus G. U. (2007) Biochemistry 46, 14018–14031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morikis D., Champion P. M., Springer B. A., Sligar S. G. (1989) Biochemistry 28, 4791–4800 [DOI] [PubMed] [Google Scholar]
- 40. Olson J. S., Soman J., Phillips G. N., Jr. (2007) IUBMB Life 59, 552–562 [DOI] [PubMed] [Google Scholar]
- 41. Schotte F., Soman J., Olson J. S., Wulff M., Anfinrud P. A. (2004) J. Struct. Biol. 147, 235–246 [DOI] [PubMed] [Google Scholar]
- 42. Shen T. J., Ho N. T., Simplaceanu V., Zou M., Green B. N., Tam M. F., Ho C. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8108–8112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shen T. J., Ho N. T., Zou M., Sun D. P., Cottam P. F., Simplaceanu V., Tam M. F., Bell D. A., Jr., Ho C. (1997) Protein Eng. 10, 1085–1097 [DOI] [PubMed] [Google Scholar]
- 44. Springer B. A., Sligar S. G. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 8961–8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carver T. E., Brantley R. E., Jr., Singleton E. W., Arduini R. M., Quillin M. L., Phillips G. N., Jr., Olson J. S. (1992) J. Biol. Chem. 267, 14443–14450 [PubMed] [Google Scholar]
- 46. Olson J. S., Foley E. W., Maillett D. H., Paster E. V. (2003) Methods Mol. Med. 82, 65–91 [DOI] [PubMed] [Google Scholar]
- 47. Salter M. D., Nienhaus K., Nienhaus G. U., Dewilde S., Moens L., Pesce A., Nardini M., Bolognesi M., Olson J. S. (2008) J. Biol. Chem. 283, 35689–35702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Perutz M. F. (1968) J. Crystal Growth 2, 54–56 [Google Scholar]
- 49. Quillin M. L., Arduini R. M., Olson J. S., Phillips G. N., Jr. (1993) J. Mol. Biol. 234, 140–155 [DOI] [PubMed] [Google Scholar]
- 50. Brucker E. A. (2000) Acta Crystallogr. D Biol. Crystallogr. 56, 812–816 [DOI] [PubMed] [Google Scholar]
- 51. Pflugrath J. W. (1999) Acta Crystallogr. D Biol. Crystallogr. 55, 1718–1725 [DOI] [PubMed] [Google Scholar]
- 52. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 53. Brucker E. A., Olson J. S., Phillips G. N., Jr., Dou Y., Ikeda-Saito M. (1996) J. Biol. Chem. 271, 25419–25422 [DOI] [PubMed] [Google Scholar]
- 54. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Park S. Y., Yokoyama T., Shibayama N., Shiro Y., Tame J. R. (2006) J. Mol. Biol. 360, 690–701 [DOI] [PubMed] [Google Scholar]
- 56. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 57. Mathews A. J., Olson J. S., Renaud J. P., Tame J., Nagai K. (1991) J. Biol. Chem. 266, 21631–21639 [PubMed] [Google Scholar]
- 58. Unzai S., Eich R., Shibayama N., Olson J. S., Morimoto H. (1998) J. Biol. Chem. 273, 23150–23159 [DOI] [PubMed] [Google Scholar]
- 59. Trent J. T., 3rd, Hvitved A. N., Hargrove M. S. (2001) Biochemistry 40, 6155–6163 [DOI] [PubMed] [Google Scholar]
- 60. Halder P., Trent J. T., 3rd, Hargrove M. S. (2007) Proteins 66, 172–182 [DOI] [PubMed] [Google Scholar]
- 61. Li T., Quillin M. L., Phillips G. N., Jr., Olson J. S. (1994) Biochemistry 33, 1433–1446 [DOI] [PubMed] [Google Scholar]
- 62. Hargrove M. S., Whitaker T., Olson J. S., Vali R. J., Mathews A. J. (1997) J. Biol. Chem. 272, 17385–17389 [DOI] [PubMed] [Google Scholar]
- 63. Srajer V., Ren Z., Teng T. Y., Schmidt M., Ursby T., Bourgeois D., Pradervand C., Schildkamp W., Wulff M., Moffat K. (2001) Biochemistry 40, 13802–13815 [DOI] [PubMed] [Google Scholar]
- 64. Aranda R., 4th, Levin E. J., Schotte F., Anfinrud P. A., Phillips G. N., Jr. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 776–783 [DOI] [PubMed] [Google Scholar]
- 65. Tian W. D., Sage J. T., Champion P. M. (1993) J. Mol. Biol. 233, 155–166 [DOI] [PubMed] [Google Scholar]
- 66. Tian W. D., Sage J. T., Champion P. M., Chien E., Sligar S. G. (1996) Biochemistry 35, 3487–3502 [DOI] [PubMed] [Google Scholar]
- 67. Lai H. H., Li T., Lyons D. S., Phillips G. N., Jr., Olson J. S., Gibson Q. H. (1995) Proteins 22, 322–339 [DOI] [PubMed] [Google Scholar]
- 68. Yang F., Phillips G. N., Jr. (1996) J. Mol. Biol. 256, 762–774 [DOI] [PubMed] [Google Scholar]
- 69. Goldbeck R. A., Pillsbury M. L., Jensen R. A., Mendoza J. L., Nguyen R. L., Olson J. S., Soman J., Kliger D. S., Esquerra R. M. (2009) J. Am. Chem. Soc. 131, 12265–12272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Goldbeck R. A., Bhaskaran S., Ortega C., Mendoza J. L., Olson J. S., Soman J., Kliger D. S., Esquerra R. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 1254–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jenkins J. D., Musayev F. N., Danso-Danquah R., Abraham D. J., Safo M. K. (2009) Acta Crystallogr. D Biol. Crystallogr. 65, 41–48 [DOI] [PubMed] [Google Scholar]
- 72. Esquerra R. M., López-Peña I., Tipgunlakant P., Birukou I., Nguyen R. L., Soman J., Olson J. S., Kliger D. S., Goldbeck R. A. (2010) Phys. Chem. Chem. Phys. 12, 10270–10278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Arredondo-Peter R., Hargrove M. S., Sarath G., Moran J. F., Lohrman J., Olson J. S., Klucas R. V. (1997) Plant Physiol. 115, 1259–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Trevaskis B., Watts R. A., Andersson C. R., Llewellyn D. J., Hargrove M. S., Olson J. S., Dennis E. S., Peacock W. J. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 12230–12234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hargrove M. S. (2000) Biophys. J. 79, 2733–2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Smagghe B. J., Hoy J. A., Percifield R., Kundu S., Hargrove M. S., Sarath G., Hilbert J. L., Watts R. A., Dennis E. S., Peacock W. J., Dewilde S., Moens L., Blouin G. C., Olson J. S., Appleby C. A. (2009) Biopolymers 91, 1083–1096 [DOI] [PubMed] [Google Scholar]
- 77. Puranik M., Nielsen S. B., Youn H., Hvitved A. N., Bourassa J. L., Case M. A., Tengroth C., Balakrishnan G., Thorsteinsson M. V., Groves J. T., McLendon G. L., Roberts G. P., Olson J. S., Spiro T. G. (2004) J. Biol. Chem. 279, 21096–21108 [DOI] [PubMed] [Google Scholar]
- 78. Mims M. P., Porras A. G., Olson J. S., Noble R. W., Peterson J. A. (1983) J. Biol. Chem. 258, 14219–14232 [PubMed] [Google Scholar]
- 79. Olson J. S., Phillips G. N., Jr. (1996) J. Biol. Chem. 271, 17593–17596 [DOI] [PubMed] [Google Scholar]
- 80. Quillin M. L., Li T., Olson J. S., Phillips G. N., Jr., Dou Y., Ikeda-Saito M., Regan R., Carlson M., Gibson Q. H., Li H. (1995) J. Mol. Biol. 245, 416–436 [DOI] [PubMed] [Google Scholar]
- 81. Springer B. A., Sligar S. G., Olson J. S., Phillips G. N. (1994) Chem. Rev. 94, 699–714 [Google Scholar]