

Abstract
Glutathione peroxidase 4 (GPx4), an abundant selenoenzyme, is ubiquitously expressed in a tissue-, cell- and differentiation-dependent manner, and it is localized in cytoplasmic, mitochondrial, and nuclear cellular compartments. Here, we report cytoplasmic and nuclear localization of GPx4 in Caco-2 intestinal epithelial cells. Enterocytic differentiation of Caco-2 cells triggers an increase in GPx4 mRNA and protein levels, mediated by enhanced promoter activity. We identified a combined cAMP response element (CREB) and CCAAT/enhancer binding protein (C/EBP) site as critical for the differentiation-triggered GPx4 promoter activity. Induction of GPx4 correlated with C/EBPα transcript levels during differentiation, suggesting a role of C/EBPα as regulator of enterocytic GPx4 expression. Consistent with the in vitro results, GPx4 protein was detected in cytoplasmic and nuclear compartments of enterocytes in human intestinal epithelia. GPx4 is uniformly expressed in colonic crypts and is differentially expressed along the crypt-to-villus axis in the small intestine with a more pronounced expression of GPx4 in the upper villi, which contain fully differentiated enterocytes. These data suggest that intestinal GPx4 expression is modulated by the enterocytic differentiation program, and the results support a direct role of nuclear GPx4 in the (selenium-dependent) prevention of oxidative damage in the gastrointestinal tract.
Keywords: Differentiation, Epithelial Cell, Intestine, Selenium, Transcription Regulation
Introduction
The essential trace element selenium is of fundamental importance to human health, acting through low molecular weight selenium compounds as well as selenocysteine-containing selenoproteins (1). Adequate selenium intake, maintaining optimized expression and/or activity of selenoproteins, has been proposed to be beneficial with respect to prevention of several oxidative stress-related neurodegenerative and cardiovascular diseases and, most notably, cancer (1, 2). Secondary end point analyses of the Nutritional Prevention of Cancer study provided compelling evidence that dietary supplementation with 200 μg selenium/day in form of high selenium yeast lowered the incidence of lung, prostate, and colorectal cancer as well as cancer mortality (3). Since the publication of this landmark trial, the anticarcinogenic capacity of selenium compounds has been further substantiated, particularly with regard to the gastrointestinal tract (4–7). Mice with genetically impaired biosynthesis of selenoproteins due to a mutant selenocysteine transfer RNA gene have been shown to be more susceptible to colon cancer than wild-type mice, thus suggesting a key role for selenoproteins as mediators of the cancer-protective effects of selenium in the colon (5). Selenoproteins that are abundantly expressed in the healthy gastrointestinal tract and putatively relevant in malignant transformation comprise thioredoxin reductases, selenoprotein P, and glutathione peroxidases (GPx)3 (8–11). To date, five selenium-dependent GPx isoenzymes have been identified in humans: GPx1 and GPx2 are highly expressed in the intestinal epithelium, having important yet redundant functions in the intracellular antioxidant defense via reduction of various hydroperoxides with overlapping substrate specificity (12). GPx3 most likely contributes to the extracellular antioxidant defense of the intestinal mucosa, as it is secreted by intestinal epithelial cells and uses various secreted thiols as reductants (14).
GPx4 activity has been detected in colonic and ileal tissue homogenates from mice (44). Among the glutathione peroxidases, GPx4 stands out in terms of its broad substrate specificity and its protective function in a variety of cellular compartments. GPx4 prevents damage of cellular membranes and subsequent cell death, based on its unique capacity (in comparison to the other GPx isoenzymes) to reduce phospholipid hydroperoxides and to repress 12/15-lipoxygenase-derived lipid peroxidation (15). Moreover, a role for GPx4 in the prevention of mutagenesis has been suggested; GPx4 repairs oxidatively damaged DNA by reduction of thymine hydroperoxide (16), and increased levels of mutagenic DNA adducts have been detected in murine embryonic fibroblast cells from GPx4+/− mice (17). A functional variant of the human GPX4 gene has been identified (18), which is associated with increased risk of colorectal adenoma and colorectal cancer (9, 11). The relevance of an adequate GPx4 expression for colon tissue homeostasis in humans notwithstanding information on protein expression, localization, and regulation of GPx4 in enterocytes is limited. Thus, we examined regulation and localization of intestinal GPx4 in differentiating Caco-2 cells, an established in vitro model of the intestinal epithelium, and verified the in vitro results by immunohistochemistry analyses of human small intestine and colon tissue.
EXPERIMENTAL PROCEDURES
Materials
Primary antibodies used in this study were as follows: rabbit anti-GPx4 (AbFrontier, Seoul, Korea); rabbit anti-HNF-4α (hepatocyte nuclear factor 4α; Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-E-cadherin (Santa Cruz Biotechnology); rabbit anti-PARP (Cell Signaling; Beverly, MA) and mouse anti-β-actin (Sigma). Secondary HRP-coupled goat anti-rabbit and anti-mouse IgG antibodies were obtained from Dianova (Hamburg, Germany). Secondary antibodies for immunofluorescence microscopy were Alexa Fluor 488-coupled goat anti-mouse and Alexa Fluor 546-coupled goat anti-rabbit IgG (Invitrogen). Reagents for SDS-PAGE were from Carl Roth GmbH (Karlsruhe, Germany); all other reagents were from Sigma, unless otherwise stated. Primers for real-time PCR and mutagenesis were synthesized by Invitrogen.
Cell Culture
Caco-2 human intestinal epithelial cells (European Collection of Cell Cultures no. 86010202) were kindly provided by Dr. R. Schins (Institut für Umweltmedizinische Forschung, Düsseldorf, Germany) and cultured as described (19). The cells were used for experiments between passage numbers 12 and 25. For differentiation studies, cells were seeded at a density of 32,000/cm2, and samples were taken at various time points ranging from day −2 to day 14 with respect to the day initially reaching confluency (day 0). Culture medium was changed every 2–3 days.
Cell Lysis and Immunoblotting
Nuclear and cytoplasmic fractions were prepared with NE-PER nuclear and cytoplasmic extraction reagents (Pierce). Whole cell lysates for immunoblotting were obtained by lysis with 1% SDS. Immunoblotting was done by standard techniques as described (19, 20): Aliquots of 20–50 μg protein were separated on SDS-PAGE gels, followed by electroblotting onto PVDF membranes (GE Healthcare). Immunodetection was carried out with SuperSignal West Pico Substrate (Pierce) and x-ray film (GE Healthcare).
Immunofluorescence Microscopy
Caco-2 cells were grown on glass coverslips in six-well plates until day 5. Medium was then replaced by serum-free medium supplemented with 200 nm sodium selenite for 24 h. Cells were fixed with methanol, and nonspecific binding sites were blocked with 3% goat serum in PBS with 0.3% Triton X-100. Cells were co-stained with GPx4 and E-cadherin antibodies diluted in PBS with 1% goat serum, followed by incubation with the secondary antibodies diluted in PBS. Coverslips were mounted with ProLong Gold antifade reagent containing DAPI (Invitrogen) and analyzed on a LSM510-Meta confocal microscope (Zeiss, Oberkochen, Germany) equipped with 40/1.3 or 63/1.4 immersion objectives.
Glutathione Peroxidase Activity Assay
Nuclear and cytoplasmic fractions were extracted from proliferating (day −1) and differentiated (day 6) Caco-2 cells. GPx activity in each fraction was assessed as described (20) with slight modifications: GPx assay buffer was supplemented with 3 mm glutathione, 600 milliunits/ml glutathione reductase and 200 μm NADPH (Roche Applied Science). Reaction was started by addition of tert-butyl hydroperoxide or cumene hydroperoxide to a final concentration of 100 μm and detected for a period of 4 min by measurement of NADPH consumption at 340 nm with a Lambda 25 spectrophotometer (PerkinElmer Life Sciences). GPx activity was calculated using the extinction coefficient of NADPH at 340 nm (ϵ = 6.2 mm−1 cm−1).
Alkaline Phosphatase Activity Assay
Caco-2 cells were collected at day −1 and day 6 in 50 mm Tris/HCl buffer (pH 7.0) containing 1% (v/v) Triton X-100 and lysed by sonication. Lysates were assessed for alkaline phosphatase activity as described elsewhere (21), using para-nitrophenyl phosphate (Sigma) as substrate.
Isolation of RNA and Real-time Reverse Transcription-PCR
Total RNA was extracted from Caco-2 cells using the RNeasy Mini kit (Qiagen; Hilden, Germany), and subjected to RT-PCR analysis using a LightCycler quantitative PCR system (version 2.0, Roche Applied Science) as described (19). Expression of β-actin was used as internal normalization control, in consideration of a comparative analysis of reference genes for Caco-2 cell differentiation studies (22). Primers were designed using the Universal ProbeLibrary Assay Design Center (Roche) and are listed in Table 1.
TABLE 1.
Primers (5′–3′) for real-time RT-PCR analysis
| Gene | Forward primer | Reverse primer |
|---|---|---|
| ACTB | CCAACCGCGAGAAGATGA | CCAGAGGCGTACAGGGATAG |
| ATF7 | ACAGAAAAAGACTCAAGGCTATTTAGA | TTGCTGAGCTGTGCTGAATC |
| CBP | TTTCCGGCAGCCTGTAGAT | TCCATGGGATTCTTTACGATG |
| C/EBPα | GGAGCTGAGATCCCGACA | TTCTAAGGACAGGCGTGGAG |
| CREB | GGAGCTTGTACCACCGGTAA | GCATCTCCACTCTGCTGGTT |
| Gpx1 | CAACCAGTTTGGGCATCAG | GTTCACCTCGCACTTCTCG |
| Gpx2 | GCTTCGCTCTGAGGCACAACCAC | GGCGCACAGGGCTCCAAATG |
| Gpx4-all | GCCTTCCCGTGTAACCAGT | GCGAACTCTTTGATCTCTTCGT |
| Gpx4-var2 | GGACACCGTCTCTCCACAGT | GGGGCAGGTCCTTCTCTATC |
| Gpx4-var3 | AGATCCACGAATGTCCCAAG | ATGTCCTTGGCGGAAAACT |
| NFYA | GGCAAGCCCGAGCTAAAC | CCGAGACTCATGCAGGTATTT |
Plasmid Constructs and Luciferase Reporter Gene Assay
A 400-bp promoter fragment of the human GPX4 gene was amplified from isolated genomic DNA of Caco-2 cells using Pfu DNA polymerase (Fermentas; St. Leon-Rot, Germany and the primers: 5′-GAATGGTGGATGAGCCTGTT-3′ (sense) and 5′-CAAGGTCACTGGAGCCTGAG-3′ (antisense). The PCR product served as template in a subsequent PCR with primers carrying recognition sites for BglII and NcoI as described elsewhere (23). After restriction digest, the product of this second PCR was cloned into the BglII/NcoI restriction sites of the firefly luciferase reporter gene plasmid pGL3basic (Promega; Madison, WI). The resulting plasmid, carrying the 400-bp GPx4 promoter fragment (−291/+107), was termed GPx4-luc. Mutant GPx4 promoter constructs with an inactivated CRE or NFYA binding element were generated from the wild-type construct GPx4-luc by site-directed in vitro mutagenesis using Pfu DNA polymerase (Fermentas) and complementary pairs of primers for PCR as well as DpnI restriction enzyme (Fermentas) for digestion of the nonmutated template plasmid. Mutation of the CRE site was achieved with the primers CCGGCCTCCCATTGGCTGAATTCGGCGCGAGCGCTCAACA and TGTTGAGCGCTCGCGCCGAATTCAGCCAATGGGAGGCCGG to generate the plasmid GPx4-CREmut-luc; whereas the NFYA site was mutated using the primers TTCCCACTCCGGCCTCCCTTTAGCTGACGTCGGCGCGAGC and GCTCGCGCCGACGTCAGCTAAAGGGAGGCCGGAGTGGGAA to yield the plasmid GPx4-NFYAmut-luc. Sequences of all plasmids were confirmed by DNA sequencing.
For luciferase reporter gene assays, Caco-2 cells grown in 12-well plates were co-transfected at day −2 or day 5 with 0.7 μg GPx4 promoter-firefly luciferase fusion construct (GPx4-luc, GPx4-CREmut-luc, or GPx4-NFYAmut-luc) and 0.1 μg Renilla luciferase expression plasmid pRL-SV40 together with 2.4 μl FuGENE 6 HD (Roche Applied Science) transfection reagent in serum-free medium. The promoter/enhancerless firefly luciferase vector pGL3basic was co-transfected with pRL-SV40 in control experiments for the determination of background promoter activity. 24 h after transfection, luciferase activities were measured in cell lysates by chemiluminescence detection in a Victor 1420 multilabel counter (PerkinElmer Life Sciences) using the Dual-Luciferase Reporter Assay (Promega) as described (19).
Immunohistochemistry
Samples of non-neoplastic human large and small intestine were collected during abdominal surgery from patients suffering from colonic and pancreatic adenocarcinoma (two men and two women; mean age 67 years, range 60–73 years). The procurement of human material during surgery was in accordance with the guidelines approved by the Ethics comittee of the Medical Faculty of the University of Düsseldorf. Oral informed consent was obtained from each patient before surgery. For immunohistochemical analysis, tissues were fixed in 4% buffered formaldehyde for 24 h, dehydrated, and embedded in paraffin. From paraffin-embedded tissue, 5-μm-thick sections were cut and mounted onto adhesive glass slides (Engelbrecht, Edermünde, Germany). After deparaffinization in xylol, slides were incubated in methanol containing 1% H2O2 for inhibition of endogenous peroxidase activity. Antigen retrieval was performed in citrate buffer (30 mm, pH 6.0) using a Dako Pascal S2800 System (DakoCytomation, Carpintera, CA), and nonspecific binding was blocked by incubation in PBS containing 0.2% saponin (Sigma) and 10% normal goat serum (Vector Laboratories, Burlingame, CA). Slides were incubated with anti-GPx4 antibody (AbFrontier), diluted in PBS containing 0.1% saponin and 2% normal goat serum. Control slides were incubated similarly, but the primary antibody was replaced by a rabbit-IgG (Sigma, I-8140). Subsequently, slides were incubated with diluted goat anti-rabbit-IgG antibody (Dianova), and GPx4 was detected by applying the VECTASTAIN Elite ABC kit (Vector Laboratories). Slides were dried submerged in xylol and cover slipped with DPX (Merck, Darmstadt, Germany). Digital images were produced using a Leica DM 4000B microscope equipped with a Leica DFC 295 camera (Leica; Bensheim, Germany).
Statistics
Values are given as means ± S.D. of three or more independent experiments. Differences between groups were tested for significance by Student's t test with *, p < 0.05; **, p < 0.01, and ***, p < 0.001 as levels of significance.
RESULTS
Differentiation Triggers an Increase in GPx4 Protein Expression and GPx Activity in Cytoplasm and Nucleus of Caco-2 Cells
Differentiation of Caco-2 cells is initiated by contact inhibition and is accompanied by morphological and phenotypical changes similar to the process of intestinal and colonic epithelial cell maturation (24, 25). As part of an extensive genetic reprogramming (26), mRNA and protein levels of antioxidant proteins such as catalase and selenoprotein P increase together with their transcriptional regulators PGC-1α (peroxisomal proliferator-activated receptor-γ coactivator 1α), FoxO1a (forkhead box protein class O1) and HNF-4α during differentiation (19, 27). We hypothesized that expression and activity of the selenoenzyme GPx4 might be regulated in the course of differentiation: Caco-2 cells were cultured over a period of 18 days, ranging from subconfluent proliferating cells to differentiated cells 14 days post confluency, and GPx4 was detected in immunoblots of whole cell lysates as a single band with an approximate molecular mass of 19 kDa. Densitometric analysis revealed a continuous increase in GPx4 expression during differentiation of Caco-2 cells. GPx4 levels were 3-fold higher at 14 days post confluency compared with proliferating cells at day −2. The most pronounced elevation was observed from day −1 to day 6, when GPx4 levels reached a plateau (Fig. 1A); therefore, we decided to compare those two time points in further experiments. Differentiation of Caco-2 cells was validated by determining the enzymatic activity of the brush border enzyme intestinal alkaline phosphatase, a marker of differentiated enterocytes. Specific intestinal alkaline phosphatase activity increased from 18 milliunits/mg (day −1) to 69 milliunits/mg (day 6). Furthermore, the transcription factor HNF-4α, another marker of enterocyte differentiation (19, 28), was robustly induced (Fig. 1B).
FIGURE 1.

GPx4 expression is stimulated in the nuclear and cytoplasmic compartment of Caco-2 cells in the course of differentiation. Caco-2 cells were cultured for the indicated periods of time with respect to the day reaching confluency (day 0). A, GPx4 protein levels were determined in whole cell lysates by immunoblotting, using β-actin as loading control (upper panel). In the densitometric analysis of GPx4 immunoblots, the data represent means ± S.D. of three independent experiments (middle panel); *, p < 0.05 and ***, p < 0.001. Alkaline phosphatase activity as marker of differentiation was measured in proliferating (day −1) and differentiated (day 6) Caco-2 cells (lower panel). ***, p < 0.001. B, detection of GPx4 protein levels in nuclear and cytoplasmic fractions of proliferating and differentiated Caco-2 cells by immunoblotting (upper panel) and densitometric analysis of three independent experiments (middle panel). *, p < 0.05 and **, p < 0.01. Fractionation was monitored by detection of nuclear proteins HNF-4α and PARP1 (lower panel). C, determination of subcellular localization of GPx4 in differentiated Caco-2 cells (day 6) by immunostaining: GPx4 (red), E-cadherin, a plasma membrane-associated protein (green), nuclei stained with DAPI (blue). mU, milliunits; rel., relative; a.u., arbitrary units.
GPx4 has been reported to be localized in nuclear, mitochondrial, and/or cytoplasmic cellular compartments (29). We determined GPx4 localization in Caco-2 cells by separation of nuclear and cytoplasmic compartments followed by immunoblot analysis. As shown in Fig. 1B, GPx4 is expressed at similar levels in the nuclear and the cytoplasmic fraction. Both fractions contained the 19-kDa GPx4 isoform that was also found in whole cell lysates. Consistent with the pattern observed in whole cell lysates, both nuclear and cytoplasmic GPx4 levels were significantly higher in differentiated (day 6) than in proliferating cells (day −1). Proper fractionation of the nuclear and cytoplasmic compartments was controlled by detection of the nuclear proteins HNF-4α and PARP1; both proteins were almost exclusively found in the nuclear fraction, except for a faint PARP1 band at day −1 in the cytoplasmic fraction. In contrast to GPx4 and HNF-4α, PARP1 expression is not affected by differentiation and thus served to confirm equal protein loading. We next analyzed the localization of GPx4 in intact methanol-fixed Caco-2 cells by means of fluorescence microscopy: GPx4 immunoreactivity was found mainly in the cytoplasm though it was co-localized with less intensity to nuclear structures. It was frequently focused around nuclei, pointing to localization of GPx4 in nuclear and/or endoplasmic membranes. In contrast, no GPx4 immunoreactivity was observed on plasma membranes as evident from the E-cadherin control staining (Fig. 1C).
By measuring glutathione peroxidase activities in nuclear and cytoplasmic fractions of Caco-2 cells at days −1 and 6, we tested whether variable GPx4 protein levels impact the degradation rate of hydroperoxides in differentiating Caco-2 cells (Fig. 2). Both tert-butyl hydroperoxide (tBHP) and cumene hydroperoxide (CuOOH) were applied as substrates because GPx4 preferably catalyzes the reduction of CuOOH over tBHP (30). Using tBHP as substrate, the GPx activity in the cytoplasmic fraction of proliferating Caco-2 cells (day −1) was 4-fold higher than in the nuclear fraction. Differentiation of Caco-2 cells caused a moderate but nonsignificant increase in GPx activity in both cellular compartments (Fig. 2A). When CuOOH was used as substrate, we measured a substantially different pattern of GPx activity. Proliferating Caco-2 cells degraded CuOOH at a similar rate as tBHP, but the GPx activity against CuOOH was markedly increased in the nuclear (+ 480%, p < 0.001) as well as in the cytoplasmic (+ 360%, p < 0.01) fraction of differentiated cells at day 6 (Fig. 2B).
FIGURE 2.

Nuclear and cytoplasmic GPx activity in differentiating Caco-2 cells. GPx activities in nuclear and cytoplasmic fractions of proliferating (day −1) and differentiated (day 6) Caco-2 cells were measured with tert-butyl hydroperoxide (A) or cumene hydroperoxide (B) as substrate. Experiments were performed in triplicate, the data representing means ± S.D. **, p < 0.01 and ***, p < 0.001.
Analysis of GPx4 mRNA Expression in Differentiating Caco-2 Cells
The structural organization of the human GPX4 gene (NCBI Gene ID 2879) has been elucidated elsewhere (31) and is depicted schematically in Fig. 3A. Three discrete human GPx4 mRNA variants may result from the use of two alternative transcription initiation sites and from alternative splicing (Fig. 3B). The relative abundance of the three mRNA variants in Caco-2 cells was analyzed by real-time RT-PCR, employing three pairs of primers that either bind to a particular (GPx4-var2 or GPx4-var3) or all (GPx4-all) variant(s). GPx4 mRNA variants 2 and 3 have unique partial sequences not shared by the other variants, but this does not apply to variant 1 (Fig. 3B). We therefore calculated the relative abundance of mRNA variant 1 by subtracting the amounts of the separately quantified variants 2 and 3 from the total amount of all three GPx4 mRNA variants quantified with primers GPx4-all. GPx4 mRNA variant 1 (NM_002085.3) was found to be by far the predominant isoform in Caco-2 cells (Fig. 3C).
FIGURE 3.

Analysis of GPx4 mRNA variants in Caco-2 cells. A, schematic representation of the exon-intron structure of the human GPX4 gene according to Ref. 31. B, schematic drawing of the composition of the three human GPx4 mRNA variants, resulting from use of two alternative transcription initiation sites and from alternative splicing. The binding sites of the three primer pairs, designed to distinguish between the GPx4 mRNA variants, are depicted. C, comparison of mRNA copy numbers (GPx4/β-actin) of total GPx4 and GPx4 variants 2 and 3 in proliferating (day −1) Caco-2 cells (left panel), and deduced calculation of the relative abundance of the three GPx4 mRNA variants (right panel). Data represent means ± S.D. of four independent experiments.
As the observed increase in GPx4 protein expression in the course of differentiation might result from transcriptional induction, we compared GPx4 mRNA expression (all variants) at days −1 and 6. Transcript levels of GPx4 were 4-fold higher in differentiated than in proliferating cells (Fig. 4). Selenoprotein transcripts are differentially affected by selenium availability, being rapidly degraded, unaffected, or even stabilized during periods of selenium deficiency (32). GPx4 mRNA levels remained unchanged whether the cells were cultured in selenium-deficient or in selenium-supplemented (200 nm sodium selenite) medium, confirming previous findings obtained by Northern blotting (33). Control experiments revealed that selenite did not affect Caco-2 cell differentiation as determined by alkaline phosphatase activity (data not shown).
FIGURE 4.
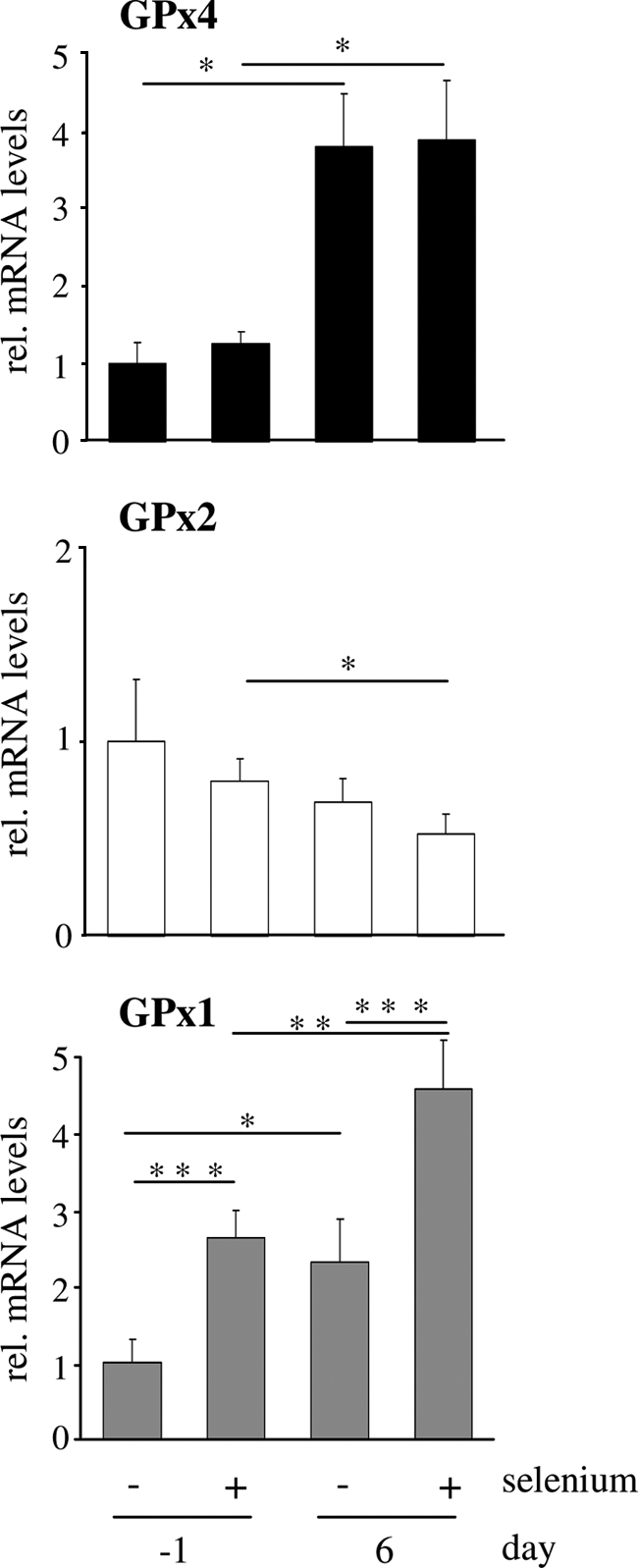
Effect of selenium status and differentiation on mRNA levels of GPx4 in comparison to GPx1 and GPx2. Proliferating (day −2) and differentiated (day 5) Caco-2 cells were cultured for 24 h in serum-free medium with or without supplementation with 200 nm sodium selenite. GPx4 mRNA levels (upper panel) as well as GPx2 (middle panel) and GPx1 mRNA levels (lower panel) were determined by quantitative PCR with normalization against β-actin. Data represent means ± S.D. of four independent experiments. *, p < 0.05; **, p < 0.01; and ***, p < 0.001. rel., relative.
We next explored how GPx4 in Caco-2 cells is regulated in comparison with the two other intracellular GPx isoenzymes, GPx1 and GPx2. Enterocytic differentiation was accompanied with significant up- and down-regulation of GPx1 and GPx2 mRNA levels, respectively. This pattern extended to the effect of selenium supplementation, which caused a marked stimulation of GPx1 and a slight attenuation of GPx2 mRNA levels (Fig. 4). The different regulation of the glutathione peroxidase isoenzymes is intriguing with regard to the opposite distribution of GPx1 and GPx2 along the crypt to villus axis in the intestine and their reciprocal expression pattern in tumorous versus nontumorous human colon tissue, as reported by others (10, 12, 44).
Regulation of GPx4 Promoter Activity in Differentiating Caco-2 Cells
The observed increase in GPx4 mRNA levels prompted us to analyze the GPx4 promoter and to identify the transcription factor binding site(s) involved in its presumed activation during Caco-2 cell differentiation. We cloned a fragment of the human GPx4 promoter, spanning 400 bp around the transcriptional start site of mRNA variant 1, into the pGL3basic luciferase reporter gene vector (Fig. 5A). This region is essential for basal GPx4 promoter activity as shown in a series of 5′-deletion constructs (23). The wild-type promoter construct (GPx4-luc) was active in Caco-2 cells, yielding >100-fold higher reporter gene activity as compared with the background activity driven from the promoter/enhancerless control vector pGL3basic. The relative promoter activity of GPx4-luc was elevated by 3-fold at day 6 versus day −1 (Fig. 5B). Analysis of the cloned GPx4 promoter fragment using the TRANSFAC database (34) revealed putative binding sites for the transcription factors SP1 (stimulating protein 1), MZF1 (myeloid zinc finger 1), GATA1/2 (GATA binding protein), CREB (cAMP response element binding protein), and NFYA (nuclear transcription factor Y α). The latter two sites are located adjacent to each other in close proximity to the GPx4 transcription initiation site (−48/−35) of exon E1, in a region critical for basal promoter activity (23, 35). The binding site for CREB (−42/−35) overlaps with a binding site for C/EBP transcription factors (35); in the following, we refer to this combined element as the CRE site (Fig. 5A). We applied site-directed in vitro mutagenesis to inactivate the CRE site and the binding site for NFYA. Inactivation of either binding site had no impact on GPx4 promoter activity in proliferating Caco-2 cells. However, mutation of the CRE site significantly impaired the induction of GPx4 promoter activity during Caco-2 cell differentiation, whereas inactivation of the NFYA site had no effect. It is concluded that the CRE site, but not the NFYA site, represents a relevant cis-regulatory element for the differentiation-triggered induction of GPx4.
FIGURE 5.

A combined CREB and C/EBP (CRE) element located near the transcriptional start site contributes to differentiation-triggered induction of GPx4 promoter activity. A, shown is a schematic drawing depicting the locations of the adjacent NFYA and CRE elements in the human GPx4 promoter. The wild-type sequence and the base exchanges (in boldface letters) in the mutated luciferase (luc) reporter gene constructs are specified. B, proliferating (day −2) and differentiated (day 5) Caco-2 cells were co-transfected with the reporter gene constructs GPx4-luc (containing a 400-bp fragment of wild-type human GPx4 promoter) or one of the mutated constructs GPx4-CREmut-luc and GPx4-NFYAmut-luc (containing the GPx4 promoter fragment mutated in either site) and the Renilla luciferase control plasmid pRL-SV40. 24 h post-transfection, relative (rel.) firefly luciferase activity was determined. Data are given as means ± S.D. of three independent experiments performed in duplicates. **, p < 0.01 and ***, p < 0.001. C, Caco-2 cells were assessed for mRNA levels of NFYA, CREB, CBP, ATF7, and C/EBPα by quantitative PCR with β-actin as normalization control. Data are given as means ± S.D. of four independent experiments and represent the mRNA levels of the transcriptional factors in differentiated cells (day 6) in relation to proliferating cells (day −1). **, p < 0.01. The dashed line indicates the mRNA expression levels of the transcription factors in proliferating Caco-2 cells (day −1) set at 1.
Next, we assessed the expression levels of NFYA and the following CRE site-specific transcription factors and coactivators in Caco-2 cells at days −1 and 6: CREB, CBP, ATF7 (activating transcription factor 7), and C/EBPα. Differentiation had no significant effect on CBP and ATF7 mRNA levels but resulted in 3-fold up-regulation of CREB and 11-fold up-regulation of C/EBPα in Caco-2 cells at day 6. In addition, NFYA mRNA levels were stimulated 3-fold in the course of differentiation (Fig. 5C).
Localization of GPx4 in Human Small Intestine and Colon
Immunohistochemistry of non-neoplastic human samples, collected during abdominal surgery, demonstrated expression of GPx4 in all cells of the intestinal epithelium, including enterocytes, goblet cells, and neuroendocrine cells. GPx4 immunoreactivity was present in both the nucleus and the cytoplasm and displayed a more pronounced expression in the upper part of the villi in the small intestine and the upper parts of the crypts in the large intestine (Fig. 6).
FIGURE 6.

Localization of GPx4 in the human small and large intestine. Analysis of paraffin sections from small intestine (A) and colon (B) by immunohistochemistry demonstrates expression of GPx4 in cytoplasmic and nuclear compartments of enterocytes in human intestinal epithelia.
DISCUSSION
Glutathione peroxidase 4 is ubiquitously expressed with highest levels found in the testis (36). In the murine intestine, GPx4 mRNA is one of the most abundant selenoprotein transcripts (8); genetic variant analysis of the human GPX4 gene links it to malignant disease risk in the gastrointestinal tract (9, 11). Investigation of intestinal GPx4 has so far been limited to the mRNA level (33, 36) and to the measurement of GPx4 activity in mouse tissue homogenates (44). To our knowledge, we provide here the first detailed characterization of GPx4 expression in human intestinal epithelia and its transcriptional induction during enterocytic differentiation of Caco-2 cells.
We applied differentiating Caco-2 cells as an in vitro model for small and large bowel-like enterocytes and demonstrated the presence of one GPx4 isoform (∼19 kDa) in the cytoplasm and nucleus of both proliferating and differentiated cells. Nuclear GPx4 expression is most prominent in spermatozoa (29), in which a testes-specific isoform with a nuclear transit peptide tag is encoded by GPx4 mRNA variant 3 (NM_001039848.1). We demonstrated the absence of this isoform in Caco-2 cells by RT-PCR analysis with primers discriminating between the three human GPx4 mRNA variants. Instead, the variant 1 (NM_002085.3) encoding mitochondrial and nonmitochondrial GPx4 is almost the only variant expressed in Caco-2 cells. Nuclear and cytoplasmic localization of the non-nuclear GPx4 isoform in Caco-2 cells is consistent with the cellular distribution of ectopically expressed homologous rat GPx4 isoforms that do not contain a nuclear leader sequence (37). Our finding of increased GPx4 mRNA and protein expression during enterocytic differentiation of Caco-2 cells adds GPx4 to a group of antioxidant enzymes, such as selenoprotein P, catalase, and superoxide dismutase (19, 27, 38), whose expression and/or activity is induced during Caco-2 cell differentiation. The elevation of GPx4 levels in differentiating Caco-2 cells was paralleled by increased degradation rates of the GPx substrates tBHP and, most notably, CuOOH. Given that GPx4 preferably reduces CuOOH over tBHP (30), we conclude that differentiation of Caco-2 cells is accompanied by an increase of GPx4 activity in both nuclear and cytoplasmic compartments.
In agreement with a previous study (33), GPx4 mRNA levels in Caco-2 cells were resistant to selenium depletion. In contrast, GPx1 and GPx2 mRNA levels were down- and up-regulated in the absence of supplemental selenium, respectively. A constant level of GPx4 mRNA during periods of limited selenium availability as against the concomitant degradation of other GPx isoforms such as GPx1 reflects the high ranking of GPx4 in the so-called selenoprotein hierarchy (32) and argues for an important role of this protein in enterocytes. Intriguingly, a function of GPx4 as a candidate mediator of selenium-dependent chemoprevention in the colon arises from studies on the association of a single nucleotide polymorphism (rs713041) in the human GPX4 gene with colorectal adenoma and colorectal cancer risk (9, 11, 18). The enzymatic capacity of GPx4 extends beyond those of other hydroperoxide-degrading GPx isoforms. GPx4 is well established as a regulator of lipid hydroperoxide and arachidonic acid metabolism, and is capable of detoxifying a broad range of hydroperoxide substrates, including complex lipid hydroperoxides in membranes and the mutagenic nucleobase lesion thymine hydroperoxide (16, 29, 39). In the colon, malignant transformation is frequently triggered by elevated levels of reactive oxygen species, causing an increased rate of epithelial cell turnover and the formation of mutagenic DNA adducts. The enterocytic expression of GPx4, as shown here by immunohistochemistry on sections of human jejunum and colon, likely contributes to the antioxidative capacity of the intestinal mucosa and might counteract colon carcinogenesis and inflammation. GPx4 exhibited the highest enzymatic activity among a number of DNA hydroperoxide-reducing enzymes tested against thymine hydroperoxide as a substrate in vitro (16). These findings gave rise to the hypothesis that GPx4 may protect DNA integrity by prevention of lipid peroxidation-induced DNA damage, e.g. etheno-nucleotide adducts, and via direct repair of oxidatively damaged DNA adducts such as thymine hydroperoxide (16, 40). The nuclear localization of GPx4, as presented in this study, supports this notion for enterocytes. We therefore propose that optimized GPx4 levels in enterocytes might be related to selenium-dependent prevention of colorectal carcinogenesis. However, the function and physiological relevance of GPx4 in enterocytes needs to be established by knockdown strategies in vitro and cell type-specific gene disruption in vivo, respectively.
Activity of the GPx4 promoter was up-regulated at day 6 compared with day −1, and we therefore presumed that transcriptional induction is a major cause for the increase in GPx4 mRNA and protein expression and GPx activity during Caco-2 cell differentiation. To identify cis-regulatory elements in the human GPx4 promoter involved in this effect, we focused on two previously described functional binding sites for transcription factors (23, 35): a NFYA site and an adjacent CREB overlapping with a C/EBP site, designated here as the CRE site. We inactivated these two cis-acting elements by introducing site-directed mutations that have been shown to effectively diminish the activity of other promoters with identical binding sites (45, 46). Neither site appeared to be relevant for basal GPx4 promoter activity in proliferating Caco-2 cells, whereas a wild-type CRE site was necessary for full promoter activation during differentiation. Mutation of the NFYA site has been demonstrated to lower GPx4 promoter activity in A431 epidermoid cells, but it had no effect in nondifferentiated HL60 cells. Opposite effects occur at the combined CREB and C/EBP (CRE) site. In A431 cells, the CRE site apparently does not bind to CRE or C/EBP transcription factors (23); however, it is capable of binding to C/EBPϵ in HL60 cells (35). Thus, it obviously depends on the cell type and likely reflects different availabilities of the transcription factors, their interacting partners and activation states, whether binding sites for the NFYA and/or CREB and C/EBP in the GPx4 promoter are functional.
To figure out which transcription factor(s) are involved in induction of GPx4 via the combined CREB and C/EBP (CRE) site, we assessed changes in the mRNA levels of CRE site-specific transcription factors and coactivators during enterocytic differentiation of Caco-2 cells. Even though we observed a 3-fold stimulation of CREB mRNA levels, total and Ser133-phosphorylated protein levels of CREB have been shown to be rather unchanged during Caco-2 cell differentiation (41). Moreover, neither the CREB coactivator CBP nor the CREB-related transcription factor ATF7 were influenced in the course of differentiation, arguing against a role for protein kinase A/CREB signaling in the differentiation-triggered induction of GPx4. Three isoforms of C/EBP have been detected in the murine intestine: C/EBPα, C/EBPβ, and C/EBPδ (42). C/EBPα is differentially expressed along the crypt-to-villus axis with highest expression in postmitotic villus-associated enterocytes in the adult mouse intestine, and it has been discussed as a mediator of enterocyte differentiation marker expression (43). We found strongly up-regulated C/EBPα mRNA levels in differentiated Caco-2 cells. The results of the promoter studies and the differentiation-induced changes in the pattern of transcription factors make it appear very likely that up-regulation of C/EBPα is responsible for transcriptional induction of GPx4 in differentiating Caco-2 cells via the combined CREB and C/EBP (CRE) binding site. Similarly, C/EBP isoforms stimulate expression of superoxide dismutase genes (13), and the elevation of superoxide dismutase enzymatic activity during early stages of Caco-2 cell differentiation (38) might be due to the up-regulation of C/EBPα.
This work was supported by Deutsche Forschungsgemeinschaft Grants STE 1782/2-1 and SFB 575/B4.
- GPx
- glutathione peroxidase
- tBHP
- tert-butyl hydroperoxide
- CREB
- cAMP-responsive element binding protein
- CBP
- CREB binding protein
- C/EBP
- CCAAT/enhancer binding protein
- CuOOH
- cumene hydroperoxide
- NFYA
- nuclear transcription factor Y α
- PARP1
- poly(ADP ribose)polymerase-1
- luc
- luciferase.
REFERENCES
- 1. Steinbrenner H., Sies H. (2009) Biochim. Biophys. Acta 1790, 1478–1485 [DOI] [PubMed] [Google Scholar]
- 2. Rayman M. P. (2009) Biochim. Biophys. Acta 1790, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 3. Clark L. C., Combs G. F., Jr., Turnbull B. W., Slate E. H., Chalker D. K., Chow J., Davis L. S., Glover R. A., Graham G. F., Gross E. G., Krongrad A., Lesher J. L., Jr., Park H. K., Sanders B. B., Jr., Smith C. L., Taylor J. R. (1996) JAMA 276, 1957–1963 [PubMed] [Google Scholar]
- 4. Davis C. D., Uthus E. O. (2002) J. Nutr. 132, 292–297 [DOI] [PubMed] [Google Scholar]
- 5. Irons R., Carlson B. A., Hatfield D. L., Davis C. D. (2006) J. Nutr. 136, 1311–1317 [DOI] [PubMed] [Google Scholar]
- 6. Jacobs E. T., Jiang R., Alberts D. S., Greenberg E. R., Gunter E. W., Karagas M. R., Lanza E., Ratnasinghe L., Reid M. E., Schatzkin A., Smith-Warner S. A., Wallace K., Martínez M. E. (2004) J. Natl. Cancer Inst. 96, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 7. Reid M. E., Duffield-Lillico A. J., Sunga A., Fakih M., Alberts D. S., Marshall J. R. (2006) Int. J. Cancer 118, 1777–1781 [DOI] [PubMed] [Google Scholar]
- 8. Hoffmann P. R., Höge S. C., Li P. A., Hoffmann F. W., Hashimoto A. C., Berry M. J. (2007) Nucleic Acids Res. 35, 3963–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Méplan C., Hughes D. J., Pardini B., Naccarati A., Soucek P., Vodickova L., Hlavatá I., Vrána D., Vodicka P., Hesketh J. E. (2010) Carcinogenesis 31, 1074–1079 [DOI] [PubMed] [Google Scholar]
- 10. Murawaki Y., Tsuchiya H., Kanbe T., Harada K., Yashima K., Nozaka K., Tanida O., Kohno M., Mukoyama T., Nishimuki E., Kojo H., Matsura T., Takahashi K., Osaki M., Ito H., Yodoi J., Murawaki Y., Shiota G. (2008) Cancer Lett. 259, 218–230 [DOI] [PubMed] [Google Scholar]
- 11. Peters U., Chatterjee N., Hayes R. B., Schoen R. E., Wang Y., Chanock S. J., Foster C. B. (2008) Cancer Epidemiol. Biomarkers Prev. 17, 1144–1154 [DOI] [PubMed] [Google Scholar]
- 12. Esworthy R. S., Swiderek K. M., Ho Y. S., Chu F. F. (1998) Biochim. Biophys. Acta 1381, 213–226 [DOI] [PubMed] [Google Scholar]
- 13. Miao L., St. Clair D. K. (2009) Free Rad. Biol. Med. 47, 344–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tham D. M., Whitin J. C., Kim K. K., Zhu S. X., Cohen H. J. (1998) Am. J. Physiol. 275, G1463–1471 [DOI] [PubMed] [Google Scholar]
- 15. Seiler A., Schneider M., Förster H., Roth S., Wirth E. K., Culmsee C., Plesnila N., Kremmer E., Rådmark O., Wurst W., Bornkamm G. W., Schweizer U., Conrad M. (2008) Cell Metab. 8, 237–248 [DOI] [PubMed] [Google Scholar]
- 16. Bao Y., Jemth P., Mannervik B., Williamson G. (1997) FEBS Lett. 410, 210–212 [DOI] [PubMed] [Google Scholar]
- 17. Ran Q., Van Remmen H., Gu M., Qi W., Roberts L. J., 2nd., Prolla T., Richardson A. (2003) Free Radic. Biol. Med. 35, 1101–1109 [DOI] [PubMed] [Google Scholar]
- 18. Bermano G., Pagmantidis V., Holloway N., Kadri S., Mowat N. A., Shiel R. S., Arthur J. R., Mathers J. C., Daly A. K., Broom J., Hesketh J. E. (2007) Genes Nutr. 2, 225–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Speckmann B., Pinto A., Winter M., Förster I., Sies H., Steinbrenner H. (2010) Free Rad. Biol. Med. 49, 777–785 [DOI] [PubMed] [Google Scholar]
- 20. Steinbrenner H., Bilgic E., Alili L., Sies H., Brenneisen P. (2006) Free Radic. Res. 40, 936–943 [DOI] [PubMed] [Google Scholar]
- 21. Nemoto E., Koshikawa Y., Kanaya S., Tsuchiya M., Tamura M., Somerman M. J., Shimauchi H. (2009) Bone 44, 805–812 [DOI] [PubMed] [Google Scholar]
- 22. Piana C., Wirth M., Gerbes S., Viernstein H., Gabor F., Toegel S. (2008) Eur. J. Pharm. Biopharm. 69, 1187–1192 [DOI] [PubMed] [Google Scholar]
- 23. Huang H. S., Chen C. J., Chang W. C. (1999) FEBS Lett. 455, 111–116 [DOI] [PubMed] [Google Scholar]
- 24. Engle M. J., Goetz G. S., Alpers D. H. (1998) J. Cell. Physiol. 174, 362–369 [DOI] [PubMed] [Google Scholar]
- 25. Pinto M., Robine-Leon S., Appay M., Kedinger M., Triadou N., Dussaulx E., Laeroix B., Simon-Assmann P., Maffen K., Fogh J., Zweibaum A. (1983) Biol. Cell 47, 323–330 [Google Scholar]
- 26. Mariadason J. M., Arango D., Corner G. A., Arañes M. J., Hotchkiss K. A., Yang W., Augenlicht L. H. (2002) Cancer Res. 62, 4791–4804 [PubMed] [Google Scholar]
- 27. Huin C., Schohn H., Hatier R., Bentejac M., Antunes L., Plénat F., Bugaut M., Dauça M. (2002) Biol. Cell 94, 15–27 [DOI] [PubMed] [Google Scholar]
- 28. Suaud L., Joseph B., Formstecher P., Laine B. (1997) Biochem. Biophys. Res. Commun. 235, 820–825 [DOI] [PubMed] [Google Scholar]
- 29. Conrad M., Schneider M., Seiler A., Bornkamm G. W. (2007) Biol. Chem. 388, 1019–1025 [DOI] [PubMed] [Google Scholar]
- 30. Ursini F., Maiorino M., Gregolin C. (1985) Biochim. Biophys. Acta 839, 62–70 [DOI] [PubMed] [Google Scholar]
- 31. Kelner M. J., Montoya M. A. (1998) Biochem. Biophys. Res. Commun. 249, 53–55 [DOI] [PubMed] [Google Scholar]
- 32. Brigelius-Flohé R. (2006) Biol. Chem. 387, 1329–1335 [DOI] [PubMed] [Google Scholar]
- 33. Wingler K., Böcher M., Flohé L., Kollmus H., Brigelius-Flohé R. (1999) Eur. J. Biochem. 259, 149–157 [DOI] [PubMed] [Google Scholar]
- 34. Heinemeyer T., Wingender E., Reuter I., Hermjakob H., Kel A. E., Kel O. V., Ignatieva E. V., Ananko E. A., Podkolodnaya O. A., Kolpakov F. A., Podkolodny N. L., Kolchanov N. A. (1998) Nucleic Acids Res. 26, 362–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hattori H., Imai H., Kirai N., Furuhama K., Sato O., Konishi K., Nakagawa Y. (2007) Biochem. J. 408, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Baek I. J., Seo D. S., Yon J. M., Lee S. R., Jin Y., Nahm S. S., Jeong J. H., Choo Y. K., Kang J. K., Lee B. J., Yun Y. W., Nam S. Y. (2007) J. Mol. Histol. 38, 237–244 [DOI] [PubMed] [Google Scholar]
- 37. Arai M., Imai H., Koumura T., Yoshida M., Emoto K., Umeda M., Chiba N., Nakagawa Y. (1999) J. Biol. Chem. 274, 4924–4933 [DOI] [PubMed] [Google Scholar]
- 38. Bestwick C. S., Milne L. (2000) Biochim. Biophys. Acta 1474, 47–55 [DOI] [PubMed] [Google Scholar]
- 39. Yoo M. H., Gu X., Xu X. M., Kim J. Y., Carlson B. A., Patterson A. D., Cai H., Gladyshev V. N., Hatfield D. L. (2010) Antioxid. Redox Signal 12, 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bartsch H., Nair J., Owen R. W. (2002) Biol. Chem. 383, 915–921 [DOI] [PubMed] [Google Scholar]
- 41. Boucher M. J., Laprise P., Rivard N. (2005) J. Cell. Physiol. 202, 178–190 [DOI] [PubMed] [Google Scholar]
- 42. Blais S., Boudreau F., Beaulieu J. F., Asselin C. (1995) Dev. Dyn. 204, 66–76 [DOI] [PubMed] [Google Scholar]
- 43. Chandrasekaran C., Gordon J. I. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 8871–8875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Florian S., Krehl S., Loewinger M., Kipp A., Banning A., Esworthy S., Chu F. F., Brigelius-Flohé R. (2010) Free Radic. Biol. Med. 49, 1694–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhong Z. D., Hammani K., Bae W. S., DeClerck Y. A. (2000) J. Biol. Chem. 275, 18602–18610 [DOI] [PubMed] [Google Scholar]
- 46. Synnestvedt K., Furuta G. T., Comerford K. M., Louis N., Karhausen J., Eltzschig H. K., Hansen K. R., Thompson L. F., Colgan S. P. (2002) J. Clin. Invest. 110, 993–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]