

Abstract
Objective
To investigate TGFβ regulation of CTGF expression in cells of the nucleus pulposus.
Methods
Real Time RT-PCR and Western blot analysis was used to measure CTGF expression in the nucleus pulposus. Transfections were used to measure the effect of Smad2/3/7 and AP1on TGFβ mediated CTGF promoter activity.
Results
CTGF expression was lower in the neonatal disc compared with the skeletally mature rat disc. An increase in CTGF expression and promoter activity was observed in nucleus pulposus cells after TGFβ treatment. Deletion analysis indicated that promoter constructs lacking smad and AP1 motifs were unresponsive to treatment. Analysis showed that full-length Smad3 and the Smad3-MH2 domain alone increased CTGF activity. Further evidence of Smad3 and AP1 involvement was seen when DN-Smad3, SiRNA-Smad3, smad7 and DN-AP1 suppressed TGFβ mediated activation of the CTGF promoter. When either Smad3 or AP1 sites were mutated, CTGF promoter induction by TGFβ was suppressed. We also observed a decrease in expression of CTGF in discs of Smad3 null mice compared to the wild type. Analysis of human nucleus pulposus indicated a trend of increasing CTGF and TGFβ expression in the degenerate state.
Conclusion
TGFβ, through Smad3 and AP1, serves as a positive regulator of CTGF expression in the nucleus pulposus. We propose that CTGF is a part of the limited reparative response of the degenerate disc.
Keywords: intervertebral disc, nucleus pulposus, CTGF, TGFβ, Smad3, AP1, disc degeneration
Introduction
The intervertebral disc is a unique structure that permits rotation as well as flexion and extension of the human spine. While sparse, cells in the nucleus pulposus secrete a complex extracellular matrix that contains fibrillar collagens and the proteoglycan aggrecan. This macromolecular assembly provides a robust hydrodynamic system that accommodates applied biomechanical forces on the spine (1-3). During degenerative disc disease, the loss of proteoglycans decreases the water binding capacity of the system resulting in a failure of the tissue to resist compressive forces. Although a great deal is known of conditions that enhance proteoglycan degeneration, there remains much to be learned concerning the regulation of matrix protein biosynthesis in cells of both the normal and degenerate disc.
Of the proteins that have been identified in the disc, only a few directly respond to factors that enhance the degradative process. CCN2/CTGF, a member of the “CCN family” of proteins has been shown to interact with, and regulate, the activity of many growth factors (4,5). This matricellular protein participates in biological functions that include extracellular matrix synthesis, angiogenesis, cellular proliferation, and migration (4-10). By binding heparin, it signals through heparan sulfate containing proteoglycans and integrins, causing substantive changes in cell function (6). In the context of the spine, CTGF has been shown to play a significant role in the development of the notochord, the embryonic analage of the intervertebral disc, and CTGF null mice die soon after birth due to severe skeletal and cartilage dysplasia (9,10). A recent histological study shows that CTGF is present in the extracellular matrix and localized to fibroblasts, inflammatory cells, and endothelial cells of the discs of patients with discogenic pain (8). In addition, CTGF expression is also regulated by HIF-1, a transcription factor that is robustly and constitutively expressed by the hypoxic tissues of the disc (11-14).
Aside from the roles defined above, CTGF is sensitive to transforming growth factor β (TGFβ) (15-17). In a number of tissues, it amplifies the multiple actions of TGFβ, often causing enhanced fibrosis (15-17); other groups have noted enhanced TGFβ levels during pathological states (18-20). The role of CTGF and its regulation within the intervertebral disc, however, remains unknown. The objective of the investigation was to examine CTGF expression by cells of the nucleus pulposus cells and to determine if its expression is regulated by TGFβ. Results of the study show for the first time that TGFβ induces CTGF expression through activation of Smad3 and AP1 signaling pathways. Furthermore, analysis of human disc samples shows a trend of increasing CTGF expression with increasing severity of degeneration.
Materials And Methods
Plasmids and Reagents
Human CTGF deletion and mutant reporter plasmids were from Dr. George Yang, Stanford University. Wild type and null Smad3 mouse embryonic fibroblasts (MEF), Flag-Smad3N (aa 1-146, MH1 domain), Flag-Smad3NL (aa 1-211, MH1 domain and linker), Flag-Smad3C (aa 199- 424, MH2 domain), Flag-Smad3LC (aa 146- 425, MH2 domain and linker) and Flag-Smad3ΔC (aa 1-381, lacks the C-terminal 43 amino acids and functions as DN-Smad3) vectors were provided by Dr. Rik Derynck, University of California at San Francisco. Plasmids were kindly provided by Dr. Bert Vogelstein, John Hopkins University [Smad2], Dr. Charles Vinson, NIH [DN-AP1; A-Fos], Dr. Silvio Gutkind, NIH [AP1 reporter], Dr. Shannon Kenney, University of Wisconsin [Gal4-c-Jun-TAD]. Smad7 (catalog #11733), Smad3 (#11742), were obtained from Addgene. As an internal transfection control, vector pRL-TK (Promega) containing Renilla reniformis luciferase gene was used. The amount of transfected plasmid, the pre-transfection period after seeding, and the post-transfection period before harvesting, have been optimized for rat nucleus pulposus cells using pSV β-galactosidase plasmid (Promega) (12). rhTGFβ3 was from R&D systems.
Isolation of nucleus pulposus cells and treatments of cells
Rat nucleus pulposus cells were isolated using an explant culture method reported earlier by Risbud et al. (12). Nucleus pulposus cells migrated out of the explant after 1 week. When confluent, the cells were lifted using a trypsin (0.25 %) EDTA (1 mM) solution and sub-cultured in 10 cm dishes. These cells expressed high levels of aggrecan and collagen type II and were used for the analysis. In some experiments cells were treated with TGFβ3 (10 ng/ml, R&D systems).
Human tissue collection and grading
Lumbar disc tissues were collected as surgical waste from individuals undergoing elective spinal surgical procedures (average age 54 years, ranging from 38-82 years). In line with Thomas Jefferson University's Institutional Review Board guidelines, informed consent for sample collection was obtained for each patient. Assessment of the disease state was performed using the modified Thompson grading (21).
Immunohistological studies
Freshly isolated spines or whole embryos were immediately fixed in 4% paraformaldehyde in PBS and then embedded in paraffin. Transverse and coronal sections, 6-8 μ in thickness, were deparaffinized in xylene, rehydrated through graded ethanol and stained with alcian blue, eosin and hematoxylin. For localizing CTGF, sections were incubated with the anti-CTGF antibody (Abcam) in 2% bovine serum albumin in PBS at a dilution of 1:100 at 4 °C overnight. After thoroughly washing the sections, the bound primary antibody was incubated with Alexa fluor-488 conjugated anti-mouse secondary antibody (Invitrogen), at a dilution of 1:100 for 45 min at room temperature. Sections were visualized using a fluorescence microscope (Olympus, Japan).
Real time RT-PCR analysis
Total RNA was extracted from nucleus pulposus cells using RNAeasy mini columns (Qiagen). Before elution from the column, RNA was treated with RNase free DNAse I (Qiagen). For human samples, total RNA was isolated from 100 to 300 mg of NP tissue. Tissue was homogenized in Trizol (Invitrogen) on ice using Omni TH Homogenizer (Omni International). Following Trizol extraction, RNA was passed through the RNAEasy mini columns. The purified, DNA-free RNA was converted to cDNA using Superscript III Reverse Transcriptase (Invitrogen). Template cDNA and gene specific primers were added (Rat CTGF F: 5′ tgacccaactatgatgcgagccaa 3′, Human CTGF F: 5′ ttgcgaagctgacctggaagagaa 3′, Human TGFβ1 F: 5′ acacactgcaagtggacatcaacg 3′) to Fast SYBR Green master mix (Applied Biosystems) and mRNA expression was quantified using the 7900HT Fast Real-Time PCR System (Applied Biosystems). 18S and GAPDH were used to normalize the expression. Melting curves were analyzed to verify the specificity of the RT-PCR reaction and the absence of primer dimer formation. Each sample is analyzed in duplicate and included a template-free control. All the primers used were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA).
Immunofluorescence microscopy
Cells were plated in flat bottom 96 well plates (4 × 103/ well) and treated with TGFβ for 6 h - 24 h. After incubation, cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% triton-X 100 in PBS for 10 min, blocked with PBS containing 5% FBS, and incubated with antibodies against CTGF (1:200) (Abcam), at 4° C overnight. As a negative control, cells were reacted with isotype IgG under similar conditions. After washing, the cells were incubated with Alexa fluor-488 conjugated anti-mouse secondary antibody (Invitrogen), at a dilution of 1:50 for 45 min at room temperature. Cells were imaged using a laser scanning confocal microscope (Olympus Fluoview, Japan).
Western blotting
Cells were placed on ice immediately following treatment and washed with ice-cold HBSS. All the wash buffers and final re-suspension buffer included 1X protease inhibitor cocktail (Roche), NaF (5 mM) and Na3VO4 (200 μM). Total cell proteins were resolved on 8-12 % SDS-polyacrylamide gels and transferred by electroblotting to PVDF membranes (Bio-Rad, CA). The membranes were blocked with 5% non-fat dry milk in TBST (50 mM Tris, pH 7.6, 150 mM NaCl, 0.1% tween 20) and incubated overnight at 4 °C in 3% non-fat dry milk in TBST with the anti-CTGF (1:2000, Abcam) or anti-β-tubulin antibody (1:2000, DSHB). Immunolabeling was detected using the ECL reagent (Amersham Biosciences). Blot intensity was determined by densitometric analysis using Kodak 1D 3.6 software (Kodak, Rochester, NY)
Transfections and dual luciferase assay
Cells were transferred to 24-well plates at a density of 4 × 104 cells/well one day before transfection. To measure the effect of TGFβ, cells were transfected with 500 ng of CTGF reporter plasmids with 500 ng pRL-TK plasmid, in some wells cells were treated with the inhibitor SB. To investigate the effect of Smad3 or AP1 on CTGF promoter activity, cells were co-transfected with 100-300 ng of A-fos (DN-AP1) or DN-Smad3 (100-300 ng) or Smad3 (100-300 ng) or backbone vector with 400 ng CTGF reporter and 300 ng pRL-TK plasmid in presence or absence of TGFβ (10 ng/ml). For the GAL4 binary assay, cells were co-transfected with 100 ng of pFR-Luc and 100 ng of GAL4dbd or GAL4dbd-cJunTAD (cJun-TAD), and treated with TGFβ (10 ng/ml). LipofectAMINE 2000 (Invitrogen) was used as the transfection reagent; for each transfection, plasmids were premixed with the transfection reagent. The next day, the cells were harvested and a Dual-Luciferase™ reporter assay system (Promega) was used for sequential measurements of firefly and Renilla luciferase activities. Quantification of luciferase activities and calculation of relative ratios were carried out using a luminometer (TD-20/20, Turner Designs, CA). At least three independent transfections were performed, and all analyses were carried out in triplicate.
Statistical analysis
All measurements were performed in triplicate, data is presented as mean ± S.E. Differences between groups were analyzed by the student t test; *p< 0.05.
Results
Expression of CTGF in the intervertebral disc
Saggital sections of the neonatal (Fig. 1 A, C) and skeletally mature rat discs (Fig. 1 B,D) were stained with an antibody to CTGF (Fig. 1A, B) or counter stained with H&E and alcian blue (Fig. 1C, D). CTGF is expressed by cells of the nucleus pulposus, annulus fibrosus and cartilaginous endplate (Fig. 1A, B). In all cases, staining is localized to the cytosol as well as to the extracellular matrix (Fig. 1A, B). There is a higher expression of CTGF in mature rat discs compared to neonatal rat tissue. Expression of CTGF in native disc tissues and cultured cells was studied using Western blot analysis. Fig. 1E indicates that nucleus pulposus tissue expresses a prominent 38 kD CTGF band. Moreover, the expression level of CTGF in nucleus pulposus tissue is higher than the annulus fibrosus (Fig. 1E).
Figure 1.
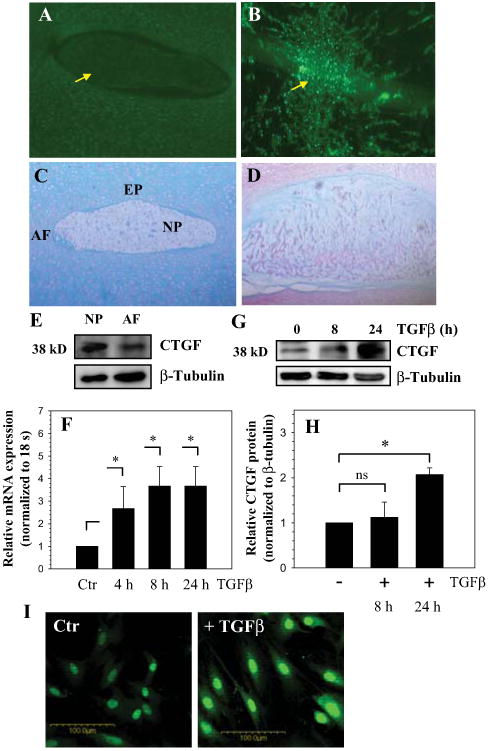
A-D. Sagittal sections of the intervertebral disc of a neonatal rat (A and C) and a mature rat (B and D). Sections were treated with CTGF antibody (A and B), or counterstained with hemotoxylin, eosin and alcian blue (C and D). CTGF was expressed at a higher level in nucleus pulposus (NP) of the mature rat compared to the neonate (see arrow). Annulus fibrosus (AF) and endplate cartilage (EP) (Mag. × 20). E. Western blot analysis of CTGF expression in rat NP and AF tissue. Expression of CTGF in tissue extracts was higher in NP than AF. F. Real time RT-PCR analysis of CTGF expression by NP cells treated with TGFβ. There was increased expression of CTGF mRNA with the TGFβ treatment. G. Western blot analysis of CTGF expression in NP cells. H. Densitometric analysis shows a significant increase in expression of CTGF protein after 24 h of TGFβ treatment. I, Immunofluorescent analysis of nucleus pulposus cells treated with TGFβ. Note, TGFβ treatment increased CTGF expression in both the nucleus and cytosol, Mag × 20. Values shown are mean ± SE from three independent experiments, * p < 0.05.
TGFβ-Smad3 signaling increases CTGF promoter activity in nucleus pulposus cells
To explore the premise that TGFβ regulates CTGF expression, nucleus pulposus cells were treated with this growth factor and expression of CTGF analyzed using real time RT-PCR. Figure 1F shows that treatment with TGFβ for 24 h results in an increase in CTGF mRNA levels in nucleus pulposus cells. In addition, we studied expression of CTGF in nucleus pulposus cells using Western blot analysis (Fig. 1G, H) and immunofluorescence microscopy (Fig. 1I). TGFβ treatment results in increased CTGF protein expression (Fig. 1G, I); the increase is pronounced 24 h after treatment (Fig. 1H).
To investigate the regulation of CTGF expression, we co-transfected nucleus pulposus cells with different size CTGF promoter constructs and measured their relative activity (Fig. 2A). Figure 2B indicates that the -736/+36 bp fragment exhibits the highest basal activity among all the promoter fragments tested (Fig. 2B). Since CTGF promoter contains a Smad responsive element and AP1 binding motif at -173/-166 bp and -294/-286 bp respectively (Fig. 2A), we tested the hypothesis that these elements were responsible for CTGF induction by TGFβ. When cells are transfected with -625/+36 bp and the shorter constructs, only the -625/+36 bp fragment elicits a significant increase in activity following treatment with TGFβ (Fig. 2C). Based on this finding, we chose the -625/+36 bp reporter plasmid for the transfection experiments described in the remainder of the study.
Figure 2.

TGFβ-Smad3 dependent regulation of CTGF promoter activity. A. Schematic of different length CTGF promoter constructs. B. NP cells transfected with each of the five reporter constructs ranging in size from -1999/+36- to -72/+36 bp. The -736/+36 bp fragment exhibited the highest level of activity. C. NP cells were transfected with -625/+36, -140/+36, -72/+36 bp CTGF reporter fragments and treated with TGFβ for 24 h. An increase in CTGF reporter activity was observed only in the promoter fragment that contains the AP1 and Smad binding sites. D. Effect of Smad3 overexpression on CTGF promoter activity. Note the dose dependent increase in CTGF reporter activity with increasing concentration of Smad3. E. Smad2 overexpression did not influence CTGF promoter activity. F. Schematic of Smad3 expression constructs used to determine the importance of different domains in mediating CTGF activity. G. Cells were transfected with different Smad3 constructs, with full-length Smad3 and the MH2 domain causing significant increase in CTGF reporter activity. H. TGFβ increased CTGF reporter activity, the addition of smad7 diminished activity in a dose-dependent manner. Values shown are mean ± SE from three independent experiments, * p < 0.05.
Next, to investigate whether induction of CTGF by TGFβ is mediated via the Smad signaling pathway, we co-transfected nucleus pulposus cells with plasmids encoding full-length Smad3 or Smad2 (Fig. 2D, E). Transfection with Smad3 increases CTGF promoter activity in a dose-dependent manner (Fig. 2D). In contrast, co-transfection with Smad2 elicits no response (Fig. 2E). To identify the domains of Smad3 that mediate CTGF promoter activity, we co-transfected cells with Smad3 domain deletion plasmids (Fig. 2F). Figure 2G shows that plasmids encoding both full-length and the MH2 domain of Smad3 cause a significant induction of the CTGF promoter activity, with the full-length Smad3 causing the greatest increase. To further examine the involvement of TGFβ-Smad signaling in CTGF induction, we co-transfected the CTGF reporter with a plasmid encoding Smad7 in presence of TGFβ. Figure 2H shows that Smad7 abolishes the TGFβ induction of CTGF promoter activity. An inhibitory effect of Smad7 on CTGF promoter activity is seen at a dose of 100 ng and is further enhanced when the concentration is increased to 300 ng (Fig. 2H).
TGFβ induces AP1 signaling activity in nucleus pulposus cells
We then determined if TGFβ modulated AP1 expression and activity in nucleus pulposus cells. Figure 3A indicates that TGFβ treatment results in a significant increase in c-Jun mRNA expression. To determine if TGFβ treatment may serve to promote cJun-TAD activity, we transfected cells with cJun-TAD or empty Gal4dbd and measured reporter activity (Fig. 3B, C). We found that in the presence of TGFβ there is a significant activation of cJun-TAD (Fig. 3C). Cells transfected with empty Gal4dbd exhibit minimal luciferase activity, which did not change after the TGFβ treatment (Fig. 3C). We also measured AP1 reporter activity in nucleus pulposus cells. Figure 3D shows that treatment with TGFβ results in a significant induction in the activity of the AP1 reporter.
Figure 3.

TGFβ increases the activity of the AP1 reporter. A. Real time RT-PCR analysis of c-Jun expression in cells treated with TGFβ. There was a significant increase in c-Jun mRNA expression after TGFβ treatment. B. Schematic of TGFβ activation of cJun-TAD. C. Cells were transfected with cJun-TAD or GAL4dbd and then treated with TGFβ. Note, TGFβ causes significant increase only in cJun-TAD reporter activity. D. Cells transfected with AP1 reporter and treated with TGFβ. An increase in AP1 reporter activity was seen following TGFβ treatment. Values shown are mean ± SE from three independent experiments, * p < 0.05.
Role of Smad and AP1 in CTGF promoter induction by TGFβ
To confirm the role of Smad3 and AP1 in TGFβ mediated induction of CTGF loss of function studies were performed. When cells were co-transfected with DN-Smad3 in the presence of TGFβ, a significant decrease in CTGF promoter induction is seen (Fig. 4A). In addition, when Smad3 expression is silenced using gene targeting siRNA, TGFβ mediated induction in CTGF promoter plasmid is suppressed (Fig. 4B). To confirm the role of AP1 in TGFβ – mediated induction of CTGF, we co-transfected NP cells with an A-fos plasmid which functions as DN-AP1. Figure 4C shows that all doses of A-fos abolish induction of CTGF promoter activity by TGFβ. Lastly, to evaluate the requirement of the Smad or the AP1 binding site in the CTGF promoter we performed site-directed mutagenesis (Fig. 4D). The results clearly show that mutation in either the Smad or AP1 binding sites completely abolishes the TGFβ responsiveness of the CTGF promoter (Fig. 4E).
Figure 4.

Smad3 and AP1 signaling regulate CTGF promoter activity. A. Cells were transfected with DN-Smad3 then treated with TGFβ. DN-Smad3 abolished TGFβ induction of CTGF reporter activity. B. Cells were transfected with Smad3 siRNA or control siRNA then treated with TGFβ. Induction of the CTGF reporter by TGFβ was blocked by the Smad3 siRNA. C. Cells were transfected with A-Fos (DN-AP1) and treated with TGFβ. A-Fos suppressed TGFβ induction of CTGF activity. D. Schematic of three different CTGF reporter constructs (WT: wild type; MT-S: mutant Smad; MT-A: mutant AP1). E. Cells transfected with WT, MT-S, and MT-A CTGF reporter constructs were treated with TGFβ. Only the WT CTGF promoter fragment with both sites intact elicited an increase in activity. Values shown are mean ± SE from three independent experiments, * p < 0.05.
To further validate the role of Smad3 in regulating CTGF expression, we examined expression of CTGF in disc tissues of Smad3 null mice. Immunohistological analysis indicates that there is a decrease in CTGF expression in discs of null mice (Fig. 5A); when compared with wild type mice, this loss of staining is most evident in the extracellular matrix (Fig. 5B). In addition, MEFs obtained from these animals indicates that there is an approximate 80% decrease in basal promoter activity of CTGF in the Smad3 null cells when compared to the wild type cells (Fig. 5E). When the Smad3 expression plasmid is co-transfected into the Smad3 KO MEFs, there is a dose-dependent restoration in CTGF promoter activity (Fig. 5F).
Figure 5.
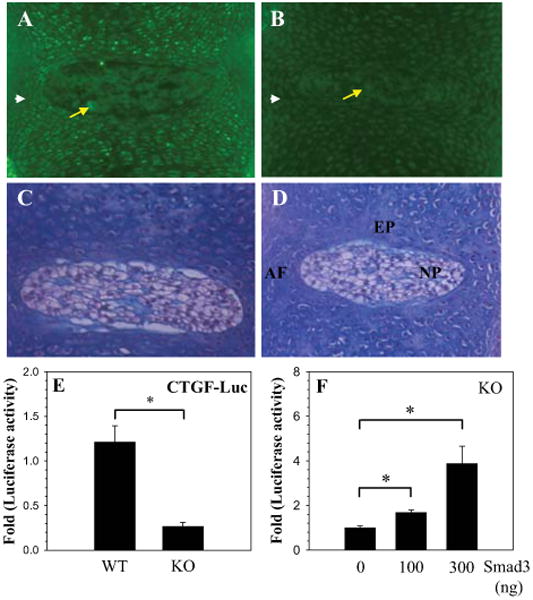
CTGF expression is dependent on Smad3. A-D. Immunohistological expression of CTGF in the intervertebral discs of Smad3 null (KO) and wild type (WT) mice. Saggital sections through the disc of WT mouse embryo (A and C) and Smad3 KO mouse embryo (B and D) were treated with anti-CTGF antibody (C and D) or counterstained with hematoxylin and eosin (C and D). Expression of CTGF was more pronounced in the NP (arrow) and AF (arrow head) of the WT (A) than in the Smad3 KO (B). Mag. × 20. E. CTGF promoter activity in MEF's isolated from smad 3 KO and WT mice. There was an 80% decrease in basal CTGF reporter activity in the Smad3 KO cells F. Smad3 KO Cells were transfected with Smad3 and CTGF reporter activity was measured. A dose dependent increase in CTGF reporter activity was seen with increasing concentration of Smad3. Values shown are mean ± SE from three independent experiments, * p < 0.05.
Expression of CTGF in human degenerate disc tissue samples increases with severity of disc degeneration
In a parallel study, we evaluated CTGF expression in human disc tissues. Real-time RT-PCR analysis shows that there is an increasing trend in expression of both CTGF and TGFβ1 mRNA in degenerate tissues when compared with the normal (Fig. 6B). We also extracted proteins from the same tissue samples and analyzed their integrity by gel electrophoresis. All tissue extracts show discrete protein bands with minimal sample deterioration (Fig. 6C). Western blot analysis was performed on equal amount of tissue protein using an antibody against CTGF. Figure 6D indicates that there is a discrete CTGF protein band (38 kD) in all samples, with minimal expression evident in normal control tissue. Inspection of the combined data set suggests a trend of increased CTGF expression with degeneration. Severely degenerate tissues (Grade IV and Grade V) generally display high level of expression of CTGF compared to less degenerate (Grade II and Grade III) and normal control sample. Not surprisingly, patient to patient variation within the same grade was also evident.
Figure 6.
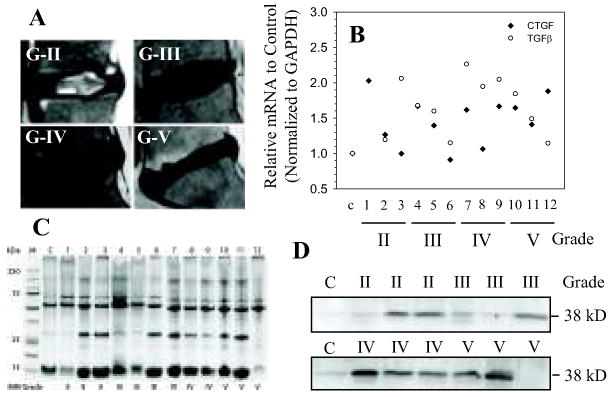
Expression of CTGF in human degenerate NP. A. Representative MRI images of patients with degenerative disc disease. (From the top left, clockwise: Grade II,III,IV,V) B. Real time RT-PCR analysis of CTGF and TGFβ mRNA expression in multiple human NP tissue samples. Samples from three different patients were used for each degenerative grade. With increased severity of disc degeneration there is a trend of increasing expression in both CTGF and TGFβ mRNA. Expression in normal control (c) sample was set at 1.0 for both CTGF and TGFβ. C. Human degenerate NP protein extracts gel electrophoresed and stained with coomasie blue. The distinct bands in each sample verifies the quality of the protein samples. D. Western blot analysis of CTGF expression in human degenerate NP samples. The 38 kD CTGF band can be seen in all samples; there was a trend towards increasing protein expression in grade IV and V samples when compared to control or possibly grade II or III.
Discussion
The experiments described in this investigation demonstrated for the first time that in the intervertebral disc, CTGF expression was regulated by TGFβ. TGFβ promoted CTGF promoter activity and gene expression through Smad3, along with AP1. The importance of Smad3 signaling was emphasized by studies of CTGF expression in the intervertebral disc of Smad3 null mice. In the disc of these mutants the expression of CTGF was low. We observed that in human degenerate discal tissues there was a trend towards increased CTGF expression. Based on these findings, we propose that an elevation in TGFβ levels, resulting from an inflammatory or degenerative insult, further upregulates expression of CTGF by the nucleus pulposus cells. This response would be expected to compensate for the loss of extracellular matrix macromolecules in disease and enhance nucleus pulposus cell survival.
We examined the expression and localization of CTGF in the intervertebral disc of animals at two different stages of skeletal maturity. Our results indicated that there was elevated CTGF expression in the extracellular matrix of both the annulus fibrosus and the nucleus pulposus discs of mature animals. This finding was not unexpected as both tissues in the adult state are rich in proteoglycans and collagen and would therefore be expected to be responsive to changes in CTGF expression. This finding would also explain why CTGF expression was low in highly cellular neonatal discs. Of course, it is also likely that the lower level of protein expression may indicate that at earlier stages of development, alternative mechanisms for the control of matrix macromolecules expression were active.
Initially, we focused on the role of TGFβ in the regulation of CTGF expression. This cytokine has been closely linked to disc function; furthermore, its deletion is known to cause fibrotic changes in a host of connective tissues (21). It was noted that TGFβ caused an increase in CTGF mRNA and protein expression levels by the cells of the nucleus pulposus, implying that regulation is mediated at the transcriptional level. Transient transfection assays using deletion constructs revealed that TGFβ increased CTGF promoter activity only when Smad and AP1 response elements were present. Indeed, when these sites were deleted, TGFβ dependent induction was lost, lending further support to the importance of both Smad and AP1 in mediating TGFβ action. Of interest, we noticed that there was a significant difference in the basal activities of the -736/+36 bp fragment when compared with the -625/+36 bp fragment. Since both fragments contained both Smad and AP1 binding sites, the results suggest that an as yet unidentified regulatory motif influenced basal CTGF expression. Whether this result is specific for the intervertebral disc, or if this difference is apparent in other connective tissues awaits further study.
Gain and loss of function experiments were performed to define the mechanism by which TGFβ influences the activity of the CTGF promoter. Smad3 overexpression enhanced CTGF promoter activity, while deletion and silencing studies revealed that there was inhibition of activity. It was noteworthy that while the full- length Smad3 maximally promoted CTGF expression, it was clear that the MH2 domain was of critical regulatory importance. Of course, it is this domain that exhibits TAD activity; the MH1 domain is concerned with DNA recognition and Smad4 binding (23). Further support for importance of Smad3 in controlling CTGF promoter activity was forthcoming from studies performed using Smad7, DN-Smad3 plasmids as well SiRNA-Smad3. We found that in the transfected cells, in the presence of TGFβ, induction in CTGF promoter activity was suppressed. Recent work indicates that Smad7 down regulated TGFβ activity by blocking TGFβ signaling at the receptor level. It either interferes with binding to type I receptors, thus preventing activation of Smad2/3, (24) or by recruiting Smurf, an E3 ubiquitin ligase, to receptors targeting them for proteasome mediated degradation (24,25). More recently Zhang et al. reported binding of Smad7 to the Smad-responsive elements by its MH2 domain, thereby disrupting the formation of the functional Smad-DNA complex (26). Thus, our results are in line with these mechanistic findings, although it remains to be determined if in nucleus pulposus cells, Smad7 functions via receptor-dependent or -independent mechanisms.
The study revealed that in nucleus pulposus cells, AP1 is a component of the TGFβ signaling pathway. Following stimulation with TGFβ there was a concomitant increase in expression and activity of c-Jun and increased activity of the AP1 responsive reporter. We also noted that co-transfection with an A-Fos plasmid blocked the inductive effects of TGFβ on CTGF promoter activity. Mutagenesis studies provided further confirmation that Smad3 and AP1 signaling complex was required for full CTGF promoter activity. Mutation in either the Smad or AP1 response elements resulted in complete inhibition of TGFβ mediated induction of CTGF promoter activity. Another important test of the hypothesis that CTGF expression was influenced by Smad3 was from analysis of cells of the Smad3 null mice. We noted that Smad3 expression in the discs of null mice was significantly lower than the wild type. Moreover, using MEF's from the null mice, we noted that basal CTGF promoter activity was suppressed, while overexpression of Smad3 caused an increase in CTGF promoter activity. Based on these gain and loss of function experiments, it is evident that Smad3 in conjunction with AP1 can mimic the effects of TGFβ on CTGF promoter function. Results of these investigations lend direct support to the notion that Smad3 is a functional modulator of CTGF promoter function and, as such, its regulation controls CTGF repression in nucleus pulposus cells in vivo.
Lastly, analysis of human disc samples revealed a trend in both CTGF and TGFβ expression in the degenerate state. While the number of control tissues were very limited (due to practical difficulties in acquiring human discs), measurement of both RNA and protein expression levels suggested that as the nucleus became degenerate there was a rise in expression of both molecules. Previous studies that documented that CTGF and TGFβ values were elevated in disc disease were conducted solely using immunohistochemistry; in contrast, our studies utilized a Western blot analysis to measure these changes (9,19). Since CTGF primarily localized to areas that evidence ingrowth of blood vessels, Peng et al. interpreted these findings as indicating that the CTGF response was angiogenic and hence promoted disc degeneration (9). This interpretation may not be entirely correct. While treatment of endothelial cells with recombinant CTGF alone causes angiogenesis (27), there was no impairment of angiogenesis in CTGF null mice (28). Moreover, there is evidence that CTGF inhibits angiogenesis mediated by VEGF, through direct binding and sequestration of the protein (29,30). Aside from promoting angiogenesis, it is now clear that VEGF also serves as a survival factor (31). In the disc, this molecule prevents nucleus pulposus cell apoptosis (31). Likewise, many recent studies indicate that CTGF may serve as a survival factor (32-34). Wahab et al. showed that CTGF promoted cell survival by maintaining the anti-apoptotic protein, Bcl-2 in its active state (34). Finally, Erwin et al. have reported that treated with rCTGF there was increased aggrecan gene expression by notochordal cells of the nucleus pulposus (35). Thus, we forward the notion that in conjunction with VEGF, the increase in the level of CTGF during degeneration is a reparative response primarily concerned with restoration of extracellular matrix molecules and nucleus pulposus cell survival (31, 36). Whether CTGF together with TGFβ can be used to signal early changes in disc function is now being subjected to intense scrutiny.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health AR050087 and AR055655. Cassie Tran is supported by NIMAS training grant T32AR052273.
Abbreviations
- CTGF
connective tissue growth factor
- TGFβ
transforming growth factor β
Contributor Information
Cassie M. Tran, Email: cassie.tran@jefferson.edu.
Dessislava Markova, Email: dessislava.markova@jefferson.edu.
Harvey E. Smith, Email: harveysmith27@gmail.com.
Bala Susarla, Email: bsusarla@usuhs.mil.
Ravi Kumar Ponnappan, Email: rkp500@gmail.com.
D Greg Anderson, Email: greg.anderson@rothmaninstitute.com.
Aviva Symes, Email: asymes@usuhs.mil.
Irving M. Shapiro, Email: irving.shapiro@jefferson.edu.
References
- 1.Feng H, Danfelter M, Stromqvist B, Heinegard D. Extracellular matrix in disc degeneration. J Bone Joint Surg Am. 2006;88:25–9. doi: 10.2106/JBJS.E.01341. [DOI] [PubMed] [Google Scholar]
- 2.Setton LA, Chen J. Mechanobiology of the intervertebral disc and relevance to disc degeneration. J Bone Joint Surg. 2006;88:52–7. doi: 10.2106/JBJS.F.00001. [DOI] [PubMed] [Google Scholar]
- 3.Ng L, Grodzinsky AJ, Patwari P, Sandy J, Plaas A, Ortiz C. Individual cartilage aggrecan macromolecules and their constituent glycosaminoglycans visualized via atomic force microscopy. J Struct Biol. 2003;143:242–57. doi: 10.1016/j.jsb.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Cicha I, Goppelt-Struebe M. Connective tissue growth factor: Context-dependent functions and mechanisms of regulation. Biofactors. 2009;35:200–8. doi: 10.1002/biof.30. [DOI] [PubMed] [Google Scholar]
- 5.Houlbourn KP, Perbal B, Acharya KR. Proteins on the catwalk: modeling the structural domains of the CCN family of proteins. J Cell Commun Signal. 2009;3:25–41. doi: 10.1007/s12079-009-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41:771–83. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grotendorst GR. Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev. 1997;8:171–9. doi: 10.1016/s1359-6101(97)00010-5. [DOI] [PubMed] [Google Scholar]
- 8.Kubota S, Takigawa M. CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis. 2007;10:1–11. doi: 10.1007/s10456-006-9058-5. [DOI] [PubMed] [Google Scholar]
- 9.Peng B, Chen J, Zhang X. Expression and Role of Connective Tissue Growth Factor in Painful Disc Fibrosis and Degeneration. Spine. 2009;34:178–82. doi: 10.1097/BRS.0b013e3181908ab3. [DOI] [PubMed] [Google Scholar]
- 10.Erwin MW. The Notochord, notochordal cell and CTGF/CCN-2: ongoing activity from development through maturation. J Cell Comm Signal. 2008;2:59–65. doi: 10.1007/s12079-008-0031-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishida T, Kawaki H, Baxter RM, Deyoung RA, Takigawa M, Lyons KM. CCN2 (Connective tissue growth factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J Cell Comm Signal. 2007;1:45–58. doi: 10.1007/s12079-007-0005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Risbud MV, Guttapalli A, Stokes DG, Hawkins D, Danielson KG, Schaer TP. Nucleus pulposus cells express HIF-1 alpha under normoxic culture conditions: a metabolic adaptation to the intervertebral disc microenvironment. J Cell Biochem. 2006;98:152–9. doi: 10.1002/jcb.20765. [DOI] [PubMed] [Google Scholar]
- 13.Higgins DF, Biju MP, Akai Y, Wutz A, Johnson RS, Haase VH. Hypoxic induction of CTGF is directly mediated by HIF-1. Am J Physiol Renal Physiol. 2004;287:F1222–32. doi: 10.1152/ajprenal.00245.2004. [DOI] [PubMed] [Google Scholar]
- 14.Hong KH, Yoo SA, Kang SS, Choi JJ, Kim WU, Cho CS. Hypoxia induces expression of connective tissue growth factor in sclerderma skin flibroblasts. Clin Exp Immunol. 2006;146:362–70. doi: 10.1111/j.1365-2249.2006.03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xia W, Kong W, Yang GP. Increased CCN2 transcription in keloid fibroblast requires cooperativity between AP-1 and SMAD binding sites. Ann of Surg. 2007;246:886–95. doi: 10.1097/SLA.0b013e318070d54f. [DOI] [PubMed] [Google Scholar]
- 16.Abreu JG, Ketpura NI, Reversade B, de Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signaling by BMP and TGF-β. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leask A, Holmes A, Abraham DJ. Connective tissue growth factor gene regulation: requirements for its induction by transforming growth factor-β2 in fibroblasts. J Biol Chem. 2003;278:13008–15. doi: 10.1074/jbc.M210366200. [DOI] [PubMed] [Google Scholar]
- 18.Yokoki H, Mukoyama M, Nagae T, Mori K, Suganami T, Sawai K, et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol. 2004;15:1430–40. doi: 10.1097/01.asn.0000130565.69170.85. [DOI] [PubMed] [Google Scholar]
- 19.Lee S, Moon CS, Sul D, Lee J, Bae M, Hong Y, et al. Comparison of growth factor and cytokine expression in patients with degenerated disc disease and herniated nucleus pulposus. Clinical Biochem. 2009 doi: 10.1016/j.clinbiochem.2009.06.017. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 20.Ren S, Babelova A, Moreth K, Xin C, Eberhardt W, Doller A, et al. Transforming growth factor-beta2 upregulates spingosine kinase-1 activity, which in turn attenuates the fibrotic response to TGF-beta2 by impeding CTGF expression. Kidney Int. 2009 doi: 10.1038/ki.2009.297. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 21.Hilibrand AS, Carlson GD, Palumbo MA, Jones PK, Bohlman HH. Radiculopathy and myelopathy at segments adjacent to the site of a previous anterior cervical arthrodesis. J Bone Joint Surg Am. 1999;81:519–528. doi: 10.2106/00004623-199904000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Uchiyama Y, Guttapalli A, Gajghate S, Mochida J, Shapiro IM, Risbud MV. SMAD3 functions as a transcriptional repressor of acid-sensing ion channel 3 (ASIC3) in nucleus pulposus cells of the intervertebral disc. J Bone Miner Res. 2008;23:1619–28. doi: 10.1359/JBMR.080502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yigong S, Massague J. Mechanisms of TGF-β signaling from the cell membrane to the nucleus. Cell Press. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 24.Hayashi H, Abdollah S, Qiu Y, Cai J, Xu YY, Grinnell BW, Richardson MA. The MAD-related protiein smad 7 associates with the TGFbeta receptor and functions as an antagonist of TGFbeta signaling. Cell. 1997;89:1165–73. doi: 10.1016/s0092-8674(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 25.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–75. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 26.Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, et al. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol. 2007;27:4488–99. doi: 10.1128/MCB.01636-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brigstock DR. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cycteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–65. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 28.Kuiper E, Roestenberg P, Ehlken C, Lambert V, van Treslong-de Groot HB, Lyons KM, et al. Angiogenesis is not impaired in connective tissue growth factor (CTGF) knock-out mice. J Histochem Cytochem. 2007;55:1139–47. doi: 10.1369/jhc.7A7258.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang CC, Lin MT, Jeng YM, Chen ST, Chu CY, Chen RJ. Effect of connective tissue growth factor hypoxia-inducible factor 1 alpha degradation and tumor angiogenesis. J Natl Cancer Inst. 2006;98:984–95. doi: 10.1093/jnci/djj242. [DOI] [PubMed] [Google Scholar]
- 30.Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K, et al. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2002;16:219–21. doi: 10.1096/fj.01-0332fje. [DOI] [PubMed] [Google Scholar]
- 31.Fujita N, Imai J, Suzuki T, Yamada M, Ninomiya K, Miyamoto K. Vascular endothelial growth factor-A is a survival factor for nucleus pulposus cells in the intervertebral disc. Biochem Biophys Res Commun. 2008;372:367–72. doi: 10.1016/j.bbrc.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 32.Gygi D, Zumstein P, Grossenbacher D, Altwegg L, Luscher TF, Gehring H. Human connective tissue growth factor expressed in Escherichia coli is a non-mitogenic inhibitor of apoptosis. Biochem Biophys Res Commun. 2003;311:685–90. doi: 10.1016/j.bbrc.2003.10.061. [DOI] [PubMed] [Google Scholar]
- 33.Croci S, Landuzzi L, Astolfi A, Nicoletti G, Rosolen A, Sartori F, et al. Inhibition of connective tissue growth factor (CTGF/CCN2) expression decreases the survival and myogenic differentiation of human rhabdomyosarcoma cells. Cancer Res. 2004;64:1730–6. doi: 10.1158/0008-5472.can-3502-02. [DOI] [PubMed] [Google Scholar]
- 34.Wahab N, Cox D, Witherden A, Mason RM. Connective tissue growth factor (CTGF) promotes activated mesangial cell survival via up-regulation of mitogen-activated protein kinase phosphatase-1 (MKP-1) Biochem J. 2007;406:131–8. doi: 10.1042/BJ20061817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erwin MW, Ashman K, Inman RD. Nucleus pulposus notochord cells secrete connective tissue growth factor and up-regulate proteoglycan expression by intervertebral disc cells. Arthritis Rheum. 2006;54:3859–67. doi: 10.1002/art.22258. [DOI] [PubMed] [Google Scholar]
- 36.Agrawal A, Gajghate S, Smith H, Anderson DG, Albert TJ, Shapiro IM. Cited2 modulates hypoxia-inducible factor-dependent expression of vascular endothelial growth factor in nucleus pulposus cells of the rat intervertebral disc. Arthritis Rheum. 2008;58:3798–808. doi: 10.1002/art.24073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.