

Abstract
Tauopathies represent a class of neurodegenerative disorders characterized by abnormal tau phosphorylation and aggregation into neuronal paired helical filaments (PHFs) and neurofibrillary tangles. AMP-activated protein kinase (AMPK) is a metabolic sensor expressed in most mammalian cell types. In the brain, AMPK controls neuronal maintenance and is overactivated during metabolic stress. Here, we show that activated AMPK (p-AMPK) is abnormally accumulated in cerebral neurons in 3R+4R and 3R tauopathies, such as Alzheimer's disease (AD), tangle-predominant dementia, Guam Parkinson dementia complex, Pick's disease, and frontotemporal dementia with parkinsonism linked to chromosome 17, and to a lesser extent in some neuronal and glial populations in the 4R tauopathies, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease. In AD brains, p-AMPK accumulation decorated neuropil threads and dystrophic neurites surrounding amyloid plaques, and appeared in more than 90% of neurons bearing pre-tangles and tangles. Granular p-AMPK immunoreactivity was also observed in several tauopathies in apparently unaffected neurons devoid of tau inclusion, suggesting that AMPK activation preceded tau accumulation. Less p-AMPK pathology was observed in PSP and CBD, where minimal p-AMPK accumulation was also found in tangle-positive glial cells. p-AMPK was not found in purified PHFs, indicating that p-AMPK did not co-aggregate with tau in tangles. Finally, in vitro assays showed that AMPK can directly phosphorylate tau at Thr-231 and Ser-396/404. Thus, activated AMPK abnormally accumulated in tangle- and pre-tangle-bearing neurons in all major tauopathies. By controlling tau phosphorylation, AMPK might regulate neurodegeneration and therefore could represent a novel common determinant in tauopathies.
Keywords: Alzheimer's disease, Tauopathies, AMPK, Tangles, Tau
Introduction
Common neurodegenerative disorders, which include amyloidoses and tauopathies, are characterized by selective and progressive loss of specific neuronal populations. The exact mechanisms triggering neurodegeneration in these disorders remain unclear, but characteristic neuronal or glial inclusion bodies implicating specific proteins have been identified and dictate the current molecular classification of the different neurodegenerative disorders [9, 11]. The amyloidoses, which include the most common form of dementia, Alzheimer's disease (AD), are characterized by the presence of amyloid deposition [14]. In AD, the core component of the amyloid plaques is amyloid-β (Aβ), a series of peptides derived from the sequential endoproteolysis of a longer precursor, the amyloid-β precursor protein (APP) [30]. APP is genetically linked to early-onset familial forms of AD and Aβ is considered to be a causative factor in AD [37]. Aβ deposits, however, are also observed in elderly non-demented individuals, suggesting that amyloid formation is not sufficient to trigger neurodegeneration. AD, like other amyloidoses, is associated with the formation of lesions containing the tau protein called neurofibrillary tangles (NFTs), a process characterized as “secondary tauopathy”. NFTs are formed by the aggregation of hyperphosphorylated tau in the form of paired helical filaments (PHFs) [3, 26].
Tau is a microtubule-associated protein involved in intracellular cargo transport via its role in microtubule polymerization and stabilization. Importantly, mutations in the gene encoding tau have been found in familial frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), demonstrating that tau per se can cause neurodegeneration [20, 27]. In this context, FTDP-17 is referred to as “primary tauopathy” like several other neurodegenerative conditions, which include progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease (AGD), Pick's disease (PiD), tangle-predominant dementia, and Guam Parkinson dementia complex (Guam PDC) [11, 38]. AGD is the most common of the primary tauopathies responsible for about 5% of late-onset dementia and may co-occur with CBD, PSP, and AD. AGD affects the limbic lobe structures and is characterized by the presence of both neuronal and glial tau inclusions. CBD is characterized by tau pathology and neurodegeneration in the cortex and basal ganglia, whereas in PSP, the regions affected include the basal ganglia, subthalamic nucleus, and the substantia nigra. PiD affects the corticolimbic regions and there is histopathological overlap with FTDP-17 because most familial PiDs are due to genetic forms of FTDP-17. Guam PDC is an endemic disorder of unknown genetic cause occurring in the Guam islands and in the Kii peninsula in Japan. The disease is characterized by a tangle pathology affecting the cortex and brainstem.
AMP-activated protein kinase (AMPK) is a sensor of cellular stress that maintains energy homeostasis by controlling the activity of several metabolic enzymes. AMPK is a heterotrimeric Ser/Thr protein kinase, consisting of a catalytic α subunit and two regulatory β and γ subunits. This kinase is activated during energy stress by different upstream kinases via Thr-172 phosphorylation within the activation loop of the catalytic α2 subunit (Thr-183 in α1) [4, 19]. AMPK is expressed in most mammalian tissues and cell types, including in cerebral neurons where it is believed to play a critical role in the control of energy homeostasis [35, 36]. Indeed, neurons, as compared to other cell types, do not efficiently store nutrients, while they are highly metabolically active by executing high-energy demanding functions, such as maintaining action potentials, making them very sensitive to energy fluctuations [35]. It is, therefore, believed that AMPK is essential for neuronal integrity and survival. In the mammalian adult brain, AMPK is predominantly expressed in neurons and strongly localizes to the nucleus [6, 40, 41]. AMPK activation has been extensively studied in hypothalamic neurons for its role in food intake [5, 33]. Little is known, however, about the constitutive levels of activation of AMPK in neurons or in glial cells in other brain regions, such as the hippocampus and cortex. Recently, we confirmed by immunohistochemistry in mouse brain that activated AMPK, phosphorylated at Thr-172 (p-AMPK), is present in the nucleus of most cortical and hippocampal neurons, whereas no significant staining for p-AMPK was found in hippocampal astrocytes [41]. Therefore, under normal conditions in the brain, AMPK is mostly active in neurons and localizes to the nucleus.
Strong evidence indicates that cerebral AMPK is overactivated during energy stress triggered by ischemia, hypoxia, or glucose deprivation, for instance. Whether this overactivation of AMPK is neuroprotective or neurotoxic is, however, a matter of debate [6, 15, 28]. AMPK activation protects primary neuronal cultures from excitotoxicity and several other insults, including exogenous Aβ treatments [6, 25]. Furthermore, mild caloric restriction, exercise, or treatments with the hormone leptin or with the natural polyphenol resveratrol (four interventions known to activate AMPK) were found to prevent different neurodegenerative mechanisms in cell culture systems or mouse models [7, 8, 16, 41]. Work conducted in animal models of stroke found, however, that AMPK activation could be detrimental to neuronal survival [29, 32].
Here, we show that activated AMPK phosphorylated at Thr-172 (p-AMPK) is abnormally and massively accumulated not only in tangle-containing neurons in AD, but also in all the major primary tauopathies, including PSP, CBD, AGD, PiD, FTDP-17, Guam PDC, and tangle-predominant dementia. Co-localization was found in nearly 90% of neurons immunoreactive for PHF1 (phospho-Ser-396/404 tau) and CP13 (phospho-Ser-202 tau). Because CP13 decorates early and mature tangles whereas PHF1 stains mature and late tangles, we concluded that p-AMPK strongly co-localized with tau in both pre-tangle- and tangle-bearing neurons in these disorders. Most regions affected by tau pathology in all the cases examined showed p-AMPK accumulation. At the subcellular level, we observed a shift of p-AMPK localization from the nucleus, observed in normal controls, to the cytoplasmic compartment where p-AMPK abnormally accumulated. Co-localization between hyperphosphorylated tau and p-AMPK was not always observed. Indeed, cytoplasmic granular p-AMPK immunoreactivity was also found in AD, tangle-predominant dementia, and Guam PDC brains in apparently normal neurons devoid of any hyperphosphorylated tau, suggesting that AMPK activation preceded tau phosphorylation. Weaker p-AMPK pathology was observed in PSP and CBD, as compared to AD, where minimal p-AMPK accumulation was also found in tangle-positive glial cells. Further, p-AMPK was not found in purified PHFs, indicating that p-AMPK did not co-aggregate with hyperphosphorylated tau in NFTs. Finally, we found that in vitro recombinant AMPK directly phosphorylated tau at residues Thr-231 and Ser-396/404. Thus, the activated form of AMPK exhibits an abnormal pattern of accumulation in tangle- and pre-tangle-bearing neurons in AD and in all major primary tauopathies. Although it remains to be determined whether AMPK activation is detrimental or neuroprotective in tauopathies, our data suggest that, by controlling tau phosphorylation, AMPK might be critical for the neurodegenerative process of these diseases.
Materials and methods
Materials
PHFs were isolated from an AD brain as described previously [23]. Polyclonal rabbit antibodies directed against phosphorylated Thr-172 αAMPK were purchased from Abcam Inc. (Cat.# ab51110; Cambridge, MA, USA) and Cell Signaling Technology (Cat.# 2535; Danvers, MA, USA). Mouse monoclonal antibodies PHF1, CP13, 2E12, MC6, PG5, CP3 and CP27 are described in Table 1. Highly specific 2E12 antibody directed against phosphorylated Thr-231 tau was generated as described before [10]. AICAR (5-aminoimidazole-4-carboxamide-1-β-riboside) was from Sigma-Aldrich (St. Louis, MO, USA). α1/2AMPK null mouse embryonic fibroblasts were obtained from Dr. Keith R. Laderoute (SRI International, Menlo Park, CA, USA).
Table 1. Primary antibodies used in this study.
| Name | Specificity | Phosphorylation sites | Dilution | Reference or source |
|---|---|---|---|---|
| p-AMPK | p-AMPK | Thr-172 (α2) | 1:200–1:1,000 | Abcam (Cat.# ab51110) |
| p-AMPK | p-AMPK | Thr-172 (α2) | 1:100–1:1,000 | Cell Signaling (Cat.# 2535) |
| CP27 | tau | – | 1:1,000 | [13] |
| CP13 | p-tau | Ser-202 | 1:200–1:500 | [13] |
| PHF1 | p-tau | Ser-396/404 | 1:200–1:1,000 | [17] |
| 2E12 | p-tau | Thr-231 | 1:200 | This study |
| MC6 | p-tau | Ser-235 | 1:200 | [22] |
| PG5 | p-tau | Ser-409 | 1:200 | [24] |
| CP3 | p-tau | Ser-214 | 1:200 | [24] |
p phospho-epitope
Human cases
Samples from the hippocampus and neighboring entorhinal and temporal isocortex were obtained from the individuals listed in Table 2. Samples of the basal ganglia were obtained from the CBD and PSP patients (Table 2). Tissue was obtained from Mayo Clinic Jacksonville brain bank for neurodegenerative disorders and neuropathological diagnoses were made according to current criteria [11].
Table 2. Human cases analyzed in this study.
| Case# | PathDx | Clinical Dx | Braak | Age | Sex | Region |
|---|---|---|---|---|---|---|
| 1 | ALS | ALS | II | 67 | M | Hp |
| 2 | Normal | Normal | 0 | 34 | F | Hp |
| 3 | Normal | Normal | II | 76 | M | Hp |
| 4 | AD | AD | VI | 82 | M | Hp |
| 5 | AD | AD | V–VI | 73 | M | Hp |
| 6 | AD | AD | V–VI | 73 | M | Temp |
| 7 | AD | AD | VI | 89 | F | Hp |
| 8 | AD | AD | VI | 89 | F | Temp |
| 9 | AD | AD | V | 72 | F | Hp |
| 10 | AD | AD | V | 72 | F | Temp |
| 11 | AD | AD | VI | 88 | M | Hp |
| 12 | AD | AD | VI | 66 | F | Hp |
| 13 | AD | AD | VI | 86 | M | Hp |
| 14 | AD | AD | VI | 69 | F | Hp |
| 15 | AD | AD | VI | 79 | M | Hp |
| 16 | FTD | FTD | 0 | 63 | M | Hp |
| 17 | PiD | PNFA | 0 | 69 | F | Hp |
| 18 | PiD | FTD | II | 65 | M | Hp |
| 19 | PiD | PiD | 0 | 72 | M | Hp |
| 20 | PiD | PiD | I | 70 | M | Hp |
| 21 | FTDP-17 (N279K) | PPND | 0 | 51 | F | Hp |
| 22 | FTDP-17 (P301L) | FTD | 0 | 53 | M | Hp |
| 23 | FTDP-17 (N279K) | PPND | 0 | 44 | F | Hp |
| 24 | FTDP-17 (P301L) | FTD | 0–I | 62 | M | Hp |
| 25 | Guam | PDC | IV–V | 70 | F | Hp |
| 26 | Guam | PDC | V | 70 | F | Hp |
| 27 | Guam | PDC | V | 76 | F | Hp |
| 28 | NFTPD | AD | IV–V | 89 | F | Hp |
| 29 | NFTPD | AD | III–IV | 80 | F | Hp |
| 30 | NFTPD | AD | III | 91 | F | Hp |
| 31 | NFTPD | AD | III–IV | 87 | F | Hp |
| 32 | CBD | CBD | 0 | 63 | M | BG |
| 33 | CBD | CBD | I | 61 | F | BG |
| 34 | CBD | PSP | 0 | 55 | F | BG |
| 35 | CBD | PSP | 0–I | 69 | M | BG |
| 36 | PSP | PSP | 0 | 61 | M | BG |
| 37 | PSP | PSP | 0 | 62 | M | BG |
| 38 | PSP | PSP | 0 | 85 | F | BG |
| 39 | PSP | PSP | 0 | 65 | F | BG |
| 40 | AGD | AD | III | 75 | M | Hp |
| 41 | AGD | ET | II–III | 83 | F | Hp |
| 42 | AGD | AD | III | 67 | F | Hp |
| 43 | AGD | Normal | III | 71 | F | Hp |
ALS amyotrophic lateral sclerosis, AD Alzheimer's disease, FTD frontotemporal dementia, PiD Pick's disease, FTDP-17 frontotemporal dementia and parkinsonism linked to chromosome 17, Guam Guam Parkinson dementia complex, NFTPD tangle-predominant dementia, CBD corticobasal degeneration, PSP progressive supranuclear palsy, AGD argyrophilic grain disease, PNFA progressive nonfluent aphasia, PPND pallidopontonigral degeneration, PDC Parkinson dementia complex, ET essential tremor, Hp hippocampus, Temp temporal cortex, BG basal ganglia, F female, M male
p-AMPK immunohistochemistry
Free floating sections (50 μm) or 5-μm-thick sections of formalin-fixed paraffin-embedded brain tissue samples were immunostained with rabbit polyclonal antibodies directed against p-AMPK (Abcam, 1:200 dilution; or Cell Signaling, 1:100 dilution). The primary antibodies used in this study and their dilutions are summarized in Table 1. For staining of free floating sections with p-AMPK (Abcam), non-specific binding was blocked by incubating the sections with 3% hydrogen peroxide, 0.25% Triton-X100 in Tris-buffered saline (TBS) for 15 min at room temperature (RT). After washing in TBS containing 0.05% Triton-X100 (TBS-T), sections were blocked in 5% normal goat serum, 1 mg/ml BSA, 1 mM NaF, 0.05% Triton-X100 in TBS for 1 h at RT. Sections were then incubated in the presence of primary antibodies diluted in TBS containing 1% goat serum, 1 mg/ml BSA, 1 mM NaF and 0.05% Triton-X100 overnight at 4°C in a humidified chamber. After washing, the sections were incubated with biotin-coupled anti-rabbit secondary antibodies (1:1,000 dilution) before incubation with streptavidin-horseradish peroxidase (1:1,000 dilution, Southern Biotech, Birmingham, AL, USA) and visualization with diaminobenzidine tetrahydrochloride. For paraffin-embedded human brain sections from AD and other tauopathies, the slides were deparaffinized by immersion in xylene and hydration through graded ethanol solutions. Antigen recovery was performed by warming the slides for 30 min at 70°C in 10 mM citrate buffer, pH 6.0. Endogenous peroxidase activity was inhibited by incubation in 3% hydrogen peroxide for 30 min at RT, after which slides were washed in TBS and stained for p-AMPK, as described above.
Double-immunofluorescence staining
Double labeling for p-AMPK (Abcam antibody) and phospho-tau (p-tau, CP13 or PHF1 antibodies) was performed in a subset of cases. After removing paraffin, antigen recovery was performed as described above. The sections were blocked with 5% normal goat serum, 1 mg/ml BSA, 1 mM NaF, 0.05% Triton-X100 in TBS for 1 h and incubated first with anti-p-AMPK antibody (1:200 dilution) at 4°C overnight. After washing in TBS-T, the sections were incubated with anti-p-tau antibodies CP13 or PHF1 (1:200 dilution) for 2 h at RT. Sections were subsequently rinsed with TBS and incubated for an additional 90 min at RT in secondary goat antibodies anti-rabbit Alexa Fluor 488 and anti-mouse IgG1 Alexa Fluor 594 (1:500, Molecular Probes-Invitrogen, San Diego, CA, USA). Sections were washed and coverslipped with Vectashield (Vector Laboratories Inc, Burlingame, CA, USA) and observed with a Zeiss Apo-Tome microscope. For quantitative analysis, three images were captured from the CA1 under a 20× objective. The number of neurons with single and double labeling for p-tau and p-AMPK was determined by manual counts from digital images.
In vitro phosphorylation assay for tau
Direct phosphorylation of tau by AMPK was examined by in vitro kinase assay. Five nanograms of active recombinant human AMPK (α1β1γ1, Sigma-Aldrich) was incubated with 5 μg of recombinant 2N4R tau (rPeptides, Bogart, GA, USA) in kinase buffer (20 mM HEPES, 1 mM MnCl2, 1 mM MgCl2, 1 mM DTT, 100 μM sodium orthovanadate, 1 mM Na2ATP) in a final volume of 50 μl for 6 h at 30°C. Recombinant human GSK3β (5 ng, R&D Systems, Minneapolis, MN, USA) was used as a positive control. Kinase reactions were terminated by addition of 3× Laemmli buffer and boiling of the samples. Phosphorylation of tau was assessed by western blotting (WB) using anti-p-tau (PHF1, CP13, CP3, MC6, PG5, 2E12) and anti-total tau antibody (CP27).
Cell culture and treatments
α1/2AMPK null fibroblasts were grown in 1:1 DMEM/Opti-MEM supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Cells were treated at confluence with AICAR for 24 h with the indicated concentrations.
Western blotting
Cell extracts (5–20 μg), purified PHFs (∼100 μg/ml), or samples obtained from the kinase assay were analyzed by SDS–PAGE using antibodies listed in Table 1. Cells were washed with phosphate-buffered saline and solubilized in ice-cold HEPES buffer (25 mM HEPES, pH 7.4, 150 mM NaCl, 1× complete proteases inhibitor mixture, Roche Applied Science, Indianapolis, IN, USA) containing 1% SDS. Samples were electrophoresed on 10–20% gels (Biorad, Hercules, CA, USA) and transferred onto 0.45-μm nitrocellulose membranes. Membranes were blocked in 5% fat-free milk in TBS, and incubated with primary antibodies overnight at 4°C. A standard ECL detection procedure was then used.
Results
Specificity of the anti-phospho-Thr-172 αAMPK antibodies
Specificity of the polyclonal antibodies directed against phospho-Thr-172 αAMPK (p-AMPK) used in this study (see Table 1) was first analyzed by WB in wild type (WT) and α1/2AMPK knockout (KO) mouse embryonic fibroblasts. As expected, immunoreactivity at the predicted molecular mass of AMPK α subunits around 62 kDa was observed in WT but not in KO cells (Fig. 1a). The observed immunoreactivity in WT cells increased upon activation of AMPK by the AMP analog AICAR, indicating that the antibodies reacted with activated AMPK. Antibody specificity was then analyzed by WB in temporal cortex brain homogenates from three AD and three normal control individuals. The antibodies revealed immunoreactivity at around 62 kDa in the brain homogenates (Fig. 1b, upper panel). No obvious difference in p-AMPK levels was observed between patients and controls in this brain region in the small group analyzed, whereas immunoreactivity for PHF1 antibody (phospho-Ser-396/404 tau epitope, Table 1) was strongly elevated in the AD patients (Fig. 1b, lower panel). These data suggested that the anti-p-AMPK antibodies are specific and do not cross-react with phospho-tau (p-tau) epitopes. This was confirmed by the observation that the anti-p-AMPK antibodies did not react with purified PHFs, which were as expected massively immunoreactive to PHF1 (Fig. 1c).
Fig. 1.
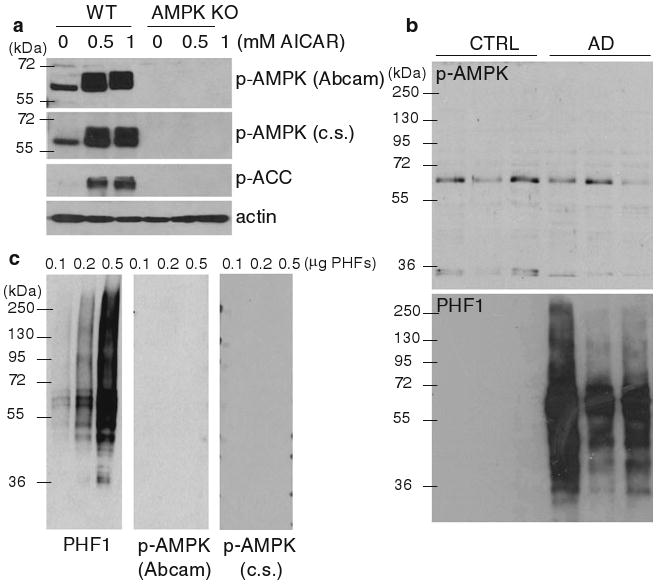
p-AMPK antibody characterization. a Wild type (WT) and α1/2AMPK knockout (AMPK KO) fibroblasts were treated for 24 h with the indicated concentrations of AMPK activator AICAR [41]. Cell extracts were then analyzed by WB for p-AMPK [using Abcam and Cell Signaling (c.s.) antibodies, Table 1], the AMPK target phospho-acetyl-CoA carboxylase (p-ACC), and actin. b WB analysis of temporal cortex sample extracts from three controls (CTRL) and three AD patients using antibodies directed against p-AMPK (Abcam, upper panel) and p-tau (PHF1, lower panel). c WB analysis of purified PHFs using antibodies directed against p-tau (PHF1, left panel) and p-AMPK [Abcam and Cell Signaling (c.s.), middle and right panels, respectively]
Abnormal accumulation of p-AMPK in cerebral neurons in AD brains
In all AD brains examined (see Table 2), we observed a strong immunoreactivity for the activated form of AMPK in different regions. p-AMPK accumulation was found in abundance in neurons of the hippocampal formation, including the CA1 region (Figs. 2a and S1), the entorhinal/transentorhinal region (Fig. 2c), and cortex (Fig. 2d), with a pattern resembling tangle-like neuropathology. p-AMPK immunoreactivity in the AD brain was confirmed using another source of polyclonal antibodies (Table 1; Fig. S1).
Fig. 2.
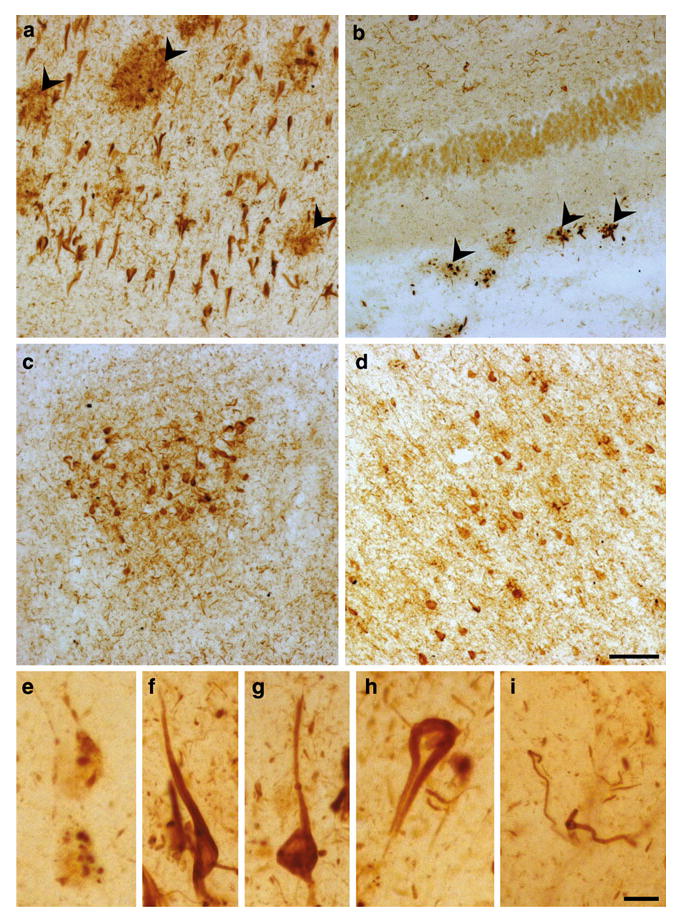
Immunostaining of p-AMPK in AD brains. p-AMPK immunostaining in the CA1 (a, e–i), dentate gyrus (b), entorhinal region (c), and temporal isocortex (d) in floating brain sections of an AD case (Braak stage VI). Arrowheads indicate p-AMPK staining in the neuritic corona of amyloid plaques (a, b). p-AMPK staining was found in intracytoplasmic vesicles (e), flame-shaped tangles (f, g), extracellular ghost tangles (h), and neuropil threads (i). Scale bar 50 μm (a–d), 10 μm (e–i)
At least three types of p-AMPK staining were observed: (a) granular intraneuronal (Fig. 2e), resembling pre-tangle p-tau aggregates; (b) tangle-like intraneuronal (Fig. 2f, g), resembling intraneuronal tau NFTs; and to a lesser extent (c) extraneuronal (Fig. 2h), resembling ghost tangles. p-AMPK immunoreactivity was also evident in dystrophic neurites surrounding amyloid plaques (Fig. 2a, b, arrowheads) and in neuropil threads (Fig. 2i), which represent two other histological abnormalities due to tau pathology in AD brains.
p-AMPK staining was predominantly nuclear in neurons of the hippocampal and cortical regions in normal control brains (Fig. 3a–c). Immunohistochemistry of p-AMPK performed in 50-μm floating sections (Fig. 2) was confirmed in 5-μm sections of formalin-fixed paraffin-embedded AD brain samples (Fig. 3d, e). This staining confirmed the strong accumulation of p-AMPK in tangle-bearing neurons and in dystrophic neurites surrounding amyloid plaques in both the hippocampus and cortex (Fig. 3d, e). As compared to normal control brains (Fig. 3a–c), p-AMPK abnormally localized in the entire cell body in numerous neurons in the AD brains (Fig. 3d, e), indicating the presence of a shift of p-AMPK from the nucleus to the cytosol in some neuronal populations in AD.
Fig. 3.
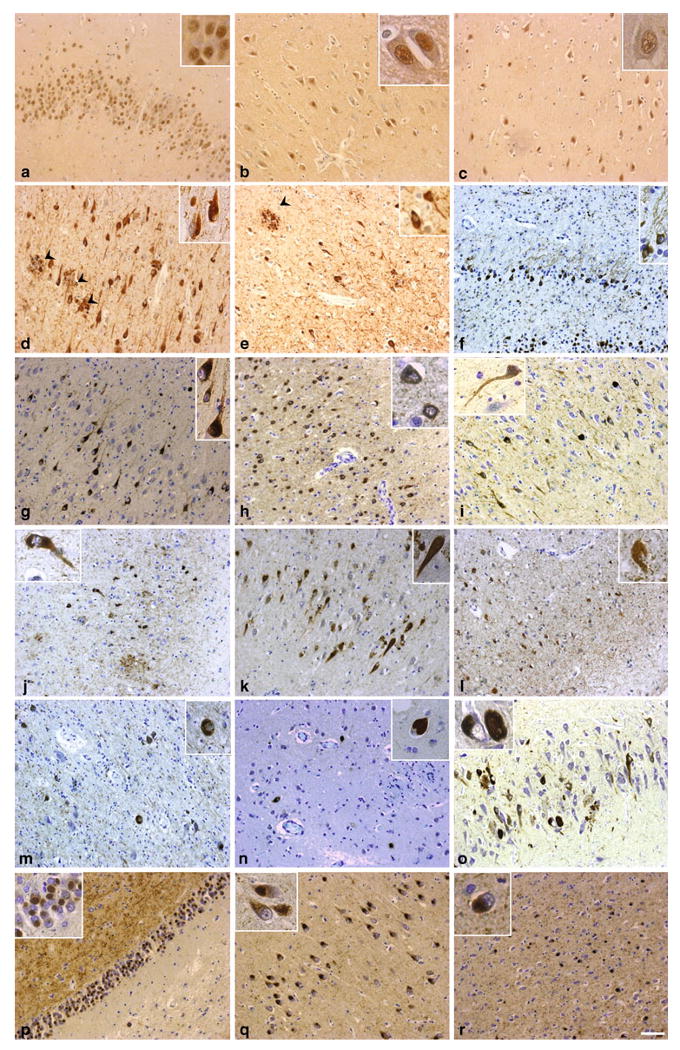
Immunostaining of p-AMPK in normal, AD, and primary tauopathy brains. p-AMPK immunoreactivity in a control individual (a–c), or in AD (d, e), FTDP-17 (f–h), Guam PDC (i, j), tangle-predominant dementia (k, l), CBD (m), PSP (n), AGD (o), and PiD (p–r) patients in different brain regions: Dentate gyrus (a, f, p), CA1 (b, d, g, i, k, o, q), temporal isocortex (c, e, h, j, l, r), basal ganglia (m, n). Arrowheads indicate p-AMPK immunoreactivity in dystrophic neurites surrounding amyloid plaques in AD (d, e). Pick bodies in PiD are strongly immunostained with p-AMPK (p–r). Paraffin sections were counterstained with toluidine blue. Scale bar 50 μm
p-AMPK accumulation in cerebral neurons in primary tauopathies
Neuronal tau accumulation in AD has been suggested to follow amyloid deposition, while this is unlikely to be the case in the primary tauopathies. In the FTDP-17 cases examined, tau accumulation is due to mutations in the tau gene (N279K or P301L, see Table 2). Abundant staining of neuronal inclusions with anti-p-AMPK antibodies was found in all four cases of FTDP-17 examined here (Fig. 3f–h), implying that activation of AMPK is associated with tau pathology even in the absence of amyloid deposition. Similarly, all cases of Guam PDC showed abundant tangle-like staining with antibodies to p-AMPK (Fig. 3i, j). Tangle-like staining was also evident in tangle-predominant dementia cases (Fig. 3k, l) as well as in CBD and PSP (Fig. 3m, n). In these latter two diseases, tau staining revealed significant numbers of “astrocytic plaques”, but these structures were not detectable in sections stained for p-AMPK (not shown). The somewhat different tau inclusions (Pick bodies) present in brains of cases of PiD (Fig. 3p–r) or AGD (Fig. 3o) were strongly positive for p-AMPK, showing that activation of this kinase appears to be associated with different types of neuronal tau inclusion, in addition to classical NFTs.
p-AMPK and p-tau co-localization in AD and primary tauopathies
Anti-tau antibodies specific for phospho-Ser-202 (CP13, Table 1) or phospho-Ser-396/404 (PHF1) were used to detect different aspects of tau pathology. CP13 is commonly used to detect tau pathology in both early (pre-tangles) and more advanced stages of NFT accumulation, whereas PHF1 is a marker for later stage tangles [1, 2]. In order to determine whether p-AMPK accumulates in both pre-tangles and tangles, we assessed p-AMPK and p-tau co-localization by double-immunofluorescence staining in the brains of AD and primary tauopathy patients using CP13 and PHF1 antibodies. In most cases, we observed a robust co-localization of p-AMPK with both phospho-Ser-202 and phospho-Ser-396/404 tau in AD brains in the different regions examined, including the CA1 (Fig. 4a–e). We estimated that more than 90% of CP13- or PHF1-positive neurons showed robust accumulation of p-AMPK in the cortex and hippocampal region. With some exceptions, granular intraneuronal staining of p-AMPK appeared in apparently normal neurons devoid of any (or with weak) p-tau immunoreactivity (Fig. 4f, g, arrowheads), indicating that p-AMPK did not always co-occur with p-tau and may precede tangle formation.
Fig. 4.
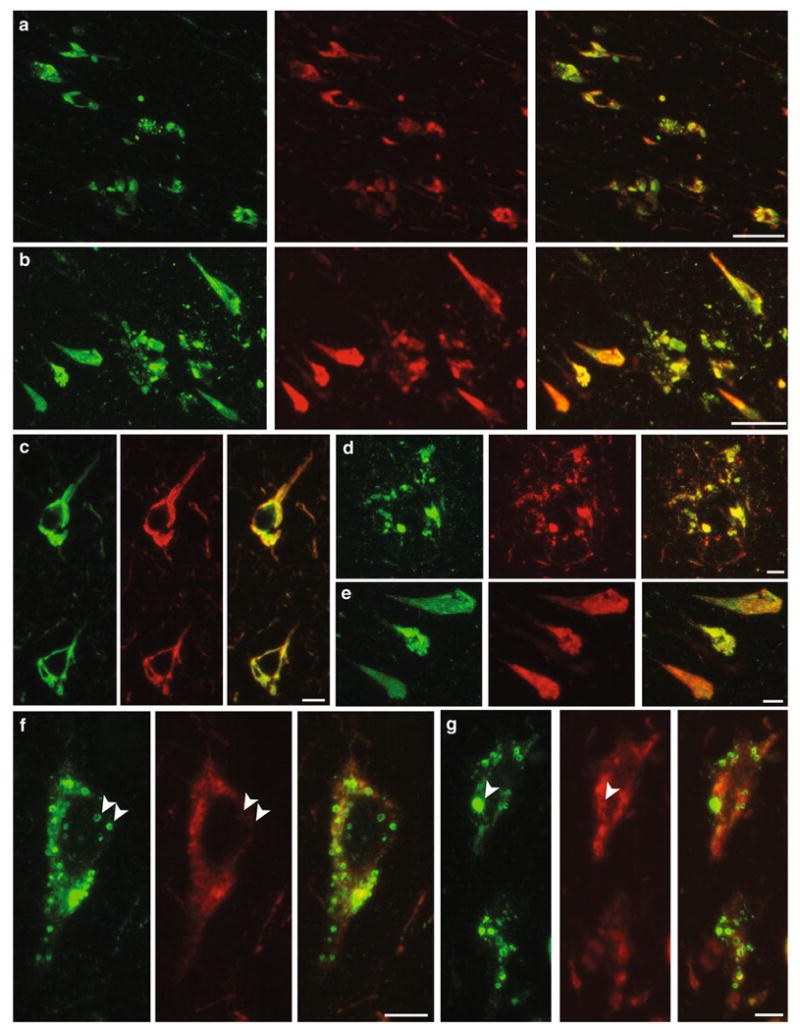
p-AMPK and p-tau co-localization in AD. Immunohistochemical co-localization of p-AMPK with p-tau, labeled with CP13 (a, c, d, f) or PHF1 (b, e, g) in AD patients (CA1 region, Braak stage V/VI). p-AMPK (green) partially or fully co-localized with p-tau (red) in tangle-bearing neurons (c, e), and in dystrophic neurites surrounding amyloid plaques (d). p-AMPK was also present in intracytoplasmic vesicles (f, g, arrowheads) where no (or very weak) staining for p-tau was present. Scale bar 50 μm (a, b), 10 μm (c–g)
In tangle-predominant dementia, FTDP-17, Guam PDC, and AGD brains, a strong co-localization between p-AMPK and phospho-Ser-202 tau was found in neurons in different regions, including in the CA1 (Fig. 5a–i). Anti-p-AMPK antibodies also decorated Pick bodies in the dentate gyrus of PiD patients (Fig. 5k). In line with what we observed in AD brains, granular intraneuronal staining of p-AMPK that was not immunoreactive for p-tau was also found in tangle-predominant dementia (Fig. 5b, c), Guam PDC (Fig. 5f), AGD (Fig. 5h), and PiD cases (Fig. 5j). As compared to AD, less p-AMPK pathology was observed in CBD and PSP. On the other hand, some neurons in several regions, including basal ganglia, showed substantial co-localization between p-AMPK and p-tau in these tauopathies (Fig. 5l, n). Minimal p-AMPK accumulation was also observed in tangle-positive glial inclusions in CBD (Fig. 5m) and PSP (Fig. 5o, p).
Fig. 5.
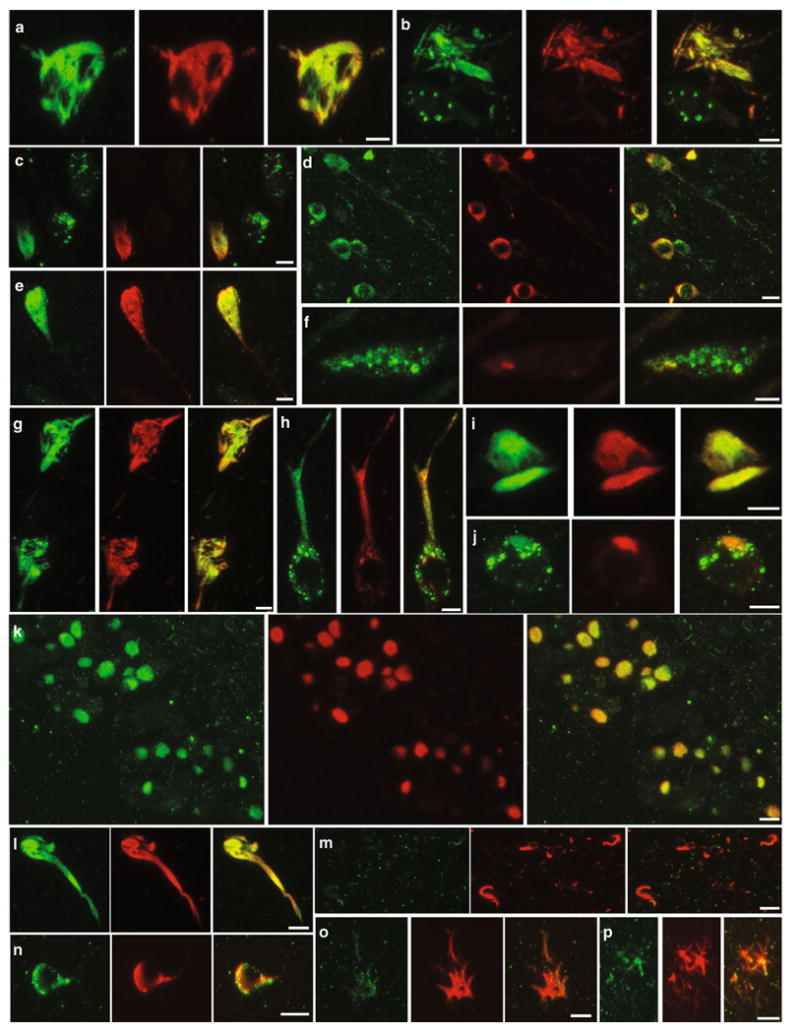
p-AMPK and p-tau co-localization in primary tauopathies. Immunohistochemical co-localization of p-AMPK (green) with p-tau, labeled with CP13 (red) in tangle-predominant dementia (a–c), FTDP-17 (d), Guam PDC (e, f), AGD (g–i), PiD (j, k), CBD (l, m), and PSP (n–p) patients. a–j CA1, k dentate gyrus, l–p basal ganglia. Scale bar 10 μm
In vitro phosphorylation of tau by AMPK
Microtubule-affinity regulating kinases (MARKs) are members of the AMPK family and were proposed to directly phosphorylate tau [31]. In this context, we asked whether recombinant AMPK could trigger phosphorylation of recombinant tau in vitro. AMPK failed to phosphorylate tau on Ser-202, Ser-214, or Ser-235, but triggered efficient phosphorylation on Thr-231 and Ser-396/404, and to a lesser extent on Ser-409 (Fig. 6).
Fig. 6.
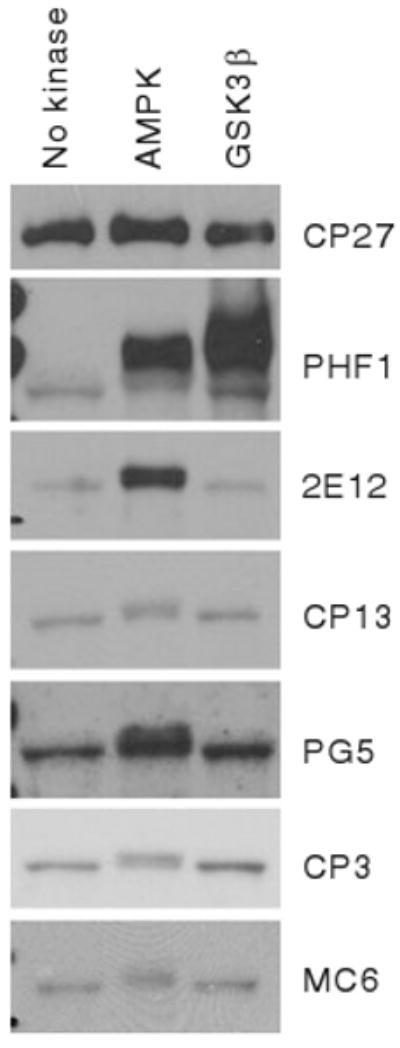
AMPK phosphorylates tau in vitro. WB analysis of recombinant tau incubated in vitro with AMPK or GSK3β using antibodies directed against total tau (CP27), or tau phosphorylated on Ser-396/404 (PHF1), Thr-231 (2E12), Ser-202 (CP13), Ser-409 (PG5), Ser-214 (CP3), or Ser-235 (MC6)
Discussion
In the present study, we assessed for the first time the levels of activated AMPK in postmortem brain samples in AD and other major tauopathies (Table 2). By immunohistochemistry using two independent polyclonal antibodies specific for phospho-Thr-172 AMPK (Table 1), we determined that p-AMPK accumulated in pre-tangle- and tangle-bearing neurons in all tauopathies examined, which include AD and the primary tauopathies FTDP-17, PSP, CBD, AGD, PiD, Guam PDC, and tangle-predominant dementia (Figs. 2, 3, S1). Strikingly, p-AMPK accumulation was found in more than 90% of neurons bearing hyperphosphorylated Ser-202 and Ser-396/404 tau epitopes (Figs. 4, 5). Furthermore, p-AMPK was found in neuropil threads and dystrophic neurites surrounding amyloid plaques, two classical PHF deposition sites in AD (Fig. 2). This pattern of p-AMPK accumulation in the entire cell body and in neurites was not observed in normal aged-matched control brains (Fig. 3a–c).
AMPK subcellular localization is controlled by its subunit composition and its level of activation [39]. We and others found that cerebral neurons predominantly express the catalytic α2 subunit and that activated AMPK preferentially localizes to the nucleus [40, 41]. In the present study, we confirmed in normal human brains that constitutively activated p-AMPK was mostly nuclear (Fig. 3a–c). Analyses by WB showed that total p-AMPK levels in temporal cortex homogenates from three AD and three normal control individuals were not significantly changed between diseased and normal brains. Although this WB analysis should be taken with caution because it was performed on total brain cell homogenates and on a small group of individuals, these results suggest that the main effect of tau pathology is not to activate AMPK but rather to trigger its translocation from the nucleus to the cytosol where the kinase may functionally interact with tau.
Our results showed that AMPK phosphorylated tau in vitro at sites that are targeted in AD and other tauopathies, suggesting that AMPK like many other kinases, such as GSK3β, cyclin-dependent protein kinase 5 (cdk5), MARK1, or ERK2, could represent a key player in tau hyperphosphorylation and aggregation in tauopathies. However, it will also be important to confirm these in vitro data by determining in cell lines and animal models whether AMPK activation (triggered or not by mutated tau) can actually promote tau phosphorylation at residues relevant to PHF formation. These results will be important because previous work has shown that AMPK activation is involved in neuroprotection upon cellular stress. Indeed, a large body of literature supports the notion that AMPK activation is beneficial not only for neuronal maintenance and survival under normal conditions but also upon metabolic stress triggered by several pathological conditions [35, 36]. Studies in drosophila have strengthened this notion by showing that AMPK deficiency causes neurodegeneration [35]. Furthermore, the AMPK activating hormone leptin was found to reduce tau phosphorylation in an AMPK-dependent manner in cell lines [16]. Previous work, including our own, also demonstrated that AMPK is neuroprotective in part by activating autophagy, an evolutionary conserved lysosomal pathway involved in the disposal of several aggregation-prone proteins, such as Aβ [21, 34, 41] and tau [12, 18, 42]. In some instances, however, AMPK activation was found to be detrimental to neuronal survival [29, 32]. It is thus tempting to speculate that tau pathology results in a similar situation of metabolic stress that may represent a trigger for AMPK cytosolic translocation. Based on our in vitro data showing that AMPK can directly phosphorylate tau, we can further hypothesize that in the specific in vivo situation of the diseased neurons, AMPK could interact with tau in the cytosol after translocating from the nucleus to enhance the phosphorylation and destabilization of the microtubule-associated protein. Further work in cell and animal models will allow us to test this hypothesis by determining whether the cytosolic accumulation of activated AMPK co-occurring with tau pathology is detrimental or neuroprotective. Additional experimental approaches will also be needed to determine how tau pathology leads to AMPK accumulation. Granular p-AMPK immunoreactivity was found in different tauopathies, including AD, in apparently unaffected neurons expressing minimal phosphorylation of tau (Figs. 4f, 5f, j), suggesting that AMPK activation preceded tau phosphorylation and aggregation and thus may be triggered by very early events in tau pathology.
In summary, we show for the first time that activated AMPK is abnormally and massively accumulated in pre-tangle- and tangle-containing neurons in AD and in all the major primary tauopathies examined. Most regions where neuronal tau pathology occurred showed p-AMPK accumulation in the different cases examined. Granular p-AMPK immunoreactivity was also observed in AD and several other tauopathies in apparently normal neurons devoid of hyperphosphorylated tau, suggesting that AMPK accumulation is an early event preceding tau pathology. Furthermore, we found that AMPK phosphorylated tau in vitro at residues relevant to PHF formation. Together, these data indicate that AMPK activation, by co-occurring with tau pathology and by mediating tau phosphorylation, might represent a key event in the control of the neurodegenerative process in tauopathies.
Supplementary Material
Acknowledgments
We thank Dr. K. R. Laderoute (SRI International, Menlo Park, CA) for kindly providing us with α1/2AMPK null fibroblasts, and Dr. A. Chan and S. Didier (The Feinstein Institute for Medical Research, Manhasset, NY) for assistance with microscopy analyses. This work was supported in part by the Institutional Clinical and Translational Science Award UL1-RR024996 (Weill Medical College of Cornell University, New York, NY, USA; CTSC Pilot Award, to P. M.) and National Institutes of Health Grant PO1 AT004511 (NCCAM Project 2, to P. M.).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00401-010-0759-x) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Valérie Vingtdeux, Litwin-Zucker Research Center for the Study of Alzheimer's Disease, The Feinstein Institute for Medical Research, North Shore-LIJ, 350 Community Drive, Manhasset, NY 11030, USA.
Peter Davies, Litwin-Zucker Research Center for the Study of Alzheimer's Disease, The Feinstein Institute for Medical Research, North Shore-LIJ, 350 Community Drive, Manhasset, NY 11030, USA; Department of Pathology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA.
Dennis W. Dickson, Neuropathology Laboratory, Mayo Clinic College of Medicine, 4500 San Pablo Road, Jacksonville, FL 32224, USA
Philippe Marambaud, Email: philippe.marambaud@einstein.yu.edu, Litwin-Zucker Research Center for the Study of Alzheimer's Disease, The Feinstein Institute for Medical Research, North Shore-LIJ, 350 Community Drive, Manhasset, NY 11030, USA; Department of Pathology, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, USA.
References
- 1.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 3.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 4.Carling D, Sanders MJ, Woods A. The regulation of AMP-activated protein kinase by upstream kinases. Int J Obes (Lond) 2008;32(Suppl 4):S55–S59. doi: 10.1038/ijo.2008.124. [DOI] [PubMed] [Google Scholar]
- 5.Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Culmsee C, Monnig J, Kemp BE, Mattson MP. AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci. 2001;17:45–58. doi: 10.1385/JMN:17:1:45. [DOI] [PubMed] [Google Scholar]
- 7.Dagon Y, Avraham Y, Magen I, Gertler A, Ben-Hur T, Berry EM. Nutritional status, cognition, and survival: a new role for leptin and AMP kinase. J Biol Chem. 2005;280:42142–42148. doi: 10.1074/jbc.M507607200. [DOI] [PubMed] [Google Scholar]
- 8.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci USA. 2007;104:7217–7222. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies P. A very incomplete comprehensive theory of Alzheimer's disease. Ann N Y Acad Sci. 2000;924:8–16. doi: 10.1111/j.1749-6632.2000.tb05553.x. [DOI] [PubMed] [Google Scholar]
- 10.Davies P. Characterization and use of monoclonal antibodies to tau and paired helical filament tau. In: Hooper NM, editor. Methods in molecular medicine, vol 32: Alzheimer's disease: methods and protocols. Humana Press Inc.; Totowa, NJ: 1998. [DOI] [PubMed] [Google Scholar]
- 11.Dickson DW. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol. 2009;3:1–23. [PMC free article] [PubMed] [Google Scholar]
- 12.Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Characterization of pathology in transgenic mice overexpressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- 14.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 15.Gadalla AE, Pearson T, Currie AJ, Dale N, Hawley SA, Sheehan M, Hirst W, Michel AD, Randall A, Hardie DG, Frenguelli BG. AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J Neurochem. 2004;88:1272–1282. doi: 10.1046/j.1471-4159.2003.02253.x. [DOI] [PubMed] [Google Scholar]
- 16.Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 18.Hamano T, Gendron TF, Causevic E, Yen SH, Lin WL, Isidoro C, Deture M, Ko LW. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- 19.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- 20.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 21.Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 23.Jicha GA, O'Donnell A, Weaver C, Angeletti R, Davies P. Hierarchical phosphorylation of recombinant tau by the paired-helical filament-associated protein kinase is dependent on cyclic AMP-dependent protein kinase. J Neurochem. 1999;72:214–224. doi: 10.1046/j.1471-4159.1999.0720214.x. [DOI] [PubMed] [Google Scholar]
- 24.Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, Horvath Z, Freeman K, Carling D, Huang L, Gonzales C, Cooper E, Smart TG, Pangalos MN, Moss SJ. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 27.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 28.Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30:480–492. doi: 10.1038/jcbfm.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38:2992–2999. doi: 10.1161/STROKEAHA.107.490904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marambaud P, Robakis NK. Genetic and molecular aspects of Alzheimer's disease shed light on new mechanisms of transcriptional regulation. Genes Brain Behav. 2005;4:134–146. doi: 10.1111/j.1601-183X.2005.00086.x. [DOI] [PubMed] [Google Scholar]
- 31.Matenia D, Mandelkow EM. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci. 2009;34:332–342. doi: 10.1016/j.tibs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 32.McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- 33.Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- 34.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 35.Poels J, Spasic MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31:944–952. doi: 10.1002/bies.200900003. [DOI] [PubMed] [Google Scholar]
- 36.Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109(Suppl 1):17–23. doi: 10.1111/j.1471-4159.2009.05916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 38.Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32:150–159. doi: 10.1016/j.tins.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27:4317–4327. doi: 10.1128/MCB.02222-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- 41.Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, Marambaud P. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285:9100–9113. doi: 10.1074/jbc.M109.060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Y, Kruger U, Mandelkow E, Mandelkow EM. Generation of tau aggregates and clearance by autophagy in an inducible cell model of tauopathy. Neurodegener Dis. 2010;7:103–107. doi: 10.1159/000285516. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.