

Abstract
Female rabbit hearts are more susceptible to torsade de pointes (TdP) in acquired long QT type 2 than males, in-part due to higher L-type Ca2+ current (ICa,L) at the base of the heart. In principle, higher Ca2+ influx via ICa,L should be balanced by higher efflux, perhaps mediated by parallel sex differences of sodium–calcium exchange (NCX) current (INCX). We now show that NCX1, like Cav1.2α, is greater at the base of female than male left ventricular epicardium and greater at the base than at the apex in both sexes. In voltage-clamp studies, inward (0, +20 mV, P < 0.04) and outward (−80, −60, −40, −20 mV, P < 0.01) INCX densities were significantly higher (1.5–2 fold) in female base compared to apex and male (base and apex) myocytes. Myocytes were incubated ±17β-oestradiol (E2 = 1 nm) and INCX was measured on days 0, 1, 2 and 3. Inward and outward INCX decreased over 2 days in female base myocytes becoming similar to INCX at the apex. E2 incubation (24 h) increased NCX1 (50%) and INCX (∼3-fold at 60 mV) in female base but not endocardium, apex or in male base myocytes. INCX upregulation by E2 was blunted by an oestrogen receptor (ER) antagonist (fulvestrant, 1 μm), and inhibition of transcription (actinomycin D, 5 μg ml−1) or translation (cycloheximide, 20 μg ml−1). Dofetilide (an IKr blocker) induced early afterdepolarizations (EADs) in female base myocytes cultured for 1 day if incubated with E2, but not without E2 or with E2+KB-R4973 (an INCX inhibitor), E2+fulvestrant or E2 with apex myocytes. Thus, E2 upregulates NCX1 by a genomic mechanism mediated by ERs, and de novo mRNA and protein biosynthesis, in a sex- and region-dependent manner which contributes to the enhanced propensity to EADs and TdP in female hearts.
Non-technical summary
Women of child bearing age are known to have longer QT intervals on their electrocardiogram recordings due to sex-differences in the heart that are mediated by the sex hormone oestrogen. This results in a higher risk of lethal arrhythmias in women when their QT interval is prolonged further through drugs or inherited congenital diseases (i.e. long QT syndrome). Using rabbit hearts, which exhibit the same sex-differences as in humans, it is here shown for the first time that oestrogen up-regulates the sodium–calcium exchanger (NCX), a critical protein that regulates calcium ions in the heart and thereby alters the force of contraction, but only at the base of the ventricles (where the heart has its widest cross section). This effect accounts for the greater risk of long QT-related arrhythmias in women.
Introduction
The long QT syndrome type 2 (LQT2) is a consequence of either gene mutations or drugs that result in a loss of function of the rapid component of the delayed rectifying K+ current, IKr. Clinical features include recurrent syncope or sudden cardiac death due to polymorphic ventricular tachycardia known as torsade de pointes (TdP) (Roden, 2004, 2008). Women have been shown to be at a greater risk than men for congenital or drug-induced forms LQT2-related arrhythmias (Makkar et al. 1993; Bednar et al. 2002). In humans and rabbits, sex differences in arrhythmia phenotype are reversed in adolescence before the surge of sex steroids such that males (boys <14 years old, male rabbits <42 days old) have the higher risk of TdP than their female counterpart (Liu et al. 2005; Goldenberg et al. 2008).
Although there is general agreement that early afterdepolarizations (EADs) initiate TdP (January & Riddle, 1989; Volders et al. 2000), controversies persist regarding the mechanisms that trigger EADs. Experimental and simulation studies proposed that in the setting of delayed repolarization (i.e. LQT2), EADs were induced by the spontaneous reactivation of L-type Ca2+ channels; moreover, the inward Ca2+ current, ICa,L, caused the EAD upstroke and did not require a spontaneous elevation of intracellular Ca2+ or Ca2+ release from the sarcoplasmic reticulum (SR) (Marban et al. 1986; January & Riddle, 1989; Viswanathan & Rudy, 1999). Subsequent studies proposed that the prolongation of action potential durations (APDs) resulted in SR Ca2+ overload, spontaneous SR Ca2+ release that enhanced the forward mode of the Na+–Ca2+ exchanger (NCX), and the depolarizing NCX current (INCX) that triggered the re-activation of ICa,L (Szabo et al. 1995; Verduyn et al. 1995; Volders et al. 2000). Simultaneous optical mapping of voltage and intracellular Ca2+ (Cai) in a rabbit model of drug-induced LQT2 showed that Cai overload and a spontaneous Cai elevation preceded EADs, and TdP was visualized as the out-of-phase firing of EADs from multiple foci (Choi et al. 2002). The blockade of IKr with E4031 caused the expected APD prolongation but also caused a reversal of the gradient of repolarization (apex to base shifted to base to apex with IKr blockade) and a marked increase in the dispersion of repolarization, which may contribute to the initiation and perpetuation of TdP (Choi et al. 2002). Optical mapping revealed that adult female and adolescent male rabbit hearts treated with an IKr blocker were highly susceptible to EADs and TdP; in contrast, hearts from the opposite sex had long APDs (2–3 s) but rarely an EAD and no TdP (Liu et al. 2005; Sims et al. 2008). Note that sex differences in the incidence of EADs and TdP risk were observed during IKr inhibition and bradycardia but without lowering K+ and Mg2+ concentrations by 50% because the latter concealed sex and age differences in arrhythmia phenotype (Liu et al. 2005; Sims et al. 2008). Sex and regional differences in the incidence of EADs were attributed to differences in L-type Ca2+ channel protein (Cav1.2α) and current (ICa,L), which was ∼30% higher at the base compared to the apex of (i) adult female and (ii) pre-pubertal male ventricular myocytes; moreover, EADs appeared first at the base of the heart (Sims et al. 2008). In principle, the higher Ca2+ influx via ICa,L at the base of the heart should be balanced by higher Ca2+ efflux, and a higher expression of NCX was suggested as a possible mechanism (Sims et al. 2008).
Besides its possible role in long QT, NCX over-expression has been linked to contractile dysfunction and enhanced arrhythmia susceptibility in human, rabbit and canine models of heart failure (Pogwizd et al. 2001; Schillinger et al. 2002; Hobai et al. 2004). Pharmacological inhibition of NCX with SEA-0400 suppressed afterdepolarizations and arrhythmias in canine (Nagy et al. 2004) and rat (Satoh et al. 2000) hearts. Sex-differences in NCX density have been controversial. One group reported that NCX protein and mRNA levels were higher in female than male rat left ventricular myocytes and that OVX reduced NCX whereas E2 replacement restored NCX levels (Chu et al. 2005, 2006). Another group reported the opposite, that left ventricular myocytes from OVX rats exhibited an increase of NCX protein, which was reversed by E2 replacement (Kravtsov et al. 2007). Still another group found no significant differences of INCX or its response to β-adrenergic activation in left ventricular myocytes of female and male pigs (Wei et al. 2007a). A possible explanation for these disparate results was the lack of attention to regional variations of NCX expression.
Several studies support the notion that sex steroids modulate the expression of cardiac ionic channels and thereby produce sex differences in arrhythmia susceptibility (Abi-Gerges et al. 2004; Coker, 2008). In adult rabbits, LQT2 elicited with dofetilide (an IKr blocker) prolonged APDs and promoted EADs more in female and castrated male myocytes than in normal male and castrated female myocytes isolated from the endocardium (Pham et al. 2001). Papillary muscles isolated from ovariectomized (OVX) rabbits exhibited a greater incidence of EADs following IKr blockade if the myocytes were treated with oestradiol (E2) compared to those treated with testosterone (Hara et al. 1998). INCX may serve as the trigger of EADs in drug-induced LQT2 by providing the depolarizing current that triggers the re-activation of ICa,L during the AP plateau. In this scenario, LQT2 prolongs APDs, resulting in a higher Ca2+ influx during each AP that can be balanced by Ca2+ efflux in most but not all regions of the heart. In adult female and pre-pubertal male myocytes from the base of the epicardium, the higher ICa,L density results in SR Ca2+ overload and spontaneous SR Ca2+ release, which increases the inward depolarizing INCX that can trigger the re-activation of ICa,L to produce EADs. Hence, the higher INCX density together with a higher ICa,L density may be important determinants of the LQT2 arrhythmia phenotype.
In this study, sex and regional differences of NCX1 (the dominant NCX isoform in the heart) and INCX were quantitatively analysed in adult rabbit ventricular myocytes. INCX density was measured from female myocytes isolated from the base and apex and tracked as a function of days after isolation (days: 0, 1, 2 and 3). Genomic effects were investigated by incubating female base myocytes in serum with or without E2 (1 nm) and with or without an oestrogen receptor (ER) antagonist, fulvestrant, and with or without actinomycin D (AD) or cycloheximide (CX) to inhibit transcription or translation, respectively. Regional genomic effects of 24 h of E2 treatment on NCX levels were investigated by comparing E2-dependent up-regulation of INCX in myocytes isolated from the base, apex and base endocardium of female hearts as well as base epicardium of male hearts. The physiological relevance of genomic oestrogen effects was assessed by measuring the incidence of EADs in apex and base myocytes after 24 h in culture media with and without E2 and before versus after the acute addition of the NCX inhibitor KB-R7943.
Methods
Ethical approval
All protocols complied with and were first approved by the University of Pittsburgh Institutional Animal Care and Use Committee and were in accordance with the current Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and the policies of The Journal of Physiology (Drummond, 2009). The rabbits were obtained from an approved commercial vendor, Myrtle's Rabbitry, and were housed in the animal facilities of the University of Pittsburgh according to Federal Regulations of the USA. In all the studies described in this article, the rabbits were first killed and the hearts were removed to isolate and culture adult ventricular myocytes for 0–3 days.
Isolation of ventricular myocytes
Ventricular myocytes were isolated as previously described, with minor modifications (Sims et al. 2008). Briefly, adult (12 weeks) New Zealand White rabbits of both sexes were killed with an intravenous injection of pentobarbital (50 mg kg−1) with a co-injection of heparin (200 U kg−1i.v.). Three-month-old rabbits were chosen because they are well past the surge of sex steroids, which starts at 42–49 days of age and peaks at ∼60 days (Berger et al. 1982; de Turckheim et al. 1983). At that age, the arrhythmia phenotype upon IKr suppression is that of adults where female hearts have an enhanced risk of TdP (Liu et al. 2005; Sims et al. 2008). After the rabbits succumbed to the lethal drug injection, the hearts were excised and perfused in a Langendorff apparatus with a Tyrode solution containing (in mmol l−1): 140 NaCl, 5.4 KCl, 1.5 CaCl2, 2.5 MgCl2, 11 glucose, and 10 Hepes (pH 7.4). All solutions were at 37°C and were oxygenated with 100% O2. Hearts were perfused with Ca2+-containing Tyrode solution for 5 min, followed by perfusion with Ca2+-free Tyrode solution for 10 min, after which collagenase type 2 (Worthington Biochemical Corp., Lakewood, NJ, USA; at 0.60 mg ml−1) was added for 20 min of digestion. The heart was then perfused with Ca2+-free Tyrode solution with 0.1 mmol l−1 CaCl2 and 0.02% albumin for 10 min (Sims et al. 2008). The left ventricle was removed and placed in a high K+ buffer containing (in mmol l−1): 110 potassium glutamate, 10 KH2PO4, 25 KCl, 2 MgSO4, 20 taurine, 5 creatine, 0.5 EGTA, 20 glucose, and 5 Hepes (pH 7.4). Sections of epicardium approximately 1 mm in depth were surgically removed from the apex and base regions of the left ventricle, and cell isolation was performed separately for each region. Myocytes from the apex were taken from 3–6 mm from the very bottom of the heart; those from the base were taken from 1–4 mm below the left atrium. The tissues were minced, and single myocytes were obtained by filtering through a 100 μm nylon mesh. Cells were allowed to settle in the high K+ solution, and the pellet was washed once and re-suspended and incubated at 37°C in phenol red-free Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum (Invitrogen no. 10082) and primocin (InvivoGen, San Diego, CA, USA; 100 μg ml−1). The sex steroids in fetal bovine serum (FBS) as analysed by Invitrogen resulted in final medium concentrations too low to have biological activity: oestradiol <3.5 pm, testosterone <20 pm and progesterone <50 pm. The sex hormone binding globulin (SHBG) concentration in FBS was <2 nm (measured by the Centre for Research in Reproduction, University of Virginia).
Electrophysiology
The whole-cell configuration of the voltage-clamp technique was used to measure the Na+/Ca2+ exchange current, INCX, at 37°C using. Membrane current was recorded with an Axopatch 200 amplifier (Molecular Devices, Sunnyvale, CA, USA). Signals were filtered at 5 kHz and sampled at 10 kHz using a Digidata 1200a interface and pCLAMP (v. 9.2) software (Molecular Devices). Patch pipettes (2–4 MΩ) were filled with (in mmol l−1): 120 glutamic acid, 120 CsOH, 0.5 MgSO4, 10 Hepes, 20 NaCl, 10 EGTA, 3 CaCl2 and 5 MgATP (pH 7.2 with CsOH). The external solution contained (in mmol l−1): 140 NaCl, 10 CsCl, 1 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes (pH 7.4 with NaOH). Nifedipine (10 μm) and ouabain (10 μm) were added to the external solution to inhibit ICa,L and Na+,K+-ATPase, respectively. Membrane currents were recorded while applying a standard voltage-ramp protocol. Cells were held at −50 mV before applying an 80 ms step depolarization to +100 mV followed by a descending voltage ramp (from +100 mV to −120 mV) lasting 1600 ms. The sweep interval was 10 s. INCX was assessed as a Ni2+-sensitive current (Hobai & O’Rourke, 2000) obtained by subtracting a residual current measured in the presence of 5 mm Ni2+ from the total current. Cell capacitance was recorded after each successful patch and used to calculate the INCX density. The INCX density is dependent on the Na+ and Ca2+ gradients, which were kept constant to minimize cell-to-cell and animal-to-animal variability.
Voltage-clamp experiments were performed 2–5 h after incubation on the day of cell isolation (day 0 or freshly prepared cells) and 1–3 days after isolation. Myocytes attached to laminin-coated plastic ware retained their rod-shape, striations and T-tubules for more than 3 days. Table 1 summarizes the mean ±s.e.m. capacitance of myocytes freshly prepared and 1–3 days old in control and in myocytes incubated with 1 nm E2. On days 2 and 3, there was a statistically significant decreases of cell capacitance of 10 and 19%, respectively. There was no significant change in capacitance between cells incubated in the presence or absence of E2 (1 nm). In studies of genomic effects of E2, cells were separated in two groups: one group was incubated in control medium plus 1:5000 dimethyl sulphoxide (DMSO), the other with the same medium plus E2 (Sigma) from a stock solution of E2 dissolved in DMSO. A genomic up-regulation of NCX by E2 would be expected to be mediated by oestrogen receptors, to require a time course of several hours and to be accompanied by a stimulation of transcription and translation. The role of oestrogen receptors in mediating the E2-dependent regulation of INCX was evaluated by incubating myocytes with E2 (1 nm) in the presence or absence of the ER antagonist fulvestrant (ICI182,780 = 1 μm, Tocris Boscience, Ellisville, MO, USA). Changes in transcription and protein synthesis associated with the up-regulation of INCX were assessed by incubating myocytes for 24 h with E2 (1 nm) in the absence and presence of inhibitors of transcription (actinomycin D, 5 μg ml−1, Calbiochem) and protein synthesis (cyclohexamide, 20 μg ml−1, Calbiochem), as previously described with rat ventricular myocytes (Benitah & Vassort, 1999).
Table 1.
Changes in membrane capacitance as a function of time
| Capacitance (pF) | Fresh | N | Day 1 | n | Day 2 | n | Day 3 | n |
|---|---|---|---|---|---|---|---|---|
| Control | 86.3 ± 5.5 | 36 | 86.2 ± 8.3 | 103 | 76.1 ± 4 | 81 | 69.1 ± 5.3 | 63 |
| Oestrogen | 87.3 ± 4.4 | 36 | 87.5 ± 8.4 | 14 | 80.5 ± 3 | 12 | 72.8 ± 5.6 | 10 |
Membrane capacitance was measured from ‘freshly’ prepared myocytes and after 1–3 days in culture, with or without oestrogen (1 nm). The mean ±s.e.m. capacitance was calculated for a large number of cells from at least 5 different hearts. By days 2 and 3 there was a statistically significant decrease in membrane capacitance of 10% and 19%, respectively (P < 0.05). Membrane capacitance was unchanged after 24 h and incubation with E2 did not change membrane capacitance on days 1–3 compared to controls.
APs were recorded under current-clamp mode (Cheng et al. 1999; Sims et al. 2008). The pipette solution contained (in mmol l−1): 130 potassium gluconate, 0.5 MgCl2, 10 NaCl, 9 KCl, 10 Hepes, 5 MgATP and 0.05 EGTA, pH 7.2 with KOH at 35°C. The external solution contained (in mmol l−1): 140 NaCl, 5.4 KCl, 2.5 CaCl2, 1 MgCl2, 11 glucose, and 10 Hepes (pH 7.4). A current pulse of 1.5 nA and 5 ms duration was injected to trigger an AP at a stimulation frequency of 0.33 Hz. To observe AP prolongation and EADs, dofetilide (400 nm) was added to the external solution after a stable AP was obtained. Fisher's exact test was used to analyse the EAD propensities among different groups.
Immunocytochemistry
NCX1 protein labelling and confocal imaging of myocytes were carried out at the Centre for Biological Imaging (http://www.cbi.pitt.edu). Briefly, myocytes were incubated on laminin-coated coverslips (BD Biosciences, Franklin Lakes, NJ, USA) for 24 h, fixed with 2% paraformaldehyde and permeabilized with 0.1% Triton X-100. NCX1 monoclonal antibody (1:200; Affinity BioReagents, Golden, CO, USA) was incubated for 60 min. Alexa 488 labelled goat anti-mouse (5 μg ml−1; Invitrogen (Molecular Probes), Carlsbad, CA, USA) was used as a secondary antibody. Immunofluorescence was measured using an Olympus 1000 Fluoview (Olympus America) confocal microscope. Metamorph (v. 7.1, Molecular Devices) was used to analyse the average fluorescence-labelled NCX1.
Western blots
Tissues were dissected from the apex and base of the epicardium and stored in liquid nitrogen. Samples were pulverized in liquid nitrogen and homogenized on ice. Lysis buffer contained (in mm): 150 NaCl, 1 EDTA, 2.5 MgCl2, 20 Hepes, 1% Triton X-100, 0.5 dithiothreitol and 0.1% SDS. Protease inhibitors and phosphatase inhibitor cocktail (PhosSTOP, Roche) were added in the lysis buffer. Supernatant resulting from centrifugation (120,000 g, 5 min) was used to measure protein concentration with the Bio-Rad assay (Bio-Rad Laboratories, Hercules, CA, USA) and run SDS-PAGE gels with 30 μg of protein per lane. NCX1 antibody was used at 1:1000 dilutions (Thermo Scientific catalogue no. MA3-926).
Data Analysis
The statistical analysis was carried out by two methods. The first method compared the INCX density at +60 mV among the groups using Student's t test, as previously described (Maack et al. 2005; Wei et al. 2007b; Ozdemir et al. 2008). Student's t test was a stringent statistical analysis where female base and female apex myocytes from different rabbit hearts were grouped and compared with male base and male apex groups of myocytes from different hearts and where P < 0.05 was considered significant. Mean INCX densities were compared between: apex vs. base for male and female myocytes, control vs. E2 treated myocytes for day 0, 1, 2, or 3 and E2 vs. E2 plus fulvestrant or actinomycin D or cycloheximide. The analysis of INCX at +60 mV can be used as an estimate of functional NCX1 proteins, with the caveat that changes in INCX represent changes in NCX protein and not cell signalling-mediated changes in INCX.
The second method analysed differences in INCX at the physiological voltages that span the cardiac action potential. INCX was separated into two components: the inward (forward mode) and outward (reverse mode) of INCX. The mean inward INCX was calculated by averaging its values at −80, −60 and −40 mV. The mean outward INCX was calculated by averaging its values at 0 and 20 mV. These voltage values were chosen to avoid errors caused by slight shifts in the reversal potential from cell to cell. First, the mean inward INCX was calculated for each heart at three voltage values (−80, −60 and −40 mV), and the mean outward INCX was calculated for each heart at two voltage values (0 and 20 mV). Then, the mean inward and outward currents were paired for each heart and voltage value to compare the currents at the base vs. apex of the same heart. For example, in Table 2, there were 12 apex–base pairs for the inward INCX (3 voltage values times 4 hearts) and eight apex–base pairs for the outward INCX (2 voltage values times 4 hearts). The same data processing algorithm was applied without pairing for comparison of male versus female groups (unpaired comparison of independent groups). The apex versus base data in the same hearts were compared using Wilcoxon's matched pairs test. The male versus female groups were compared using the Mann–Whitney U test. Serial changes between days 0, 1, 2 and 3 were evaluated using Friedman's ANOVA for repeated measurements. The myocytes harvested from the base and apexes of the heart are referred to the base- and apex-myocytes, respectively. Data are presented as means ± standard deviation, unless otherwise indicated in the text; P≤ 0.05 was considered significant. Data distributions were examined using the Shapiro–Wilk W test for normality. Non-parametric tests were applied because most distributions deviated significantly from normal.
Table 2.
Inward and Outward INCX densities at the base and apex of adult female and male hearts
| N/P | Mean ±s.d. | Median (25–75% range) | P values | ||
|---|---|---|---|---|---|
| Female Base | Outward INCX | 4/8 | 0.83 ± 0.44 | 0.81 | †0.03 |
| (0.54 ÷ 1.00) | #0.009 | ||||
| Inward INCX | 4/12 | −0.55 ± 0.36 | −0.46 | †<0.01 | |
| (−0.82 ÷−0.25) | #0.33 | ||||
| Female Apex | Outward INCX | 4/8 | 0.31 ± 0.32 | 0.26 | #0.03 |
| (0.04 ÷ 0.58) | |||||
| Inward INCX | 4/12 | −0.38 ± 0.38 | −0.20 | #0.95 | |
| (−0.67 ÷−0.06) | |||||
| Male Base | Outward INCX | 4/8 | 0.31 ± 0.27 | 0.25 | †0.04 |
| (0.14 ÷ 0.48) | |||||
| Inward INCX | 4/12 | −0.64 ± 0.29 | −0.63 | †<0.01 | |
| (−0.85 ÷−0.41) | |||||
| Male Apex | Outward INCX | 4/8 | 0.72 ± 0.27 | 0.66 | |
| (0.52 ÷ 1.01) | |||||
| Inward INCX | 4/12 | −0.33 ± 0.23 | −0.37 | ||
| (−0.51 ÷−0.15) |
Base versus Apex for each heart using Wilcoxon matched pairs test;
Female versus Male groups using Mann–Whitney U test (unpaired).
Four groups of myocytes were investigated: female base and apex and male base and apex (N= 4 hearts of each sex). The mean inward INCX density (±s.d.) was calculated at three voltages (−80, −60, and −40 mV) from base myocytes of each heart and compared to measurements from apex myocytes of the same heart. Similarly, the mean outward INCX density (±s.d.) was calculated at two voltage values (0 and 20 mV) from base myocytes of each heart and compared to measurements from apex myocytes from the same heart. In some hearts, several myocytes (n= 1–3) were used to calculate the mean inward and outward INCX.
Results
Regional distribution of NCX1 and INCX in adult rabbit hearts
We previously reported that L-type Ca2+ channel protein (Cav1.2α) and its current (ICa,L) are elevated at the base of adult female ventricles compared to the apex and to values found in male hearts (Sims et al. 2008). Figure 1A shows a Western blot of NCX1 distribution from three male and three female rabbit hearts where epicardial tissue samples were taken from the base and apex and were run together on the same gel. The data show that NCX1 channel protein levels with relative molecular mass (Mr) of ∼120,000 were significantly higher at the base of female hearts compared to the base of male hearts and the apex of both sexes. Western blots were repeated on four more male and four more female hearts on separate gels to ascertain the validity of these findings. NCX1 protein densities were normalized with respect to β-actin and revealed a statistically significant increase of NCX1 density (38%, P < 0.01, n= 7) at the base of female hearts compared to males and compared to the apex of both sexes. A summary bar graph of NCX1 protein density from the apex and base of seven male and seven female hearts is shown in Fig. 1B. The higher levels of NCX1 density matched the previously reported distribution of Cav1.2α density (Sims et al. 2008). INCX density was measured from myocytes isolated from the base and apex of female and male hearts by applying a voltage ramp from +100 to −120 mV in voltage-clamp experiments (Fig. 2A) and taking INCX as the Ni2+-sensitive current (Fig. 2B) (Hobai & O’Rourke, 2000). To compare INCX density among different myocytes, the current was divided by the cell's capacitance, and INCX density (pA pF−1) was measured for 12 points along the ramp protocol (in mV, at +100, + 80, +60, +40, +20, 0, −20, −40, −60, −80, −100 and −110) and plotted as a function of membrane potential. Figure 2C plots the mean ±s.e.m. of INCX density as a function of voltage for male base and apex myocytes as well as female base and apex myocytes, from the epicardium of adult hearts. All I–V plots were recorded in 2–5 h after cell isolation. As summarized in Fig. 2D, INCX (at +60 mV) was similar for male base (1.76 ± 0.27 pA pF−1), male apex (1.70 ± 0.30 pA pF−1) and female apex myocytes (1.30 ± 0.50 pA pF−1) but was significantly higher in female base myocytes (2.59 ± 0.32 pA pF−1). Based on both Student's t test and two-factor ANOVA without replication, INCX density was higher at the base of female myocytes compared to male myocytes (P < 0.05) and to the apex of both sexes P < 0.032 and P < 0.017). The reversal potential was −14.3 ± 3.2 in male and −18.2 ± 4.6 mV in female myocytes, which was not statistically significant (P= 0.68).
Figure 1. Distribution of NCX1 protein in male and female rabbit heart tissue samples were dissected from the epicardium of male and female hearts and processed as described in Methods.
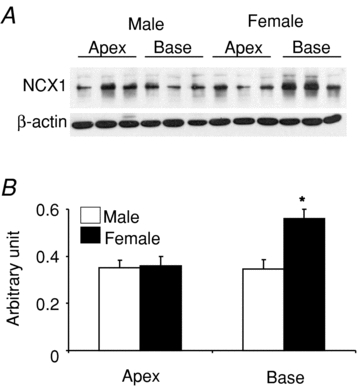
A, Western blot analysis using an NCX1 antibody were compared for protein samples from 3 male and 3 female hearts from the base and the apex of the left ventricular epicardium. The samples were run on the same gel and normalized with respect to their β-actin content. B, summary bar graph of NCX1 protein density from a total of 7 male and 7 female hearts. The data show a statistically significant increase in NCX1 channel protein (P < 0.02) at Mr∼120,000 at the base of female compared to male hearts and the apex of both sexes.
Figure 2. Distribution of INCX density in male and female rabbit hearts.
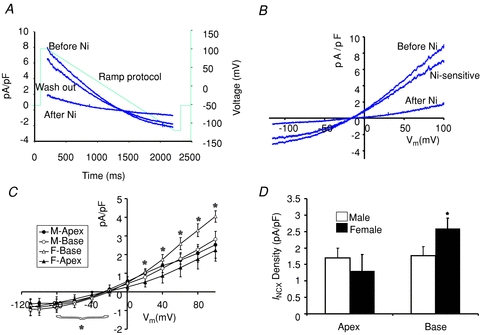
A, a voltage ramp protocol from −100 to +100 mV was applied in 2 s to ventricular myocytes in the whole-cell configuration of the patch clamp technique. Representative traces of current densities (pA pF−1) as a function of time (ms) before, after and after washout of 5 mm NiCl2. Dotted line indicates the ramp protocol. B, I–V plot of INCX, the Ni2+-sensitive current (pA pF−1). C, the mean ±s.e.m.INCX was plotted as a function of voltage for every 20 mV for male base (n= 6 cells/3 hearts) and apex (n= 6/3) and for female base (6/4) and apex (6/4) myocytes. *P < 0.05 for Inward INCX at F-Base compared to F-Apex, M-Base and M-apex. D, summary bar graphs of INCX densities measured at +60 mV from the apex and base of male and female hearts. Based on Student's t test and ANOVA 2 paired factors, there was no statistical difference between INCX male base and apex myocytes (P < 0.353 t test and 0.118, ANOVA) but there was a statistically significant difference between female base and female apex (P < 0.032, t test and P < 0.017 ANOVA).
The physiological significance of these sex and regional differences in NCX1 protein and INCX density was evaluated by measuring the inward and outward NCX currents in the voltage range that spans the cardiac action potential (Table 2). For each heart, the mean ±s.d. of the inward INCX was calculated for base myocytes at four voltages, −80, −60 and −40 cmV, and compared at each voltage with values measured from apex myocytes. Similarly, the mean outward INCX was calculated for base myocytes at 0 and 20 mV and compared at each voltage with values measured from apex myocytes. Statistical significance was tested using Wilcoxon's matched pairs test, which gave us 12 matched pairs (P) to compare inward INCX at the base and apex (P= three voltage values ×N where N= four hearts) and gave us eight matched pairs to compare outward INCX (P= two voltage values ×N where N= 4 hearts). As summarized in Table 2, the mean outward INCX density at the base of female myocytes was 2.3-fold greater than at the apex (P < 0.001) and the mean inward INCX density at the base was 2-fold greater than at the apex (P < 0.01). The mean outward INCX density at the base of female myocytes was 2.3-fold greater than at the base of male base myocytes (P < 0.04) and 1.3-fold greater than male apex myocytes (P= 0.04). The mean inward INCX at the base of female myocytes was 1.1-fold greater and 2-fold greater than male base and apex myocytes, respectively (P < 0.01). It is reasonable to expect that these significant sex differences in INCX density in both the forward (inward INCX) and reverse (outward INCX) modes of the exchanger during an action potential will have a considerable impact on Ca2+ handling.
Time-dependent changes in INCX
To elucidate the mechanisms whereby INCX density is greater at the base of female hearts, myocytes were isolated from the base and apex and incubated in serum free of sex steroids, and INCX was measured from freshly isolated myocytes (2–5 h), and then at days 1, 2 and 3. In freshly prepared myocytes, INCX density was consistently greater in myocytes from the base compared to the apex (Fig. 3A). At the base, INCX density decreased between day 0 and day 2 and levelled off by day 3. In contrast, INCX at apex myocytes from the same hearts did not significantly change (Fig. 3A–D). At +60 mV, the mean ±s.e.m. of the INCX density was compared for female myocytes from the base vs. apex on day 0 (fresh: 2.17 ± 0.28 vs. 0.68 ± 0.28 pA pF−1, n= 6/4, P < 0.03), day 1 (1.87 ± 0.45 vs. 0.68 ± 0.36 pA pF−1, n= 6/4, P < 0.04), day 2 (1.39 ± 0.63 vs. 0.88 ± 0.28 pA pF−1, n= 5/4, P < 0.445) and day 3 (1.39 ± 0.63 vs. 0.80 ± 0.28 pA pF−1, n= 6/4, P < 0.366) (Fig. 3E). At the base, INCX density decreased by 14.3% on day 1 and by 35.9% on day 2. Figure 4 compares the changes of outward (Fig. 4A) and inward (Fig. 4B) INCX at days 1 and 2 compared to day 0. For outward INCX, the 3-day trend showed a significant decrease (Friedman's ANOVA, P= 0.006; *in day 3, P < 0.05 compared to day 0) for myocytes at the base but not for myocytes at the apex (P= 0.1). For inward INCX, the 3-day trend was highly significant at the base (P= 0.0006, for days 0, 1 and 2) and just reached significance at the apex (P= 0.04). There were no significant changes in male INCX as a function of time. In male myocytes from the base of the heart, INCX (at 60 mV) was 2.23 ± 0.67 on day 0 (n= 6/4 hearts; P < 0.01 compared to female myocytes), 2.27 ± 0.64 on day 1 (n= 5/4 hearts, P < 0.05), 2.89 ± 0.47 on day 2 (n= 7/4 hearts) and 3.47 ± 0.77 pA pF−1 on day 3 (n= 10/4 hearts). Hence, INCX at the base of the female epicardium decreased over 2 days when incubated in serum at 37°C. The decrease in current density could not be attributed to run-down since it was not observed at the apex or in male myocytes.
Figure 3. Apex–base changes in INCX as a function of time in culture.
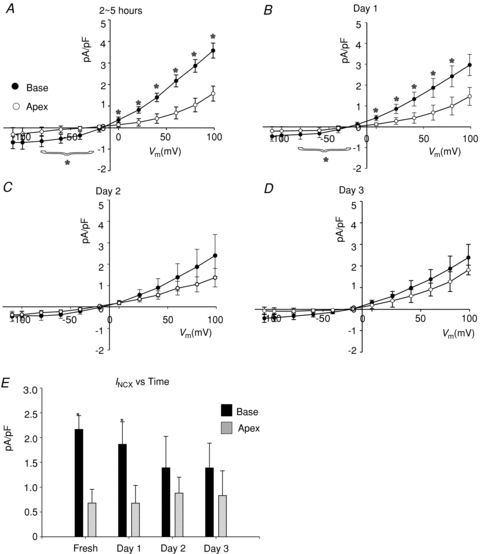
A–D, I–V plots of INCX density recorded from base and apex female myocytes recorded from freshly isolated cells (2–5 h) (A), and myocytes after 1 (B), 2 (C) or 3 (D) days in culture. E, summary bar graph of mean INCX recorded at +60mV from the base and apex as a function of time in culture. *P < 0.05. for base compared to apex.
Figure 4. Changes of outward and inward INCX at base and apex vs. time.
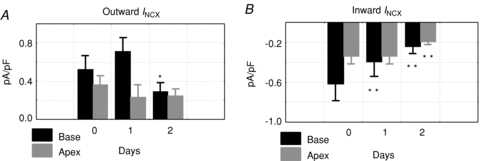
A, statistical analysis of outward INCX for myocytes from the base and apex for days 0, 1 and 2. The 3-day (0, 1 and 2) trend of mean outward INCX showed a highly significant decrease (Friedman's ANOVA, P= 0.006) at the base but not at the apex (P= 0.1); *P < 0.05 compared to day 0. The increase on day 2 at the base was not significant. B, statistical analysis of inward INCX for myocytes from the base and apex for days 0, 1, and 2. The 3-day (0, 1 and 2) trend of mean inward INCX showed a highly significant decrease (Friedman's ANOVA, P= 0.0006) at the base and just reached significance at the apex (P= 0.04); **P < 0.01 compared to day 0.
Role of oestrogen in the up-regulation of INCX
The down-regulation of INCX in female but not in male myocytes as a function of time could be due to the altered cellular environment as myocytes were transferred from the female rabbit heart to the culture medium. Numerous factors changed from a blood perfused heart to the culture medium, among them circulating levels of sex steroids, and here we tested the hypothesis that oestrogen plays a major role in the regulation of INCX. The role of oestrogen (E2) regulation on INCX was tested by incubating female myocytes from the base and apex in serum with and without E2 (1 nm) and by measuring INCX on days 1–3. After 24 h of incubation, E2 caused a marked up-regulation of INCX in base myocytes (Fig. 5A) but had no effect on myocytes from the apex (Fig. 5B). At the base, there were statistically significant differences between control (CTL: 1.20 ± 0.25 pA pF−1) and E2 treated (E2: 3.5 ± 0.86 pA pF−1, n= 6/4 hearts) myocytes on day 1 (Fig. 5B and C), day 2 (CTL: 1.9 ± 0.88 pA pF−1 and E2 treated: 3.60 ± 0.45 pA pF−1, n= 6/4 hearts, in both sexes) and day 3 (CTL: 1.95 ± 0.64 pA pF−1 and E2 treated: 4.75 ± 0.92 pA pF−1, n= 6/4 hearts, Fig. 5C). At the apex, there were no significant differences of INCX at 60 mV between control (CTL: 0.78 ± 0.31 pA pF−1) and E2 treated (E2: 0.56 ± 0.24 pA pF−1, n= 6/4 hearts, both sexes) myocytes on day 1 (Fig. 5B and D) or on day 2 (CTL: 1.16 ± 0.58 pA pF−1 and E2 treated: 0.90 ± 0.40 pA pF−1, n= 6/4 hearts, in both sexes) and day 3 (CTL: 0.81 ± 0.42 pA pF−1 and E2 treated: 0.87 ± 0.29 pA pF−1, n= 6/4 hearts, Fig. 5D). Figure 6 shows the analysis of the inward and outward components of INCX for base (Fig. 6A and B) and apex (Fig. 6C and D) myocytes incubated in the absence and presence of E2 (1 nm). There was a statistically significant E2-dependent up-regulation of the inward and outward INCX densities in myocytes from the base but not the apex. After 1 day of incubation in E2, at day 1, 2 and 3 both components of INCX were significantly greater in base myocytes incubated in E2 (1 nm) compared to control myocytes incubated in vehicle only. Note that control myocytes in the presence of DMSO had higher INCX currents (Fig. 6) than controls in the absence of DMSO (Fig. 4) and that this effect was even more pronounced on day 3 (Fig. 6A and B). Myocytes from the apex showed no significant increases of either inward or outward INCX over the same 3 day incubation. The effect of E2 incubation was also tested in female myocytes isolated from the base of the endocardium (Fig. 7A) and male myocytes isolated from the base of the epicardium (Fig. 7B). In both cases, E2 had no significant effect on the I–V relationship of INCX after 24 h (Fig. 7) and 48 h (not shown). Hence, incubation with E2 raised the INCX density in female base myocytes (Fig. 6A and B) in a region specific manner as E2 caused no significant up-regulation in female apex myocytes (Fig. 6C and D), or female base endocardial (Fig. 7A and B) or male base epicardial cells (Fig. 7C and D).
Figure 5. Effect of oestrogen on INCX in female base cardiac myocytes.
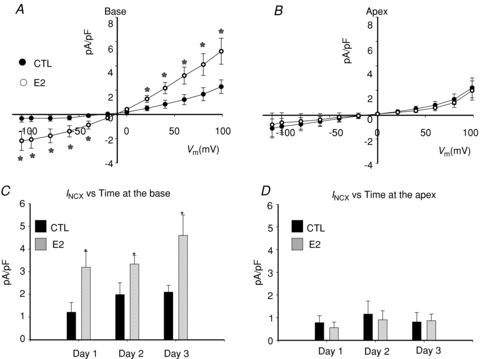
A, INCX in female base myocytes incubated for 1 day in the absence (Control, CTL) or presence of 17β-estrodial (E2, 1 nm). B, same as in (A) but with myocytes from the apex. C, summary of mean INCX±s.d. density at 60 mV, in control female base myocytes (CTL) compared to myocytes incubated in 1 nm oestrogen (E2). D, summary of mean INCX±s.d. density at 60 mV, in control female apex myocytes (CTL) compared to myocytes incubated in 1 nm oestrogen (E2). INCX with and without E2 was compared for days 1, 2 and 3 because E2 added acutely on day 0 had no effect on INCX. In these experiments, control myocytes were also exposed to DMSO (1 part in 10,000).
Figure 6. Effects of E2 on inward and outward INCX, base and apex.
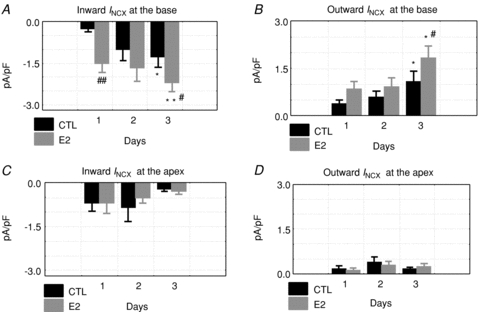
Inward and outward INCX were compared as a function of time (days 1–3) for control (CTL: incubated in DMSO alone) and oestrogen treated myocytes (E2: incubated in 1 nm E2/DMSO). Friedman's ANOVA for repeated measurements was used to test for statistically significant changes in current. At the base, the inward (A) and outward (B) currents changed significantly with *P < 0.05 and **P < 0.01 for day 1 vs. 3 and #P < 0.05 and ##P < 0.01 for CTL vs. E2 on the same day. In myocytes from the apex, the currents did not exhibit statistically significant changes (C and D). Friedman's ANOVA for repeated measurements yielded P= 0.89 for apex controls for day 1 vs. 3 and P= 0.37 for apex E2 vs. control for inward INCX (C), and P= 0.22 for apex control for day 1 vs. 3 and P= 0.61 for apex E2 vs. control for outward INCX (D).
Figure 7. Effects of E2 on female base endo- and male base epimyocytes.
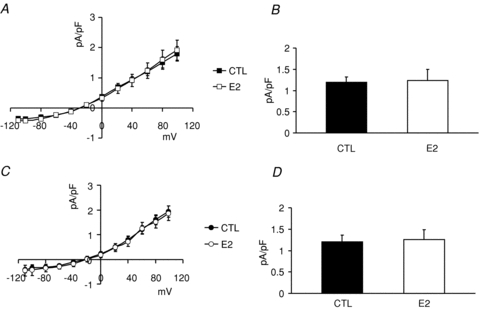
Myocytes were isolated from female base endocardial and male base epicardial regions of rabbit hearts and were incubated with or without E2 (1 nm) for 24 h. A, I–V plots of INCX measured from female base endocardial cells which were incubated in vehicle (DMSO: 1/10,000, n= 5 cells, 3 hearts) or E2 (1 nm, n= 5 cells, 3 hearts). B, INCX (mean ±s.e.m. at 60 mV) was 1.97 ± 0.12 in controls (CTL) and 1.24 ± 0.26 pA pF−1 in oestrogen (E2) treated cells. E2 did not significantly change INCX in female base endocardial myocytes. C, I–V plots of INCX measured from male base epicardial cells which were incubated in vehicle (DMSO: 1/10,000, n= 5 cells, 3 hearts) or E2 (1 nm, n= 5 cells, 3 hearts). D, INCX (mean ±s.e.m. at 60 mV) was 1.20 ± 0.156 in controls (CTL) and 1.25 ± 0.24 pA pF−1 in oestrogen (E2) treated cells. E2 did not significantly change INCX in male base epicardial myocytes.
Genomic regulation of INCX by E2
A genomic regulation of INCX by E2 implies that E2 increases INCX by increasing NCX1 protein levels mediated by oestrogen receptors (ER) and an increase in mRNA levels. Female myocytes from the base and apex were incubated with and without E2 (1 nm) for 24 h to measure changes in NCX1 protein by immuno-histochemistry. As shown in Fig. 8A and B, myocytes from the apex had similar levels of NCX1 whether incubated without (Fig. 8A) or with E2 (Fig. 8B). In contrast, myocytes from the base had higher levels of NCX1 when incubated with E2 (Fig. 8D) compared to control medium (Fig. 8C). For a quantitative analysis of NCX1 protein in myocytes treated with and without E2, all myocytes were handled in an identical manner and were analysed 24 h after isolation by immuno-histochemistry using confocal microscopy. In controls (no E2), the mean fluorescence intensity of NCX1 label was measured from apex myocytes (n= 18 myocytes from 3 hearts, 18/3) and was set arbitrarily to 100% with a s.e.m. of 1.4%. When incubated with E2, apex myocytes (n= 16/3 hearts) had intensities of 104 ± 7.8%. Control base myocytes (n= 26/3 hearts) had a mean intensity of 113 ± 3.6% (P= 0.014 CTL apex vs. CTL base) which increased to 131 ± 9.43% when incubated with E2 (P < 0.04 CTL base vs. E2 base). Figure 8E summarizes the statistical analysis of NCX1 protein density in the four groups of myocytes; female base myocytes treated with E2 expressed a significantly greater density of NCX1 than the other three groups of myocytes. Note that the NCX1 staining in the apex was primarily located on the cell surface, with sparse labelling of T-tubules, consistent with the findings by Kieval et al. (1992). When the cells from the base were treated with E2 for 24 h, there was a significant increase of NCX1 labelling.
Figure 8. Effect of oestrogen on NCX1 protein levels.
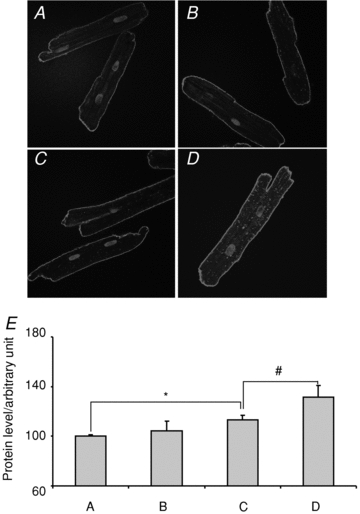
A and B, representative immuno-histochemistry of NCX1 in apex female myocytes incubated for 1 day in the absence and presence of E2 (1 nm), respectively. C and D, as for A and B but with myocytes from the base. E, summary of relative NCX1 intensity from control apex (A), oestrogen treated apex (B), control base (C) and E2-treated base (D) myocytes. *P < 0.05, #P < 0.01.
The role of oestrogen receptors in the up-regulation of NCX1 and INCX was investigated by incubating female base myocytes in serum containing E2 with or without the oestrogen receptor antagonist fulvestrant (ICI182,780; ICI; 1 μm)(Robertson, 2001; Osborne et al. 2004) followed by measurements of INCX density at days 1, 2 and 3. ICI (1 μm) alone did not alter INCX compared to controls without ICI (not shown). on day 1, there was no significant difference in INCX in myocytes incubated with and without ICI (Fig. 9A) but on days 2, and 3 ICI caused marked reductions of INCX (Fig. 9B and C). As summarized in Fig. 9D, the mean ±s.d. of INCX density (at 60 mV) was not statistically different on day 1 (2.69 ± 0.45 for E2 + ICI, n= 6/4 hearts and 3.13 ± 0.49 pA pF−1 for E2, n= 9/4 hearts). on day 2, ICI significantly depressed INCX from 3.81 ± 0.56 pA pF−1 (E2, n= 6/5 hearts) to 1.58 ± 0.47 pA pF−1 (E2 + ICI, n= 7/5 hearts, P < 0.05) and on day 3 from 4.45 ± 0.98 pA pF−1 (E2, n= 9/5 hearts) to 1.91 ± 0.61 pA pF−1 (E2 + ICI, n= 7/3 hearts, P < 0.05), respectively. Figure 9 summarizes Wilcoxon's paired test of outward (Fig. 9E) and inward (Fig. 9F) INCX indicating that E2 up-regulates INCX in its physiological range in both the forward and reverse modes.
Figure 9. Effect of E2 receptor antagonist ICI 182,780.
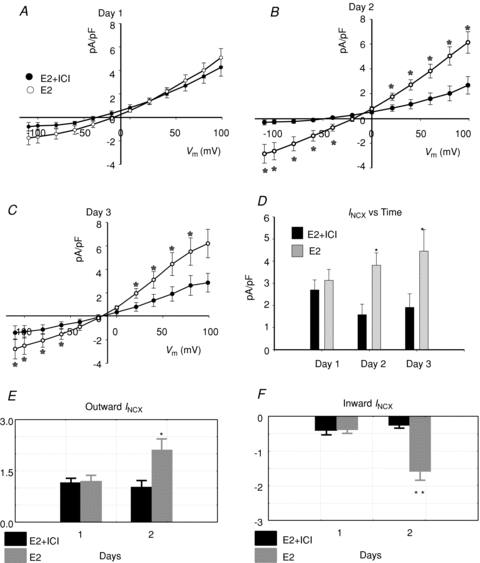
INCX density was measured from myocytes treated with E2 (1 nm) in the presence or absence of the ER antagonist ICI 182,780 (ICI, 1 μm) for 1 (A), 2 (B) or 3 (C) days. I–V plots from controls myocytes and myocytes incubated in ICI alone were not significantly different from myocytes incubated with E2 plus ICI. D, summary histograms of mean ±s.e.m. of INCX (at 60 mV) at female base myocytes treated with E2 or E2 + ICI, at days 1–3. *P < 0.05. E, Wilcoxon's paired test of outward INCX; for E2 treated cells *P= 0.03, day 1 vs. day 2 but for fulvestrant treated cells, E2 + ICI: P= 0.8, day 1 vs. day 2. F, Wilcoxon's paired test of inward INCX; for E2 treated cells **P= 0.008, day 1 vs. day 2, but for fulvestrant treated cells, E2 + ICI P= 0.2, day 1 vs. day 2.
Female base myocytes were treated with and without cycloheximide (CX) to determine if the up-regulation of INCX by E2 required protein biosynthesis. As shown in Fig. 10A, I–V plots of INCX were superimposed from four groups of myocytes: control (CTL), E2 (E2), cycloheximide plus E2 (CX+E2) and cycloheximide only (CX) treated myocytes. I–V plots show that cycloheximide alone had no effect on INCX, but blocked its up-regulation by E2. Figure 10C summarizes the effect of cycloheximide on E2-dependent up-regulation of INCX.
Figure 10. Effects of Cycloheximide and actinomycin D onINCX up-regulation by E2.
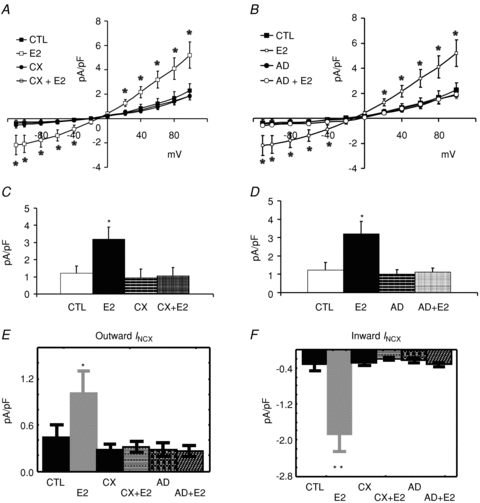
A, I–V plots of INCX were measured from female base-myocytes which were incubated in serum (CTL) (n= 7 cells/4 hearts), E2 (1 nm) (n= 6/3), cycloheximide (CX) (n= 6/3) or CX + E2 (n= 7/3). E2 treated myocytes had higher INCX densities than controls and CX blunted the up-regulation of INCX by E2. CX treatment alone had no effect on INCX. B, I–V plots of INCX were measured from female base myocytes which were incubated in serum (CTL) (n= 7 cells/4 hearts), E2 (1 nm) (n= 6/3), actinomycin D (AD) (n= 7/3) or AD + E2 (n= 7/3). E2 treated myocytes had higher INCX densities than controls and AD blunted the up-regulation of INCX by E2. AD treatment alone had no effect on INCX. C, summary of mean INCX±s.d. measured at 60 mV for all four groups of myocytes. E2 treated myocytes had statistically significant higher INCX, an effect suppressed by CX (*P < 0.05 compared to CTL). D, summary of mean INCX±s.d. measured at 60 mV for all four groups of myocytes. E2 treated myocytes had statistically significant higher INCX, an effect suppressed by AD. E, Wilcoxon's paired test of outward INCX (data taken from panels A and B) showed that CX and AD suppressed the up-regulation of outward INCX by E2; *P < 0.05 compared to CTL. F, Wilcoxon's paired test of inward INCX (data taken from panels A and B) showed that CX and AD suppressed the up-regulation of inward INCX by E2; **P < 0.01 compared to CTL.
Alternatively, female base myocytes were treated with actinomycin D (AD) to determine if enhanced transcription of NCX1 genes was required for the up-regulation of INCX by E2. As shown in Fig. 10B, I–V plots of INCX were superimposed from four groups of myocytes: control (CTL), E2 (E2), actinomycin plus E2 (AD+E2) and actinomycin D alone (AD). The data show that AD alone had no effect on INCX but blunted the INCX up-regulation elicited by E2. Figure 10D summarizes the inhibition of AD on E2 up-regulation of INCX. Wilcoxon's paired test of outward (Fig. 10E) and inward (Fig. 10F) INCX showed that CX and AD suppressed the E2-dependent up-regulation of INCX.
These data show that E2 (1 nm) increased the expression of NCX1 and the INCX density in female base epicardial myocytes by a mechanism that involves gene regulation since it is mediated by oestrogen receptors, enhanced transcription and protein biosynthesis.
E2 promotes EADs
We previously showed that in freshly prepared female myocytes, the addition of an IKr blocker elicited EADs in myocytes from the base but not from the apex and not from male myocytes (Sims et al. 2008). Here we assessed the effect of oestrogen on EAD susceptibility by incubating base and apex myocytes with E2 (1 nm) with or without the ER antagonist ICI (1 μm) for 24 h, then measuring the action potential under current-clamp conditions before and after the addition of the IKr blocker dofetilide (400 nm).
As shown in Fig. 11, female base (Fig. 11A) myocytes (n= 5/3 hearts) did not exhibit EADs after adding dofetilide unless incubated with E2, where prominent EADs were observed (n= 6 out of 7 myocytes, from 3 hearts) (Fig. 11C). In myocytes incubated with the ER antagonist ICI plus E2, dofetilide failed to elicit EADs (Fig. 11D) (n= 4 out of 5 myocytes had no EAD, 3 hearts). In contrast, female apex myocytes incubated with E2 did not exhibit EADs in the presence of dofetilide (no EADs in 5 out of 5 myocytes, 3 hearts) (Fig. 11B). Fisher's exact test showed a significant base–apex differences in dofetilide-induced EADs for myocytes incubated with E2 (two-tail P= 0.008) and in female base myocytes treated with E2 ± ICI (two-tail P= 0.031).
Figure 11. Genomic effect of E2 and arrhythmia susceptibility.
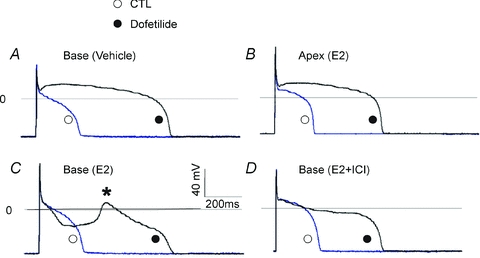
A and B, action potentials (APs) were measured from female myocytes after 1 day in culture while pacing the cells at 0.5 Hz, before (○, control) and after bathing the cells with 400 nm dofetilide (•, dofetilide). APs recorded from a myocyte from the base incubated with vehicle (A) and apex with E2 (B). Dofetilide is known to selectively block IKr causing the expected AP prolongation (>1 s), but there were no early afterdepolarizations (EADs). C, female base myocytes as in A were incubated with 1 nm E2; APs were recorded before (○, control) and after the addition of dofetilide (400 nm) (•, dofetilide). Dofetilide elicited EADs (*) in 15–25 beats. D, APs recorded from female base myocytes treated with E2 plus the ER antagonist (ICI) (○, control). The addition of dofetilide prolonged APs (•, dofetilide) but there were no EADs; hence the ER antagonist blocked E2-dependent EADs.
Up-regulation of INCX contributes to EAD risk
To evaluate the role of INCX in the initiation of early afterdepolarizations (EADs), the inhibition of INCX by the drug KB-R7943 was tested in rabbit female base myocytes. As shown in Fig. 12A, KB-R7943 reduced the outward component (reverse mode) of INCX in a concentration-dependent manner, at 1, 3 and 10 μm. The inhibition by 1 μm was not statistically significant but inhibition was highly significant at 3 and 10 μm (n= 4 cells, 2 hearts). KB-R7943 at 1 μm reduced the AP plateau potential and AP durations, but at 3 and 10 μm, KB-R7943 caused a triangulation of the AP suggesting that the drug is less selective at the higher concentrations (n= 6 cells, N= 2 hearts). When female base myocytes were incubated in the absence (n= 6 cells, 2 hearts) and presence of E2 (1 nm, n= 6 myocytes, N= 2 hearts), the subsequent addition of dofetilide (100 nm) to the bathing solution prolonged AP durations but only myocytes treated with E2 exhibited EADs (n= 6 out of 6 cells). The subsequent addition of KB-R7943 (1 μm) to the bathing solution reduced AP durations in all cells and suppressed EADs in all cells incubated with E2.
Figure 12. Effect of INCX inhibition on E2-induced EADs.
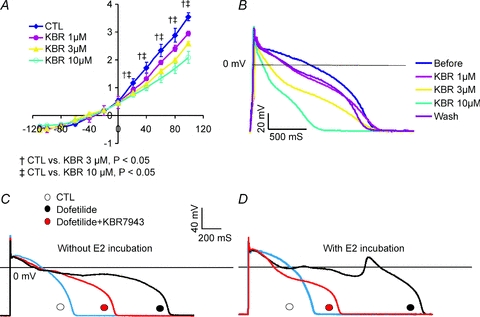
A, I–V plot of INCX measured from a female base myocyte (n= 3 myocytes, 2 hearts) before and after perfusion with 1, 3 or 10 μm KB-R7943. As expected, KB-R7943 inhibited the reverse mode of NCX; at 1 μm, the inhibitor tended to reduce the outward NCX current but at 3 and 10 μm there was a statistically significant inhibition of INCX at voltages ≥0 mV. B, in normal Tyrode solution, KB R7943 reduced the AP plateau potential and AP durations as a function of concentrations 1, 3 and 10 μm. The inhibition of outward INCX by 1 and 3 μm KB-R7943 could be partly reversed by washing out the drug. C, in a female base myocyte incubated for 24 h without E2, dofetilide (100 nm) prolonged AP durations and reduced the plateau potential and did not elicit EADs (n= 6 myocytes, 3 hearts). The subsequent addition of KB-R7943 (1 μm) reduced AP durations by ∼50% (n= 6 myocytes, 3 hearts). D, in female base myocytes incubated with E2 (1 nm) for 24 h, dofetilide (100 nm) prolonged APDs and induced EADs. The addition of KB R7943 (1 μm) + dofetilide (100 nm) to the bathing medium suppressed EADs (n= 6 out of 6 myocytes, 3 hearts).
Discussion
The main findings are that NCX1 channel protein and INCX are significantly higher at the base of female left ventricles than those of males and are heterogeneously distributed being higher at the base than apex of female left ventricles. The regional distribution of INCX density parallels the ICa,L density reported for female and male rabbit hearts. The regional distribution of NCX1 protein measured in ventricular muscles was retained in myocytes isolated from the base and apex of male and female hearts since INCX density was statistically greater at the base of female compared to apex ventricles and to male myocytes. Adult ventricular myocytes could be cultured in serum for a period of 3 days and were shown to exhibit stable INCX densities. However, INCX in female base myocytes decreased over 48 h to become similar to INCX measured from male and apex female myocytes. The latter gradual decrease of INCX was blunted by incubation of myocytes with oestrogen (E2, 1 nm), which also increased NCX1 channel proteins. E2 increased INCX in a region-dependent manner by a genomic mechanism because it was correlated with an increase in NCX1 protein expression, did not change the properties of INCX, was mediated by oestrogen receptors and was blunted by inhibitors of transcription and translation. Functionally, E2 promoted EADs in myocytes from the base of female ventricles, whereas female apex myocytes did not respond to E2 and had no EADs. These findings bring out new mechanistic insights on the impact of oestrogen on sex differences in the arrhythmia phenotype by regulating NCX in a region-dependent manner.
Cultured cardiac myocytes
The ability to culture cardiac myocytes for 3 days provided a critical tool to study the genomic effects of E2. A major challenge was to establish a technique to allow us to measure reproducible current densities from cell to cell and from animal to animal. There is a growing consensus that cultures of adult ventricular myocytes can be kept several days to weeks with negligible de-differentiation (Mitcheson et al. 1998). Key features of the protocol include (a) the selection of attached myocytes with preserved rod shape and T-tubule organization, (b) the supplementation of the medium with a serum of known composition and in particular, with levels of sex steroids too low to have biological activity, and (c) the careful monitoring of cell capacitance (see Table 1) over the 3 day time course of the study. Others showed that mouse cardiac myocytes could be kept in culture for 72 h with no significant changes in NCX or β-adrenergic signalling (Sambrano et al. 2002). Rabbit ventricular myocytes in serum-free medium for 6 days had reduced ICa,L but no significant changes in ICa,L density or responses to isoprenaline (Mitcheson et al. 1996, 1997). Feline myocytes cultured in medium supplemented with serum (5% FBS) were found to maintain stable ICa,L current for 14 days (Schackow et al. 1995), which suggested that serum suppressed the de-differentiation of adult myocytes compared to serum-free media (Mitcheson et al. 1996). In this study, myocytes incubated in DMEM plus 5% FBS for 72 h maintained their rod shape and total membrane capacitance, and INCX density did not change when measured from male (base or apex) or female (apex) myocytes (Fig. 2C and D, Fig. 3). The decrease in INCX density as a function of time (day 0 to day 2) was statistically significant but only at female base myocytes (Fig. 3). The decrease of INCX was reversed by treating female base myocytes with E2 and most important E2 did not alter INCX density in female apex (Fig. 5), endocardial and male myocytes (Fig. 7). These findings indicate that the cultured cardiac myocytes were stable for 3 days under our experimental condition and allowed us to detect possible gene regulation by E2 with a high degree of confidence.
Regional heterogeneities of INCX are mediated by oestrogen
In drug induced LQT2, adult female rabbits are more prone to EADs and TdP than males. However, this sex difference in arrhythmia phenotype is reversed in adolescent rabbits where pre-pubertal male are more prone to E4031-induced EAD and TdP than pre-pubertal female rabbits (Liu et al. 2005). We previously showed that E4031 promoted EADs in freshly isolated base myocytes from adult female rabbits but not in cells from the apex or from adult males (Sims et al. 2008). Here, we showed that the IKr blocker dofetilide failed to induce a large EAD in base myocytes from adult females if they were incubated in E2-free medium for 24 h; such an interval in an E2-free environment was sufficient to down-regulate INCX (Fig. 3E). However, when myocytes were treated with E2, dofetilide induced large EADs and when treated with E2 plus an ER antagonist (ICI) EADs were suppressed (Fig. 11C and D). These results are consistent with findings that E4031in Langendorff hearts induced EADs that occurred exclusively at the base of adult female and pre-pubertal male hearts (Sims et al. 2008). Computational and experimental studies showed that the increase in INCX and ICa,L at the base of the female heart promoted EADs and explained the origins of EAD, under a conditions of prolonged APD due to IKr/IKs blockade (Sims et al. 2008). APD prolongation alone is not sufficient to induce EADs, as was shown in pre-pubertal female hearts, which have longer APDs than those of males yet males have EADs and TdP (Liu et al. 2005). Consistent with these observations in intact hearts, 1-day-old female myocytes from the apex and the base (treated with E2 and ER antagonist) had APDs >1 s yet no EADs, unless treated with E2 (Figs 11C and 12D).
Selectivity of KB-R7943
As shown in Fig. 12A, KB-R7943 inhibited the reverse mode of INCX (1 Ca2+ in vs. 3 Na+ out) with little effect on the forward mode (3 Na+ in vs. 1 Ca2+ out) in agreement with most reports (Iwamoto et al. 1996; Amran et al. 2003) and in contrast with the finding that the drug inhibited the forward and reverse mode with an IC50 of ∼1 μm for both directions (Kimura et al. 1999). KB-R7943 was shown to inhibit NCX with variable potency; in rat myocytes IC50 was 1.2–2.4 μm and >30 μm, respectively, for the reverse and forward mode, and in guinea pig myocytes, IC50 values were 0.32 and 17 μm for the reverse and forward modes, respectively (Kimura et al. 1999). At higher concentrations of KB-R7943, the drug inhibits several currents in guinea pig myocytes, namely INa, ICa,L and IK1 with IC50 values of 14, 8 and 7 μm, respectively (Kimura et al. 1999). KB-R7943 was found to inhibit the high affinity binding of [3H]ryanodine to the skeletal and cardiac isoforms of ryanodine receptors with IC50 values of ∼5 and 13 μm, respectively (Barrientos et al. 2009). In HEK and HELA cell expression systems, KB-R7943 was shown to suppress the canonical transient receptor potential (TRPC) channel and mitochondrial Ca2+ uniporter, respectively, both with IC50 values in the micromolar range (Kraft, 2007; Santo-Domingo et al. 2007). The wide range of possible KB-R7943 targets clouds the mechanism of action of the drug as an inhibitor of EADs through a selective NCX inhibition. For these reasons, the current experiments were intentionally carried out at low concentrations (1 μm), following a brief exposure to the drug (∼5 min) to reduce off-target actions of KB-R7943.
Genomic regulation of cardiac NCX by oestrogen
Oestrogen, like other steroid hormones, is thought to impart its effects by binding to intracellular oestrogen receptors (ERs) which are then able to modulate transcription of selected genes by classical and non-classical genomic pathways. In addition, rapid effects of steroids may result from non-transcriptional action via signal transduction (Levin, 2008). The time course of E2 mediated up-regulation of INCX is unlikely to be mediated by a signal transduction, but by genomic effects. The blockade of oestrogen effects by the ER antagonist fulvestrant is often used as evidence in support of a genomic mechanism (Osborne et al. 2004). There are two ER subtypes, α and β, and either or both may be involved in altering cardiac ion channel expression (Levin, 2002). The enhanced expression of NCX1 protein and its current, INCX, by oestrogen appears to be mediated by a genomic mechanism involving the binding of oestrogen to its receptors (Fig. 9) and a gene regulation requiring an activation of transcription and protein synthesis (Fig. 10). The current findings reveal that these oestrogen effects on cardiac ion channel expression depend on the sex of the animal and on the region of ventricular myocardium. Ongoing studies are identifying the ER subtypes that are involved in the regional genomic regulation of INCX, and possible regulation of ICa,L, SERCA2, auxiliary subunits and cardiac ryanodine receptors (RyR2). Future studies will be needed to characterize the genomic effects of androgens and the synergism between sex steroids. The investigation of ion channel expression in isolated myocytes was effective in bringing new insights to an old problem because our approach considered the location of the myocytes in the heart and the source (sex and age of the rabbits) of the hearts. Otherwise, the highly quantitative voltage-clamp measurements would not have discerned statistically significant differences of INCX density mediated by E2.
Isolated myocyte experiments allowed us to measure the E2 concentration needed to alter INCX; titrations showed that 0.5 to 10 nm concentrations of E2 were effective at increasing INCX but with different time courses of INCX elevation. Lower concentrations (< 0.2 nm) did not up-regulate INCX and higher (>10 nm) did not have further effects on INCX. In female base myocytes incubated in the absence of E2, INCX density (at +60 mV) decreased gradually on days 1 and 2 then remained stable between days 2 and 3 (Fig. 3). The latter suggests that INCX decreased due to the lack of oestrogen in the cell culture environment. When cells were cultured with physiological oestrogen concentration, INCX density (Fig. 5) and NCX1 protein (Fig. 8) levels increased and stabilized by day 2 compared to myocytes cultured in E2-free medium. Hence, the up-regulation of INCX by oestrogen took ∼1.5 days to reach equilibrium.
NCX and arrhythmia phenotype
Our findings show that in female hearts, NCX and Cav1.2α (Sims et al. 2008) are up-regulated at the base of the epicardium, which contributes to sex differences in the arrhythmia phenotype in LQT2 (Fig. 11). In normal conditions, the female heart expresses more ICa,L at the base, which is balanced by a higher expression of INCX such that Ca2+ influx is balanced with Ca2+ efflux. The following mechanisms link the higher Ca2+ currents (ICa,L and INCX) to the enhanced propensity to EADs in LQT2: a higher Ca2+ influx during prolonged APs occurs via ICa,L, which results in sarcoplasmic reticulum (SR) Ca2+ overload, spontaneous SR Ca2+ release, and larger depolarizing INCX that trigger the re-activation of ICa,L. Thus, the higher level of INCX provides the depolarizing pulse that re-activates ICa,L during the plateau phase of the AP that triggers EADs and a higher incidence of TdP (Sims et al. 2008). The higher expression levels of INCX in heart failure are thought to be highly arrhythmogenic because of enhanced propensity to EADs by a similar mechanism (Armoundas et al. 2003). The apex–base heterogeneity of INCX and ICa,L may be a contributing arrhythmogenic factor that promotes spatially discordant APD alternans, which predict the occurrence of polymorphic ventricular tachycardia (PVT) (Bers, 2007).
Limitations
The study provides compelling evidence in support of regional genomic regulation of NCX by oestrogen at the base of the female epicardium. Although endocardial (Fig. 7) and apical myocytes did not respond to E2 incubation, a detailed transmural and apex–base distribution of INCX is lacking due to technical limitations. Further studies will be required to investigate the possible genomic regulation of other Ca2+ channels and transporters, namely ICa,L, SERCA2 and cardiac ryanodine, receptors by oestrogen.
Acknowledgments
This work was supported by NIH-NHLBI grants: HL57929 and HL70722 to G.S.
Glossary
Abbreviations
- AD
actinomycin D
- CX
cycloheximide
- E2
17β-oestradiol
- EAD
early afterdepolarization
- ICI
fulvestrant or ICI182,780
- INCX
sodium–calcium exchange current
- LQT2
long QT syndrome type 2
- NCX
sodium–calcium exchanger (NCX1)
- TdP
torsade de pointes
Author contributions
All authors approved the final version for publication. G.C. contributed to the collection, analysis and interpretation of data and to drafting the article. X.Y. was an equal contributor, contributed to the collection and analysis oft data and to drafts of the manuscript. S.A. made significant contributions to the collection of immuno-histochemistry and analysis of data. V.S. made critical contributions to the statistical analysis of the data and tests of various statistical approaches to select the most suitable analysis. G.S. contributed to the conception and design of the experiments, the interpretation of data, drafted and revised the article for important intellectual content and provided the financial support for the study. Conflicts of interest: none.
Author's present address
X. Yang: Department of Pharmacology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China.
References
- Abi-Gerges N, Philp K, Pollard C, Wakefield I, Hammond TG, Valentin JP. Sex differences in ventricular repolarization: from cardiac electrophysiology to Torsades de Pointes. Fundam Clin Pharmacol. 2004;18:139–151. doi: 10.1111/j.1472-8206.2004.00230.x. [DOI] [PubMed] [Google Scholar]
- Amran MS, Homma N, Hashimoto K. Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovasc Drug Rev. 2003;21:255–276. doi: 10.1111/j.1527-3466.2003.tb00121.x. [DOI] [PubMed] [Google Scholar]
- Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O’Rourke B. Role of sodium-calcium exchanger in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos G, Bose DD, Feng W, Padilla I, Pessah IN. The Na+/Ca2+ exchange inhibitor 2-(2-(4-(4-nitrobenzyloxy)phenyl)ethyl)isothiourea methanesulfonate (KB-R7943) also blocks ryanodine receptors type 1 (RyR1) and type 2 (RyR2) channels. Mol Pharmacol. 2009;76:560–568. doi: 10.1124/mol.109.057265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednar MM, Harrigan EP, Ruskin JN. Torsades de pointes associated with nonantiarrhythmic drugs and observations on gender and QTc. Am J Cardiol. 2002;89:1316–1319. doi: 10.1016/s0002-9149(02)02337-8. [DOI] [PubMed] [Google Scholar]
- Benitah JP, Vassort G. Aldosterone upregulates Ca2+ current in adult rat cardiomyocytes. Circ Res. 1999;85:1139–1145. doi: 10.1161/01.res.85.12.1139. [DOI] [PubMed] [Google Scholar]
- Berger M, Jean-Faucher C, de Turckheim M, Veyssiere G, Blanc MR, Poirier JC, Jean C. Testosterone, luteinizing hormone (LH) and follicle stimulating hormone (FSH) in plasma of rabbit from birth to adulthood. Correlation with sexual and behavioural development. Acta Endocrinol (Copenh) 1982;99:459–465. doi: 10.1530/acta.0.0990459. [DOI] [PubMed] [Google Scholar]
- Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2007;70:23–49. doi: 10.1146/annurev.physiol.70.113006.100455. [DOI] [PubMed] [Google Scholar]
- Cheng J, Kamiya K, Liu W, Tsuji Y, Toyama J, Kodama I. Heterogeneous distribution of the two components of delayed rectifier K+ current: a potential mechanism of the proarrhythmic effects of methanesulfonanilideclass III agents. Cardiovasc Res. 1999;43:135–147. doi: 10.1016/s0008-6363(99)00061-9. [DOI] [PubMed] [Google Scholar]
- Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and Torsade de Pointes in rabbit hearts with type 2 long QT syndrome. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SH, Goldspink P, Kowalski J, Beck J, Schwertz DW. Effect of estrogen on calcium-handling proteins, β-adrenergic receptors, and function in rat heart. Life Sci. 2006;79:1257–1267. doi: 10.1016/j.lfs.2006.03.037. [DOI] [PubMed] [Google Scholar]
- Chu SH, Sutherland K, Beck J, Kowalski J, Goldspink P, Schwertz D. Sex differences in expression of calcium-handling proteins and β-adrenergic receptors in rat heart ventricle. Life Sci. 2005;76:2735–2749. doi: 10.1016/j.lfs.2004.12.013. [DOI] [PubMed] [Google Scholar]
- Coker SJ. Drugs for men and women – How important is gender as a risk factor for TdP? Pharmacol Ther. 2008;119:186–194. doi: 10.1016/j.pharmthera.2008.03.005. [DOI] [PubMed] [Google Scholar]
- de Turckheim M, Berger M, Jean-Faucher C, Veyssiere G, Jean C. Changes in ovarian oestrogens and in plasma gonadotrophins in female rabbits from birth to adulthood. Acta Endocrinol (Copenh) 1983;103:125–130. doi: 10.1530/acta.0.1030125. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, Robinson JL, Locati EH, Ackerman MJ, Benhorin J, Kaufman ES, Napolitano C, Priori SG, Qi M, Schwartz PJ, Towbin JA, Vincent GM, Zhang L. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation. 2008;117:2184–2191. doi: 10.1161/CIRCULATIONAHA.107.701243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Danilo P, Jr, Rosen MR. Effects of gonadal steroids on ventricular repolarization and on the response to E4031. J Pharmacol Exp Ther. 1998;285:1068–1072. [PubMed] [Google Scholar]
- Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–299. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai IA, O’Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- Kieval RS, Bloch RJ, Lindenmayer GE, Ambesi A, Lederer WJ. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol Cell Physiol. 1992;263:C545–550. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- Kimura J, Watano T, Kawahara M, Sakai E, Yatabe J. Direction-independent block of bi-directional Na+/Ca2+ exchange current by KB-R7943 in guinea-pig cardiac myocytes. Br J Pharmacol. 1999;128:969–974. doi: 10.1038/sj.bjp.0702869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–236. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Kravtsov GM, Kam KW, Liu J, Wu S, Wong TM. Altered Ca2+ handling by ryanodine receptor and Na+-Ca2+ exchange in the heart from ovariectomized rats: role of protein kinase A. Am J Physiol Cell Physiol. 2007;292:C1625–1635. doi: 10.1152/ajpcell.00368.2006. [DOI] [PubMed] [Google Scholar]
- Levin ER. Estrogen receptor-β and the cardiovascular system. Trends Endocrinol Metab. 2002;13:184–185. doi: 10.1016/s1043-2760(02)00604-5. [DOI] [PubMed] [Google Scholar]
- Levin ER. Rapid signaling by steroid receptors. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1425–1430. doi: 10.1152/ajpregu.90605.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Choi BR, Drici MD, Salama G. Sex modulates the arrhythmogenic substrate in prepubertal rabbit hearts with Long QT 2. J Cardiovasc Electrophysiol. 2005;16:516–524. doi: 10.1046/j.1540-8167.2005.40622.x. [DOI] [PubMed] [Google Scholar]
- Maack C, Ganesan A, Sidor A, O’Rourke B. Cardiac sodium-calcium exchanger is regulated by allosteric calcium and exchanger inhibitory peptide at distinct sites. Circ Res. 2005;96:91–99. doi: 10.1161/01.RES.0000151334.48676.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- Marban E, Robinson SW, Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–1192. doi: 10.1172/JCI112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC, Levi AJ. Action potentials, ion channel currents and transverse tubule density in adult rabbit ventricular myocytes maintained for 6 days in cell culture. Pflugers Arch. 1996;431:814–827. doi: 10.1007/s004240050073. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC, Levi AJ. Cultured adult rabbit myocytes: effect of adding supplements to the medium, and response to isoprenaline. J Cardiovasc Electrophysiol. 1997;8:1020–1030. doi: 10.1111/j.1540-8167.1997.tb00626.x. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC, Levi AJ. Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc Res. 1998;39:280–300. doi: 10.1016/s0008-6363(98)00128-x. [DOI] [PubMed] [Google Scholar]
- Nagy ZA, Virag L, Toth A, Biliczki P, Acsai K, Banyasz T, Nanasi P, Papp JG, Varro A. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed after depolarization in canine heart. Br J Pharmacol. 2004;143:827–831. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90(Suppl 1):S2–S6. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A, Sipido KR. Pharmacological inhibition of Na/Ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. 2008;102:1398–1405. doi: 10.1161/CIRCRESAHA.108.173922. [DOI] [PubMed] [Google Scholar]
- Pham TV, Sosunov EA, Gainullin RZ, Danilo P, Jr, Rosen MR. Impact of sex and gonadal steroids on prolongation of ventricular repolarization and arrhythmias induced by IK-blocking drugs. Circulation. 2001;103:2207–2212. doi: 10.1161/01.cir.103.17.2207. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure: Roles of sodium-calcium exchange, inward rectifier potassium current, and residual β-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Robertson JF. ICI 182,780 (Fulvestrant)–the first oestrogen receptor down-regulator – current clinical data. Br J Cancer. 2001;85(Suppl 2):11–14. doi: 10.1054/bjoc.2001.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426. [DOI] [PubMed] [Google Scholar]
- Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513. [DOI] [PubMed] [Google Scholar]
- Sambrano GR, Fraser I, Han H, Ni Y, O’Connell T, Yan Z, Stull JT. Navigating the signalling network in mouse cardiac myocytes. Nature. 2002;420:712–714. doi: 10.1038/nature01306. [DOI] [PubMed] [Google Scholar]
- Santo-Domingo J, Vay L, Hernandez-Sanmiguel E, Lobaton CD, Moreno A, Montero M, Alvarez J. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br J Pharmacol. 2007;151:647–654. doi: 10.1038/sj.bjp.0707260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. KB-R7943 block of Ca2+ influx via Na+/Ca2+ exchange does not alter twitches or glycoside inotropy but prevents Ca2+ overload in rat ventricular myocytes. Circulation. 2000;101:1441–1446. doi: 10.1161/01.cir.101.12.1441. [DOI] [PubMed] [Google Scholar]
- Schackow TE, Decker RS, Ten Eick RE. Electrophysiology of adult cat cardiac ventricular myocytes: changes during primary culture. Am J Physiol Cell Physiol. 1995;268:C1002–1017. doi: 10.1152/ajpcell.1995.268.4.C1002. [DOI] [PubMed] [Google Scholar]
- Schillinger W, Schneider H, Minami K, Ferrari R, Hasenfuss G. Importance of sympathetic activation for the expression of Na+-Ca2+ exchanger in end-stage failing human myocardium. Eur Heart J. 2002;23:1118–1124. doi: 10.1053/euhj.2001.3044. [DOI] [PubMed] [Google Scholar]
- Sims C, Reisenweber S, Viswanathan PC, Choi BR, Walker WH, Salama G. Sex, age, and regional differences in L-type calcium current are important determinants of arrhythmia phenotype in rabbit hearts with drug-induced long QT type 2. Circ Res. 2008;102:e86–100. doi: 10.1161/CIRCRESAHA.108.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo B, Kovacs T, Lazzara R. Role of calcium loading in early afterdepolarizations generated by Cs+ in canine and guinea pig Purkinje fibers. J Cardiovasc Electrophysiol. 1995;6:796–812. doi: 10.1111/j.1540-8167.1995.tb00356.x. [DOI] [PubMed] [Google Scholar]
- Verduyn SC, Vos MA, Gorgels AP, Van Der Zande J, Leunissen JD, Wellens HJ. The effect of flunarizine and ryanodine on acquired torsades de pointes arrhythmias in the intact canine heart. J Cardiovasc Electrophysiol. 1995;6:189–200. doi: 10.1111/j.1540-8167.1995.tb00770.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan PC, Rudy Y. Pause induced early afterdepolarizations in the long QT syndrome: a simulation study. Cardiovasc Res. 1999;42:530–542. doi: 10.1016/s0008-6363(99)00035-8. [DOI] [PubMed] [Google Scholar]
- Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels AP, Wellens HJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- Wei SK, McCurley JM, Hanlon SU, Haigney MC. Gender differences in Na/Ca exchanger current and β-adrenergic responsiveness in heart failure in pig myocytes. Ann N Y Acad Sci. 2007a;1099:183–189. doi: 10.1196/annals.1387.026. [DOI] [PubMed] [Google Scholar]
- Wei SK, Ruknudin AM, Shou M, McCurley JM, Hanlon SU, Elgin E, Schulze DH, Haigney MC. Muscarinic modulation of the sodium-calcium exchanger in heart failure. Circulation. 2007b;115:1225–1233. doi: 10.1161/CIRCULATIONAHA.106.650416. [DOI] [PubMed] [Google Scholar]